

Abstract
Apoptosis repressor with caspase recruitment domain (ARC) is a highly potent and multifunctional inhibitor of apoptosis that is physiologically expressed predominantly in post-mitotic cells such as cardiomyocytes, skeletal muscle cells and neurons. ARC was also found to be up-regulated in many forms of malignant tumours. ARC impairs the cellular apoptotic responsiveness to a wide range of stresses and insults, including extrinsic apoptosis initiation via death receptor ligands, dysregulation of cellular Ca2+ homeostasis and endoplasmatic reticulum (ER) stress, genotoxic drugs, ionizing radiation, oxidative stress and hypoxia. ARC is subject to both transcriptional and post-translational regulation and exhibits its function through a multitude of molecular interactions with upstream transducers of apoptosis signals. This review summarizes, structures and comments on the published knowledge regarding ARC and its roles in modulating apoptotic cell death responsiveness in physiological and pathophysiological contexts.
Keywords: apoptosis repressor with caspase recruitment domain (ARC), apoptosis, Bax, cancer, cardiophysiology, caspases, cell death
Introduction
Counteracting cell proliferation by apoptotic cell death is indispensable to maintain tissue homeostasis. Apoptosis also eliminates superfluous cells during developmental tissue- and organ sculpting, as well as damaged and/or mutated cells. The signalling networks of apoptosis are subject to complex regulation by an interplay of pro- and anti-apoptotic signalling molecules. In healthy cells, anti-apoptotic proteins provide a tolerance threshold to mild apoptotic stress and thereby prevent excessive, unwanted cell death. This tolerance is particularly pronounced in long-lived, highly specialized post-mitotic cells such as neurons, cardiomyocytes or skeletal muscle cells. However, up-regulation or over-activation of anti-apoptotic proteins can render cells insensitive to physiological cell death signals, thereby contributing to the development of cancer. The protein apoptosis repressor with caspase recruitment domain (ARC) was first described more than a decade ago [1] and was independently identified as muscle-enriched cytoplasmic protein in a later study [2]. In various signalling pathways leading to apoptosis, ARC is believed to exert its function through multiple protein–protein interactions and was also shown to be subject to significant transcriptional and post-translational regulation. Here, we structure the disparate published knowledge on ARC and discuss the functions of ARC in the context of cellular physiology and pathophysiology.
ARC identification and tissue-specific expression
ARC was identified by Koseki and colleagues during a database search of expressed sequence tags with the aim to identify human cDNAs with homologies to the caspase recruitment domain (CARD) of casapse-9 [1]. A year later, conducting a yeast two hybrid screen for proteins involved in RNA processing, Stoss et al. independently discovered ARC, termed here as muscle-enriched cytoplasmic protein [2]. The human ARC gene (deposited under the name Nol3 in the human genome database) is located on chromosome 16q21-q23 and spans 4 exons and 3 short introns (Fig. 1A). ARC is expressed from exons 2 to 4, yielding a 208 amino acid protein with a calculated molecular mass of 22.6 kD [1, 2]. Stoss et al. also suggested the existence of an alternative splicing product, termed nucleolar protein of 30 kD (Nop30) [2]. Nop30 likewise is believed to be expressed from exons 2 to 4, albeit transcription from exon 2 is frameshifted. Nop30 is a protein of 219 aa with a calculated molecular mass of 24.3 kD and contains an alternative COOH-terminal domain which localizes it exclusively in the nucleoli, where it interacts with splicing factor SRp30c [2]. However, the spliced transcript has so far not been conclusively shown to produce a protein as all data were gathered from experiments employing ectopic Nop30 expression.
fig 1.

Organization of the Nol3 gene and ARC sequence similarities. (A) Organization of the Nol3 locus. ARC is expressed from exons 2 to 4 of the Nol3 gene. Coding regions are shaded in grey. ATG and TGA indicate start and stop codons. Separating introns are shown as lines. (B) Alignment of mammalian ARC protein sequences. Mammalian ARC sequences were aligned using Clustal W software. Asterisks indicate identical amino acids, ‘:’ indicates conserved substitutions and ‘.’ indicates semi-conserved substitutions. The CARD region (aa 1–98) is highly conserved between all five species shown. The P/E rich region likewise presents with a high sequence similarity. (C) Domain organization of ARC. ARC comprises an NH2-terminal CARD, followed by a region rich in proline and glutamate (P/E).
Orthologues of human ARC were experimentally also identified on gene or protein level in rattus norvegicus, mus musculus and ovis aries [3–5], with the identification of rat ARC cDNA actually predating the identification of the human ARC protein [3]. Predicted sequences were deposited also for bos taurus and canis lupus familiaris. The ARC gene in mouse (chromosomal location 8 51.0 cM) codes for a 220 aa protein with a calculated mass of 24.5 kD, while no evidence could be found for the expression of additional splice variants, suggesting that a Nop30-like product is unlikely to exist in mice [4]. Rat ARC (chromosomal location 19q11) is composed of 221 aa and has a calculated molecular weight of 24.6 kD [3]. Like in mouse, alternative splice variants of ARC have not been reported. A sequence alignment highlights that the ARC protein sequences are highly conserved between species (Fig. 1B).
Northern blot analysis of mRNA expression in human tissue showed that ARC is expressed in skeletal muscle and heart tissue, while the transcript was not found in brain, placenta, lung, liver, kidney, pancreas or various lymphoid-haematopoietic tissues [1]. ARC transcripts were also found in rat brain cDNA libraries, and corresponding mRNA species were very prominent in skeletal and cardiac muscle tissue [3]. Correspondingly, high amounts of murine ARC mRNA were found in heart and skeletal muscle, lower amounts in brain and testis, and only residual amounts in lung and kidney tissue, while ARC mRNA was absent in liver and spleen [4].
