

Abstract
Although overexpression of cyclin A2 is reportedly an indicator of a poor prognosis of various malignancies including endometrial carcinoma, its molecular mechanism remains undetermined. To address this issue, we examined the effect of cyclin A2 on the development of resistance to chemotherapeutic drugs. The expression of cyclin A2 protein was increased in advanced-stage and chemotherapy-refractory stage endometrial carcinomas compared with that in early-stage tumours. The expression levels of cyclin A2 in endometrial carcinoma cell lines correlated positively with the IC50 for cisplatin. Endometrial carcinoma HHUA cells that overexpressed cyclin A2 showed increased resistance to cisplatin in vitro and in vivo, via the activation of a survival pathway, the inositol-3 phosphate kinase (PI3K) cascade. The use of a cDNA microarray identified an Akt-binding protein, periplakin, as a novel target of cyclin A2. The cyclin A2-induced up-regulation of periplakin was mediated via direct binding of Sp1 to the promoter that was activated by cyclin A2 along with chromatin remodelling involving CBP/p300, and the siRNA-mediated silencing of periplakin suppressed the PI3K pathway. These results indicate cyclin A2 to be involved in the acquisition of aggressive behaviour of tumour cells through the activation of PI3K by cyclin A2-induced periplakin, and to be a promising therapeutic target.
Keywords: chemotherapy, cyclin A2, endometrial carcinoma, periplakin, PI3K
Introduction
Cyclin A2 is unique among cyclins in that it is involved in both the G1/S and G2/M phases of the cell cycle and can activate key cyclin-dependent kinases (CDKs), such as CDK1 and CDK2. The cyclin A2–CDK complex plays an important role in the initiation of DNA replication and progression from S to M phase [1]. Overexpression of cyclin A2 has been reported in various malignant tumours such as lung, liver and stomach cancer [2–4], and was an indicator of a poor prognosis. We previously reported that cyclin A2 overexpression was correlated with a poor prognosis in cases of endometrial carcinoma [5]. The association of cyclin A overexpression with aggressive clinicopathological phenotypes has been attributed mainly to enhanced growth potential. Although several studies have identified new functions of cyclin A2 other than regulation of the cell cycle, for example, DNA repair [6] and p53-independent anti-apoptosis [7], the molecular mechanisms that explain these findings have not been fully elucidated.
Cisplatin, commonly used in combination with drugs such as adriamycin and taxanes, is a key drug for treating endometrial carcinoma [8]. Resistance to these drugs is a major therapeutic barrier. Recent studies have revealed that cisplatin-resistant carcinoma cells escaped from cisplatin-induced apoptosis by activating various survival mechanisms [9]. The activation of the phosphatidyl inositol-3-kinase (PI3K)-Akt pathway, a major constituent of the mitochondrial anti-apoptotic pathway, was reportedly one of the mechanisms by which carcinoma cells survive the effect of chemotherapeutic agents such as cisplatin [10] and paclitaxel [11]. In addition, the activation of the PI3K pathway is of great significance in endometrial carcinoma because this particular malignancy has frequent mutations in the phosphatase and tensin homologue deleted on chromosome ten (PTEN) gene, which encodes a protein that inactivates the PI3K pathway [12].
Therefore, the present study was undertaken to elucidate the functions of cyclin A2 other than growth stimulation that lead to the aggressive behaviour of cyclin A2-positive tumours, with special reference to the effect on sensitivity to chemotherapeutic drugs.
Materials and methods
Collection of cancer and normal endometrial tissues
Nineteen endometrial carcinoma and four normal endometrial tissues were obtained from patients who underwent hysterectomy at Shinshu University Hospital. All specimens were collected from the uterus immediately after resection and kept at −80°C until protein extraction. The study was approved by the hospital ethics committee, and all patients gave informed consent before surgery.
Agent, antibody and vector
Cisplatin and adriamycin were provided by Nippon Kayaku Co. (Tokyo, Japan) and Shionogi Seiyaku Co. (Osaka, Japan), respectively. Paclitaxel was purchased from Calbiochem (Darmstadt, Germany). Wortmannin was obtained from Sigma-Aldrich (St. Louis, MO, USA). U0126 was purchased from Promega (Madison, WI, USA). WST-1 cell proliferation assay reagent was purchased from Roche (Mannheim, Germany). Mouse monoclonal anti-phospho-Akt ser-473 antibody, rabbit anti-Akt antibody, mouse monoclonal anti-phospho-Erk1/2 antibody, mouse monoclonal Erk1/2 antibody, rabbit polyclonal anti-phospho-Bad at serine 112 residue and that at serine 136 residue, rabbit polyclonal anti-cleaved caspase-3 antibody were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). Mouse monoclonal anti-cyclin A2 antibody, mouse monoclonal anti-cyclin E1 antibody, rabbit polyclonal anti-AP2 antibody, rabbit polyclonal anti-Sp1 antibody, goat polyclonal anti-periplakin antibody, rabbit polyclonal anti-CBP/p300 antibody, rabbit polyclonal anti-cellar retinoic acid-binding protein 2 antibody and goat polyclonal anti-stanniocalcin1 antibody were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Rabbit polyclonal anti-acetyl-H3 antibody, mouse monoclonal anti-Aven antibody, and mouse monoclonal anti-sorbin and SH3-domains containing 1 antibody were obtained from Upstate Biotechnology (Lake Placid, NY, USA). Rabbit polyclonal anti-phosphoserine antibody was obtained from Abcam (Cambridge, MA, USA). pRC/CMV/cyclin A2 was kindly provided by Dr M. Nakanishi at Nagoya City University (Nagoya, Japan). The pGL3 basic, control vector and pRL/TK vectors were purchased from Promega.
Immunohistochemistry
Immunostaining for periplakin was performed as described previously [5]. In brief, three micrometer-thick sections were deparaffinized and boiled in 0.01 M citrate buffer (pH 6.0) for 25 min in a microwave oven. They were then treated with 0.1% hydrogen peroxide and incubated with 10% normal rabbit serum. The sections were incubated with an anti-periplakin antibody, which was used at a dilution of 1:100, at 4°C overnight. After washing in phosphate-buffered saline, they were incubated with biotinylated rabbit anti-goat immunoglobulin G, treated with peroxidase-conjugated streptavidin, and stained with diaminobenzidine and 0.15% hydrogen peroxidase. Counterstaining was performed with haematoxylin.
Immunofluorescent staining
Cultured cells in a two-well Laboratory-Tek Chamber Slide were fixed in cold acetone for 15 min; then, 0.3% hydrogen peroxide was applied to block endogenous peroxide activity, and the cells were incubated with normal rabbit serum. The cells were incubated with an anti-periplakin antibody (1:100) at room temperature for 1 hr, and Alexa Fluor 594 rabbit anti-goat immunoglobulin G (Invitrogen, Carlsbad, CA, USA) was used; 4′,6′-diamidino-2-phenylindole (DAPI, Roche) was used to stain DNA. All specimens were examined using a laser-scanning spectral confocal microscope (Leica TCS SP2; Leica Microsystems, Wetzlar, Germany).
Cell culture and transfection
The endometrial carcinoma cell lines Ishikawa, and Hec-1A and Hec-1B were gifts from Dr H. Nishida (Kasumigaura Medical Center, Tsuchiura, Japan) and Dr H. Kuramoto at Kitazato University (Sagamihara, Kanagawa), respectively. HHUA was purchased from the Riken Cell Bank (Saitama, Japan) with the permission of Dr Ishiwata at the Ishiwata Laboratory (Mito, Japan). KLE and RL95-2 were purchased from American Type Culture Collection (Rockville, MD, USA).
