

Abstract
We describe a simple but sensitive fluorescence method to accurately detect the esterification activity of lecithin:cholesterol acyltransferase (LCAT). The new assay protocol employs a convenient mix, incubate and measure scheme. This is possible by using the fluorescent sterol, dehydroergosterol (DHE) in place of cholesterol as the LCAT substrate. The assay method is further enhanced by incorporation of an amphiphilic peptide in place of apolipoprotein A-I as the lipid emulsifier and LCAT activator. Specific fluorescence detection of DHE ester synthesis is achieved by employing cholesterol oxidase to selectively render unesterified DHE non-fluorescent. The assay accurately detects LCAT activity in buffer and in plasma that is depleted of apolipoprotein B lipoproteins by selective precipitation. Analysis of LCAT activity in plasmas from control subjects and sickle cell disease (SCD) patients confirms previous reports of reduced LCAT activity in SCD and demonstrates a strong correlation between plasma LCAT activity and LCAT content. The fluorescent assay combines the sensitivity of radiochemical assays with the simplicity of non-radiochemical assays to obtain accurate and robust measurement of LCAT esterification activity.
Keywords: dehydroergosterol, dehydroergosteryl ester, cholesterol oxidase, apolipoprotein A-I mimetic, amphipathic peptide, sickle cell disease
The plasma enzyme lecithin:cholesterol acyltransferase (EC 2.3.1.43) is essential for the efficient transit of cholesterol through the plasma compartment. The enzyme facilitates the process of reverse-cholesterol transport by potentiating the migration of excess cholesterol from tissues throughout the body towards the liver hepatocytes. The hepatocytes guide excess cholesterol and cholesterol-derived bile acids to the bile ducts for elimination [1]. The enzyme circulates in plasma, predominantly in association with high-density lipoproteins (HDL) where its principal mechanism of action is the transacylation of a fatty acid from phosphatidylcholine within HDL to cholesterol within the same HDL to form cholesteryl ester (CE) [1]. The CE product accumulates in the HDL interior until it is cleared by hepatic lipoprotein receptors, either directly through selective CE uptake from HDL particles captured by HDL-specific receptors or by an indirect route comprised of CE transfer to the apolipoprotein B lipoproteins via cholesteryl ester transfer protein followed by clearance of the recipient lipoproteins through the hepatic apolipoprotein B/E-receptors [2]. Intracellular lipases subsequently de-esterify the CE to liberate cholesterol for further processing.
The benefits of LCAT for sustaining health are evident in situations where reduced LCAT levels and activity are linked to diseases. This is most apparent in persons with deleterious mutations in both alleles of the LCAT gene resulting in fish eye disease when partial LCAT activity remains and familial LCAT deficiency when LCAT activity is essentially absent. Persons with fish eye disease have low levels of HDL cholesterol and develop lipid-rich, corneal opacities. Those with familial LCAT deficiency are hypocholesterolemic with very low HDL cholesterol levels, exhibit corneal opacities and, in addition, develop anemia and kidney disease typified by fatty deposits in the glomeruli [3]. Persons with normal LCAT alleles are also reported to experience reductions in LCAT activity in conjunction with certain diseases including coronary artery disease [4], diabetes [5], kidney disease [6;7]; rheumatoid arthritis [8] and anemia [9;10]. The impact of reduced LCAT activity on the disease processes in otherwise normal persons remains to be determined.
Acquiring evidence of the involvement of LCAT in disease, disease screening, monitoring treatments, development of recombinant forms of LCAT for research and therapy, and the study of LCAT enzymology in general is dependent on the availability of manageable, accurate and reproducible assays of LCAT enzymatic activity. The most commonly-used assays employ substrates composed of radioisotope-labeled cholesterol carried in HDL-like complexes formed from combinations of apolipoprotein A-I (ApoA-I), cholesterol and phosphatidylcholine [11;12]. In another variation, substrate is provided by endogenous plasma lipoproteins pre-equilibrated with radioisotope-labeled cholesterol [13;14]. The radiochemical-based assays have provided the means to obtain the majority of knowledge of LCAT mechanisms and function. They enable sensitive detection of the action of LCAT on authentic substrates.
Non-radioactive assay schemes have also been developed. One type relies on the direct determination of the decrease in unesterified cholesterol in endogenous lipoproteins as a result of LCAT activity during incubation of whole plasma [15;16]. A variation of this assay scheme supplements plasma with cholesterol in phosphatidylcholine vesicles [17]. The cholesterol content of plasma before and after incubation is determined with common colorimetric assay methods. Although quite simple to perform, the colorimetric method is less sensitive than the radiochemical assays and prone to greater variation due to its dependence on measurements of small decrements in unesterified cholesterol within a large pool of plasma cholesterol. Gas chromatography has been used in place of colorimetry to obtain greater sensitivity but it is more procedurally complicated [18;19].
Fluorescence-based assays have also been described. One type employs a water soluble, fluorescently-labeled phosphatidylcholine derivative which serves to detect the phospholipase activity of LCAT [20]. The assay is uncomplicated and the fluorescence-based analysis provides much greater sensitivity than the colorimetric assays. However, the enzymatic activity assayed is not esterification and the lipolysis data do not fit standard Michaelis-Menten enzyme kinetics, which makes data interpretation and comparisons challenging.