These patterns largely correspond to protein expression levels, with high ARC levels having been reported in human heart and skeletal muscle tissue, but also reported in brain samples and primary granulosa cells [6–10]. In mice, ARC was found in heart, skeletal muscle, diaphragm and brain tissue [4, 7, 11], while only residual amounts could be detected in kidney samples [12]. Likewise, ARC is highly expressed in rat heart and skeletal muscle as well as in brain tissue, vascular smooth muscle and adrenal gland [9, 13–16], but not in liver, spleen or lung [13, 15, 17].
The molecular structure of ARC
Although structural data based on X-ray crystallography or NMR spectroscopy have not yet been reported for ARC, sequence homologies to known motifs and domains of other proteins allowed associating ARC with the cell death signalling processes. ARC comprises two functional regions – a CARD followed by a region rich in proline/glutamate (P/E) tandem repeats (Fig. 1C). The ARC CARD was found to be highly homologous to the CARDs of caspase-2 and caspase adaptor proteins RIP associated Ich-1/CED homologous protein with death domain (RAIDD) and Apaf-1 [1]. RAIDD and Apaf-1 are part of the large multi-protein activation platforms for caspase-2 (PIDDosome) or caspase-9 (Apoptosome) [18, 19]. From the outset this strengthened the notion that ARC could be involved in modulating apoptosis signalling by interaction with caspases or adaptor proteins. Indeed it was shown that ARC can bind to procaspases-8 and, at least in overexpression scenarios, to procaspase-2 by CARD interaction [1, 11]. ARC can also bind to itself via the CARD, with the dimer largely losing its anti-apoptotic potential [11].
The P/E rich region comprises the last 110 aa of human ARC and is highly acidic [2]. The P/E rich region can bind considerable amounts of calcium, which was shown to negatively regulate the interaction with procaspase-8 [20]. The interaction with procaspase-8 is further modulated by phosphorylation at threonine 149 within the P/E rich region: Phosphorylation by protein kinase CK2 is a requirement for ARC to bind procaspase-8 and apparently also to associate with the outer mitochondrial membrane [14, 21].
Discrepancies become evident in the published literature when comparing the detected molecular weights of ARC and Nop30 to calculated molecular weights when using immunoblotting procedures. During classical SDS gel electrophoresis, Nop30 presents with a larger than expected molecular weight of approximately 30 kD rather than displaying as a band at the expected 24 kD. This was suggested to arise from its strong bipolar charge distribution between the CARD and the COOH-terminal region of Nop30 [2]. Likewise, post-translational modifications of ARC such as the mentioned phosphorylation, calcium binding but also ubiquitination [22, 23] may contribute to higher apparent molecular weights. Recombinantly expressed His-tagged human ARC migrates at approximately 27 kD [24], while native ARC as found expressed in human cells can be detected also at larger sizes. For example, ARC expressed in human HCT-116, MDA-MB-231, HeLa or DU145 cancer cell lines was shown to migrate at approximately 34 kD, while ARC in BJAB and DG-75 lymphoma cells migrates as a consistently larger band of 38 kD [9]. In comparison rat ARC can yield product sizes ranging from 25–33 kD. In the widely used H9c2 rat embryonic heart cells, ARC runs at a slightly smaller size due to 12 aa deletion in the P/E rich region [9, 15, 17]. Whether this deletion has a functional consequence is however not known [17]. Because the apparent molecular weight might carry information on cell line or tissue-specific modifications of ARC, we collated a table listing the apparent molecular weights of ARC as described for different tissues, cell lines and species (Table S1). For human samples information on whether the antibodies used were specific in the detection of ARC over Nop30 was also added, even though, as mentioned before, the endogenous expression of Nop30 has not yet been convincingly shown.
(Patho)physiology and ARC expression
Physiological role and expression of ARC in cardiac and skeletal muscle tissue
Because ARC is most prominently expressed in cardiac and skeletal muscle tissue, it was extensively investigated whether ARC contributes to the high apoptosis resistance of post-mitotic muscle cells and plays a role in maintaining cardiophysiological function. ARC-deficient mice were shown to develop normally, and morphological or histological differences in mice hearts or skeletal muscle could not be detected [7]. ARC-deficient mice exert normal cardiac function when resting. However, under conditions of cardiovascular ischemia or pressure overload, significantly accelerated cardiomyopathy was observed in these animals. Cardiomyopathy was associated with increased numbers of apoptotic cardiomyocytes, highlighting a physiological role for ARC in elevating the apoptosis resistance of heart muscle cells [7]. Indeed, the cardio-specific overexpression of ARC in mice results in increased heart sizes due to surplus cells and significantly improves contractile recovery during reperfusion [12]. In line with this finding, ARC overexpressing cardiomyocytes were also found to be more resistant to apoptosis signalling induced by calcium overload and hypoxic stress in an in vitro model of ischaemia [12]. An increased apoptosis resistance of post-mitotic myocytes can also be achieved by exercise training: Both x-linked inhibitor of apoptosis protein (XIAP) and ARC protein levels in the rat soleus and heart muscles increase with intensive training [25]. In contrast to the transcriptional up-regulation of XIAP, elevated ARC protein levels seem to be established by protein stabilization rather than increased gene transcription. Significantly reduced ARC protein levels were detected in heart, aorta and skeletal muscle of hypertensive rats, and the reduction in ARC expression correlated with increased apoptosis of skeletal muscle tissues when compared to samples from normotensive rats [16, 26, 27]. This indicates that prolonged hypertensive stress affects the apoptosis resistance of cardiac tissue via depletion of ARC. In addition, ARC from hypertensive animals also migrated as a slightly larger protein in immunoblotting experiments, indicating that post-translational modification might further alter its functionality in this scenario. A significant loss in ARC protein was likewise observed in human cardiac tissue from patients with end-stage heart failure [7]. Importantly, ARC mRNA levels were not affected in this scenario, indicating that ARC protein destabilization rather than a down-regulation of gene transcription contributes to increased cell death susceptibility of human cardiac tissue [7]. ARC levels were also shown to drop following hypoxia/reoxygenation in cultured rat cardiomyocytes, a loss which can, however, be counteracted by repeated short post-conditioning cycles of hypoxia/reoxygenation [28]. Interestingly, a recent study conducted in rabbits suggested that endogenous levels of oestrogen mediate a constitutively higher ARC expression in the heart tissue of female individuals, which confers elevated apoptosis resistance in response to ischaemia/reperfusion and might explain the enhanced myocardial recovery of female individuals [29].