To establish endometrial carcinoma cells that stably express cyclin A2, HHUA cells were transfected with pRC/CMV/cyclinA2 or pRC/CMV by lipofection according to the supplier’s instruction (Lipofectamine 2000, Invitrogen). At 48 hrs after transfection, the medium was changed to selection medium and a stable expression clone was selected.
Unless otherwise stated, cells were seeded in 60-mm dishes with 4 ml of growth medium and cisplatin was added on the third day after 16 hr of serum starvation. Inhibitors such as wortmannin and U0126 were added an hour before cisplatin treatment.
Western blot analysis
Proteins extracted from cells of sub-confluent cultures or surgically resected tissues were subjected to a Western blot analysis as described previously [13]. Blocking was performed with 5% non-fat milk or 3% bovine serum albumin in PBS-T for 1 hr at room temperature. The membranes were blotted with primary antibody at 4°C overnight and then incubated with a peroxidase-conjugated secondary antibody. Bound antibodies were visualized using the ECL Western Blot Detection Reagent (Amersham, Piscataway, NJ, USA).
Immunoprecipitation
Non-confluent cells were lysed in 1.0 ml of RIPA buffer (20 mM Tris pH 8.0, 1% Triton X-100, 0.5% Na-DOC, 0.1% SDS, 10 mM EDTA, 150 mM NaCl, 1 mM NaF, 1 mM, 1 μg/ml leupeptin and 2 μg/ml aprotinin). Five hundred micrograms of precleared protein was incubated for 2 hrs at 4°C with 4 μg of specific antibody or normal rabbit IgG and 60 μl of pre-washed protein-A/G beads. The precipitate was washed three times with the lysis buffer before being taken up in Laemmli buffer and was resolved using SDS-PAGE.
WST-1 assay
Cells were plated at a density of 2.0 × 103 cells into 96-well plates and cultured under optimal condition (37°C in a 5% CO2 incubator) for measurement. The cell viability was measured by a modified 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay (WST-1 assay) according to the manufacturer’s instructions. A450 was measured using a microplate reader (Multiskan JX, Thermo Bioanalysis, Tokyo, Japan).
Cytotoxicity assay
Cells were plated at a density of 2.0 × 103 cells into 96-well plates. After 24 hrs at 37°C in a 5% CO2 incubator, the cells were exposed to serial dilutions of chemotherapeutic agents for 2 hrs. Then, the medium was replaced with fresh medium. Cell viability was measured 5 days later using the WST-1 assay as described above. The results were expressed as the IC50, the drug concentration of inducing 50% growth suppression compared to control cells. IC50 values were read from a graph expressing the concentration of drug against the percentage of relative growth. All concentrations were tested in 12 replicated wells and each experiment was performed three times independently.
Assessment of apoptosis
Cell lysates were collected 48 hrs after cisplatin (20 μM) treatment and analysed by Western blotting using antibody against the active form of caspase-3. The intensity of the signal for cleaved caspase-3 was normalized to the β-actin signal and quantified using a densitometric analysis.
To demonstrate DNA fragmentation, cells treated with cisplatin were analysed using an ApoStrand ELISA apoptosis detection kit (BIOMOL International Inc., Plymouth Meeting, PA, USA) according to the manufacturer’s directions. Briefly, 5000 fixed cells were transferred into 96-well plate and they were incubated with 50 μl of formamide at room temperature for 10 min and heated at 56°C for 30 min to denature the DNA. To detect DNA fragmentation, cells were incubated with a mixture of a single-stranded DNA antibody and peroxidase-conjugated anti-mouse IgM for 1 hr. Then, the colour was developed by adding 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid). Absorbance was read in an ELISA plate reader at 405 nm.
Small interfering RNA
Cyclin A2-specific siRNAs, periplakin-specific siRNAs and control siRNA were purchased from Ambion Inc. (Austin, TX, USA). For transfection, 2 × 105 cells/dish plated in a 35-mm dish and transfected with siRNA 24 hrs later with JET-SI reagent (Ambion Inc.) according to the manufacturer’s instructions. To assess the effect of cyclin A2’s silencing on chemosensitivity, cyclin A2 or control siRNA-transfected cells were seeded in 96-well plates and subjected to cytotoxicity assay described above.
Oligonucleotide array
Gene expression was assessed using Affymetrix GeneChIP Human Genome U133 plus 2.0 arrays. The gene ChIP contained 54,675 probe sets, representing more than 47,000 transcripts, including 38,500 genes. Total RNA samples were labelled according to the Affymetrix GeneChIP one-cycle target labelling protocol (Affymetrix, Santa Clara, CA, USA). After fragmentation, biotinylated cRNA was hybridized to the GeneChIP for 16 hrs at 45°C and scanned according to the manufacturer’s specifications. All intensity values were scaled to an average of 150 per GeneChIP according to the GeneChIP Operating Software available in the Affymetrix Microarray Suite. We used a statistical analysis including both a GeneSpring P-value cut-off (P < 0.05) and at least a 2-fold change to further increase the stringency of the analysis.
Validation of gene expression using real-time quantitative PCR
Total RNA was extracted from cultured cells using RNeasy columns (Quiagen, Valencia, CA, USA) and treated with DNase I. cDNA was synthesized from mRNA samples using PrimeScript RT reagent (Takara Bio Inc., Otsu, Japan). Reactions were carried out using the SYBR Green method. SYBR Green PCR master mix (Takara Bio. Inc.) was used in 25-μl reactions set-up in triplicate with each primer. Reactions were run at 95°C for 1 min followed by 40 cycles of 95°C for 15 sec and 60°C for 30 sec, and followed by a dissociation curve analysis to ensure that the fluorescence signal was not derived from prime-dimer formation. The threshold cycle was determined. Quantitation of gene expression was calculated by the comparative CT method using β-actin as the internal control gene.
Chromatin immunoprecipitation assay
Cells were seeded at a density of 10 × 106 in 150-mm culture dishes. Chromatin fixation and purification were performed as described previously [14]. In brief, cellular proteins and DNA were fixed by adding formaldehyde to 1% (w/v) final concentration for 10 min at room temperature. Cells were then sonicated to prepare a chromatin suspension of 200–500 bp DNA. Twenty microlitres of the soluble chromatin was set aside as the input fraction, and the remainder was diluted 1:10 in dilution buffer (2 mM EDTA, 20mM Tris pH 8, 150 mM NaCl and 1% Triton X-100). Lysates were then incubated at 4°C overnight with rotation with protein A (50% slurry in 10 mM Tris, pH 8, 1 mM EDTA) or protein G-agarose with 2 μg of each antibody or normal IgG. Complexes were precipitated and serially washed three times each with low-salt buffer (20 mM Tris pH 8, 150 mM NaCl, 2 mM EDTA, 0.1% SDS and 1% Triton X-100), high-salt buffer (20 mM Tris pH 8, 500 mM NaCl, 2 mM EDTA, 01% SDS and 1% Triton X-100), LiCl wash buffer (10 mM Tris pH 8, 250 mM LiCl, 1 mM EDTA, 1% deoxycholate and 1% Nonidet P-40) and TE buffer (10 mM Tris pH 8 and 1 mM EDTA). Washed complexes were eluted with 100 μl of elution buffer (1% SDS and 100 mM NaHCO3), and the cross-links were reversed by incubation at 65°C for 4 hrs. DNA was purified utilizing a Qiaquick PCR purification kit (Qiagen, Valencia, CA). One microlitre of the purified DNA was then amplified using real-time PCR described previously [15] with primers for the periplakin promoter region (5′-tagcgcagaaccaggtgaa-3′ and 5′-gtaagcaccaccttctcggg-3′).