We present a new kind of LCAT assay to detect and quantitate sterol esterification. The assay possesses the sensitivity of radiochemical and fluorescence-based assays combined with the simplicity of protocols requiring only aliquot mixing, incubation and measurement. This is possible by employing dehydroergosterol (DHE), a naturally occurring fluorescent sterol, as the sterol substrate for LCAT and by using cholesterol oxidase (COx) to enable exclusive detection of fluorescence by the DHE ester product [21]. A further advantage is obtained in terms of substrate preparation and cost by substituting a synthetic, LCAT-activating peptide for ApoA-I. The peptide, which has previously been studied under various identifications for its anti-atherosclerotic properties [22;23], forms stable lipid complexes that are reported to react with LCAT at least as well as reconstituted HDL preparations formed from ApoA-I, PC and cholesterol [24].
MATERIALS AND METHODS
Materials and reagents
Dehydroergosterol (ergosta-5,7,9,(11),22-tetraen-3β-ol), cholesterol oxidase (Streptomyces sp) and bovine serum albumin (fatty acid free) were from Sigma-Aldrich (St. Louis, MO). 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) was obtained from Avanti Polar Lipids (Alabaster, AL). Human HDL came from Athens Research & Technology (Athens, GA). LCAT activating peptide (LAP642) was obtained from GenScript (Piscataway, NJ) as a custom synthesis of L-amino acids in the sequence PVLDLFRELLNELLEALKQKLK. Other peptides were obtained from the same source. The ELISA kits for quantitating LCAT mass in plasma were from ALPCO (Salem, NH). Cholesterol assay kits were from Wako Diagnostics (Richmond, VA). Recombinant human LCAT (rhLCAT) was purified from the medium of a HEK293F cell clone that was stably transfected with the human LCAT gene as described [25]. The purified rhLCAT was stored frozen at −80° C at 1 to 2 mg/ml protein concentration in phosphate buffered saline with 10% glycerol. Under such conditions, the rhLCAT retained its original activity for at least 6 months. The rhLCAT was thawed in wet ice for use and was stable to repeated freeze-thaw cycles.
Preparation of HDL-mimetic substrate
A concentrated solution of HDL-mimetic substrate (HMS), designated 10xHMS, was prepared by combining a solution of 5 mM LAP642 in methanol with a chloroform solution containing 45mM POPC and 5mM DHE to obtain a final mole ratio composition of 2:9:1 for LAP642:POPC:DHE. Solvents were removed by evaporation under a stream of nitrogen gas followed by high vacuum. The dried mixture was dispersed in buffer (150 mM NaCl, 20 mM sodium phosphate, 1mM EDTA, pH 7.4) to obtain a final DHE concentration of 1 mM. The solution was sealed under N2 and dispersed by brief vortexing and sonication (≤ 1minute) in a water bath sonicator to obtain a clear solution. The solution was protected from light and stored under N2 at 4° C.
LCAT activity assay
Solutions of rhLCAT for assay were prepared by dilution of an aliquot of the 1 to 2 mg/ml stock solution into buffer containing at least 60 μM albumin to prevent loss of rhLCAT to the walls of the polypropylene vials that otherwise occurred. Assay incubations were performed in triplicate in 96-well black polystyrene plates containing between 2 μL to 5 μL of rhLCAT solution and 100 μL of HMS, prepared by dilution of 1 volume of 10xHMS in 9 volumes of buffer containing 5 mM β-mercaptoethanol and 60 μM albumin, unless noted otherwise. Wells for background fluorescence determination contained HMS with an aliquot of buffer in place of rhLCAT solution. Wells to determine instrument response contained a range of HMS concentrations in 100 μL. Plates were on ice during additions. Plates were incubated at 37° C for 30 or 60 minutes. Reactions were stopped by returning the plates to ice and adding 25 μL of stop solution composed of 5 units/ml COx in buffer containing 7% Triton X100, which blocks LCAT activity. Plates were incubated at 37° C for a further 30 to 60 minutes to complete oxidation of unesterified DHE. Plates were equilibrated to ambient temperature and fluorescence yield, which is temperature sensitive, was detected in a SpectraMax Gemini fluorescence plate reader (Molecular Devices, LLC, Sunnyvale CA) set at 325 nm excitation and 425 nm emission with a 420 nm cutoff filter. Due to the low signal/noise performance of the plate reader, emission could not be adequately measured in the preferred wavelength range of 375 nm to 390 nm, which spans the maximum emission of DHE in a lipid matrix [26]. The nmol of DHE ester formed in each well was calculated by subtracting the background fluorescence from the fluorescence detected in the well after COx treatment. The resulting net fluorescence was divided by the slope (fluorescence/nmol) obtained from a linear regression fit of a plot of nmol DHE versus the fluorescence of a series of serially diluted samples of HMS in buffer to which 7% Triton X100 was added without COx.
Assay of LCAT activity and content in plasma
Blood was drawn in EDTA tubes from volunteers (control) and from patients with sickle cell disease (SCD) in steady-state, which is defined as normal baseline status without acute pain. The SCD patients were at least 18 years of age and identified to be homozygous for the SS hemoglobin mutation. Blood specimens were obtained under NIH Clinical Center protocol 03-H-0015, which is registered at web site http://clinicalstudies.info.nih.gov. All subjects signed informed consent documents in accordance with the Declaration of Helsinki. Plasma was prepared from freshly-drawn blood by centrifugation and stored at −80° C until use. The plasma content of LCAT protein was determined by ELISA. The plasma samples were prepared for the LCAT activity assay by first removing the apolipoprotein B-containing lipoproteins through selective precipitation with phosphotungstic acid and magnesium chloride, as previously described [14]. The LCAT activity was determined by combining 2 μL of apolipoprotein B-depleted plasma with 100 μL HMS and incubating for 2 hours. The remainder of the assay followed the standard protocol described above.