Physiological role and expression of ARC in brain tissue
Low levels of ARC protein were reported to be present in brain tissue samples from human, rat and mouse [6, 7, 9, 30, 31]. Even though ARC deficiency does not result in behavioural or brain histological differences in mice [7], ARC was suggested to mediate neuronal cell death resistance. In particular it was found that with increasing age, ARC expression levels in the mouse cortex might decline [30]. This loss in ARC could be abrogated by keeping mice on calorie restricted diets. A similar effect of calorie restriction on ARC protein abundance was also reported in skeletal muscle tissue of male rats [32]. It was furthermore shown in mouse models that chronic alcohol intake reduces ARC expression in cerebellar tissue and that this loss in ARC expression might be linked with alcohol intake-associated increases in apoptotic cell death in the cerebellum [31]. Because many of the above observations were made in experimentally rather minimalistic studies, more comprehensive and functional data are required to unequivocally answer whether ARC plays a physiological role in preventing apoptosis in brain tissues.
ARC expression and localization in cancer
Even though physiologically ARC expression is absent in most tissues, malignant tumours frequently express ARC at high levels. ARC expression might therefore be a contributing factor in tumour development and progression through enhancing cellular apoptosis resistance. ARC was found expressed in diverse types of cancers, including pancreatic, colorectal, breast, lung, cervical and prostate cancers, as well as in glioblastoma, melanoma, lymphoma and others. For example, a screen of tissue samples of 44 primary human colon adenocarcinomas and matching samples of adjacent benign colonic epithelium found significantly higher levels of ARC expressed in the cancerous tissue [8]. Similarly, ARC was detected in cervical and ovarian primary carcinomas but not in associated samples of normal tissue [8]. The expression of ARC has also been analysed in a large number of human cancer cell lines, as well as in primary cultures of rat and mouse cells, and it was found that the abundance of ARC in these experimental model systems can vary significantly. Presence or absence of ARC expression and, where comparisons were made, the relative abundance of ARC in various tissues and cell lines are listed in Table S2. This table allows to navigate through widely used model systems and can assist in experimental planning by helping in the selection of tissues, cells or cell lines presenting with different levels of endogenous ARC expression.
Discrepancies were also found in the subcellular localization of ARC, which initially raised hope that the spatial protein distribution might correlate with tumour malignancy and could thus be used as a marker for disease progression or therapy responsiveness. High amounts of ARC in the cytosolic compartment were found in most invasive mammary ductal carcinomas, while benign breast tissue only stained positive for ARC expression in the nucleus [33]. Similarly, ARC levels were shown to selectively increase in the cytosols of colon cancer tissues [8]. In contrast, subcellular fractionation in human HCT-116, MD-BM-231 and MIA PaCa-2 cancer cell lines indicated a higher abundance of ARC in the nuclei [9]. Likewise, ARC was reported to be enriched in the nuclear fraction of PC12 rat pheochromocytoma cells [15], while in human melanoma cell lines ARC was found predominantly in the cytosol or at mitochondria [34]. Endogenous ARC in rat H9c2 cells resides largely in the soluble cytosolic fraction, whereas ectopically overexpressed ARC tended to associate with membranes [13]. Even when taking into account that the immunofluorescence or subcellular fractionation analyses employed in some of these studies suffered from limited resolution, lacked counter-stains or specificity controls, no consistent picture of ARC distribution patterns has emerged yet. However, besides cytosolic interactions with apoptosis regulating proteins, an additional role of ARC in the nucleus has been described (see below). This nuclear function of ARC might therefore explain differential subcellular distribution patterns of ARC.
Regulation of ARC protein levels by gene transcription and protein degradation
In tissues or cells presenting with ARC expression, little is known about the regulation of ARC transcription. It was recently shown that activated N-Ras and H-Ras can drive ARC expression, with the promoter being activated in a mitogen-activated protein kinase kinase (MEK)/extracellular-signal-regulated kinase (ERK)-dependant manner [35]. ARC expression in cancer cells correlated with Ras expression, and Ras expression was sufficient to drive ARC expression in otherwise ARC– epithelial cells [35]. The transcription factors driving ARC expression were however not yet identified. It was also found that the ARC promoter contains an optimal binding site for tumour suppressor p53 and evidence has been provided that p53 can negatively regulate ARC expression in cardiomyocyte-derived rat H9c2 cells [36]. p53 also drives the expression of various Bcl-2 homology 3 (BH3)-only proteins, of which p53 up-regulated modulator of apoptosis (Puma) is a major cytosolic inducer of apoptosis in response to DNA damage [37] and also antagonizes the anti-apoptotic function of ARC in the cytosol (see later section).
In reverse, endogenous ARC can also directly antagonize the transcription factor activity of p53. ARC can bind to tumour suppressor p53 via a region between amino acids 125–175 located within the P/E rich region [24]. The corresponding binding site within p53 is located within the p53 tetramerization domain (amino acids 332–356). ARC impairs p53 tetramerization and exposes a nuclear export signal. Indeed, a nuclear accumulation of ARC was particularly pronounced in breast cancer cell lines and tumours which express functional wild-type p53, whereas nuclear ARC was rarely detected in breast cancers expressing mutant p53 [24]. This finding thus highlighted that ARC can also exert an important anti-apoptotic function within the nucleus.
