

Abstract
Induction of tumour necrosis factor-α (TNF-α) expression leads to myocardial depression during sepsis. However, the underlying molecular mechanisms are not fully understood. The aim of this study was to investigate the role of Rac1 in TNF-α expression and cardiac dysfunction during endotoxemia and to determine the involvement of phosphoinositide-3 kinase (PI3K) in lipopolysaccharide (LPS)-induced Rac1 activation. Our results showed that LPS-induced Rac1 activation and TNF-α expression in cultured neonatal mouse cardiomyocytes. The response was inhibited in Rac1 deficient cardiomyocytes or by a dominant-negative Rac1 (Rac1N17). To determine whether PI3K regulates Rac1 activation, cardiomyocytes were treated with LY294002, a PI3K selective inhibitor. Treatment with LY294002 decreased Rac1 activity as well as TNF-α expression stimulated by LPS. Furthermore, inhibition of PI3K and Rac1 activity decreased LPS-induced superoxide generation which was associated with a significant reduction in ERK1/2 phosphorylation. To investigate the role of Rac1 in myocardial depression during endotoxemia in vivo, wild-type and cardiomyocyte-specific Rac1 deficient mice were treated with LPS (2 mg/kg, i.p.). Deficiency in Rac1 significantly decreased myocardial TNF-α expression and improved cardiac function during endotoxemia. We conclude that PI3K-mediated Rac1 activation is required for induction of TNF-α expression in cardiomyocytes and cardiac dysfunction during endotoxemia. The effect of Rac1 on TNF-α expression seems to be mediated by increased NADPH oxidase activity and ERK1/2 phosphorylation.
Keywords: sepsis, tumour necrosis factor-α, lipopolysaccharide, cardiac dysfunction, PI3 kinase, Rac1 GTPase
Introduction
Sepsis is a major consequence of infectious diseases and one of the leading causes of death in the intensive care unit [1]. Myocardial dysfunction induced by endotoxins or lipopolysaccharides (LPS) of Gram-negative bacteria is a common complication of septic shock and renders septic patients at high risk of developing multi-organ failure, which is associated with high mortality [2, 3]. The inhibitory effect of LPS on cardiac function is mediated through the production of pro-inflammatory cytokines [4]. Tumour necrosis factor-α (TNF-α) is a major cytokine responsible for cardiac dysfunction during sepsis [5–11]. However, the molecular mechanisms underlying myocardial TNF-α production during sepsis are not fully understood.
Rac GTPases are a subfamily of Ras-homologous (Rho) GTPases and act as molecular switches, cycling between active guanosine 5′-triphosphate (GTP)-bound and inactive guanosine 5′-diphosphate (GDP)-bound states [12, 13]. The switch is activated when an upstream signal activates a guanine nucleotide exchange factor (GEF), which then acts to facilitate the release of GDP from the Rac GTPase and the subsequent binding of GTP. Rac activity is terminated by hydrolysis of GTP to GDP, a process which is accelerated by GTPase-activating proteins (GAPs). Rac is an intracellular transducer of signalling and can interact with specific effectors that regulate diverse cellular functions, such as cytoskeletal remodelling, microtubule stability, gene transcription and superoxide (O2−) production [12, 13].
There are three different Rac proteins: the ubiquitously expressed Rac1, the haematopoietic cell-specific Rac2 and Rac3 that is expressed in the brain, liver, lung and pancreas [14]. Previous studies have shown that LPS increases Rac1 activity in phagocytes; however, the effect of Rac1 on TNF-α expression in these cells remains controversial [15–17]. Rac1 is the predominant Rac proteins in cardiomyocytes [18, 19]. Furthermore, Rac1 is activated during LPS stimulation and contributes to myocardial TNF-α expression [20]. However, regulation of Rac1 activation during LPS stimulation is not fully understood. Phosphoinositide-3 kinases (PI3K) are a family of evolutionary conserved signalling molecules that mediate many cellular responses. The production of phosphatidylinositol (3,4,5)-triphosphate (PtdIns(3,4,5)P3) from PI3K activates Rac via a PtdIns(3,4,5)P3-sensitive GEF. However, Rac can also be activated by PI3K-independent mechanisms [21]. Whether Rac activation in cardiomyocytes during LPS stimulation is mediated by PI3K remains to be determined.
NADPH oxidase is an enzyme system that catalyses the NADPH-dependent reduction of oxygen to O2− and consists of multi-subunits including Nox2 (gp91phox), p22phox, p40phox, p47phox, p67phox and Rac. It has been shown that NADPH oxidase is a major source of O2− in cardiomyocytes under pathophysiological conditions and activation of Rac is essential for NADPH oxidase activation [22]. We have demonstrated that Nox2-containing NADPH oxidase plays a pivotal role in LPS-induced cardiac TNF-α production [23]. However, the role of Rac1 in cardiac dysfunction during sepsis remains unknown.
In the present study, we hypothesized that Rac1 was necessary for LPS-induced TNF-α expression and myocardial dysfunction via NADPH oxidase activation. To test this hypothesis, a cardiac-specific Rac1-deficient mouse was generated. Our results demonstrated that LPS-induced Rac1 activation in cardiomyocytes is PI3K dependent. Rac1 deficiency blocked cardiomyocyte TNF-α expression and decreased LPS-induced O2− generation. Furthermore, cardiac-specific deficiency of Rac1 improved myocardial function in endotoxemia.