Detection of cholesterol and DHE esterification by HPLC
A 3 ml solution of HMS prepared as described above and diluted to 60 μM DHE plus a second similar preparation of HMS in which cholesterol was substituted for DHE, were incubated with 0.8 μg/ml rhLCAT at 37° C. At various times during the incubation, duplicate 0.2 ml aliquots were transferred from each to separate glass tubes containing 0.2 ml buffer and a 2 ml solvent mixture composed of ethyl acetate/acetone/methanol (70/30/0.5 V/V). Each solvent volume also contained 2 μg 1,2 di-O-hexadecylglycerol (Sigma-Aldrich) as a recovery standard. Lipids were extracted by brief shaking of capped tubes, centrifugation to resolve phases, transfer of the upper solvent phase to clean tubes, and removal of solvent by evaporation under a nitrogen stream followed by high vacuum. Dried extracts were dissolved in 0.2 ml trimethylpentane/methanol/tetrahydrofuran (95/5/3) for determination of sterol and steryl ester content by HPLC with an Agilent 1100 HPLC system (Santa Clara CA) equipped with a SEDEX 75 evaporative light-scattering detector (Shimadzu Scientific Instruments, Columbia, MD) [27].
Statistical analysis and curve fitting
Student’s two-tailed t-test for unpaired data was used to calculate p-values. Significant differences were accepted when the p-value was less than 0.05. Linear and nonlinear curve fits were performed with SigmaPlot 12 (Systat Software, San Jose, CA).
RESULTS AND DISCUSSION
Substrate design
The rate of sterol esterification by LCAT is highly dependent on the form in which the sterol and phosphatidylcholine substrates are presented to the enzyme. These hydrophobic substrates will form phospholipid-bilayer vesicles in the aqueous phase with cholesterol intercalated throughout the bilayer, but LCAT reacts relatively slowly with such vesicular substrates. The reactivity of the substrate preparation is greatly enhanced by the inclusion of ApoA-I, the natural activator of LCAT [11;28].
We used LAP642, an ApoA-I mimetic peptide, in place of ApoA-I in the assay. The peptide forms stable complexes with phosphatidylcholine and sterol that react suitably well with LCAT [24]. Several advantages are obtained by employing the peptide. First, the preparation of substrate complexes is greatly simplified, compared to the detergent dialysis or sonication procedures required to complex ApoA-I with lipids [12]. Second, synthetic peptides are more readily available than ApoA-I in terms of preparation or cost, at current rates.
The assay takes advantage of the fact that the sterol selectivity of LCAT is quite broad (Fig. 1). Sterols of a diverse range of structures and composition are accepted as substrates for esterification by LCAT [29;30]. One critical structural requirement is that the 3-hydroxyl group is in the β conformation [29]. Dehydroergosterol was selected for the assay because of its fluorescence properties with the expectation that as a 3β-hydroxy sterol it should be a suitable substrate. Cholestatrienol (cholesta-5,7,9(11)-trien-3β-ol) could also be a candidate for these reasons but, unlike DHE, it is not readily available from commercial sources [31].
Figure 1.
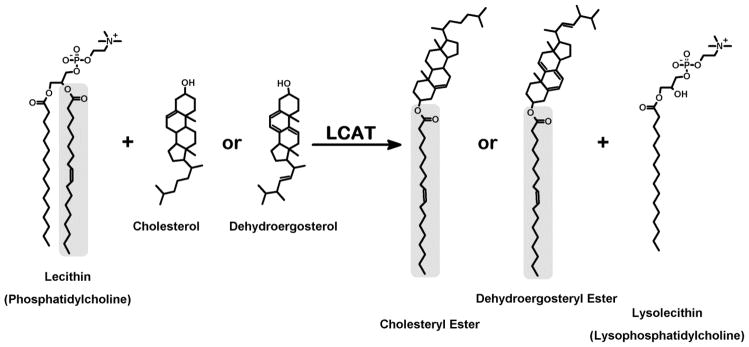
Substrates and products of LCAT. The principal reaction catalyzed by LCAT is transfer of an acyl group from the sn-2 position of phosphatidylcholine to the 3β-hydroxyl group of cholesterol. The broad sterol specificity of LCAT allows dehydroergosterol to be substituted for cholesterol in the assay and enable detection of dehydroergosteryl ester by fluorescence of its conjugated triene fluorophore.
To verify that DHE is a sufficient LCAT substrate and to learn how DHE compares to cholesterol as a substrate in the HMS formulation, sterol esterification by rhLCAT was compared in HMS containing DHE with HMS containing cholesterol. The formation of steryl ester product and depletion of sterol substrate was monitored by HPLC with mass detection (Fig. 2). The results confirm DHE is esterified by LCAT, although at a slower rate than esterification of cholesterol. The specific activity of rhLCAT was 13 nmol/μg/hr for DHE ester synthesis and 52 nmol/μg/hr for CE synthesis. Specific activities derived from the sterol depletion results were essentially the same. The results verify that HMS prepared with DHE is satisfactory for the detection of LCAT esterification activity.
Figure 2.

Substrate specificity of rhLCAT for dehydroergosterol versus cholesterol in HDL-mimetic substrate. Separate HDL-mimetic substrates, each composed of LAP642, 1-palmitoyl-2-oleoyl-sn-glycerophosphocholine and sterol in mole portions of 2:9:1, respectively, were prepared with dehydroergosterol (A) and with cholesterol (B) as the sterol component. The substrates were reacted with 0.8 μg/ml rhLCAT at a sterol concentration of 59 μM. Periodically, aliquots were removed from the respective reaction volumes and extracted with solvents to isolate the sterols and steryl esters for determination of mass by HPLC with evaporative light-scattering detection (Materials and Methods). The data for steryl ester synthesis (●) were fit by nonlinear regression to the equation for a mono-exponential rise to a maximum (solid line). The data for sterol depletion (▲) were fit by nonlinear regression to the equation for a mono-exponential decay (dashed line).