Besides the transcriptional regulation of ARC, protein levels of ARC are regulated by degradation through the ubiquitin–proteasome system. In untreated cells, ARC was shown to be a rather long-lived protein with half times ranging from 16 or 35 hrs, as reported for human cancer cells such as MCF-7, HeLa or rat H9c2 cells [22, 23]. ARC is targeted for proteasomal degradation through ubiquitylation by E3-ubiquitin ligase Mdm2 [22], and an ARC triple mutant that cannot be ubiquitylated (K17R, K68R, K163R) correspondingly shows increased stability and enhanced anti-apoptotic potential [23]. Because Mdm2 also regulates the ubiquitylation and degradation of p53 [38, 39], ARC expression and degradation therefore are tightly interlinked with the p53/Mdm2 interplay. In addition to the role of active Ras in driving ARC expression, Ras was also shown to impair the ubiquitylation and proteasomal degradation of endogenous ARC [35]. This mode of ARC stabilization however seems independent of Mdm2 because concomitant changes in Mdm2 levels were not observed [35]. ARC can be post-translationally stabilized in rats subjected to exercise training [25], and ARC protein levels drop in response to hypoxia/ischaemia and oxidative stress in rat H9c2 cells as well as human HeLa and MCF-7 cancer cells, without mRNA levels being affected [22, 23]. These studies therefore indicate that modulations in ARC stability and degradation are physiologically relevant. The regulation of ARC expression and degradation is summarized and visualized in Figure 2.
fig 2.
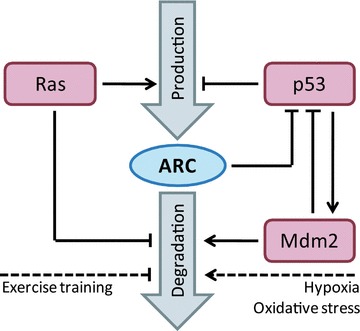
Regulation of ARC expression by gene transcription and protein degradation. Steady state ARC protein levels are established by the balance of transcription/translation and protein degradation. Protein production is promoted by Ras signalling and negatively regulated by p53. Mdm2 enhances ARC degradation, while Ras signalling stabilizes ARC. The regulatory interplay between ARC, p53 and Mdm2 was visualized as well. Conditions positively or negatively affecting the stability of ARC are displayed as dashed lines.
Multi-functionality of ARC in apoptosis inhibition
Elevated expression of ARC was shown to inhibit various modalities of apoptotic cell death signalling, while loss of ARC conversely sensitizes to apoptosis. In the following sections we summarize the knowledge on the ARC-dependent intracellular signalling in various pathways (Fig. 3).
fig 3.
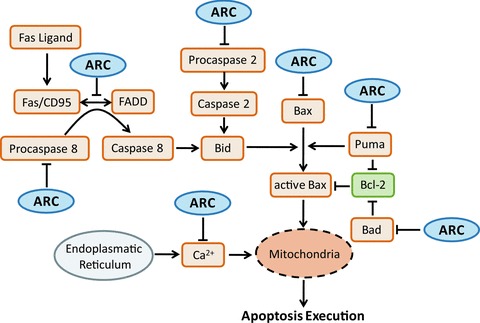
Multifunctionality of ARC in regulating apoptosis signalling. The multifunctionality of ARC in regulating apoptosis signalling was visualized using a simplified pathway diagram. The diagram displays the core components and reactions of apoptosis signalling pathways that are affected by ARC.
Extrinsic apoptosis initiation
Extrinsic apoptosis initiated via death ligands CD95L/FasL and tumour necrosis factor (TNF)-α can efficiently be inhibited by ARC overexpression [1, 20]. Inhibition was shown to depend on the interaction of the ARC CARD with the death effector domain (DED) of procaspase-8, but not with other caspases such as procaspases-1, -3 or -9 [1]. As the DED is not part of the fully processed mature caspase-8, these data suggest that the interaction of ARC with procaspase-8 competes with recruitment of procaspase-8 to the death-inducing signalling complex (DISC), which comprises activated death receptors, Fas-associated death domain (FADD) and other adaptor proteins. Of note, the interaction of ARC with procaspase-8 in cellulo but not in vitro was shown to depend on ARC being phosphorylated by protein kinase CK2 at T149 [14]. This phosphorylation localizes ARC to mitochondria, and only there it is able to bind to procaspase-8. ARC therefore might impair death receptor mediated apoptosis also by limiting the amount of soluble procaspase-8 available for DISC-mediated activation. However, recently an alternative, death receptor independent mode of mitochondria-localized caspase-8 activation was reported that crucially depends on cardiolipin [40]. Whether ARC plays a modulating role in this process has not been investigated yet. CK2-dependent phosphorylation of ARC was also shown to be required to counteract cardiomyocyte hypertrophy in response to phenylephrine, angiotensin II or TNF-α[41]. In these scenarios, ARC phosphorylation dropped as a consequence of reactive oxygen species-driven inactivation of CK2. ARC inactivation might therefore assist in mediating the reported synergies between reactive oxygen species and death receptor-mediated apoptosis initiation [42]. Besides hypothetical phosphorylation sites that were predicted for the Nop30 splice variant of ARC [2], it was not yet investigated whether other kinases can phosphorylate ARC at this or other positions and thereby modulate apoptotic cellular responsiveness.
Apart from binding to procaspase-8, ARC was also shown to mediate extrinsic apoptosis more immediately on the level of the DISC. Endogenous ARC can inhibit the assembly of the Fas/CD95 DISC by binding to Fas/CD95 and FADD [11]. These associations relied on the ARC CARD, which interacted with the death domain of Fas/CD95 or FADD, thereby posing additional potential of modulating death receptor-mediated signalling. Importantly, ARC did not bind to other proteins containing DEDs or death domains such as flice-like inhibitory protein (FLIP), receptor-interacting serine/threonine-protein kinase (RIP) or tumour necrosis factor receptor type 1-associated death domain protein (TRADD) [11], indicating that interaction with procaspase-8, FADD and Fas/CD95 is highly specific. Whether ARC can inhibit the DISC formation during tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) or TNF-α-induced extrinsic apoptosis remains to be shown.
ARC was also identified to interact with procaspase-2 [1], albeit so far only at conditions of protein overexpression. Although it has not yet been investigated whether ARC interferes with PIDDosome-dependent caspase-2 activation, it is conceivable that ARC might influence the unusual activation of caspase-2 at the DISC: DISC-dependent caspase-2 activation has been reported in response to CD95 stimulation in human T- and B-cell lines [43], as well as in response to DNA damage-induced formation of the CD95 DISC [44].