Reporter construction and luciferase reporter gene assay
The periplakin reporter plasmid containing the Sp1-binding site was obtained by cloning into the KpnI and XhoI restriction sites of the pGL3 basic vector, a 226-bp fragment of the human periplakin promoter (bp −408 to −182 relative to the most 3′ transcription initiation site), which was generated by PCR with the following oligonucleotides: 5′ggggtacctagcgcagaaccaggtgaag3′ and 5′ccgctcgagagaaggtggtgcttaccttcc3′. Cells were transfected in 6-well plates with 4 μg of either pRC/CMV or pRC/CMV/cyclinA2, 400 ng of pGL3 reporter plasmid and 30 ng of pRL/TK Renilla luciferase expression vector. The assay was performed with the dual-luciferase reporter assay system (Promega) according to the manufacturer’s instructions.
Animal experiment
Five-week-old female BALB/c nude mice (Nihon CLEA, Tokyo, Japan) were used. Procedures involving animals and their care are conducted in conformity with the institutional guidelines that are in compliance with national (D.L. n.116, G. U., suppl. 40, 18 Febbraio 1992, Circolare No. 8, G.U., July 14 1994) and international laws and policies (EEC Council Directive 86/609, OJ L 358,1, 12 December 1987; Guide for the Use of Laboratory Animals, U.S. National Research Council, 1996). In serum-free medium 5 × 106 cells were implanted subcutaneously in the back of each animal. Cisplatin (4 mg/kg) dissolved in PBS was intraperitoneally injected once a week for 4 weeks. Visible tumours were measured using digital calipers and tumour volume was calculated according to a commonly used approximated formula: width2× length × 0.5.
Statistical analysis
All data are represented as mean ± SEM. The Mann–Whitney U-test or Kruscal–Wallis test was used to assess differences. Spearman’s rank correlation was used for correlation analysis between the expression of cyclin A and the IC50 for chemotherapeutic drugs, and the significance of the correlation was evaluated with a paired t-test. Differences were considered significant if P < 0.05. All data presented were representative of at least three independent experiments. These analyses were made using the StatView System (Abacus, Berkeley, CA, USA).
Results
Cyclin A2 protein expression correlated with resistance to cisplatin in endometrial carcinoma tissues and cell lines
The expression of cyclin A2 protein in tissue samples from normal endometrium, early-stage, advanced-stage and chemotherapy-refractory endometrial carcinoma was examined. The tumours refractory to chemotherapy were defined as uterine tumours surgically resected after multiple courses of platinum-based chemotherapy. The amount of cyclin A2 protein was larger in advanced-stage tumours than in normal tissues and early-stage tumours (Fig. 1A and B). Importantly, although the difference was not significant, the expression of cyclin A2 was stronger in chemotherapy-resistant tumours than in advanced-stage tumours (Fig. 1A and B). The expression of cyclin E, another partner of CDK2, was also increased in carcinoma tissues, but was decreased in chemotherapy-refractory tumours as compared with advanced-stage tumours (Fig. 1B).
Fig 1.
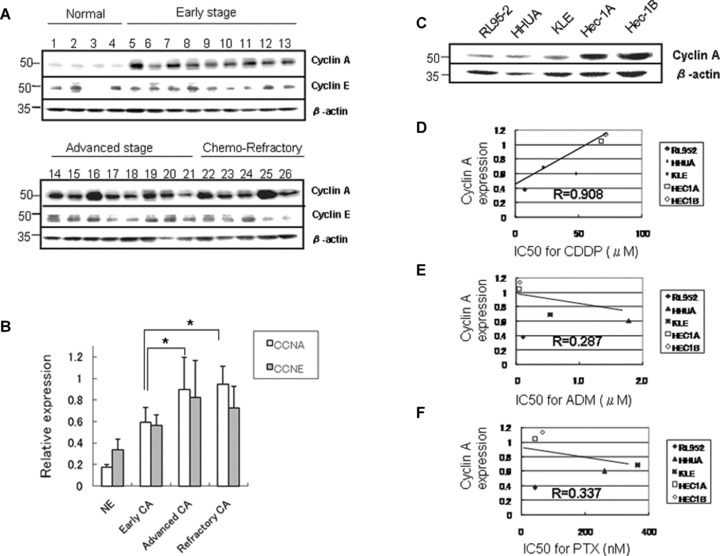
Overexpression of cyclin A2 was associated with resistance to chemotherapy in endometrial carcinoma tissue and cell lines. (A) The expression of cyclin A2 and cyclin E1 protein extracted from normal endometrium (case no. 1–4), and early-stage (stage I, case no. 5–13), advanced-stage (stages II–III, case no. 14–21) and chemotherapy-refractory (case no. 22–26) endometrial carcinoma specimens using Western blotting. (B) The densitometric quantification of cyclin A2 and cyclin E1 protein expression relative to β-actin (relative expression) as shown in Fig. 1A. The expression of cyclin A2 was increased, especially in advanced-stage tumours and refractory tumours. In contrast, the expression of cyclin E1 was decreased in refractory tumours (NE, normal endometrium; CA, carcinoma; *P < 0.05) (C) The expression of endogenous cyclin A2 protein in various endometrial carcinoma cell lines. (D) The expression of endogenous cyclin A2 was strongly (R= 0.908) correlated with the median inhibitory concentration (IC50) for cisplatin (CDDP, P < 0.05). (E) The correlation of endogenous cyclin A2 expression with the IC50 for adriamycin (ADM). (F) The correlation of endogenous cyclin A2 expression with the IC50 for paclitaxel (PTX). There was no significant correlation between cyclin A2 and IC50 for adriamycin or paclitaxel.
The expression of endogenous cyclin A2 varied in different endometrial carcinoma cell lines (Fig. 1C). The IC50 values for cisplatin, adriamycin and paclitaxel in these lines were compared with the expression of cyclin A2 protein. The expression of cyclin A2 was strongly correlated with the IC50 for cisplatin (P= 0.0374, correlation coefficient (R) = 0.908; Fig. 1D), but not with that of paclitaxel or adriamycin (Fig. 1E and F).
Cyclin A2 promotes cisplatin resistance and growth activity in HHUA cells in vitro and in vivo
We introduced a cyclin A2 expression plasmid into a human endometrial carcinoma cell line, HHUA, showing weak cyclin A2 expression (Fig. 1C), and established a stable transfectant (HHUA-cycA) with elevated cyclin A2 levels compared with control vector-transfected HHUA (HHUA-vec) cells (Fig. 2A, left panel). Silencing of cyclin A2 was also accomplished using cyclin A2-specific siRNA, which reduced the amount of protein by 78% (Fig. 2A, right panel). HHUA-cycA cells exhibited good proliferative activity compared with HHUA-vec cells, and this effect was reversed by introducing siRNA for cyclin A2 in a dose-dependent manner (Fig. 2B). The HHUA-cycA cells were significantly more resistant to cisplatin and adriamycin than the HHUA-vec cells; the IC50 for cisplatin was 80.1 and 46.1 μM, respectively (Fig. 2C). Likewise, the IC50 for adriamycin was 1.91 and 0.88 μM, respectively (Fig. 2D). However, cyclin A2 did not have a significant effect on paclitaxel (Fig. 2E). The silencing of cyclin A2 using siRNA reduced the cell viability after cisplatin treatment by 53% compared with the control HHUA-cycA cells (Fig. 2F, left). To confirm this result, we performed the same silencing experiments using another endometrial carcinoma cell line, Hec-1A, which showed strong endogenous cyclin A2 expression (Fig. 1C). Cisplatin reduced the viability of Hec-1A cells by 38% by abrogating cyclin A2 expression (Fig. 2F, right).
Fig 2.