The specific activity obtained with cholesterol substrate in this study is several-fold higher than those reported for reactions of purified plasma LCAT with reconstituted HDL substrate, where specific activities ranged from 4 to 13 nmol/μg/hr [11;32;33]. The large difference between this and previous studies may be due to substrate types, substrate composition, LCAT preparation or any number of other reasons that will require further investigation to resolve.
Assay design and characteristics
A critical feature of the fluorescence assay is the ability to exclusively detect fluorescence of esterified DHE without interference from the excess, unesterified DHE that remains when the LCAT reaction is stopped. This is accomplished by rendering unesterified DHE non-fluorescent by selective oxidation with COx. Dehydroergosterol is known to be a substrate for COx [34].
How this feature is used to detect LCAT activity is shown in Fig. 3. Addition of COx to a solution of HMS causes a time-dependent loss of fluorescence due to the oxidation of DHE (Fig. 3a). In HMS samples incubated with rhLCAT before COx addition, the terminal fluorescence following COx treatment is higher due to the prior synthesis of DHE ester, which is not a COx substrate and therefore remains fluorescent. The magnitude of additional terminal fluorescence is directly proportional to the amount of DHE ester in the sample.
Figure 3.

Unmasking of dehydroergosteryl ester fluorescence by oxidation of excess dehydroergosterol substrate with cholesterol oxidase. For each curve, separate 100 μL volumes of HDL-mimetic substrate containing 100 μM dehydroergosterol in the assay were combined with rhLCAT to final concentrations of 0 μg/ml (A), 1 μg/ml (B), 2.4 μg/ml (C) or 4.5 μg/ml (D) and incubated for 1 hour at 37° C (Materials and Methods). Next, at 0 minutes in the figure, 25 μL of stop solution containing 5 units cholesterol oxidase per ml of 7% Triton X100 in buffer was added to each sample. The fluorescence at 325 nm excitation and 425 nm emission was monitored over time at 37° C. The higher terminal fluorescence at 20 min in (B), (C) and (D) relative to (A) is due to dehydroergosteryl ester fluorescence.
Hydrogen peroxide is a product of the COx reaction and has the potential to promote oxidation of the DHE ester, which could cause nonspecific loss of fluorescence. We, however, found no evidence of this in experiments showing no gain in DHE ester fluorescence when catalase was included during the COx incubation step to scavenge peroxide (results not shown).
Application of this methodology enables determination of the time-dependent formation of DHE ester as a function of LCAT concentration (Fig. 4A). These data provide the information needed to calculate specific activity for LCAT. The data for this calculation are obtained from the pseudo-linear region of the reaction curve where less than 10% of the DHE is esterified. The specific activity derived from those data was 16 nmol/μg/hr (Fig. 4B).
Figure 4.

Time-course of dehydroergosteryl ester synthesis as a function of rhLCAT concentration. (A) Volumes of HDL-mimetic substrate of the composition described in Fig. 2 were incubated with rhLCAT at concentrations of 0.1 μg/ml (●), 0.25 μg/ml (○), 0.5 μg/ml ((▲) and 1 μg/ml (△). At the times indicated, two 100 μL aliquots were removed from each sample for fluorescence quantitation of dehydroergosteryl ester (Materials and Methods). (B) The data for amount of dehydroergosteryl ester formed at 30 minutes incubation (circumscribed by the ellipse (A)) are plotted as a function of rhLCAT concentration. The line indicates the linear regression fit. The slope, which corresponds to the specific activity of rhLCAT, is 16 nmol/μg/hr.
We examined the substrate saturation kinetics of the assay and found they are well fit by the Michaelis-Menten kinetic equation (Fig. 5). The results yielded an apparent Km of 6 μM in terms of LAP642 concentration and 3 μM and 27 μM in terms of DHE and phosphatidylcholine concentration, respectively. The saturation data confirm that using 60 μM or greater DHE (i.e. ≥ 120 μM LAP642) in the assay is sufficiently greater than the Km and therefore avoids the rate limiting effects of insufficient substrate content.
Figure 5.

Substrate saturation curve for HDL-mimetic substrate with rhLCAT. The composition of the HDL-mimetic substrate containing dehydroergosterol is presented in Fig. 2. The rhLCAT concentration was 0.5 μg/ml and the reaction time was 30 minutes. The initial reaction rates (Vo) are plotted as a function of the concentration of LCAT-activating peptide (LAP642). The data were fit by nonlinear regression to the Michaelis–Menten kinetic equation (solid line). The apparent Km (appKm) derived from the fit was 6 μM for LAP642. Similarly, the appKm was 3 μM and 27 μM based on DHE and POPC concentration, respectively. The Vmax was 5 μM/hr.
To apply the assay to the detection of LCAT activity in plasma samples, the apolipoprotein B lipoproteins should be removed from the plasma prior to assay. This is done to lower the amount of cholesterol that could otherwise compete with DHE. An example of the benefit of precipitation is shown in Fig. 6A where the slope of activity versus rhLCAT concentration (4 nmol/μg/hr) in whole plasma is less than in precipitated plasma or in buffer (5 nmol/μg/hr). In contrast, the slope of activity versus rhLCAT concentration in precipitated plasma is identical to that of rhLCAT in buffer, which suggests interference by lipoproteins or other co-precipitating plasma components is eliminated. The identical slopes also indicate the precipitation procedure does not cause loss of LCAT and the precipitation reagents do not interfere with activity. The positive offset in activity values for precipitated plasma compared to the results in buffer is due to the activity of endogenous plasma LCAT. A specific activity of 7 nmol/μg/hr for plasma LCAT is derived from the plasma activity detected in the absence of rhLCAT and the plasma LCAT content determined by ELISA.