Materials and methods
Animals and preparation of neonatal mouse cardiomyocytes
The investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institute of Health (NIH Publication #85–23, revised 1996) and experimental protocols were approved by Animal Use Subcommittee at the University of Western Ontario. C57BL/6 wild-type (WT) and Rac1 floxed (Rac1f/f) mice [24] were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). In Rac1f/f mice, LoxP sites were inserted at both sides of exon 1 of the Rac1 gene. Cre transgenic mice (CreTG/+) with overexpression of Cre recombinase under the control of α-myosin heavy-chain promoter were provided by Dr. E. Dale Abel (University of Utah, UT, USA). This Cre recombinase can excise the region between the loxP sites and is specifically expressed in cardiomyocytes because is it under the control of the α-myosin heavy-chain promoter. The generation of cardiomyocyte-specific Rac1 knockout mice (Rac1–/–) was achieved by breeding Rac1f/f mice with CreTG/+ mice as we have recently described [25]. Neonatal cardiomyocytes were prepared and cultured according to methods we have previously described [26]. Cells were treated with LPS (1 μg/ml; Sigma-Aldrich, Oakville, Ontario, Canada), apocynin (400 μM; Sigma-Aldrich), LY294002 (10 μM; Sigma-Aldrich) and U0126 (10 μM; Sigma-Aldrich) or infected with adenoviruses.
Adenoviral infection of neonatal cardiomyocytes
Cardiomyocytes were infected with adenoviruses carrying a dominant-negative form of Rac1 (Ad-Rac1N17, Vector Biolabs, Philadelphia, PA, USA), Cre recombinase (Ad-Cre, Vector Biolabs) or green fluorescence protein (Ad-GFP, a gift from Dr. J. Lipp, Medical University of Vienna, Austria) as a control, at a multiplicity of infection of 10 plaque forming units/cell. Adenovirus-mediated gene transfer was implemented as previously described [5]. All experiments were performed after 24 hrs of adenoviral infection.
Rac1 activity assay
Rac1 activity was measured using the EZ-Detect Rac1 activation kit (Pierce, Rockford, IL, USA) according to the manufacturer’s protocol. Briefly, cell or tissue lysates (1 mg protein) were incubated with 20 μg of glutathione S-transferase (GST)-human Pak1-p21 binding domain at 4°C for 1 hr. The beads were washed three times to remove the unbound material and were then boiled in 2× SDS sample buffer for 5 min. to elute Rac1-GTP. Rac1-GTP and total Rac1 protein levels were detected by Western blot analysis using an anti-Rac1 antibody included in the kit.
PI3K activity assay
PI3K activity in cultured cardiomyocytes or myocardial tissue lysates was determined using a competitive ELISA kit (Echelon Biosciences, Salt Lake City, UT, USA) according to the manufacture’s protocol with modifications [27]. Briefly, cell or tissue lysates (25 μg protein) were incubated with phosphatidylinositol (4,5)-bisphosphate [PI(4,5)P2] substrate (100 pmol) in 100 μl kinase reaction buffer. The reaction products were incubated with a PI(3,4,5)P3 detector protein, and then added to the PI(3,4,5)P3-coated microplate for competitive binding. A peroxidase-linked secondary detection reagent and colorimetric detection at 450 nm was used to detect PI(3,4,5)P3 detector protein binding to the plate.
Measurement of TNF-α mRNA
Total RNA was extracted from cardiomyocytes using the Trizol Reagent (Invitrogen, Burlington, ON, Canada) as per manufacturer’s instructions. TNF-α mRNA levels were determined by real-time RT-PCR using the same primer as in our previous report [5].
Measurement of TNF-α protein
TNF-α protein levels were measured using a mouse TNF-α ELISA kit (eBioscience, San Diego, CA, USA), according to the manufacturer’s instructions. The measurements were standardized with cell numbers or expressed as TNF-α levels to total proteins.
Western blot analysis
Thirty micrograms of protein lysates were subjected to separation on a 10% SDS-PAGE gel, followed by electrotransfer to nitrocellulose membranes. Blots were probed with specific antibodies against ERK1/2 and phospho-ERK1/2, p38 and phospho-p38 (Cell Signaling Technology, Danvers, MA, USA), respectively, as previously reported [5]. Signals were detected by the chemiluminescence detection method and quantified by densitometry.
Lucigenin assay
NADPH-dependent superoxide (O2−) generation was measured in cell lysates by lucigenin-enhanced chemiluminescence (40 μg of protein, 100 μM β-NADH, 5 μM lucigenin; Sigma-Aldrich). The chemiluminescence was detected by a mutilabel counter (SpectraMax M5; Molecular Devices, Sunnyvale, CA, USA). Replicates were incubated in the presence of the flavoprotein inhibitor (diphenyleneiodonium, 10 μM) to ensure O2− was generated from NADPH oxidase, as previously described [23]. The light signal was monitored for 1.5 sec. and counts per second were presented as NADPH oxidase activity that was diphenyleneiodonium inhibitable.
Isolated mouse heart preparations
Adult Rac1f/f and Rac1–/– (male, 3 months old) mice were treated with LPS (2mg/kg, i.p.) or saline. After 2 hrs, mouse hearts were isolated and perfused in a Langendorff-system with Krebs-Henseleit buffer at 3 ml/min. constant flow. The perfusion buffer was kept at 37°C and consistently bubbled with a mixture of 95% O2 and 5% CO2. Myocardial function was assessed as previously described with modifications [23]. Briefly, a 6–0 silk suture was passed through the apex of the left ventricle and threaded through a light-weight rigid coupling rod, which was connected to a force-displacement transducer (FT03) to record tension and heart rate. The heart work was calculated by multiplying the force (g) by the heart rate (bpm). Maximal and minimal first derivatives of force (+dF/dtmax and –dF/dtmin) which represent the rate of contraction and relaxation, respectively, were analysed by PowerLab Chart program (AD Instruments, Colorado Springs, CO, USA).
Statistical analysis
Results are presented as mean ± S.E.M. from at least three independent experiments. Differences between two groups were analysed by a standard Student t-test. For multigroup comparisons, one or two-way ANOVA followed by Student–Newman–Keuls or Bonferroni post-test was performed. P < 0.05 was considered statistically significant.