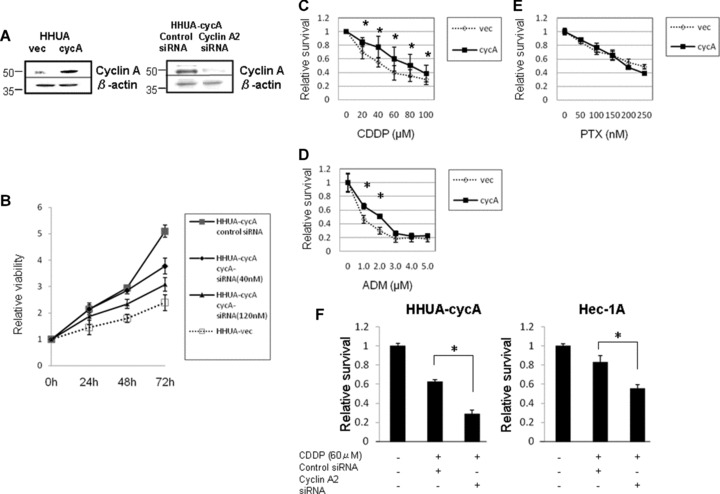
Cyclin A2 enhanced the proliferation and cisplatin resistance in endometrial carcinoma HHUA cells in vitro. (A) The expression of cyclin A2 was increased in HHUA cells transfected with RC/CMV/cyclin A2 (HHUA-cycA) compared with vector-transfected HHUA cells (HHUA-vec) (left panel). Cyclin A2 silencing using cyclin A2-specific siRNA (40 nM) in HHUA-cycA cells resulted in reduced expression of cyclin A2 protein (right panel). (B) HHUA-cycA cells (black line) showed increased proliferative activity compared with HHUA-vec cells (dashed line). Transfection of cyclin A2-specific siRNA (40 or 120 nM) reduced the proliferation of HHUA-cycA cells in a dose-dependent fashion in the WST-1 assay. (C–E) Dose-inhibitory curve for cisplatin (CDDP), adriamycin (ADM) and paclitaxel (PTX) in HHUA-vec and HHUA-cycA cells. Cyclin A2 increased the resistance to cisplatin and adriamycin, but not paclitaxel (*P < 0.05). (F) Silencing of cyclin A2 using siRNA (40 nM) reduced cisplatin resistance in HHUA-cycA (left panel) and Hec-1A (high endogenous cyclin A2 expression, right panel) cells. Viability was measured 5 days after cisplatin treatment using WST-1 assay. The viability was normalized with the value of control cells and expressed as relative survival (*P < 0.05).
HHUA-cycA cells and HHUA-vec cells were injected subcutaneously into nude mice and tumours appeared 2 weeks later. Subcutaneous tumours from HHUA-cycA cells without cisplatin (Fig. 3A, CyACDDP(−)) were 1.2 times larger than those from HHUA-vec cells (Fig. 3A, VecCDDP(−)). Then, cisplatin was administered intraperitoneally once a week for 4 weeks. Four weeks after the injection, tumours of HHUA-cycA cells (Fig. 3A, CycACDDP(+), and B, right) exhibited a significantly higher rate of growth, 1.6 times that of HHUA-vec cells (Fig. 3A, VecCDDP(+), and B, left). Haematoxylin–eosin staining of the HHUA-vec tumour 1 week after the last cisplatin injection revealed massive necrosis (Fig. 3C, lower left). In contrast, the HHUA-cycA tumour showed focal areas of necrosis (Fig. 3C, lower right). These results indicated that cyclin A2 expression provided endometrial carcinoma cells with increased cisplatin resistance in vitro and in vivo.
Fig 3.
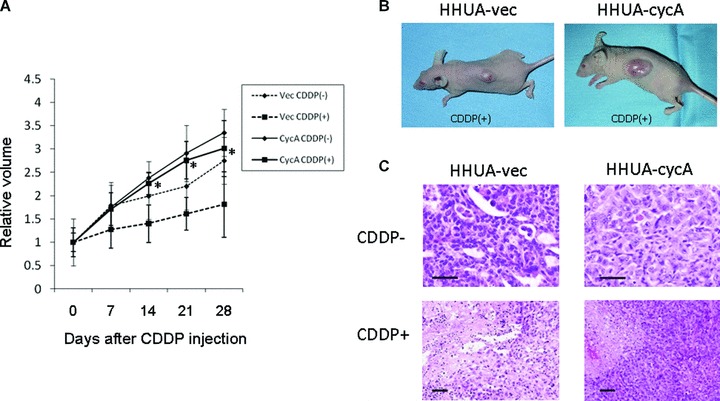
The effects of cyclin A2 on cisplatin resistance in vivo. (A) Graphic demonstration of tumour growth from HHUA-vec (dashed line) and HHUA-cycA cells (black line) with or without cisplatin (CDDP) treatment (4 mg/kg, intraperitoneally once a week for 4 weeks). Tumours from HHUA-cycA cell (CycACDDP(−), n= 6) grew 1.2 times faster than those from HHUA-vec cells (VecCDDP(−), n= 6) without CDDP treatment. The difference in tumour growth after CDDP treatment between HHUA-cycA (CycACDDP(+), n= 7) and HHUA-vec (VecCDDP(+), n = 7) cells was larger (1.6) than that without CDDP (*P < 0.05). (B) Subcutaneous inoculation of HHUA-cycA cells into nude mice followed by CDDP injection produced a larger mass (right) than that of HHUA-vec cells (left). (C) Haematoxylin and eosin staining of the mouse tumours from HHUA-vec (left) and HHUA-cycA cells (right). Cisplatin treatment resulted in extensive necrosis in HHUA-vec cell tumours (lower left) compared with HHUA-cycA cell tumours (lower right). Scale bars: 15 μm.
Cyclin A2 suppresses cisplatin-induced apoptosis by activating the PI3K pathway
We added various concentrations of cisplatin to HHUA-cycA and HHUA-vec cells, and evaluated apoptosis. Cisplatin-treated HHUA-cycA cells showed a decrease in DNA fragmentation by 29% compared with the control cells (Fig. 4A, leftmost two bars), as well as in the expression of cleaved caspase-3 by 38% (Fig. 4B). Moreover, transfection of cyclin A2-specific siRNA prior to the cisplatin treatment significantly increased apoptosis (Fig. 4A, rightmost two bars).
Fig 4.
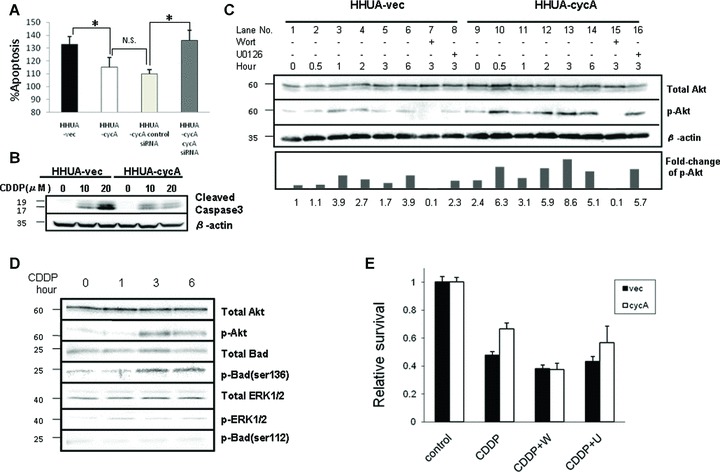
Cyclin A2 suppressed cisplatin-induced apoptosis via PI3K pathway. (A) Cisplatin-induced apoptosis (cisplatin 20 μM) detected by the ApoStrand method was decreased in HHUA-cycA cells compared with HHUA-vec cells. Transfection of siRNA for cyclin A2 into HHUA-cycA recovered the apoptosis to the level in HHUA-vec cells. The percentage of apoptotic cells was calculated using the value for control cells (N.S.: not significant; *P < 0.05). (B) Cisplatin treatment (20 μM) resulted in reduced expression of cleaved caspase-3 in HHUA-cycA cells after 48 hrs compared with vector-transfected cells. (C) Forced expression of cyclin A2 resulted in increased expression of p-Akt before the addition of cisplatin (20 μM) (lanes 1 and 9), and for 6 hrs after the addition (lanes 6 and 14) in HHUA-cycA cells compared with HHUA-vec cells. Wortmannin (W, 1 μM), but not UO126 (U, 1 μM), markedly reduced the expression of p-Akt. Five individual experiments indicated comparable results, and representative blots are shown. The lower graph indicates the relative intensities of p-Akt protein against total Akt protein quantified using densitometry. The value was mean from five individual experiments. (D) Cisplatin-induced Akt activation was associated with Bad phosphorylation at Ser 136 in HHUA-cycA cells, but not with ERK activation and Bad Ser 112 phosphorylation. (E) Cisplatin treatment reduced the number of viable cells among both HHUA-cycA2 cells and HHUA-vec cells, but the number of viable cells was larger for HHUA-cycA cells. Wortmannin (1 μM) treatment prior to cisplatin treatment abrogated the survival advantage against cisplatin in HHUA-cycA cells, but no such effect was observed on UO126 (1 μM) treatment (N.S.: not significant; *P < 0.05).