Figure 6.

The effect of plasma and HDL on the reaction of HDL-mimetic substrate with endogenous plasma LCAT and rhLCAT. (A) Removal of apolipoprotein B lipoproteins from plasma prior to assay reduces plasma interference with the rate of dehydroergosterol esterification. Two sets of human plasma samples were prepared in which sample volumes contained 95% human plasma and 5% rhLCAT solution (0.1% bovine serum albumin in buffer) to obtain final rhLCAT concentrations in plasma of 0, 5, 10 and 20 μg/ml. The endogenous LCAT content of the human plasma, determined by ELISA, was 10 μg/ml. The apolipoprotein B-containing lipoproteins were selectively precipitated (Materials and Methods) from one set of plasma samples. Two microliters from untreated (□) and apolipoprotein B-deficient plasma (■) were incubated in triplicate with 100 μL of HDL-mimetic substrate (100 μM dehydroergosterol) for 2 hours (Materials and Methods). For comparison, 2 μL aliquots of the same rhLCAT solutions were diluted into buffer and assayed (●). The error bars indicate standard deviation and are shown where standard deviation is greater than the symbol size. Note that the volume used to obtain the X and Y data is the aliquot size of the test sample (i.e. 2 μL). The lines represent linear regression fits of the data. (B) The reactivity of HMS with endogenous plasma LCAT and with rhLCAT is relatively insensitive to the amount of HDL. The reactivity of HMS with 2 μL of rhLCAT in buffer (10 μg/ml) (△), or with 2 μL of plasma (□) or with 2 μL of plasma supplemented with rhLCAT (10 μg/ml) (○) was tested without (left-most data points) or with added amounts of purified HDL (plus within symbol). The DHE esterification rates are plotted as function of the amount of free cholesterol in HDL (HDL-FC).
To determine whether assay variability between plasma samples would result from variable HDL content and the associated free cholesterol that could compete with DHE, endogenous, plasma LCAT activity was assayed in normal plasma and in plasma supplemented with increasing amounts of HDL (Fig. 6B). The highest HDL concentration tested was 0.5 mM free cholesterol, which corresponds to approximately 2.5 mM total HDL cholesterol and is near the upper limit of the HDL-FC concentration that might be encountered in a normal population [35]. The mole ratio of DHE to free cholesterol at the highest HDL concentration tested was 10. The results indicate there was no significant effect of HDL content on the rates of DHE esterification.
The influence of HDL content on activity of rhLCAT in buffer and in plasma was also tested (Fig. 6B). Here also, the presence of HDL did not cause a significant modification of the activity of rhLCAT with HMS.
Applications
The importance of HDL in preserving cardiovascular health has been an impetus in efforts to discover peptide mimetics of ApoA-I that embody the critical features of ApoA-I and, consequently, can be used as a treatment to supplement or enhance HDL functions [36;37]. The new protocol provides an easy method to screen peptides for LCAT activation. We used the assay to compare LCAT activation by LAP642 with that of other ApoA-I mimetic peptides, some of which were previously shown in unrelated assay formats to activate LCAT [38]. The results are presented in Table 1 and reveal a broad degree of activation potential among the peptides, even though all the peptides are comparable in their abilities to form stable complexes with lipids (data not shown). The lipid solubilizing properties of the peptides derive from the high degree of amphiphilicity (i.e. large hydrophobic moment) which is characteristic of all the peptides tested. Amphiphilicity, which results from segregation of polar and non-polar amino acids to opposite sides of the peptide in an alpha-helical conformation, enables the peptides to interact strongly with phospholipids in an aqueous environment [39]. The data imply the strength of peptide interaction with lipid is not the only peptide feature required to activate LCAT.
Table 1.
Dependence of LCAT activity on amino acid sequence of peptide in the substrate.
| Peptide | Amino Acid Sequence | <H> a | <μH>b | Relative Activity c |
|---|---|---|---|---|
| LAP642 | P V L D L F R E L L N E L L E A L K Q K L K | −1.08 | 0.91 | 100 |
| LAP24 | P T L D L F R E L L N E L L E A L K Q K L K | −1.12 | 0.88 | 84 |
| LAP25 | P S L D L F R E L L N E L L E A L K Q K L K | −1.13 | 0.87 | 70 |
| LAP2FE | Ac-E W L K A F Y E K V L E K L K E L F -NH2d | −0.94 | 0.77 | 43 |
| LAP5Fe | Ac-D W L K A F Y D K V F E K F K E F F -NH2 | −1.07 | 0.83 | 28 |
| PEP76 | D V F Q A L K E L F A Q L L E K W K Q V | −1.11 | 1.03 | 0 |
Note: HDL-mimetic substrates composed of amphiphilic peptide, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine and dehydroergosterol in mole portions of 2:9:1, respectively, were reacted at 63 μM dehydroergosterol with 0.6 μg/ml rhLCAT as described in Materials and Methods.
Mean hydrophobicity (kcal/mol/residue) calculated as the sum of amino acid hydrophobicities [40] divided by the number of residues.
Mean hydrophobic moment [41].
Percent rhLCAT esterification activity relative to LAP642.