Results
Myocardial Rac1 activation by LPS
To examine the effect of LPS on Rac1 activity, neonatal cardiomyocytes isolated from C57BL/6 mice were treated with LPS (1 μg/ml) for 5, 15, 30 and 60 min. As shown in Figure 1A, Rac1 activity in these cells peaked at 15 min. and declined to about control levels by 60 min. after LPS stimulation. To verify the in vitro results, Rac1f/f mice were treated with LPS (2 mg/kg, i.p.) or saline for 30 min. and myocardial Rac1 activity was measured. In response to LPS, myocardial Rac1 activity was significantly increased (P < 0.05, Fig. 1B). These data show that LPS activates Rac1 in cardiomyocytes in vitro and in the myocardium in vivo.
fig 1.
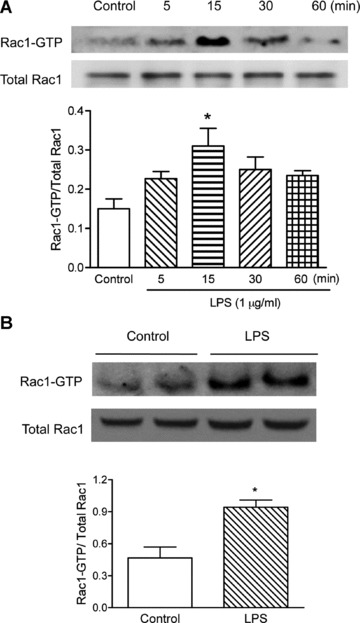
Effects of LPS on Rac1 activity in neonatal cardiomyocytes and in the adult myocardium. (A) Cardiomyocytes were isolated from WT mice, cultured for 48 hrs, and then treated with vehicle or LPS (1 μg/ml) for 5, 15, 30 and 60 min. Rac1 activity was measured using the EZ-Detect Rac1 activation kit. (B) Adult male Rac1f/f mice were treated with LPS (2 mg/kg, i.p.) for 30 min. Rac1 activity in the left ventricular myocardium was measured as described above. Data are means ± S.E.M. from three to four mice or independent experiments. *P < 0.05 versus control.
Rac1 activation and cardiomyocyte TNF-α expression during LPS stimulation
To elucidate the role of Rac1 in LPS-induced TNF-α expression, neonatal cardiomyocytes isolated from WT mice were infected with an adenovirus carrying a dominant-negative form of the Rac1 gene (Ad-Rac1N17), which selectively inhibits Rac1 activity. As shown in Figure 2A and B, overexpression of Rac1N17 significantly decreased LPS-induced TNF-α mRNA and protein levels by 60% and 56%, respectively (P < 0.01). This result was further confirmed using Rac1 deficient cardiomyocytes. Cultured neonatal cardiomyocytes from Rac1f/f mice were infected with Ad-Cre. Expression of Cre recombinase in Rac1f/f cells decreased Rac1 protein levels by 70% (Fig. 2C). LPS-induced TNF-α mRNA and protein expression were reduced by 67% and 41% in Ad-Cre infected Rac1f/f cells, respectively (P < 0.05, Fig. 2D and E). These data show that LPS-induced TNF-α expression requires Rac1 activity.
fig 2.
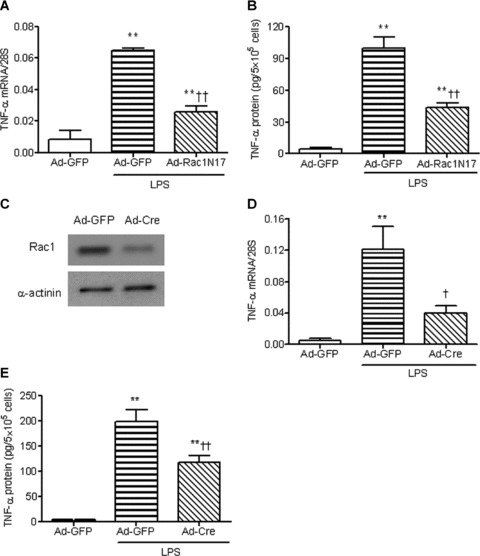
Rac1 enhances LPS-induced TNF-α expression in neonatal cardiomyocytes. WT cardiomyocytes were infected with Ad-GFP or Ad-Rac1N17 for 24 hrs. Cardiomyocytes were treated with LPS (1 μg/ml) for 3 or 5 hrs. TNF-α mRNA (A) and TNF-α protein in culture medium (B) were measured by real-time RT-PCR and ELISA, respectively. Neonatal cardiomyocytes from Rac1f/f mice were infected with Ad-GFP and Ad-Cre for 24 hrs. Rac1 protein was measured by Western blot analysis (C). Rac1f/f cells, infected with Ad-GFP or Ad-Cre, were treated with LPS for 3 or 5 hrs. TNF-α mRNA (D) and TNF-α protein in culture medium (E) were measured as described above. Data are means ± S.E.M. from three to seven independent experiments. **P < 0.01 versus Ad-GFP; †P < 0.05, ††P < 0.01 versus Ad-GFP+LPS.
