

Abstract
Heme oxygenase-1 (HO-1), also known as heat shock protein 32 (hsp-32) is a stress-induced cytoprotective protein. The present investigation evaluated the capacity of HO-1 to reduce the incidence of reperfusion-induced ventricular fibrillation (VF) and infarct size. HO-1 transgenic (Tg) mice were generated using a rat HO-1 genomic transgene. Isolated mouse hearts obtained from Tg and non-transgenic (NTg) groups were exposed to 20 min. of global ischemia and 120 min. of reperfusion. Epicardial electrocardiogram was recorded to monitor the incidence of reperfusion-induced VF and at the end of the reperfusion period, detection of HO-1 by immunohistochemistry and measurement of infarct size using the tetrazolium chloride method were carried out. Results shown here provide additional support for cardioprotective effects of HO-1 as demonstrated by the reduced infarct size. Moreover, overexpression of the HO-1 efficiently reduced the incidence of ischemia/reperfusion induced VF in HO-1 Tg mice.
Keywords: ischemia/reperfusion, isolated mouse hearts, HO-1 transgenic, HO-1 knockout
Introduction
In the past decades, extensive characterization of heme oxygenase-1 heat shock protein 32 (HO-1; hsp-32) and its isozymes have been conducted. HO-1 may be induced by various stressors such as ultraviolet irradiation, hypoxia and ischemia, providing protection against tissue damage [1, 2]. The enzymatic function of HO-1 is to degrade heme, which produces iron, the antioxidant bilirubin and the endogen carbon monoxide (CO). Enhanced levels of these degradation products were shown to play a major role in the cytoprotective effects of HO-1 [3].
Previously, experiments conducted in our laboratory demonstrated that low levels of HO-1 mRNA and protein along with reduced enzyme activity correlated with poor recovery of the ischemic/reperfused myocardium. Moreover, increased incidence of ventricular arrhythmias and infarct size were detected in hearts of HO-1 knockout (KO)–/– mice [4, 5]. Furthermore, Varadi et al. demonstrated reduced infarct size and ventricular fibrillation (VF) after the administration of CORM-3, a CO-releasing molecule [6]. To explain the cause and development of the ventricular arrhythmias, a range of different mechanisms have been suggested, but relatively few studies have investigated the mechanism(s) of VF at the level of gene expression. As an example, the long QT syndrome and idiopathic VF, as currently understood, are cardiac disorders based on mutations for genes encoding potassium channels, which cause sudden cardiac death from ventricular arrhythmias [7, 8]. The major objective of the present study was to investigate the role of HO-1 on the incidence of reperfusion-induced VF and infarct size in Tg mouse hearts.
Methods
Transgenic mice
Transgenic (Tg) mice were generated as described by Araujo et al.[9]. In brief, CFY mouse eggs were injected with 52 kb rat HO-1 construct containing 27.7 kb of the 5′ upstream region, 8.3 kb of HO-1 gene and 16 kb of the 3′ downstream region. Cloning was carried out by PCR of rat genomic P1 library with two sets of PCR primers corresponding to the first exon (5′-GCT-TCG-GTG-GGT-TAT-CTG-CCG-TTA-T-3′ and 5′-CAG-TCT-TAC-AGG-CGG-GGA-ATG-TGA-G-3′), and the fifth exon of rat HO-1 gene (5′-GAG-ACG-CCC-CGA-GGA-AAA-TCC-CAG-AT-3′ and 5′-CCC-AAG-AAA-AGA-GAG-CCA-GGC-AAG-AT-3′). Two clones were identified as positives, and restriction mapping showed that both of them included the same insert. The 52 kb band was equivalent to the HO-1 gene and flanking regions. This 52 kb fragment, including the native HO-1 gene and its promoter, was excised and digested with β-agarase and then microinjected. Mice were genotyped by PCR using the set of primers specific for rat HO-1 exon 5.