Peptide terminal modifications are indicated by Ac- for acetylated N-terminus and by –NH2 for amidated C-terminus.
5F peptide [38].
To demonstrate the potential of the activity assay to be used as a screening test and clinical study tool, we measured and compared LCAT activity and LCAT content in plasma from control subjects with that in plasma from SCD patients. It has previously been shown by conventional methods that LCAT activity is diminished in SCD patients [9;10]. When we applied the new protocol, we also detected reduced LCAT activity in SCD patients compared to control subjects (Fig. 7B). Moreover, the decline in activity was reflected in a significant decrease in LCAT content in SCD plasma (Fig. 7A). There was a strong correlation between LCAT activity and LCAT content (Fig. 7C). The results suggest the new protocol could serve to gauge LCAT content in plasma.
Figure 7.

Detection of differences in plasma LCAT activity and mass between control subjects and sickle cell disease (SCD) patients. (A) LCAT concentration, determined by ELISA, is significantly lower in plasma from SCD patients (□) relative to control subjects (●) (p < 0.0001). The solid lines indicate the means. (B) LCAT activity is significantly lower in plasma from SCD patients relative to control subjects (p = 0.002). Activity was determined as described in Materials and Methods. (C) Correlation between plasma LCAT activity and concentration. The LCAT activity in each plasma sample is plotted against the corresponding LCAT concentration. The best fit of the data to a line was determined by linear regression (solid line). The 95% confidence interval (dashed lines) and prediction interval (dotted lines) is also shown. The Pearson correlation coefficient (r) p-value are shown.
The assay design is well suited for high-throughput screening. No washes or separations are required. The assay follows a simple mix, incubate and measure scheme. Measurement involves a standard spectroscopy that has high sensitivity. The assay reagents are relatively stable, readily available and easily prepared in large batches for large-scale screening.
There are limitations to what can be done with the fluorescent assay. For instance, the activity of LCAT in plasma is a function of the amount and quality of LCAT present and the amounts and qualities of the plasma lipoprotein substrates. The assay can be used to detect LCAT concentration and quality (e.g. mutation effects) if HMS is used in sufficient excess over endogenous lipoproteins. The assay can’t, however, be used to detect the reactivity of LCAT with endogenous lipoproteins. Normally, such LCAT activity is detected by equilibrating radiolabeled cholesterol into endogenous lipoproteins before reaction followed by detection of radiolabeled ester product formation. This is not possible with DHE in place of radiolabeled cholesterol since the cholesterol in the lipoproteins would strongly compete with DHE and prevent sufficient DHE ester formation for detection during the initial (i.e. linear) stages of the reaction. Presently the assayed is limited to detection of esterification in HMS.
Conclusion
A new approach for assay of LCAT enzymatic activity is presented in this report. The assay is robust and conforms to standard Michaelis-Menten enzyme kinetics. The assay has the sensitivity of radiochemical assays but it is simpler to execute and more rapid to perform. The assay is also suitable for laboratories lacking certification to use radioisotopes. These features should make it a useful tool for the study of LCAT in the research laboratory and may make it suitable and informative as a diagnostic assay for the clinical laboratory.
Acknowledgments
Portions of this work were funded by an SBIR grant to AlphaCore Pharma from the National Heart, Lung, and Blood Institute, National Institutes of Health (1 R43 HL092656-01A1). GJK and LM are supported by the National Institutes of Health Division of Intramural Research (1 ZIA HL006013-04). The authors thank Bruce Auerbach and Brian Krause for helpful discussions and ideas and for facilitating the production of rhLCAT. The authors thank research nurse Marlene Peters-Lawrence, RN for recruiting subjects, and thank all the patients who provided blood samples.
Abbreviations used
- ApoA-I
apolipoprotein A-I
- DHE
dehydroergosterol
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- HMS
HDL-mimetic substrate
- rhLCAT
recombinant human LCAT
- LAP
LCAT-activating peptide
- HEK293F
human embryonic kidney 293F cell line
- SCD
sickle cell disease
Footnotes
Disclosures
RH and NE are employed by AlphaCore Pharma which is developing rhLCAT for therapeutic purposes. RH is a shareholder in AlphaCore Pharma.