Involvement of PI3K in Rac1 activation in cardiomyocytes during LPS stimulation
To investigate the involvement of PI3K in Rac1 activation in cardiomyocytes during LPS stimulation, PI3K activity was determined. In response to LPS (1 μg/ml), PI3K activity was significantly increased in cultured neonatal cardiomyocytes (P < 0.05, Fig. 3A). In vivo treatment of LPS (2 mg/kg, i.p.) also activated PI3K in the myocardium in the adult mice (P < 0.05, Fig. 3B). To further study the contribution of PI3K in LPS-induced Rac1 activation, cardiomyocytes were treated with LY294002, a selective inhibitor of PI3K. Our data showed that LY294002 decreased LPS-induced Rac1-GTP by 41% in cardiomyocytes (Fig. 3C). Similarly, LPS-stimulated Rac1-GTP levels in the myocardium were also blocked by 71% after LY294002 treatment (Fig. 3D). In addition, LY294002 significantly decreased LPS-induced TNF-α mRNA and protein levels (P < 0.05, Fig. 3E and F). These results indicate that Rac1 activation in cardiomyocytes is mediated by PI3K during LPS stimulation.
fig 3.
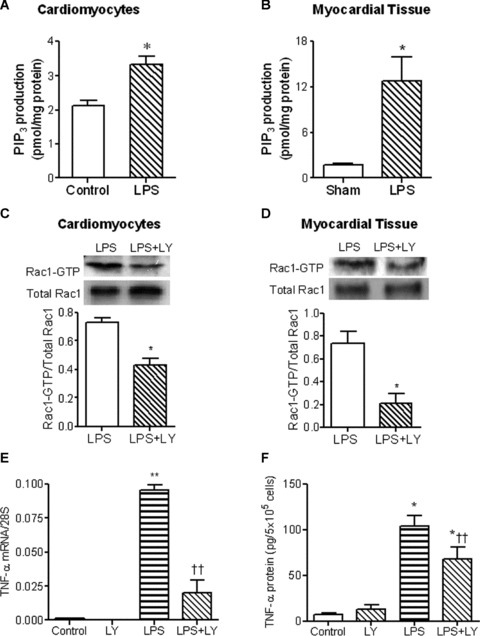
Role of PI3K in LPS-induced Rac1 activation and TNF-α expression in neonatal cardiomyocytes. Cultured WT neonatal cardiomyocytes was stimulated with LPS at 1 μg/ml for 30 min. WT mice were treated with LPS (2 mg/kg, i.p.) for 30 min. PI3K activities in cardiomyocytes (A) and myocardium (B) were determined by competitive ELISA. Rac1 activities in WT cardiomyocytes (C) and myocardium (D) stimulated with LPS for 30 min. in the presence or absence of the PI3K inhibitor LY294002 (10 μM and 7.5 mg/kg, i.p.) were determined by EZ-Detect Rac1 activation kit. WT cardiomyocytes were treated with LPS (1 μg/ml) in the presence or absence of the PI3K inhibitor LY294002 for 3 or 5 hrs. TNF-α mRNA (E) and TNF-α protein in culture medium (F) were measured by real-time RT-PCR and ELISA, respectively. Data are means ± S.E.M. from three to five independent experiments. *P < 0.05, **P < 0.01 versus control or sham; †P < 0.05, ††P < 0.01 versus LPS.
Role of PI3K and Rac1 in NADPH oxidase activation during LPS stimulation
Our lab has recently demonstrated that Nox2-containing NADPH oxidase contributes to LPS-induced TNF-α expression in cardiomyocytes [23]. Consistent with this notion, the present study demonstrated that LPS increased NADPH oxidase-mediated O2− generation (P < 0.05, Fig. 4A), which was blocked by apocynin, a selective NADPH oxidase inhibitor. Moreover, apocynin, significantly reduced TNF-α mRNA levels by 46% and protein levels by 43% in response to LPS (P < 0.01, Fig. 4D and E). To detect if PI3K activation mediates LPS-induced NADPH oxidase activity, cardiomyocytes were treated with LY294002. Figure 4A showed that O2− production stimulated by LPS was significantly blocked by LY294002. To determine whether Rac1 is involved in regulating NADPH oxidase activity, cardiomyocytes were treated with Ad-Rac1N17 to specifically block Rac1 activation. Our data showed that LPS-induced O2− generation in these cells was significantly inhibited by Ad-Rac1N17 (P < 0.05, Fig. 4B). Similarly, O2− production was also significantly reduced in Rac1 deficient cardiomyocytes (P < 0.05, Fig. 4C). These results suggest that PI3K and Rac1 are critical for NADPH oxidase activation in cardiomyocytes during LPS stimulation.
fig 4.
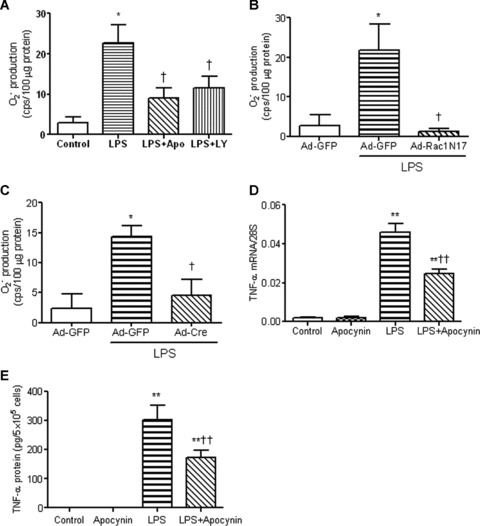
PI3K and Rac1 enhance LPS-induced superoxide (O2−) generation and TNF-α expression in neonatal cardiomyocytes. (A) Effects of apocynin and LY294002 on O2− production. WT cells were treated with LPS (1 μg/ml) for 2 hrs with or without apocynin (400 μM) and LY294002 (10 μM). O2− production was measured by the lucigenin assay. (B) Effects of Ad-Rac1N17 on O2− production. WT cells were infected with Ad–GFP or Ad-Rac1N17. O2− production was measured 2 hrs after LPS treatment (1 μg/ml). (C) O2− production in Rac1 deficient cardiomyocytes. Rac1f/f cardiomyocytes were infected with Ad-GFP or Ad-Cre for 24 hrs followed by treatment with LPS for 2 hrs. O2− production was measured. WT cardiomyocytes were treated with LPS (1 μg/ml) in the presence or absence of 400 μM apocynin for 3 and 5 hrs. TNF-α mRNA (D) and TNF-α protein in culture medium (E) were measured by real-time PCR analysis and ELISA, respectively. Data are means ± S.E.M. from three to five independent experiments. *P < 0.05, **P < 0.01 versus control and Ad-GFP; †P < 0.05, ††P < 0.01 versus LPS and Ad-GFP+LPS.