RT-PCR
RNAs were isolated from the left and right ventricles (LV and RV), and septum (about 50 mg) of mouse hearts by guanidine isothiocyanate acid/phenol method, and 5 μg of total RNA was used to synthesize first strand cDNA by the SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen, San Diego, CA, USA). The cDNA product was amplified by PCR with specific primers for rat HO-1 exon 5 (CCC-TTC-CTG-TGT-CTT-CCT-TTG and ACA-GCC-GCC-TCT-ACC-GAC-CAC-A). The RT-PCR product was separated using 1.5% agarose gel, and visualized by ethidium bromide.
Animals and heart preparation
Male wild-type, HO-1 Tg and HO-1 KO–/– mice (25–35 g) were used for all studies. Animals received humane care in compliance with the ‘Principles of Laboratory Animal Care’ formulated by the National Society for Medical Research and the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institute of Health (NIH Publication No. 86–23, revised 1996). Mice were anaesthetized with 80 mg/kg of pentobarbital sodium; heparin was used as anticoagulant (intraperitoneally 1000 IU/kg). After the anaesthesia, the chest was opened; the heart was rapidly excised and mounted to a ‘working’ perfusion apparatus [5]. The perfusion was established with a modified oxygenated Krebs–Henseleit buffer with the following concentrations (in mM): 118.4 NaCl, 4.1 KCl, 2.5 CaCl2, 25 NaHCO3, 1.17 KH2PO4, 1.46 MgCl2 and 11.1 glucose. The perfusion buffer was previously saturated with a mixture of 95% O2 and 5% CO2, pH 7.4 at 37°C. In all experiments global ischemia was induced for 20 min. followed by 2 hrs of reperfusion.
Registration of VF and measurement of cardiac function
Epicardial electrocardiograms (ECGs) were recorded throughout the experimental period by two silver electrodes attached directly to the heart and connected to a data acquisition system (ADInstruments, Powerlab, Castle Hill, Australia). ECGs were analysed to determine the presence or absence of VF. Hearts were considered to be in VF if an irregular undulating baseline was apparent on ECGs. If the duration of VF was longer than 2 min., the VF was defined as sustained VF, otherwise the VF was non-sustained. If VF developed and the sinus rhythm did not spontaneously return within the first 2 min. of reperfusion, hearts were electrically defibrillated by a defibrillator using two silver electrodes and 15 V square-wave pulse of 1 msec. duration and reperfused [5].
Measurement of infarct size
Infarct size was measured, at the end of each experiment, with 10 ml of 1% triphenyl tetrazolium chloride (TTC) solution in phosphate buffer (Na2HPO4 88 mM, NaH2PO4 1.8 mM) injected via the side arm of the aortic cannula then stored at –70°C for later analysis. Frozen hearts were sliced transversely in a plane perpendicular to the apico-basal axis into 1–2-mm-thick sections, weighted, blotted dry, placed in between microscope slides and scanned on a Hewlett-Packard Scanjet 5p single pass flat bed scanner (Hewlett-Packard, Palo Alto, CA, USA). Using the NIH Image 1.61 image processing software, each digitized image was subjected to equivalent degrees of background subtraction, brightness and contrast enhancement for improved clarity and distinctness. Infarct zones of each slice were traced and the respective areas were calculated in terms of pixels. The areas were measured by computerized planimetry software and these areas were multiplied by the weight of each slice, then the results summed up to obtain the weight of the risk zone (total weight of the LV, mg) and the infarct zone (mg). Infarct size was expressed as the ratio, in percentage, of the infarct zone to the risk zone [5].