Moreover, ARC can serve to mediate crosstalk from intrinsic death pathways (reviewed below) to extrinsic apoptosis initiation. ARC can bind significant amounts of Ca2+ via its P/E rich region. The ectopic expression of ARC significantly reduces cytosolic Ca2+ transients in response to thapsigargin and A23187 [20]. Interestingly, Ca2+ loading negatively modulates the interaction of ARC with procaspase-8 [20], indicating that calcium ionophores or sarco/endoplasmic reticulum Ca2+ ATPases inhibitors might prove useful in sensitizing cells expressing elevated amounts of ARC to DISC-dependent caspase-8 activation. It was furthermore shown that the BH3-only proteins Puma and Bad can bind to the CARD of ARC via their BH3 regions in adenovirally transduced H9c2 cells, thereby interfering with the binding of ARC to procaspase-8 [36]. Puma and Bad have been implemented in initiating apoptosis in response to a wide range of insults, including genotoxic stress, proteasome inhibition, ER stress, growth factor withdrawal or kinase inhibition [37, 45]. Puma expression and Bad activation in ARC expressing cells thus could contribute to additional amounts of procaspase-8 becoming available for DISC recruitment. Synergistic responses can be observed between death ligands such as TRAIL and various classes of other drugs which are currently investigated in clinical trials of TRAIL combination treatments [46]. Alongside the activation of parallel stress response pathways and other complex alterations in the interplay of apoptosis regulators during such combination treatments, ARC might act as an additional contributor to TRAIL sensitization in multi drug-treatment paradigms.
Intrinsic apoptosis initiation and the mitochondrial apoptosis pathway
Many stressors inducing the intrinsic cell death pathway, such as genotoxic drugs, proteasome inhibitors, ionizing radiation or radical stress, entirely or partially depend on the transcriptional induction of (subsets of) BH3-only proteins. BH3-only proteins belong to the Bcl-2 protein family and activate the pro-apoptotic members Bax and Bak and/or antagonize anti-apoptotic family members such as Bcl-2, Bcl-xL or Mcl-1 [47]. Activated Bax/Bak proteins form pores through which cytochrome-c and Smac are released into the cytosol, where they facilitate effector caspase activation and apoptosis execution [48–50]. As mentioned before, ARC was shown to bind BH3-only proteins Puma and Bad in a study using rat H9c2 cardiomyocytes ectopically expressing ARC [36]. The binding to Puma and Bad requires the region of the ARC CARD stretching from amino acids 31 to 70 and the Puma or Bad BH3 domains, as was shown by comprehensive immunoprecipitation experiments. Functionally, ARC would therefore behave similar to anti-apoptotic Bcl-2 like proteins in eliminating excess levels of Bad and Puma.
Bad exclusively binds to anti-apoptotic Bcl-2 family members and in absence of cellular stress is maintained in a phosphorylated, inactive state [45]. Apoptosis in response to broad spectrum kinase inhibitor staurosporine is believed to be at least partially dependent on the inhibition of Bad phosphorylation [51]. However, ARC overexpression is not able to inhibit or reduce staurosporine-induced apoptosis in human Jurkat cells [20]. Puma is thought to be capable of both directly activating Bax/Bak as well as antagonizing anti-apoptotic Bcl-2 family members. Puma is a major transducer of p53-dependent apoptosis in response to genotoxic stress as was shown in response to various DNA-damaging chemotherapeutics as well as γ irradiation [37]. ARC indeed was shown to protect rat cardiomyocytes, human HeLa cervical and SGC-7901 gastric cancer cells from apoptosis induced by doxorubicin or ionizing radiation [52–54] and it was suggested that the doxorubicin resistance is directly dependent on the ARC-Puma interaction [54]. However, ARC overexpression in Jurkat cells did not protect from etoposide-induced apoptosis [20]. Taken together these reports indicate that the Puma antagonizing function of ARC might be significantly more cell protective than the sequestration of Bad, but also that the potency of ARC in restricting apoptotic signalling in response to genotoxic stress may vary between different cell types, cell lines and treatment paradigms.
The anti-apoptotic function of ARC in the mitochondrial pathway is not limited to interactions with BH3-only proteins. Experiments carried out in native extracts from mouse hearts and human HEK293 cells showed that ARC binds to inactive cytosolic Bax via a CARD interaction with the Bax COOH-terminal region [11]. Physiological expression of ARC in rat H9c2 cardiomyocytes was required to prevent spontaneous Bax activation, and ectopic ARC expression could protect from apoptosis induced by Bax overexpression [11]. A direct CARD-dependent interaction of endogenous ARC with Bax was also confirmed in an independent study using H9c2 cells, where ARC provided significant protection against cell death induced by ischaemia/reperfusion and hydrogen peroxide [55]. Interestingly, in vitro experiments suggested that the interaction of ARC and Bax is not direct but may require additional interaction partners to mediate ARC–Bax association [20]. It was also shown that cytosolic p53 can induce the mitochondrial apoptosis pathway by activating Bax and that Puma is required to release cytosolic p53 from Bcl-xL [56, 57]. Because ARC can interact with both Puma and p53, it would clearly warrant further investigation of whether ARC can modulate the interplay of p53, Bax, Puma and Bcl-xL. Bax activation has also been shown to promote mitochondrial fission, a function of Bax that is independent of its role in facilitating cytochrome c release from the mitochondria [58]. Recent data obtained from ARC-overexpressing HeLa cells and cardiomyocytes showed that ARC can significantly reduce mitochondrial fission during doxorubicin and hydrogen peroxide induced apoptosis [54, 59], which may indicate that ARC can antagonize both the pore formation activity of Bax as well as its role in driving mitochondrial fission. It was, however, also suggested that the interaction between ARC and Puma can impair the mitochondrial targeting of dynamin-related protein-1, a promoter of mitochondrial fission [54]. Even though these functions may need to be further investigated in additional model systems, it seems to emerge that the anti-apoptotic role of ARC is also coupled with the prevention mitochondrial fission.