References
- 1.Rousset X, Vaisman B, Amar M, Sethi AA, Remaley AT. Lecithin:cholesterol acyltransferase--from biochemistry to role in cardiovascular disease. Curr Opin Endocrinol Diabetes Obes. 2009;16:163–171. doi: 10.1097/med.0b013e328329233b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwartz CC, VandenBroek JM, Cooper PS. Lipoprotein cholesteryl ester production, transfer, and output in vivo in humans. J Lipid Res. 2004;45:1594–1607. doi: 10.1194/jlr.M300511-JLR200. [DOI] [PubMed] [Google Scholar]
- 3.Santamarina-Fojo S, Hoeg JM, Assmann G, Brewer HB. Lecithin Cholesterol Acyltransferase Deficiency and Fish Eye Disease. In: Scriver CR, Beaudet D, Valle D, Sly W, Childs B, Kinzler K, Vogelstein B, editors. Metabolic & Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2006. pp. 2817–2833. [Google Scholar]
- 4.Smuts CM, Weich HF, Weight MJ, Faber M, Kruger M, Lombard CJ, Benade AJ. Free cholesterol concentrations in the high-density lipoprotein subfraction-3 as a risk indicator in patients with angiographically documented coronary artery disease. Coron Artery Dis. 1994;5:331–338. doi: 10.1097/00019501-199404000-00009. [DOI] [PubMed] [Google Scholar]
- 5.Weight MJ, Coetzee HS, Smuts CM, Marais MP, Maritz JS, Hough FS, Benade AJ, Taljaard JJ. Lecithin:cholesterol acyltransferase activity and high-density lipoprotein subfraction composition in type 1 diabetic patients with improving metabolic control. Acta Diabetol. 1993;30:159–165. doi: 10.1007/BF00572861. [DOI] [PubMed] [Google Scholar]
- 6.McLeod R, Reeve CE, Frohlich J. Plasma lipoproteins and lecithin:cholesterol acyltransferase distribution in patients on dialysis. Kidney Int. 1984;25:683–688. doi: 10.1038/ki.1984.74. [DOI] [PubMed] [Google Scholar]
- 7.Gillett MP, Obineche EN, El-Rokhaimi M, Lakhani MS, Abdulle A, Sulaiman M. Lecithin: cholesterol acyltransfer, dyslipoproteinaemia and membrane lipids in uraemia. J Nephrol. 2001;14:472–480. [PubMed] [Google Scholar]
- 8.Charles-Schoeman C, Watanabe J, Lee YY, Furst DE, Amjadi S, Elashoff D, Park G, McMahon M, Paulus HE, Fogelman AM, Reddy ST. Abnormal function of high-density lipoprotein is associated with poor disease control and an altered protein cargo in rheumatoid arthritis. Arthritis Rheum. 2009;60:2870–2879. doi: 10.1002/art.24802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jain SK, Shohet SB. Red blood cell [14C]cholesterol exchange and plasma cholesterol esterifying activity of normal and sickle cell blood. Biochim Biophys Acta. 1982;688:11–15. doi: 10.1016/0005-2736(82)90572-7. [DOI] [PubMed] [Google Scholar]
- 10.Emokpae MA, Uadia PO, Osadolor HB. Lecithin:cholesterol acyltransferase, lipoprotein lipase and lipoproteins in adult Nigerians with sickle cell disease. Afr J Biochem Res. 2010;4:17–20. [Google Scholar]
- 11.Chen CH, Albers JJ. Characterization of proteoliposomes containing apoprotein A-I: a new substrate for the measurement of lecithin: cholesterol acyltransferase activity. J Lipid Res. 1982;23:680–691. [PubMed] [Google Scholar]
- 12.Albers JJ, Chen CH, Lacko AG. Isolation, characterization, and assay of lecithin-cholesterol acyltransferase. Meth Enz. 1986;129:763–783. doi: 10.1016/0076-6879(86)29103-x. [DOI] [PubMed] [Google Scholar]
- 13.Stokke KT, Norum KR. Determination of lecithin: cholesterol acyltransfer in human blood plasma. Scand J Clin Lab Invest. 1971;27:21–27. doi: 10.3109/00365517109080184. [DOI] [PubMed] [Google Scholar]
- 14.Dobiasova M, Stribrna J, Pritchard PH, Frohlich JJ. Cholesterol esterification rate in plasma depleted of very low and low density lipoproteins is controlled by the proportion of HDL2 and HDL3 subclasses: study in hypertensive and normal middle-aged and septuagenarian men. J Lipid Res. 1992;33:1411–1418. [PubMed] [Google Scholar]
- 15.Patsch W, Sailer S, Braunsteiner H. An enzymatic method for the determination of the initial rate of cholesterol esterification in human plasma. J Lipid Res. 1976;17:182–185. [PubMed] [Google Scholar]
- 16.Dieplinger H, Kostner GM. The determination of lecithin:cholesterol acyl transferase in the clinical laboratory: a modified enzymatic procedure. Clin Chim Acta. 1980;106:319–324. doi: 10.1016/0009-8981(80)90316-2. [DOI] [PubMed] [Google Scholar]
- 17.Manabe M, Abe T, Nozawa M, Maki A, Hirata M, Itakura H. New substrate for determination of serum lecithin:cholesterol acyltransferase. J Lipid Res. 1987;28:1206–1215. [PubMed] [Google Scholar]
- 18.Marcel YL, Vezina C. A method for the determination of the initial rate of reaction of lecithin: cholesterol acyltransferase in human plasma. Biochim Biophys Acta. 1973;306:497–504. doi: 10.1016/0005-2760(73)90188-4. [DOI] [PubMed] [Google Scholar]
- 19.Wallentin L, Vikrot O. Evaluation of an in vitro assay of lecithin:cholesterol acyl transfer rate in plasma. Scand J Clin Lab Invest. 1975;35:661–667. [PubMed] [Google Scholar]
- 20.Bonelli FS, Jonas A. Continuous fluorescence assay for lecithin:cholesterol acyltransferase using a water-soluble phosphatidylcholine. J Lipid Res. 1992;33:1863–1869. [PubMed] [Google Scholar]