Immunohistochemistry
Paraffin sections (7 μm) of tissue were incubated in the presence of polyclonal antibody and purified liver HO-1 obtained from rats (Stress Gen Biotech., Victoria, BC, Canada). Reactions were visualized by immunoperoxidase colour reaction in NTg, HO-1 Tg and HO-1 KO ischemic/reperfused mouse myocardium. Formalin fixed tissues were paraffin embedded and 7 μm sections were placed on poly-l-lysine coated glass slides (Sigma, St. Louis, MO, USA). Following deparaffinization and rehydration, samples were used for quenching endogenous peroxidases and blocking non-specific binding sites by 3% H2O2 in normal goat serum. Sections were incubated for another 2 hrs with HO-1 antibody at a dilution of 1:750 as described in the manufacturer’s manual. After 10 min. of phosphate-buffered solution washing, slides were incubated for additional 30 min. in the presence of purified biotinylated anti-rabbit IgG (Vector, Burlingame, CA, USA) at a dilution of 1:200. Slides were rewashed in phosphate-buffered solution and incubated with peroxidase conjugated streptavidin (Zymed, Carlsbad, CA, USA) for 30 min. followed by red colour development using 3-amino-9-ethylcarbazole in 0.1 M of acetate buffer (pH 5.2). Sections were counterstained with Gill’s haematoxylin for 15–20 sec., rinsed with deionized water and dipped in 1% of lithium carbonate. After draining off water slides were placed in oven for 30 min. at 80°C, and then covered with permount cover slips. Sections were photographed using a Zeiss (Göttingen, Germany) light microscope.
Statistics
The data for infarct size were expressed as the mean ± S.E.M. One-way analysis of variance was first carried out to test for any differences between the mean values of groups. If differences were established the values of NTg group were compared to those of HO-1 Tg, and HO-1 KO–/– groups by multiple t-test followed by Bonferroni test. Because of the non-parametric distribution of the incidence of VF (sustained and non-sustained), the chi-square test was used to compare individual groups. A change of P < 0.05 was considered to be statistically significant.
Results
The expression of the rat HO-1 transgene was confirmed in the mouse heart at mRNA levels. Total RNA was isolated from the non-ischemic LV, RV and septum (S) of Tg and NTg littermates and RT-PCR was carried out using a specific primer corresponding to the rat HO-1. Figure 1 shows the intense rat HO-1 signal detected in the LV, RV and S in hearts obtained from HO-1 Tg animals (Fig. 1. upper part, right), while the signal was absent in RNA samples obtained from NTg mice (Fig. 1, upper part, left). The results were expressed as relative ratio and shown in Fig. 1 (lower part).
Fig 1.
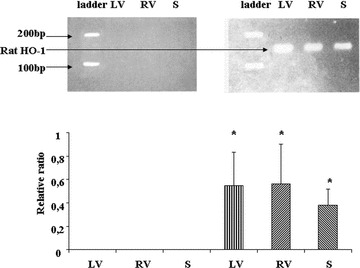
The detection of rat HO-1 transgene by PCR in the mouse myocardium. Total RNA was obtained from mouse LV, RV and septum (S), respectively, and was used to synthesize first strand cDNA. Rat HO-1 was amplified by PCR exon 5-specific primer and visualized with ethidium bromide as a 166 bp band by agarose gel electrophoresis in the mouse heart. Representative pictures of HO-1 RT-PCR results are shown in the upper part of the figure, the specific bands of Tg mouse myocardium depicted in the upper right, while the results of NTg littermates shown in the upper left. Lower part of the figure shows the calculated values of HO-1 Tg signal intensity in NTg and Tg tissues. n= 4 in each group, mean ± S.E.M., *P < 0.05, comparisons were made to the corresponding NTg values.
To confirm the HO-1 expression at protein level, cardiac samples were obtained from NTg, HO-1 Tg and HO-1 KO mouse myocardium and immunohistochemical staining was performed. Figure 2A shows that HO-1 in NTg myocardium was stained in blue, the staining was intensified in Tg myocardium, and was diminished in KO myocardium after 20 min. of ischemia followed by 120 min. of reperfusion.
Fig 2.