Apoptosis induced by ER Ca2+ release could be inhibited by ARC, as was shown in response to calcium ionophore A23187 or thapsigargin, an inhibitor of the sarco/endoplasmic reticulum Ca2+ ATPases [20]. ARC was shown to act as an efficient cytosolic Ca2+ buffering protein, and Ca2+ binding is facilitated by the P/E rich domain [20]. In agreement, depleting ARC expression in Mel-RM and MM200 melanoma cells by RNA interference sensitized these cells to thapsigargin- or tunicamycin-induced apoptosis [34]. As ARC expression seems elevated in most melanomas [34], antagonizing ARC therefore might prove helpful in sensitizing these otherwise highly resistant cells to apoptosis.
Concluding comments
We have collated the published knowledge on the expression, interactions and regulation of ARC and their physiological consequences, and showed that the current literature points out a high functional diversity of ARC in modulating cellular apoptosis responsiveness. ARC plays a prominent role in elevating the resistance threshold to various cellular stresses in post-mitotic cells, which in fact are required to survive for a life time, as well as in many types of cancers through multiple protein–protein interactions. Of note, for each individual function of ARC other proteins were already described that can fulfil similar roles. Redundancy of the individual functions of ARC with other proteins should however not distract from the relevance of ARC as an apoptosis modulator. Functional redundancies between proteins rather indicate that the processes which are being regulated are of high importance and require robust control mechanisms [60]. The functional diversity of ARC exceeds that of the majority of other proteins involved in cell death and survival signalling and contributes to increasing apoptosis resistance economically on a broad scale.
Acknowledgments
This research was supported by NBIP Ireland funded under the Irish Higher Education Authority Programme for Third Level Institutions (HEA PRTLI) Cycle 4, co-funded by the Irish Government and the European Union ‘Investing in your future’, as well as by grants from the Irish Health Research Board (RP/2006/258) and Science Foundation Ireland (07/RFP/BICF601). A.H.L.-G. is a recipient of an NBIP Career Enhancement and Mobility Fellowship co-funded by Marie Curie Actions (EU FP7), the Irish HEA PRTLI cycle 4 and the Italian National Research Council.
Conflict of interest
The authors declare no competing interests.
Supporting Information
Discrepancies in the apparent molecular weight of ARC.
Table S2 ARC expression and relative abundance.
References
- 1.Koseki T, Inohara N, Chen S, et al. ARC, an inhibitor of apoptosis expressed in skeletal muscle and heart that interacts selectively with caspases. Proc Natl Acad Sci USA. 1998;95:5156–60. doi: 10.1073/pnas.95.9.5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stoss O, Schwaiger FW, Cooper TA, et al. Alternative splicing determines the intracellular localization of the novel nuclear protein Nop30 and its interaction with the splicing factor SRp30c. J Biol Chem. 1999;274:10951–62. doi: 10.1074/jbc.274.16.10951. [DOI] [PubMed] [Google Scholar]
- 3.Geertman R, McMahon A, Sabban EL. Cloning and characterization of cDNAs for novel proteins with glutamic acid-proline dipeptide tandem repeats. Biochim Biophys Acta. 1996;1306:147–52. doi: 10.1016/0167-4781(96)00036-x. [DOI] [PubMed] [Google Scholar]
- 4.Abmayr S, Crawford RW, Chamberlain JS. Characterization of ARC, apoptosis repressor interacting with CARD, in normal and dystrophin-deficient skeletal muscle. Hum Mol Genet. 2004;13:213–21. doi: 10.1093/hmg/ddh018. [DOI] [PubMed] [Google Scholar]
- 5.Ekhterae D, Hinmon R, Matsuzaki K, et al. Infarction induced myocardial apoptosis and ARC activation. J Surg Res. 2011;166:59–67. doi: 10.1016/j.jss.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Engidawork E, Gulesserian T, Yoo BC, et al. Alteration of caspases and apoptosis-related proteins in brains of patients with Alzheimer’s disease. Biochem Biophys Res Commun. 2001;281:84–93. doi: 10.1006/bbrc.2001.4306. [DOI] [PubMed] [Google Scholar]
- 7.Donath S, Li P, Willenbockel C, et al. Apoptosis repressor with caspase recruitment domain is required for cardioprotection in response to biomechanical and ischemic stress. Circulation. 2006;113:1203–12. doi: 10.1161/CIRCULATIONAHA.105.576785. [DOI] [PubMed] [Google Scholar]
- 8.Mercier I, Vuolo M, Jasmin JF, et al. ARC (apoptosis repressor with caspase recruitment domain) is a novel marker of human colon cancer. Cell Cycle. 2008;7:1640–7. doi: 10.4161/cc.7.11.5979. [DOI] [PubMed] [Google Scholar]
- 9.Wang M, Qanungo S, Crow MT, et al. Apoptosis repressor with caspase recruitment domain (ARC) is expressed in cancer cells and localizes to nuclei. FEBS Lett. 2005;579:2411–5. doi: 10.1016/j.febslet.2005.03.040. [DOI] [PubMed] [Google Scholar]
- 10.Sasson R, Rimon E, Dantes A, et al. Gonadotrophin-induced gene regulation in human granulosa cells obtained from IVF patients. Modulation of steroidogenic genes, cytoskeletal genes and genes coding for apoptotic signalling and protein kinases. Mol Hum Reprod. 2004;10:299–311. doi: 10.1093/molehr/gah041. [DOI] [PubMed] [Google Scholar]