Role of PI3K and Rac1 in ERK1/2 and p38 activation during LPS stimulation
We have previously shown that activation of ERK1/2 and p38 MAPK was essential for NADPH oxidase signalling and LPS-induced TNF-α expression in cardiomyocytes [23]. The effects of LPS on ERK1/2 activation were also determined in the present study. As shown in Figure 5A, LPS rapidly increased phosphorylation of ERK1/2 which peaked at 30 min. and returned to control levels after 2 hrs. LPS-induced ERK1/2 phosphorylation was completely blocked by a PI3K inhibitor, LY294002 (Fig. 5B), suggesting that LPS regulated ERK1/2 activity via PI3K. To examine whether Rac1 activity leads to ERK1/2 phosphorylation, ERK1/2 activation was measured in Ad-Rac1N17 infected cardiomyocytes. Overexpression of Rac1N17 significantly decreased LPS-induced phosphorylation of ERK1/2 compared with Ad-GFP infected group (Fig. 5C). Furthermore, U0126, a selective ERK1/2 inhibitor, decreased LPS-stimulated TNF-α mRNA and protein levels by 46% and 69%, respectively (Fig. 5D and E). Conversely, inhibition of Rac1 activation by Ad-Rac1N17 had no effect on p38 phosphorylation induced by LPS (Fig. 6). Taken together, these results suggest that the effects of PI3K and Rac1 on cardiomyocytes are mediated by ERK1/2 but not by p38 MAPK signalling.
fig 5.
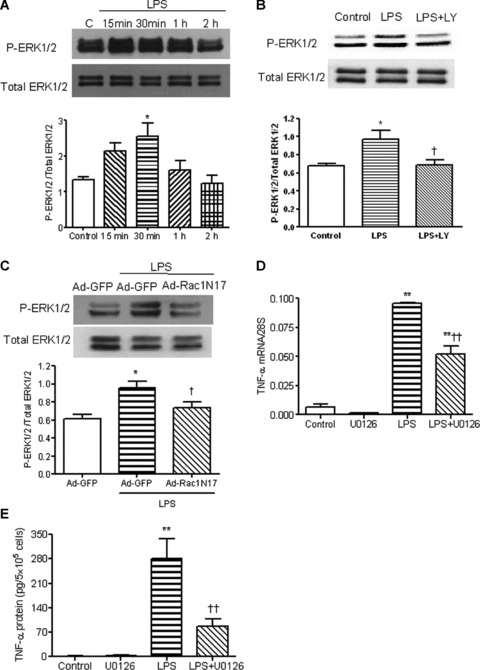
Effects of PI3K and Rac1 on LPS-induced ERK1/2 phosphorylation in neonatal cardiomyocytes. (A) WT cardiomyocytes were treated with vehicle or LPS (1 μg/ml) for 15 min., 30 min., 1 and 2 hrs. ERK1/2 phosphorylation in these cells was measured by Western blot analysis. (B) WT cardiomyocytes were treated with LPS (1 μg/ml) for 30 min. with or without LY294002. ERK1/2 phosphorylation in these cells was measured. (C) Neonatal cardiomyocytes were infected with Ad-GFP or Ad-Rac1N17 for 24 hrs. Cardiomyocytes were treated with LPS (1 μg/ml) for 30 min. ERK1/2 phosphorylation was measured as described above. (D) and (E) Cardiomyocytes were treated with LPS with or without the ERK1/2 inhibitor U0126 (10 μM) for 3 and 5 hrs. TNF-α mRNA (D) and protein in culture medium (E) were measured by real-time RT-PCR and ELISA, respectively. Data are means ± S.E.M. from three to five independent experiments. *P < 0.05, **P < 0.01 versus control; †P < 0.05, ††P < 0.01 versus Ad-GFP+LPS and LPS.
fig 6.
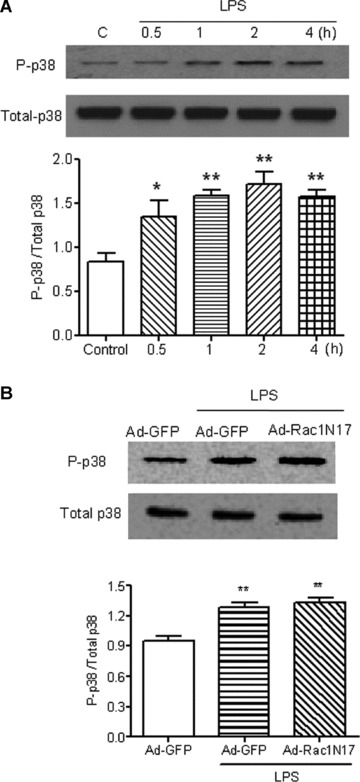
Effect of Rac inhibition on LPS-induced p38 phosphorylation in neonatal cardiomyocytes. Phosphorylation of p38 was measured by Western blot analysis. (A) WT cardiomyocytes were treated with vehicle or LPS (1 μg/ml) for 0.5, 1, 2 and 4 hrs. (B) Cardiomyocytes were infected with Ad-GFP or the dominant-negative Rac, Ad-Rac1N17 for 24 hrs at which point the cells were treated with LPS for 2 hrs. Data are means ± S.E.M. from four to five independent experiments. *P < 0.05, **P < 0.01 versus control.