- 21.McIntosh AL, Atshaves BP, Huang H, Gallegos AM, Kier AB, Schroeder F. Fluorescence techniques using dehydroergosterol to study cholesterol trafficking. Lipids. 2008;43:1185–1208. doi: 10.1007/s11745-008-3194-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anantharamaiah GM, Mishra VK, Garber DW, Datta G, Handattu SP, Palgunachari MN, Chaddha M, Navab M, Reddy ST, Segrest JP, Fogelman AM. Structural requirements for antioxidative and anti-inflammatory properties of apolipoprotein A-I mimetic peptides. J Lipid Res. 2007;48:1915–1923. doi: 10.1194/jlr.R700010-JLR200. [DOI] [PubMed] [Google Scholar]
- 23.Busseuil D, Shi Y, Mecteau M, Brand G, Kernaleguen AE, Thorin E, Latour JG, Rheaume E, Tardif JC. Regression of aortic valve stenosis by ApoA-I mimetic peptide infusions in rabbits. Br J Pharmacol. 2008;154:765–773. doi: 10.1038/bjp.2008.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vaisman B, Remaley AT. Measurement of lecithin:cholesterol acyltransferase activity with the use of a peptide-proteoliposome substrate. In: Freeman LA, editor. Lipoproteins and Cardiovascular Disease. Humana Press; Totowa: 2013. pp. 343–352. [DOI] [PubMed] [Google Scholar]
- 25.Rousset X, Vaisman B, Auerbach B, Krause BR, Homan R, Stonik J, Csako G, Shamburek R, Remaley AT. Effect of recombinant human lecithin cholesterol acyltransferase infusion on lipoprotein metabolism in mice. J Pharmacol Exp Ther. 2010;335:140–148. doi: 10.1124/jpet.110.169540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fischer RT, Cowlen MS, Dempsey ME, Schroeder F. Fluorescence of delta 5,7,9(11),22-ergostatetraen-3 beta-ol in micelles, sterol carrier protein complexes, and plasma membranes. Biochemistry. 1985;24:3322–3331. doi: 10.1021/bi00334a037. [DOI] [PubMed] [Google Scholar]
- 27.Homan R, Anderson MK. Rapid separation and quantitation of combined neutral and polar lipid classes by high-performance liquid chromatography and evaporative light-scattering mass detection. J Chromatography B. 1998;708:21–26. doi: 10.1016/s0378-4347(97)00651-8. [DOI] [PubMed] [Google Scholar]
- 28.Jonas A. Synthetic substrates for lecithin:cholesterol acyltransferase. J Lipid Res. 1986;27:689–698. [PubMed] [Google Scholar]
- 29.Piran U, Nishida T. Utilization of various sterols by lecithin-cholesterol acyltransferase as acyl acceptors. Lipids. 1979;14:478–482. doi: 10.1007/BF02533465. [DOI] [PubMed] [Google Scholar]
- 30.Lavallee B, Provost PR, Belanger A. Formation of pregnenolone- and dehydroepiandrosterone-fatty acid esters by lecithin-cholesterol acyltransferase in human plasma high density lipoproteins. Biochim Biophys Acta. 1996;1299:306–312. doi: 10.1016/0005-2760(95)00222-7. [DOI] [PubMed] [Google Scholar]
- 31.Wustner D. Fluorescent sterols as tools in membrane biophysics and cell biology. Chem Phys Lipids. 2007;146:1–25. doi: 10.1016/j.chemphyslip.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 32.Pownall HJ, Pao Q, Massey JB. Acyl chain and headgroup specificity of human plasma lecithin:cholesterol acyltransferase. Separation of matrix and molecular specificities. J Biol Chem. 1985;260:2146–2152. [PubMed] [Google Scholar]
- 33.Sparks DL, Anantharamaiah GM, Segrest JP, Phillips MC. Effect of the cholesterol content of reconstituted LpA-I on lecithin:cholesterol acyltransferase activity. J Biol Chem. 1995;270:5151–5157. doi: 10.1074/jbc.270.10.5151. [DOI] [PubMed] [Google Scholar]
- 34.Hale JE, Schroeder F. Asymmetric transbilayer distribution of sterol across plasma membranes determined by fluorescence quenching of dehydroergosterol. Eur J Biochem. 1982;122:649–661. doi: 10.1111/j.1432-1033.1982.tb06488.x. [DOI] [PubMed] [Google Scholar]
- 35.Heiss G, Tamir I, Davis CE, Tyroler HA, Rifkand BM, Schonfeld G, Jacobs D, Frantz ID., Jr Lipoprotein-cholesterol distributions in selected North American populations: the lipid research clinics program prevalence study. Circulation. 1980;61:302–315. doi: 10.1161/01.cir.61.2.302. [DOI] [PubMed] [Google Scholar]
- 36.Sethi AA, Amar M, Shamburek RD, Remaley AT. Apolipoprotein AI mimetic peptides: possible new agents for the treatment of atherosclerosis. Curr Opin Investig Drugs. 2007;8:201–212. [PubMed] [Google Scholar]
- 37.Hovingh GK, Bochem AE, Kastelein JJ. Apolipoprotein A-I mimetic peptides. Curr Opin Lipidol. 2010;21:481–486. doi: 10.1097/MOL.0b013e3283404507. [DOI] [PubMed] [Google Scholar]
- 38.Datta G, Chaddha M, Hama S, Navab M, Fogelman AM, Garber DW, Mishra VK, Epand RM, Epand RF, Lund-Katz S, Phillips MC, Segrest JP, Anantharamaiah GM. Effects of increasing hydrophobicity on the physical-chemical and biological properties of a class A amphipathic helical peptide. J Lipid Res. 2001;42:1096–1104. [PubMed] [Google Scholar]
- 39.Fernandez-Vidal M, Jayasinghe S, Ladokhin AS, White SH. Folding amphipathic helices into membranes: amphiphilicity trumps hydrophobicity. J Mol Biol. 2007;370:459–470. doi: 10.1016/j.jmb.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hessa T, Kim H, Bihlmaier K, Lundin C, Boekel J, Andersson H, Nilsson I, White SH, von HG. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature. 2005;433:377–381. doi: 10.1038/nature03216. [DOI] [PubMed] [Google Scholar]
- 41.Pownall HJ, Knapp RD, Gotto AM, Jr, Massey JB. Helical amphipathic moment: application to plasma lipoproteins. FEBS Letters. 1983;159:17–23. [Google Scholar]