The effect of cyclin A2 on the PI3K pathway based on the detection of phosphorylated Akt (p-Akt) at different time points after cisplatin treatment was evaluated. HHUA-vec cells showed weak expression of p-Akt before cisplatin treatment (Fig. 4C, lane 1). In contrast, HHUA-cycA cells exhibited strong basal expression of p-Akt prior to (2.4-fold, lane 9) and after (lanes 10–14) cisplatin treatment. Pretreatment with wortmannin (lanes 7 and 15), but not U0126 (lanes 8 and 16), abolished the phosphorylation of Akt.
Next, we examined the involvement of the pro-apoptotic protein Bad, which is a downstream substrate of Akt in this cascade. The phosphorylation of Akt was observed 3 hrs after cisplatin treatment, and was associated with the phosphorylation (inactivation) of Bad at serine 136 (Fig. 4D). In contrast, phosphorylated Erk1/2, which is a hallmark of MAPK’s activation, showed only a slight increase after cisplatin treatment and Bad phosphorylated at serine 112, a target residue of Erk1/2, remained unchanged.
We then examined the effect of a specific PI3K inhibitor, wortmannin, on cell viability. Addition of cisplatin alone reduced the viability of HHUA-cycA cells and HHUA-vec cells by 34 and 53%, respectively (Fig. 4E, leftmost two bars). Pretreatment with wortmannin before the addition of cisplatin further reduced the viability of both HHUA-cycA and HHUA-vec cells; however importantly, the cyclin A2-induced cisplatin resistance in HHUA-cycA cells disappeared. In contrast, pretreatment with U0126 did not influence the cyclin A2-induced cisplatin resistance (Fig. 4E, rightmost bars), suggesting that cyclin A2 specifically activated the anti-apoptotic PI3K pathway.
Cyclin A2 up-regulated an Akt-binding protein, periplakin, which enhances Akt activity
Affymetrix U133 plus 2.0 microarrays using total RNA extracted from HHUA-cycA and HHUA-vec cells revealed a total of 1024 genes that were up-regulated more than 2-fold in HHUA-cycA cells. Of the 1024 genes, those showing a weak basal signal were eliminated, and we eventually selected 12 candidate genes as shown in Table 1. The up-regulation of all 12 genes in HHUA-cycA cells was further validated using real-time PCR (Fig. 5A). To the extent that antibodies were commercially available, Western blotting was used to ascertain the expression of the protein transcripts of 5 of the 12 genes (Fig. 5B). The 12 included several genes for anti-apoptotic proteins such as AVEN[17], EMP1[24] and CALB1[26]. FGF9[16], PPL[21] and SORBS1[23] are reportedly linked to the PI3K cascade. Among these candidate genes, we focused on PPL encoding periplakin, which was revealed to bind directly to Akt [21]. We demonstrated periplakin expression in all endometrial carcinoma cell lines (Fig. 5C, upper panel) and the expression of periplakin paralleled that of cyclin A2 (Fig. 5C, lower graph). Immunostaining demonstrated that periplakin was expressed in the cytoplasm of endometrial carcinoma cells (Fig. 5D, upper right and lower left). Periplakin is reportedly localized in cytoplasm or cell surface [21]. We further demonstrated cytoplasmic localization of periplakin, where Akt exists, by immunofluorescent staining (Fig. 5D, lower right). Immunoprecipitation revealed that periplakin was directly associated with Akt (Fig. 5E, upper panel) and the Akt-bound fraction of periplakin in HHUA-cycA cells also increased significantly (Fig. 5E, lower panel). The siRNA for periplakin decreased the expression of periplakin protein by 61% (Fig. 5F, top panel), resulting in a reduction in basal p-Akt and cisplatin-treated p-Akt activities by 67 and 71%, respectively (Fig. 5F).
Table 1.
The names and function of up-regulated genes in cyclin A2 transfectant (HHUA-cycA) compared with HHUA-vec cell analysed with the Affymetrix oligonucleotide array
| Gene symbol | UniGene number | Fold change | Possible functions | References |
|---|---|---|---|---|
| FGF9 | 130679 | 2.06 | Endometrial epithelial cell proliferation | [16] |
| AVEN | 1632771 | 2.16 | Anti-apoptosis | [17] |
| PTGER4 | 161867 | 2.25 | Tumour progression | [18] |
| RB1CC1 | 161505 | 2.4 | Inhibits proliferation | [19] |
| PLAUR | 682030 | 2.57 | Tumour progression | [20] |
| PPL | 160815 | 3.05 | Akt binding | [21] |
| CRABP2 | 219620 | 3.08 | Mediates retinoic acid signalling | [22] |
| SORBS1 | 136646 | 3.73 | Adaptor protein of insulin receptor | [23] |
| EMP1 | 231335 | 3.84 | Chemoresistance | [24] |
| STC1 | 135066 | 5.14 | Regulates Ca homeostasis | [25] |
| CALB1 | 139098 | 11.4 | Anti-apoptosis | [26] |
| SNTB1 | 2724554 | 12.5 | Unknown |
Fig 5.
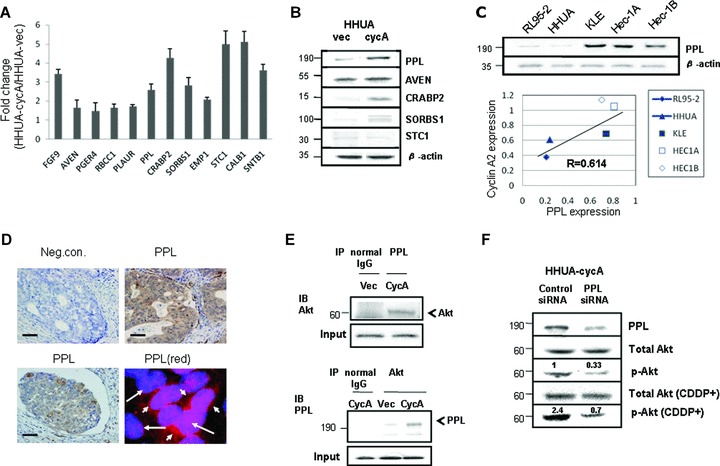
Verification of microarray analysis and the expression of a novel cyclin A2 target, periplakin. (A) The elevated levels of mRNA for 12 candidate genes in Table 1 were quantitatively confirmed using real-time PCR. (B) The expression of periplakin (PPL), aven (AVEN), cellular retinoic acid-binding protein 2 (CRABP2), sorbin and SH3-domains containing 1 (SORBS1) and stanniocalcin 1 (STC1) proteins was increased in HHUA-cycA cells. (C) The expression of periplakin protein was increased in endometrial carcinoma cell lines with elevated cyclin A2 protein expression (top). There was strong and positive correlation (R= 0.614, P < 0.05). (D) Immunohistochemical staining for periplakin was detected in the cytoplasm of endometrial carcinoma cells (upper right and lower left). Upper left micrograph shows staining with normal rabbit IgG instead of periplakin antibody as a negative control. Scale bars: 15 μm. Immunofluorescent staining for periplakin (red, lower right) indicated cytoplasmic staining, and DAPI was used for nuclear staining (blue). Long arrow, nucleus; short arrow, cytoplasm. (E) Immunoprecipitation revealed the direct binding of periplakin to Akt in HHUA-cycA cells (top and bottom). Akt-binding fraction was increased in HHUA-cycA cells compared with HHUA-vec cells (bottom) (IP, immunoprecipitation; IB, immunoblotting). (F) Transfection of periplakin-specific siRNA (40 nM) to HHUA-cycA cells resulted in reduced periplakin protein expression and p-Akt with and without cisplatin (20 μM) treatment. The signal for p-Akt was quantified by densitometry and normalized to total Akt. The value was mean from five individual experiments.