Role of Rac1 in myocardial dysfunction during endotoxemia
To study the role of Rac1 in myocardial depression during endotoxemia in vivo, we generated cardiac-specific Rac1 knockout mice using Cre-loxP recombination as described in the methods. Our data showed that the Rac1 protein was selectively knocked down in the heart but not in the skeletal muscle and lungs in Rac1–/– mice (Fig. 7A). Rac1–/– and Rac1f/f mice were treated with vehicle or LPS (2 mg/kg, i.p.). Our data demonstrated that LPS-induced myocardial TNF-α mRNA and protein levels were significantly decreased (P < 0.01, Fig. 7B and C). After 2 hrs of LPS in vivo treatment, cardiac function was determined using the Langendorff preparation. The rate of contraction and relaxation, heart rate and heart work were significantly reduced in both Rac1f/f and Rac1–/– mice after endotoxemia (P < 0.05, Fig. 8). However, compared with Rac1f/f mice, heart work and rate of contraction (+dF/dtmax) were significantly increased in Rac1–/– mice (P < 0.05, Fig. 8).
fig 7.
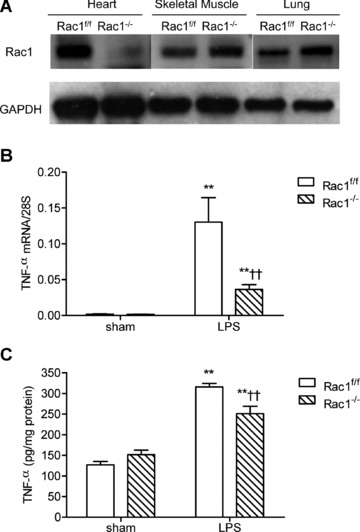
TNF-α expression in Rac1f/f and Rac1–/– adult mouse myocardium during endotoxemia. (A) Rac1 protein expression in heart, skeletal muscle and lungs in Rac1f/f and Rac1–/– mice as determined by Western blot analysis. TNF-α mRNA (B) and protein (C) levels in Rac1f/f and Rac1–/– heart tissues were measured after 2 and 4 hrs of LPS treatment (2 mg/kg, i.p.). Data are means ± S.E.M., n = 3 to 10 per group. **P < 0.01 versus sham Rac1f/f, ††P < 0.01 versus LPS Rac1f/f.
fig 8.
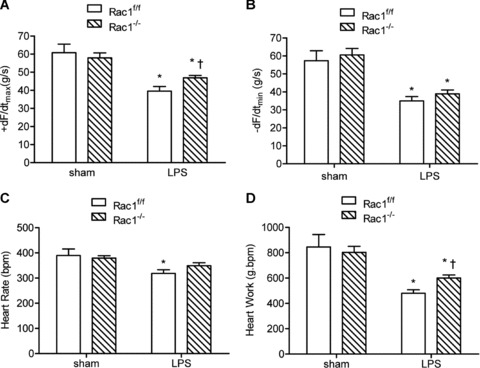
Cardiac function in Rac1f/f and Rac1–/– mice after 2 hrs of LPS treatment (2 mg/kg, i.p.). Mouse hearts were isolated and perfused using the Langendorff system. Contractile function of heart was determined. Changes in contraction (+dF/dtmax, A), relaxation (–dF/dtmin, B), heart rate (C) and heart work (D) are presented. Data are means ± S.E.M., n = 5 to 7 per group. *P < 0.05 versus sham Rac1f/f, †P < 0.05 versus LPS Rac1f/f.
Discussion
The present study demonstrated for the first time that Rac1-mediated TNF-α expression following LPS stimulation occurs downstream of PI3K signalling in cardiomyocytes. Rac1 activation increased O2− generation, and ERK1/2 MAPK phosphorylation leading to increased TNF-α expression in cardiomyocytes. More importantly, our study provided evidence that cardiac-specific Rac1 deficiency improved cardiac function during endotoxemia. PI3K-mediated activation of Rac1 represents a novel signalling pathway by which LPS induces cardiomyocyte TNF-α expression and cardiac dysfunction (Fig. 9).
fig 9.
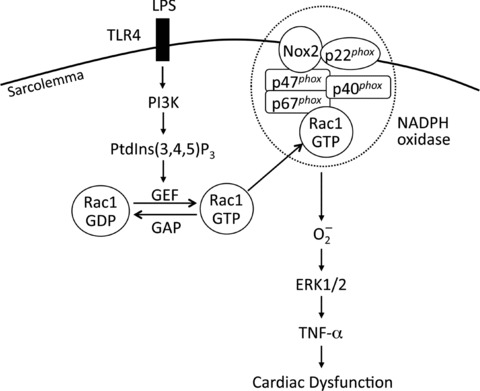
Schematic Rac1 signalling pathway leading to cardiomyocyte TNF-α expression and cardiac dysfunction during LPS stimulation. LPS activates Rac1 via TLR4/PI3K/PtdIns(3,4,5)P3/GEF signalling. Activation of Rac1 activates NADPH oxidase leading to production of O2−, phosphorylation of ERK1/2 MAPK, and expression of TNF-α. Increased myocardial TNF-α production results in cardiac dysfunction during endotoxemia. TLR4: toll-like receptor 4; GEF: guanine nucleotide exchange factor; GAP: GTPase-activating proteins.