- 11.Nam YJ, Mani K, Ashton AW, et al. Inhibition of both the extrinsic and intrinsic death pathways through nonhomotypic death-fold interactions. Mol Cell. 2004;15:901–12. doi: 10.1016/j.molcel.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 12.Pyo JO, Nah J, Kim HJ, et al. Protection of cardiomyocytes from ischemic/hypoxic cell death via Drbp1 and pMe2GlyDH in cardio-specific ARC transgenic mice. J Biol Chem. 2008;283:30707–14. doi: 10.1074/jbc.M804209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ekhterae D, Lin Z, Lundberg MS, et al. ARC inhibits cytochrome c release from mitochondria and protects against hypoxia-induced apoptosis in heart-derived H9c2 cells. Circ Res. 1999;85:e70–7. doi: 10.1161/01.res.85.12.e70. [DOI] [PubMed] [Google Scholar]
- 14.Li PF, Li J, Muller EC, et al. Phosphorylation by protein kinase CK2: a signaling switch for the caspase-inhibiting protein ARC. Mol Cell. 2002;10:247–58. doi: 10.1016/s1097-2765(02)00600-7. [DOI] [PubMed] [Google Scholar]
- 15.Dowds TA, Sabban EL. Endogenous and exogenous ARC in serum withdrawal mediated PC12 cell apoptosis: a new pro-apoptotic role for ARC. Cell Death Differ. 2001;8:640–8. doi: 10.1038/sj.cdd.4400855. [DOI] [PubMed] [Google Scholar]
- 16.Quadrilatero J, Bloemberg D. Apoptosis repressor with caspase recruitment domain is dramatically reduced in cardiac, skeletal, and vascular smooth muscle during hypertension. Biochem Biophys Res Commun. 2010;391:1437–42. doi: 10.1016/j.bbrc.2009.12.084. [DOI] [PubMed] [Google Scholar]
- 17.Neuss M, Monticone R, Lundberg MS, et al. The apoptotic regulatory protein ARC (apoptosis repressor with caspase recruitment domain) prevents oxidant stress-mediated cell death by preserving mitochondrial function. J Biol Chem. 2001;276:33915–22. doi: 10.1074/jbc.M104080200. [DOI] [PubMed] [Google Scholar]
- 18.Tinel A, Tschopp J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science. 2004;304:843–6. doi: 10.1126/science.1095432. [DOI] [PubMed] [Google Scholar]
- 19.Zou H, Li Y, Liu X, et al. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem. 1999;274:11549–56. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]
- 20.Jo DG, Jun JI, Chang JW, et al. Calcium binding of ARC mediates regulation of caspase 8 and cell death. Mol Cell Biol. 2004;24:9763–70. doi: 10.1128/MCB.24.22.9763-9770.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tan WQ, Wang JX, Lin ZQ, et al. Novel cardiac apoptotic pathway: the dephosphorylation of apoptosis repressor with caspase recruitment domain by calcineurin. Circulation. 2008;118:2268–76. doi: 10.1161/CIRCULATIONAHA.107.750869. [DOI] [PubMed] [Google Scholar]
- 22.Foo RS, Chan LK, Kitsis RN, et al. Ubiquitination and degradation of the anti-apoptotic protein ARC by MDM2. J Biol Chem. 2007;282:5529–35. doi: 10.1074/jbc.M609046200. [DOI] [PubMed] [Google Scholar]
- 23.Nam YJ, Mani K, Wu L, et al. The apoptosis inhibitor ARC undergoes ubiquitin-proteasomal-mediated degradation in response to death stimuli: identification of a degradation-resistant mutant. J Biol Chem. 2007;282:5522–8. doi: 10.1074/jbc.M609186200. [DOI] [PubMed] [Google Scholar]
- 24.Foo RS, Nam YJ, Ostreicher MJ, et al. Regulation of p53 tetramerization and nuclear export by ARC. Proc Natl Acad Sci USA. 2007;104:20826–31. doi: 10.1073/pnas.0710017104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siu PM, Bryner RW, Murlasits Z, et al. Response of XIAP, ARC, and FLIP apoptotic suppressors to 8 wk of treadmill running in rat heart and skeletal muscle. J Appl Physiol. 2005;99:204–9. doi: 10.1152/japplphysiol.00084.2005. [DOI] [PubMed] [Google Scholar]
- 26.Quadrilatero J, Rush JW. Increased DNA fragmentation and altered apoptotic protein levels in skeletal muscle of spontaneously hypertensive rats. J Appl Physiol. 2006;101:1149–61. doi: 10.1152/japplphysiol.00194.2006. [DOI] [PubMed] [Google Scholar]
- 27.Quadrilatero J, Rush JW. Evidence for a pro-apoptotic phenotype in skeletal muscle of hypertensive rats. Biochem Biophys Res Commun. 2008;368:168–74. doi: 10.1016/j.bbrc.2008.01.067. [DOI] [PubMed] [Google Scholar]
- 28.Li Y, Ge X, Liu X. The cardioprotective effect of postconditioning is mediated by ARC through inhibiting mitochondrial apoptotic pathway. Apoptosis. 2009;14:164–72. doi: 10.1007/s10495-008-0296-4. [DOI] [PubMed] [Google Scholar]
- 29.Bouma W, Noma M, Kanemoto S, et al. Sex-related resistance to myocardial ischemia-reperfusion injury is associated with high constitutive ARC expression. Am J Physiol Heart Circ Physiol. 2010;298:H1510–7. doi: 10.1152/ajpheart.01021.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shelke RR, Leeuwenburgh C. Lifelong caloric restriction increases expression of apoptosis repressor with a caspase recruitment domain (ARC) in the brain. FASEB J. 2003;17:494–6. doi: 10.1096/fj.02-0803fje. [DOI] [PubMed] [Google Scholar]
- 31.Ren J, Babcock SA, Li Q, et al. Aldehyde dehydrogenase-2 transgene ameliorates chronic alcohol ingestion-induced apoptosis in cerebral cortex. Toxicol Lett. 2009;187:149–56. doi: 10.1016/j.toxlet.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dirks AJ, Leeuwenburgh C. Aging and lifelong calorie restriction result in adaptations of skeletal muscle apoptosis repressor, apoptosis-inducing factor, X-linked inhibitor of apoptosis, caspase-3, and caspase-12. Free Radic Biol Med. 2004;36:27–39. doi: 10.1016/j.freeradbiomed.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 33.Mercier I, Vuolo M, Madan R, et al. ARC, an apoptosis suppressor limited to terminally differentiated cells, is induced in human breast cancer and confers chemo- and radiation-resistance. Cell Death Differ. 2005;12:682–6. doi: 10.1038/sj.cdd.4401631. [DOI] [PubMed] [Google Scholar]