Cyclin A2 promotes the transcription of periplakin via the Sp1-binding site in the periplakin promoter
The periplakin promoter reportedly spans from −382 bp to +135 bp with three AP2 and one Sp1 (−277 bp) binding sites [27] (Fig. 6A). To examine the expression mechanisms of periplakin, we performed a luciferase assay using a reporter that contained the region spanning from −409 bp to −182 bp, and possessed one crucial Sp1-binding site at −277 (Fig. 6A). The results showed that co-transfection of the cyclin A2 expression plasmid enhanced the promoter’s activity 3.8-fold compared with co-transfection of the vector plasmid (Fig. 6B).
Fig 6.
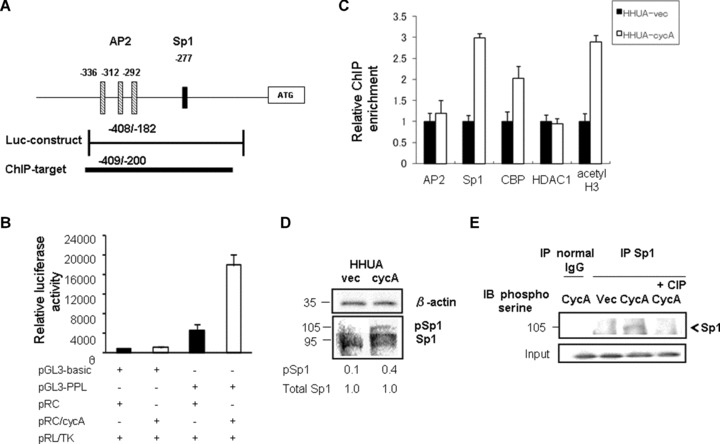
Cyclin A2 enhanced the periplakin promoter activity via phosphorylation of Sp1. (A) Geographic demonstration of the promoter region of the human periplakin (PPL) gene. The different boxes indicate previously characterized binding sites for the AP2 and Sp1 transcription factors. The bar below the diagram represents the insert for the luciferase reporter construct (Luc-construct) and the amplicon used for ChIP assay (ChIP target). (B) The strongest luciferase activity was observed when the PPL (−409/−182)-luciferase reporter construct (PGL3-PPL) was co-transfected with the cyclin A2 expression plasmid (pRC/cycA) to HHUA cells. All luciferase activities were normalized with Renilla luciferase activity to account for variability in transfection efficiency. The value represented as mean ± SEM from five individual experiments. (C) ChIP analysis indicated the recruitment of Sp1, CBP/p300 and acetylated histone H3 (acetyl H3), but not AP2 and histone deacetylase 1 (HDAC1), to the PPL promoter in HHUA-cycA cells. Immunoprecipitated chromatin was subjected to real-time PCR with periplakin-specific primers. Fold enrichment represents the recovery of chromatin-associated DNA fragments immunoprecipitated by each antibody compared to input control. Each value was obtained from five independent experiments. (D) HHUA-cycA cells had stronger expression of phosphorylated (activated) Sp1 protein (105 kD) and reduced non-phosphorylated Sp1 (95 kD) compared with vector-transfected cells. Each value indicates the relative intensity of total Sp1 or phosphor-Sp1 (pSp1) protein against β-actin quantified using densitometry from five individual experiments. (E) Immunoprecipitation revealed that Cyclin A2 increased the phosphorylation of Sp1 at serine residues, which was abolished by adding calf intestinal phosphatase into cell lysate (rightmost lane) (IP, immunoprecipitation; IB, immunoblotting).
Cyclin A2-induced up-regulation of periplakin expression is mediated via the binding of Sp1, which is phosphorylated by cyclin A2, to the periplakin promoter
We then examined how cyclin A2 induced the expression of periplakin. Studies indicated that the cyclin A2–CDK complex phosphorylate Sp1, and the phosphorylated (activated) Sp1 binds to the promoter of target genes, resulting in elevated levels of transcription [14, 28]. The periplakin promoter reportedly possessed an Sp1 consensus sequence at bp −277 [27]. We developed a primer set targeting the Sp1-binding site (Fig. 6A), and performed a ChIP (chromatin immunoprecipitation) assay using an antibody directed against Sp1 and chromatin obtained from HHUA-cycA and control cells. The ChIP assay showed that the recruitment of Sp1 to the promoter containing the Sp1-binding site at −277 was increased 3-fold in HHUA-cycA cells compared with control cells (Fig. 6C). Furthermore, increased recruitment of Sp1 in HHUA-cycA cells was associated with enhanced recruitment of CBP/p300 and acetylated core histone H3, but not with histone deacetylase 1, in this region (Fig. 6C). We could not detect significant differences in the binding of AP2 in this locus between the HHUA-cycA and HHUA-vec cells (Fig. 6C).
As cyclin A2 and CDK are reported to induce the phosphorylation of Sp1 [14, 28–30], we investigated the effect of cyclin A2 on the phosphorylation of Sp1. The overall expression of Sp1 protein did not differ between HHUA-cycA and control cells; however, the HHUA-cycA cells showed a 4-fold increase in the phosphorylated (105-kD form) Sp1 (Fig. 6D). Because Sp1 has a site for serine phosphorylation by cyclin A2–CDK complex [29], we further evaluated the serine phosphorylation of Sp1 in HHUA-cycA using immunoprecipitation with anti-Sp1 antibody. Sp1 protein in the HHUA-cycA cells was more phosphorylated than that from control cells, which was confirmed by the treatment with calf alkaline phosphatase (Fig. 6E).
Discussion
In the present study, we demonstrated that (i) endometrial carcinoma tissues refractory to chemotherapy had higher levels of cyclin A2 than other tissue types, (ii) the increased expression of endogenous cyclin A2 protein was correlated with resistance to cisplatin in endometrial carcinoma cell lines and (iii) forced expression of cyclin A2 in endometrial carcinoma HHUA cells increased not only growth activity but also cisplatin resistance in vitro and in vivo. These results clearly indicated that the expression of cyclin A2 conferred the resistance to cisplatin on endometrial carcinoma cells. Cyclins are known to be expressed in the respective phase of the cell cycle, and cells expressing cyclin D, E or A are generally considered to be in the G1/S phase. In addition, platinum agents are thought to target cells in the G1/S phase [31]. Therefore, it seems unreasonable for cyclin A to provide resistance rather than sensitivity to platinum. In this context, cyclin E, another partner of CDK2, reportedly enhanced proliferation and sensitized cancer cells to cisplatin [32]. To our knowledge, this is the first report to elucidate that cyclin A2 confers cisplatin resistance to human carcinoma cells.