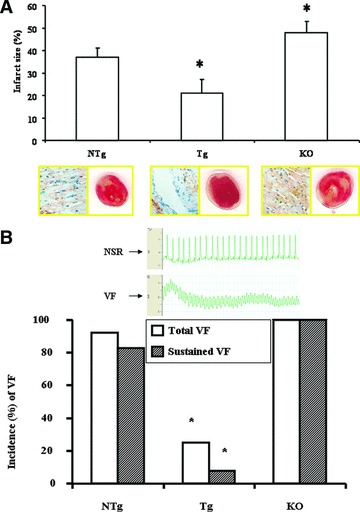
Infarct size and HO-1 staining in isolated NTg, Tg and HO-1 KO mouse hearts subjected to 20 min. of normotermic global ischemia followed by 120 min. of reperfusion. Infarct is represented by white area surrounding by the living red tissues (A, right lower part). Mean ± S.E.M., *P < 0.05 compared to the NTg control values. Immunochemical localization of HO-1 is stained by blue in NTg, Tg and KO myocardium (A, left lower part). (B) Representative ECG records corresponding to sinus rhythm and VF, respectively, are given in the upper part of (B). The incidence (%) of total (open bars) and sustained (hatched bars) reperfusion-induced VF in isolated NTg, Tg and KO mouse hearts are shown in the lower part of (B). The incidence of reperfusion-induced VF was registered, and comparisons were made to the values of NTg group. n= 12 in each group, *P < 0.05. Because of the non-parametric distribution in the incidence of total and sustained VF, the chi-square non-parametric test was used to compare individual groups.
The results of the infarct size studies are also presented in Fig. 2A. We have detected a significantly reduced infarct size in hearts obtained from HO-1 Tg mouse in comparison with the NTg control value of 37 ± 4%versus 20 ± 6% (*P < 0.05). In agreement with our previous study the infracted area was markedly increased in the HO-1 KO hearts to 47 ± 5% (*P < 0.05) in comparison with NTg group.
Figure 2B shows the incidence (%) of reperfusion-induced VF as total (sustained and non-sustained) and sustained VF in NTg, HO-1 Tg and HO-1 KO–/– mouse groups. Thus, the incidence of reperfusion-induced total and sustained VF (Fig. 2B) was significantly reduced in the HO-1 Tg group to 25% (*P < 0.05) and 8% (*P < 0.05) in comparison with the NTg control values of 92% and 83%, respectively. In hearts obtained from HO-1 KO–/– mice (Fig. 2B), the incidence of reperfusion-induced total and sustained VF were 100% and 100%, respectively, showing the important role of HO-1 in arrhythmogenesis.
Discussion
Myocardial ischemia/reperfusion (I/R) induced damage is one of the leading causes of morbidity and mortality in industrialized societies. Consequently, extensive effort has been devoted to characterization of the underlying mechanisms contributing to this spectrum of disorders and to develop new strategies for avoiding I/R-induced damage. Increasingly, this field of medical research is exploring approaches involving modification of the regulation of genes contributing to cardiovascular homeostasis. It has been shown by other investigators that induction of various survival genes has the capability to reduce the I/R-induced damage and enhance the viability of the myocardium after I/R [10]. Consistent with this observation, the reduction of the levels of various cell death-related proteins also protects the heart from I/R-induced injury.
Other studies have shown that cardioprotection against I/R may also be enhanced by changes in the levels of redox-related and detoxifying proteins. One of the potential candidates for this phenomenon is the HO-1 system, an essential enzyme responsible for the cleavage of heme to biliverdin, endogenous CO and iron. Previously, we have found a reduced level of HO-1 mRNA and protein expression in fibrillated ischemic/reperfused rat myocardium [4]. Consistent with this observation, experiments conducted in our laboratory demonstrated that reduced HO-1 enzyme activity ultimately led to reduced level of endogenous CO production. In a subsequent study also in our laboratory, HO-1 KO mice were used to demonstrate the role of HO-1 and endogenous CO in protection against I/R-induced arrhythmias and injury [5]. These studies demonstrated that eliminating HO-1 causes a reduced level of endogenous CO production, leading to enlarged infarct size and poor recovery of post-ischemic cardiac function. Furthermore, VF was detected in all of the HO-1 KO–/– hearts, demonstrating the important role of HO-1 and endogenous CO formation in the development of reperfusion-induced VF. These findings led to further investigations of the crucial role of HO-1 in the development of I/R-induced VF, in which anti-arrythmogenic effect was studied using Tg mice overexpressing rat HO-1. Heart tissue harvested from this Tg strain contained elevated HO-1 levels and the animals exhibited reduced infarct size and incidence of VF in comparison with wild-type control mice. Results of the present investigation further confirm the cardioprotective role of the HO-1 in ischemic/reperfused myocardium.