Rho GTPases are a large family of proteins that include the Rac proteins (Rac1, 2 and 3) as well as RhoA and Cdc42. The role of Rho GTPases on TNF-α production during LPS stimulation has been studied, but the results differ depending on cell types and experimental conditions [15–17, 28]. For example, inhibition of Rho GTPase including Rac, RhoA and Cdc42 increased LPS-stimulated TNF-α production in macrophages [17]. In resting neutrophils, RhoA suppresses TNF-α production by inhibiting NF-κB activity [28]. Upon LPS stimulation, RhoA is activated and increases TNF-α expression, suggesting a dual role of RhoA in TNF-α production in human neutrophils [28]. Additionally, studies have shown that Rac1 activation promoted LPS-induced TNF-α expression in macrophages and Kupffer cells [15, 16]. Furthermore, Rac1 mediates myocardial TNF-α expression during LPS stimulation [20]. However, the role of Rac1 in cardiac dysfunction during endotoxemia remains unknown. In the present study, we demonstrated that Rac1 is rapidly and transiently activated by LPS in cultured neonatal cardiomyocytes and in the adult myocardium. Rac1 inhibition or deficiency blocked LPS-induced TNF-α production. Furthermore, myocardial TNF-α expression was decreased and cardiac function was significantly improved in cardiac-specific Rac1–/– mice during endotoxemia. Thus, both in vitro and in vivo evidence from our study demonstrated that LPS activates Rac1 and promotes cardiomyocyte TNF-α expression leading to cardiac dysfunction.
Rac GTPases are regulated by GEFs that promote the exchange of GDP for GTP, and GAPs that accelerate the hydrolysis of GTP. Available evidence suggests that Rac activation depends mainly on the activation of GEFs [21]. In this regard, Rac-activating GEFs such as Vav, Sos, PAK-interacting exchange factor (PIX), Switch-associated protein (SWAP)-70 and P-Rex, can be activated directly by PtdIns(3,4,5)P3, a lipid product of PI3K [21]. In the present study, PI3K activity, as determined by the production of PtdIns(3,4,5)P3, was significantly increased in cultured cardiomyocytes in vitro and in the myocardium in vivo during LPS stimulation. Inhibition of PI3K activity by a selective inhibitor, LY294006, significantly decreased Rac1-GTP levels and resulted in a concomitant decrease in TNF-α expression stimulated by LPS. These results indicate that PI3K is required for Rac1 activation in cardiomyocytes during LPS stimulation.
Activated Rac1 interacts with specific effectors that regulate diverse physiological functions. One of these effectors is p67phox, a subunit of NADPH oxidase [29]. A critical step for NADPH oxidase assembly and activation is the hetero-dimerization of Nox2 with p67phox[30]. Interestingly, the interaction between p67phox and Rac1 results in increased affinity of p67phox for Nox2 [31]. In addition, recent studies involving p67phox–Rac1 chimeras have reported that Rac1 induced an ‘activating’ conformational change in p67phox[32, 33]. Thus, Rac proteins are required for NADPH oxidase activation and superoxide production. NADPH oxidase is an important source of reactive oxygen species in the heart and activation of NADPH oxidase has been shown to contribute to the pathogenesis of cardiovascular diseases including: cardiac hypertrophy [34], hypertension [35], atherosclerosis [36] and heart failure after myocardial infarction [37, 38]. We have recently demonstrated that NADPH oxidase is activated in cardiomyocytes which results in myocardial TNF-α expression and cardiac dysfunction during endotoxemia [23]. In agreement with these data, the present study demonstrated that LPS increases O2− generation. Inhibition of NADPH oxidase activity significantly decreased LPS-induced TNF-α expression in cardiomyocytes. Furthermore, we showed that inhibition of PI3K activity decreased LPS-induced O2− production. Similarly, deficiency in Rac1 or overexpression of dominant-negative Rac1 significantly suppressed O2− generation and TNF-α expression in response to LPS stimulation. These results suggest that PI3K-mediated Rac1 activity promotes neonatal cardiomyocyte TNF-α expression induced by LPS via activation of NADPH oxidase.
MAPKs (p38, ERK1/2 and JNKs) are key signalling molecules involved in the regulation of many biological processes including inflammatory responses and the expression of pro-inflammatory cytokines. Indeed, activation of ERK1/2 and p38 MAPK regulates the expression of TNF-α in phagocytes during sepsis [15, 28, 39]. We have recently demonstrated that ERK1/2 and p38 MAPKs are downstream of NADPH oxidase signalling in LPS-induced TNF-α expression [23]. In the present study, we showed that LPS increased the phosphorylation of ERK1/2 and p38 in cultured neonatal cardiomyocytes. Inhibition of PI3K and Rac1 activity blocked LPS-stimulated ERK1/2 phosphorylation but had no effect on p38 activity. Moreover, inhibition of ERK1/2 activity decreased LPS-induced TNF-α production. Thus, PI3K-mediated Rac1 activity regulates LPS-induced TNF-α expression in cardiomyocytes via ERK1/2 activation.
In summary, the present study provides strong evidence that Rac1 activation is required for cardiomyocyte TNF-α expression and cardiac dysfunction during endotoxemia. Activation of Rac1 through PI3K increases NADPH oxidase and ERK1/2 activity, leading to increased myocardial TNF-α expression during LPS stimulation (Fig. 9). Our study suggests that Rac1 may represent a novel therapeutic target for TNF-α expression and myocardial dysfunction in sepsis.
Acknowledgments
This study was supported by an operating grant from the Canadian Institutes of Health Research (MOP-14653, Q.F.). Dr. Q.F. is a Career Investigator of the Heart and Stroke Foundation of Ontario. We thank Dr. Lamis Hammoud for a critical review of the manuscript, and Dr. Houxiang Hu and Mrs. Murong Liu for excellent technical support.