The present study also revealed that cyclin A2-induced cisplatin resistance is caused by the suppression of drug-induced (i.e. cisplatin-induced) apoptosis. In addition, we elucidated that the cyclin A2-mediated reduction in apoptosis was because of activation of the PI3K/Akt pathway. In endometrial carcinoma, PI3K is of particular interest because PTEN mutations are very common in endometrial carcinogenesis [12], and a study revealed that p-Akt levels were elevated in cisplatin-resistant endometrial cancer cell lines harbouring PTEN gene mutations [33]. Akt’s activation reportedly enhanced cisplatin resistance via inactivation of the pro-apoptotic Bad protein at serine 136 in an ovarian carcinoma cell line [34]. Consistent with this report, phosphorylation of Akt after cisplatin treatment in HHUA-cycA cells was involved in the phosphorylation of Bad at serine 136. Although a report showed that the MAPK cascade, another constituent of the mitochondrial pathway, was also involved in cisplatin resistance in ovarian carcinoma [34], we could not detect the cyclin A2-induced phosphorylation of Erk1/2 and Bad at serine 112.
Periplakin is a member of the plakin family and the autoantigen of neoplastic pemphigus [27]. Periplakin is known to bind to intermediate filaments and Akt [21], although its precise effects on the PI3K cascade remain undetermined. In the present study, we demonstrated a direct association of periplakin with Akt in HHUA cells. In addition, silencing of periplakin using siRNA significantly reduced basal p-Akt expression, suggesting that the binding of periplakin positively regulated the Akt activity. Although the reason why periplakin reinforced the Akt cascade is not fully understood, periplakin may act as a kind of intracellular scaffold protein for the stable expression and activation of Akt, because the present study indicated the direct binding of Akt to periplakin and a previous report indicated the association of periplakin with cytoskeleton proteins including intermediate filaments.
Regarding the mechanisms of cyclin A2-induced up-regulation of periplakin, the luciferase assay in the present study using reporters containing the periplakin promoter indicated cyclin A2 to be involved in the transcriptional up-regulation of periplakin via the Sp1-binding site. In addition, another transcription factor, AP2, located close to the Sp1-binding site did not have a significant role in the cyclin A2-induced up-regulation. The present study also demonstrated that the serine residue of Sp1 was phosphorylated in association with increased recruitment of Sp1 to the periplakin promoter as shown using the ChIP assay. Collectively, these results demonstrated that cyclin A2-activated Sp1 and its binding to the Sp1-binding sequence had a pivotal role in the cyclin A2-induced expression of periplakin. A previous study demonstrated that cyclin A2 directly bound to Sp1 and phosphorylated it at serine 59, resulting in enhanced DNA-binding capacity [29]. Haidweger et al.[30] also demonstrated that the binding of cyclin A to Sp1 in U2OS cells enhanced Sp1-responsive promoter activity. In addition, Sp1 activated by cyclin A actually accelerated the transcription of the metalloproteinase-2 [14] and cytidylyltransferase-α genes [28]. We also demonstrated that the enhanced recruitment of Sp1 to the specific promoter was associated with increased recruitment of CBP/p300. The CBP/p300 coactivator protein, which enhances transcription through histone acetyltransferase activity, was reportedly associated with Sp1 and promoted transcription of various genes such as the p21 [35] and tumour necrosis factor-α[36] genes. Our result was consistent with these reports that Sp1 promotes transcription via chromatin remodelling with CBP/p300.
Cyclin–CDK complexes have been considered promising therapeutic targets, and preclinical or clinical trials of small molecular CDK inhibitors have been ongoing [37]. However, no CDK inhibitor has been approved for commercial use mainly because of an insufficient anti-tumour effect. In this regard, Tetsu et al.[38] attributed the results to mutual compensatory mechanisms among CDKs. Our clinical data regarding the increased expression of cyclin A2 in patients with advanced or chemotherapy-refractory stage disease, which is associated with a poor prognosis, suggest cyclin A2 to be a new therapeutic target in endometrial carcinoma. Interestingly, cyclin A2 is the only cyclin that binds CDKs involved in both G1/S and G2/M of the cell cycle (CDK2 and CDK1, respectively); thus, cyclin A2 expression might lead to decreases in both CDK1 and CDK2 activities, which subsequently overcome the compensatory mechanisms of CDKs. Although the cyclin A2 inhibitor has not yet developed, an application of micro-RNA may be a potential way to suppress the expression of cyclin A2.
In summary, the present study is the first to identify an anti-apoptotic function of cyclin A2 with increased activation of the PI3K pathway. In addition, we found that periplakin was the molecular target of cyclin A2 and an enhancer of the PI3K cascade. Further investigation is warranted in order to clarify the role of cyclin A2 in cancer cells and to develop gene therapy that targets cyclin A2.
Acknowledgments
This work was supported in part by Grant-in-Aid for Scientific Research from the Ministry of Education, Science and Culture (No. 06454468 and No. 07807154), Japan.
References
- 1.Girard F, Strausfeld U, Fernandez A, et al. Cyclin A is required for the onset of DNA replication in mammalian fibroblasts. Cell. 1991;67:1169–79. doi: 10.1016/0092-8674(91)90293-8. [DOI] [PubMed] [Google Scholar]
- 2.Dobashi Y, Shoji M, Jiang SX, et al. Active cyclin A-CDK2 complex, a possible critical factor for cell proliferation in human primary lung carcinomas. Am J Pathol. 1998;153:963–72. doi: 10.1016/S0002-9440(10)65638-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chao Y, Shih YL, Chiu JH, et al. Overexpression of cyclin A but not Skp 2 correlates with the tumor relapse of human hepatocellular carcinoma. Cancer Res. 1998;58:985–90. [PubMed] [Google Scholar]
- 4.Mrena J, Wiksten JP, Kokkola A, et al. Prognostic significance of cyclin A in gastric cancer. Int J Cancer. 2006;119:1897–901. doi: 10.1002/ijc.21944. [DOI] [PubMed] [Google Scholar]
- 5.Shih HC, Shiozawa T, Kato K, et al. Immunohistochemical expression of cyclins, cyclin-dependent kinases, tumor-suppressor gene products, Ki-67, and sex steroid receptors in endometrial carcinoma: positive staining for cyclin A as a poor prognostic indicator. Hum Pathol. 2003;34:471–8. doi: 10.1016/s0046-8177(03)00124-2. [DOI] [PubMed] [Google Scholar]
- 6.Muller-Tidow C, Ji P, Diederichs S, et al. The cyclin A1-CDK2 complex regulates DNA double-strand break repair. Mol Cell Biol. 2004;24:8917–28. doi: 10.1128/MCB.24.20.8917-8928.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsieh JK, Yap D, O’Connor DJ, et al. Novel function of the cyclin A binding site of E2F in regulating p53-induced apoptosis in response to DNA damage. Mol Cell Biol. 2002;22:78–93. doi: 10.1128/MCB.22.1.78-93.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Susumu N, Sagae S, Udagawa Y, et al. Randomized phase III trial of pelvic radiotherapy versus cisplatin-based combined chemotherapy in patients with intermediate- and high-risk endometrial cancer: a Japanese Gynecologic Oncology Group study. Gynecol Oncol. 2008;108:226–33. doi: 10.1016/j.ygyno.2007.09.029. [DOI] [PubMed] [Google Scholar]