- 34.Chen LH, Jiang CC, Watts R, et al. Inhibition of endoplasmic reticulum stress-induced apoptosis of melanoma cells by the ARC protein. Cancer Res. 2008;68:834–42. doi: 10.1158/0008-5472.CAN-07-5056. [DOI] [PubMed] [Google Scholar]
- 35.Wu L, Nam YJ, Kung G, et al. Induction of the apoptosis inhibitor ARC by Ras in human cancers. J Biol Chem. 2010;285:19235–45. doi: 10.1074/jbc.M110.114892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li YZ, Lu DY, Tan WQ, et al. p53 initiates apoptosis by transcriptionally targeting the antiapoptotic protein ARC. Mol Cell Biol. 2008;28:564–74. doi: 10.1128/MCB.00738-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu J, Zhang L. PUMA, a potent killer with or without p53. Oncogene. 2008;27:S71–83. doi: 10.1038/onc.2009.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haupt Y, Maya R, Kazaz A, et al. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–9. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 39.Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 40.Gonzalvez F, Schug ZT, Houtkooper RH, et al. Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J Cell Biol. 2008;183:681–96. doi: 10.1083/jcb.200803129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murtaza I, Wang HX, Feng X, et al. Down-regulation of catalase and oxidative modification of protein kinase CK2 lead to the failure of apoptosis repressor with caspase recruitment domain to inhibit cardiomyocyte hypertrophy. J Biol Chem. 2008;283:5996–6004. doi: 10.1074/jbc.M706466200. [DOI] [PubMed] [Google Scholar]
- 42.Izeradjene K, Douglas L, Tillman DM, et al. Reactive oxygen species regulate caspase activation in tumor necrosis factor-related apoptosis-inducing ligand-resistant human colon carcinoma cell lines. Cancer Res. 2005;65:7436–45. doi: 10.1158/0008-5472.CAN-04-2628. [DOI] [PubMed] [Google Scholar]
- 43.Lavrik IN, Golks A, Baumann S, et al. Caspase-2 is activated at the CD95 death-inducing signaling complex in the course of CD95-induced apoptosis. Blood. 2006;108:559–65. doi: 10.1182/blood-2005-07-007096. [DOI] [PubMed] [Google Scholar]
- 44.Olsson M, Vakifahmetoglu H, Abruzzo PM, et al. DISC-mediated activation of caspase-2 in DNA damage-induced apoptosis. Oncogene. 2009;28:1949–59. doi: 10.1038/onc.2009.36. [DOI] [PubMed] [Google Scholar]
- 45.Danial NN. BAD: undertaker by night, candyman by day. Oncogene. 2008;27:S53–70. doi: 10.1038/onc.2009.44. [DOI] [PubMed] [Google Scholar]
- 46.Johnstone RW, Frew AJ, Smyth MJ. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat Rev Cancer. 2008;8:782–98. doi: 10.1038/nrc2465. [DOI] [PubMed] [Google Scholar]
- 47.Brenner D, Mak TW. Mitochondrial cell death effectors. Curr Opin Cell Biol. 2009;21:871–7. doi: 10.1016/j.ceb.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 48.Wei MC, Zong WX, Cheng EH, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–30. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li P, Nijhawan D, Budihardjo I, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–89. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 50.Du C, Fang M, Li Y, et al. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 51.Zha J, Harada H, Yang E, et al. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14–3-3 not BCL-X(L) Cell. 1996;87:619–28. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 52.Yamane K, Kinsella TJ. CK2 inhibits apoptosis and changes its cellular localization following ionizing radiation. Cancer Res. 2005;65:4362–7. doi: 10.1158/0008-5472.CAN-04-3941. [DOI] [PubMed] [Google Scholar]
- 53.An J, Li P, Li J, et al. ARC is a critical cardiomyocyte survival switch in doxorubicin cardiotoxicity. J Mol Med. 2009;87:401–10. doi: 10.1007/s00109-008-0434-z. [DOI] [PubMed] [Google Scholar]
- 54.Wang JX, Li Q, Li PF. Apoptosis repressor with caspase recruitment domain contributes to chemotherapy resistance by abolishing mitochondrial fission mediated by dynamin-related protein-1. Cancer Res. 2009;69:492–500. doi: 10.1158/0008-5472.CAN-08-2962. [DOI] [PubMed] [Google Scholar]
- 55.Gustafsson AB, Tsai JG, Logue SE, et al. Apoptosis repressor with caspase recruitment domain protects against cell death by interfering with Bax activation. J Biol Chem. 2004;279:21233–8. doi: 10.1074/jbc.M400695200. [DOI] [PubMed] [Google Scholar]
- 56.Chipuk JE, Bouchier-Hayes L, Kuwana T, et al. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science. 2005;309:1732–5. doi: 10.1126/science.1114297. [DOI] [PubMed] [Google Scholar]
- 57.Chipuk JE, Kuwana T, Bouchier-Hayes L, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–4. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- 58.Sheridan C, Delivani P, Cullen SP, et al. Bax- or Bak-induced mitochondrial fission can be uncoupled from cytochrome c release. Mol Cell. 2008;31:570–85. doi: 10.1016/j.molcel.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 59.Li J, Li Y, Qin D, et al. Mitochondrial fission leads to Smac/DIABLO release quenched by ARC. Apoptosis. 2010;15:1187–96. doi: 10.1007/s10495-010-0514-8. [DOI] [PubMed] [Google Scholar]
- 60.Kitano H. Biological robustness. Nat Rev Genet. 2004;5:826–37. doi: 10.1038/nrg1471. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Discrepancies in the apparent molecular weight of ARC.
Table S2 ARC expression and relative abundance.