The endogenous HO-1/CO system can protect myocardial cells via different signalling mechanisms against doxorubicin-induced mitochondrial damage [11]. Thus, interventions influencing the function of HO-1/CO signalling in connection with mitochondrial gene expression or repression such as cytochrome oxidase B subunit III and ATP synthase subunit 6 [12] should be effective in reducing targeted mitochondrial damage related to VF-induced sudden cardiac death. Ongoing investigations are planned to assess the impact of the HO-1/CO system on mitochondrial gene regulation and to clarify the involvement of mitochondrial genes or other mechanisms in the anti-arrythmogenic effect of HO-1 system. The results of the present study can be translated to an actual clinical situation with some caution, if mitochondria, as the primary energy source of the myocardium, are involved in various diseases connected to myocardial ischemia; however, further studies need to be done to investigate how this genomic gene (HO-1) affects the function of the aforementioned mitochondrial genes.
Acknowledgments
This study was supported by grants from OTKA 72315, OTKA 78223, GVOP-3.2.1.-2004–04-0269/3.0, TAMOP-4.2.2–08/1–2008-0007 and TAMOP-4.2.1.B-09/1/KONV-2010–0007.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Idriss NK, Blann AD, Lip GY. Hemoxygenase-1 in cardiovascular disease. J Am Coll Cardiol. 2008;52:971–8. doi: 10.1016/j.jacc.2008.06.019. [DOI] [PubMed] [Google Scholar]
- 2.Bach FH. Heme oxygenase-1 as a protective gene. Wien Klin Wochenschr. 2002;114:1–3. [PubMed] [Google Scholar]
- 3.Kirkby KA, Adin CA. Products of heme oxygenase and their potential therapeutic applications. Am J Physiol Renal Physiol. 2006;290:F563–71. doi: 10.1152/ajprenal.00220.2005. [DOI] [PubMed] [Google Scholar]
- 4.Bak I, Papp G, Turoczi T, et al. The role of heme oxygenase-related carbon monoxide and ventricular fibrillation in ischemic/reperfused hearts. Free Radic Biol Med. 2002;33:639–48. doi: 10.1016/s0891-5849(02)00913-9. [DOI] [PubMed] [Google Scholar]
- 5.Bak I, Szendrei L, Turoczi T, et al. Heme oxygenase-1-related carbon monoxide production and ventricular fibrillation in isolated ischemic/reperfused mouse myocardium. FASEB J. 2003;17:2133–5. doi: 10.1096/fj.03-0032fje. [DOI] [PubMed] [Google Scholar]
- 6.Varadi J, Lekli I, Juhasz B, et al. Beneficial effects of carbon monoxide-releasing molecules on post-ischemic myocardial recovery. Life Sci. 2007;80:1619–26. doi: 10.1016/j.lfs.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 7.Splawski I, Timothy KW, Sharpe LM, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 8.Splawski I, Timothy KW, Decher N, et al. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc Natl Acad Sci USA. 2005;102:8089–96. doi: 10.1073/pnas.0502506102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Araujo JA, Meng L, Tward AD, et al. Systemic rather than local heme oxygenase-1 overexpression improves cardiac allograft outcomes in a new transgenic mouse. J Immunol. 2003;171:1572–80. doi: 10.4049/jimmunol.171.3.1572. [DOI] [PubMed] [Google Scholar]
- 10.Ferdinandy P, Schulz R, Baxter GF. Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol Rev. 2007;59:418–58. doi: 10.1124/pr.107.06002. [DOI] [PubMed] [Google Scholar]
- 11.Piantadosi CA, Carraway MS, Babiker A, et al. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ Res. 2008;103:1232–40. doi: 10.1161/01.RES.0000338597.71702.ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szendrei L, Turoczi T, Kovacs P, et al. Mitochondrial gene expression and ventricular fibrillation in ischemic/reperfused nondiabetic and diabetic myocardium. Biochem Pharmacol. 2002;63:543–52. doi: 10.1016/s0006-2952(01)00913-3. [DOI] [PubMed] [Google Scholar]