- 9.Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265–79. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 10.Yang X, Fraser M, Moll UM, et al. Akt-mediated cisplatin resistance in ovarian cancer: modulation of p53 action on caspase-dependent mitochondrial death pathway. Cancer Res. 2006;66:3126–36. doi: 10.1158/0008-5472.CAN-05-0425. [DOI] [PubMed] [Google Scholar]
- 11.Clark AS, West K, Streicher S, et al. Constitutive and inducible Akt activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol Cancer Ther. 2002;1:707–17. [PubMed] [Google Scholar]
- 12.Tashiro H, Blazes MS, Wu R, et al. Mutations in PTEN are frequent in endometrial carcinoma but rare in other common gynecological malignancies. Cancer Res. 1997;57:3935–40. [PubMed] [Google Scholar]
- 13.Shiozawa T, Miyamoto T, Kashima H, et al. Estrogen-induced proliferation of normal endometrial glandular cells is initiated by transcriptional activation of cyclin D1 via binding of c-Jun to an AP-1 sequence. Oncogene. 2004;23:8603–10. doi: 10.1038/sj.onc.1207849. [DOI] [PubMed] [Google Scholar]
- 14.Wang CH, Chang HC, Hung WC. p16 inhibits matrix metalloproteinase-2 expression via suppression of Sp1-mediated gene transcription. J Cell Physiol. 2006;208:246–52. doi: 10.1002/jcp.20660. [DOI] [PubMed] [Google Scholar]
- 15.Casalino L, Bakiri L, Talotta F, et al. Fra-1 promotes growth and survival in RAS-transformed thyroid cells by controlling cyclin A transcription. EMBO J. 2007;26:1878–90. doi: 10.1038/sj.emboj.7601617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsai SJ, Wu MH, Chen HM, et al. Fibroblast growth factor-9 is an endometrial stromal growth factor. Endocrinology. 2002;143:2715–21. doi: 10.1210/endo.143.7.8900. [DOI] [PubMed] [Google Scholar]
- 17.Chau BN, Cheng EH, Kerr DA, et al. Aven, a novel inhibitor of caspase activation, binds Bcl-xL and Apaf-1. Mol Cell. 2000;6:31–40. [PubMed] [Google Scholar]
- 18.Kleivi K, Lind GE, Diep CB, et al. Gene expression profiles of primary colorectal carcinomas, liver metastases, and carcinomatoses. Mol Cancer. 2007;6:2. doi: 10.1186/1476-4598-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chano T, Ikegawa S, Kontani K, et al. Identification of RB1CC1, a novel human gene that can induce RB1 in various human cells. Oncogene. 2002;21:1295–98. doi: 10.1038/sj.onc.1205178. [DOI] [PubMed] [Google Scholar]
- 20.Memarzadeh S, Kozak KR, Chang L, et al. Urokinase plasminogen activator receptor: prognostic biomarker for endometrial cancer. Proc Natl Acad Sci U S A. 2002;99:10647–52. doi: 10.1073/pnas.152127499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Den Heuvel AP, De Vries-Smits AM, Van Weeren PC, et al. Binding of protein kinase B to the plakin family member periplakin. J Cell Sci. 2002;115:3957–66. doi: 10.1242/jcs.00069. [DOI] [PubMed] [Google Scholar]
- 22.Schug TT, Berry DC, Shaw NS, et al. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell. 2007;129:723–33. doi: 10.1016/j.cell.2007.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ahn MY, Katsanakis KD, Bheda F, et al. Primary and essential role of the adaptor protein APS for recruitment of both c-Cbl and its associated protein CAP in insulin signaling. J Biol Chem. 2004;279:21526–32. doi: 10.1074/jbc.M307740200. [DOI] [PubMed] [Google Scholar]
- 24.Jain A, Tindell CA, Laux I, et al. Epithelial membrane protein-1 is a biomarker of gefitinib resistance. Proc Natl Acad Sci U S A. 2005;102:11858–63. doi: 10.1073/pnas.0502113102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yeung HY, Lai KP, Chan HY, et al. Hypoxia-inducible factor-1-mediated activation of stanniocalcin-1 in human cancer cells. Endocrinology. 2005;146:4951–60. doi: 10.1210/en.2005-0365. [DOI] [PubMed] [Google Scholar]
- 26.Christakos S, Liu Y. Biological actions and mechanism of action of calbindin in the process of apoptosis. J Steroid Biochem Mol Biol. 2004;89–90:401–4. doi: 10.1016/j.jsbmb.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 27.Aho S, Rothenberger K, Tan EM, et al. Human periplakin: genomic organization in a clonally unstable region of chromosome 16p with an abundance of repetitive sequence elements. Genomics. 1999;56:160–8. doi: 10.1006/geno.1998.5704. [DOI] [PubMed] [Google Scholar]
- 28.Banchio C, Schang LM, Vance DE. Phosphorylation of Sp1 by cyclin-dependent kinase 2 modulates the role of Sp1 in CTP: phosphocholine cytidylyltransferase alpha regulation during the S phase of the cell cycle. J Biol Chem. 2004;279:40220–6. doi: 10.1074/jbc.M406468200. [DOI] [PubMed] [Google Scholar]
- 29.Fojas de Borja P, Collins NK, Du P, et al. Cyclin A-CDK phosphorylates Sp1 and enhances Sp1-mediated transcription. EMBO J. 2001;20:5737–47. doi: 10.1093/emboj/20.20.5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haidweger E, Novy M, Rotheneder H. Modulation of Sp1 activity by a cyclin A/CDK complex. J Mol Biol. 2001;306:201–12. doi: 10.1006/jmbi.2000.4406. [DOI] [PubMed] [Google Scholar]
- 31.Donaldson KL, Goolsby GL, Wahl AF. Cytotoxicity of the anticancer agents cisplatin and taxol during cell proliferation and the cell cycle. Int J Cancer. 1994;57:847–55. doi: 10.1002/ijc.2910570614. [DOI] [PubMed] [Google Scholar]
- 32.Bedrosian I, Lee C, Tucker SL, et al. Cyclin E-associated kinase activity predicts response to platinum-based chemotherapy. Clin Cancer Res. 2007;13:4800–6. doi: 10.1158/1078-0432.CCR-07-0142. [DOI] [PubMed] [Google Scholar]
- 33.Gagnon V, Mathieu I, Sexton E, et al. AKT involvement in cisplatin chemoresistance of human uterine cancer cells. Gynecol Oncol. 2004;94:785–95. doi: 10.1016/j.ygyno.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 34.Hayakawa J, Ohmichi M, Kurachi H, et al. Inhibition of BAD phosphorylation either at serine 112 via extracellular signal-regulated protein kinase cascade or at serine 136 via Akt cascade sensitizes human ovarian cancer cells to cisplatin. Cancer Res. 2000;60:5988–94. [PubMed] [Google Scholar]
- 35.Owen GI, Richer JK, Tung L, et al. Progesterone regulates transcription of the p21(WAF1) cyclin-dependent kinase inhibitor gene through Sp1 and CBP/p300. J Biol Chem. 1998;273:10696–701. doi: 10.1074/jbc.273.17.10696. [DOI] [PubMed] [Google Scholar]
- 36.Barthel R, Tsytsykova AV, Barczak AK, et al. Regulation of tumor necrosis factor alpha gene expression by mycobacteria involves the assembly of a unique enhanceosome dependent on the coactivator proteins CBP/p300. Mol Cell Biol. 2003;23:526–33. doi: 10.1128/MCB.23.2.526-533.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol. 2006;24:1770–83. doi: 10.1200/JCO.2005.03.7689. [DOI] [PubMed] [Google Scholar]
- 38.Tetsu O, McCormick F. Proliferation of cancer cells despite CDK2 inhibition. Cancer Cell. 2003;3:233–45. doi: 10.1016/s1535-6108(03)00053-9. [DOI] [PubMed] [Google Scholar]