Conflict of interest
The authors confirm that there are no conflicts of interest
References
- 1.Angus DC, Wax RS. Epidemiology of sepsis: an update. Crit Care Med. 2001;29:S109–16. doi: 10.1097/00003246-200107001-00035. [DOI] [PubMed] [Google Scholar]
- 2.Court O, Kumar A, Parrillo JE. Clinical review: myocardial depression in sepsis and septic shock. Crit Care. 2002;6:500–8. doi: 10.1186/cc1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ward PA, Gao H. Sepsis, complement, and the dysregulated inflammatory response. J Cell Mol Med. 2009;13:4154–60. doi: 10.1111/j.1582-4934.2009.00893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parrillo JE, Burch C, Shelhamer JH, et al. A circulating myocardial depressant substance in humans with septic shock. Septic shock patients with a reduced ejection fraction have a circulating factor that depresses in vitro myocardial cell performance. J Clin Invest. 1985;76:1539–53. doi: 10.1172/JCI112135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peng T, Lu X, Lei M, et al. Inhibition of p38 MAPK decreases myocardial TNF-alpha expression and improves myocardial function and survival in endotoxemia. Cardiovasc Res. 2003;59:893–900. doi: 10.1016/s0008-6363(03)00509-1. [DOI] [PubMed] [Google Scholar]
- 6.Natanson C, Eichenholz PW, Danner RL, et al. Endotoxin and tumor necrosis factor challenges in dogs simulate the cardiovascular profile of human septic shock. J Exp Med. 1989;169:823–32. doi: 10.1084/jem.169.3.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tracey KJ, Beutler B, Lowry SF, et al. Shock and tissue injury induced by recombinant human cachectin. Science. 1986;234:470–4. doi: 10.1126/science.3764421. [DOI] [PubMed] [Google Scholar]
- 8.Grandel U, Fink L, Blum A, et al. Endotoxin-induced myocardial tumor necrosis factor-alpha synthesis depresses contractility of isolated rat hearts: evidence for a role of sphingosine and cyclooxygenase-2-derived thromboxane production. Circulation. 2000;102:2758–64. doi: 10.1161/01.cir.102.22.2758. [DOI] [PubMed] [Google Scholar]
- 9.Bozkurt B, Kribbs SB, Clubb FJ, Jr, et al. Pathophysiologically relevant concentrations of tumor necrosis factor-alpha promote progressive left ventricular dysfunction and remodeling in rats. Circulation. 1998;97:1382–91. doi: 10.1161/01.cir.97.14.1382. [DOI] [PubMed] [Google Scholar]
- 10.Meng X, Brown JM, Ao L, et al. Myocardial gene reprogramming associated with a cardiac cross-resistant state induced by LPS preconditioning. Am J Physiol. 1998;275:C475–83. doi: 10.1152/ajpcell.1998.275.2.C475. [DOI] [PubMed] [Google Scholar]
- 11.Oral H, Dorn GW, 2nd, Mann DL. Sphingosine mediates the immediate negative inotropic effects of tumor necrosis factor-alpha in the adult mammalian cardiac myocyte. J Biol Chem. 1997;272:4836–42. doi: 10.1074/jbc.272.8.4836. [DOI] [PubMed] [Google Scholar]
- 12.Hordijk PL. Regulation of NADPH oxidases: the role of Rac proteins. Circ Res. 2006;98:453–62. doi: 10.1161/01.RES.0000204727.46710.5e. [DOI] [PubMed] [Google Scholar]
- 13.Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–79. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 14.Haataja L, Groffen J, Heisterkamp N. Characterization of RAC3, a novel member of the Rho family. J Biol Chem. 1997;272:20384–8. doi: 10.1074/jbc.272.33.20384. [DOI] [PubMed] [Google Scholar]
- 15.Thakur V, Pritchard MT, McMullen MR, et al. Chronic ethanol feeding increases activation of NADPH oxidase by lipopolysaccharide in rat Kupffer cells: role of increased reactive oxygen in LPS-stimulated ERK1/2 activation and TNF-alpha production. J Leukoc Biol. 2006;79:1348–56. doi: 10.1189/jlb.1005613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanlioglu S, Williams CM, Samavati L, et al. Lipopolysaccharide induces Rac1-dependent reactive oxygen species formation and coordinates tumor necrosis factor-alpha secretion through IKK regulation of NF-kappa B. J Biol Chem. 2001;276:30188–98. doi: 10.1074/jbc.M102061200. [DOI] [PubMed] [Google Scholar]
- 17.Monick MM, Powers LS, Butler NS, et al. Inhibition of Rho family GTPases results in increased TNF-alpha production after lipopolysaccharide exposure. J Immunol. 2003;171:2625–30. doi: 10.4049/jimmunol.171.5.2625. [DOI] [PubMed] [Google Scholar]
- 18.Satoh M, Ogita H, Takeshita K, et al. Requirement of Rac1 in the development of cardiac hypertrophy. Proc Natl Acad Sci USA. 2006;103:7432–7. doi: 10.1073/pnas.0510444103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pracyk JB, Tanaka K, Hegland DD, et al. A requirement for the rac1 GTPase in the signal transduction pathway leading to cardiac myocyte hypertrophy. J Clin Invest. 1998;102:929–37. doi: 10.1172/JCI2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu H, Shan L, Peng T. Rac1 mediates sex difference in cardiac tumor necrosis factor-alpha expression via NADPH oxidase-ERK1/2/p38 MAPK pathway in endotoxemia. J Mol Cell Cardiol. 2009;47:264–74. doi: 10.1016/j.yjmcc.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 21.Welch HC, Coadwell WJ, Stephens LR, et al. Phosphoinositide 3-kinase-dependent activation of Rac. FEBS Lett. 2003;546:93–7. doi: 10.1016/s0014-5793(03)00454-x. [DOI] [PubMed] [Google Scholar]
- 22.Moldovan L, Mythreye K, Goldschmidt-Clermont PJ, et al. Reactive oxygen species in vascular endothelial cell motility. Roles of NAD(P)H oxidase and Rac1. Cardiovasc Res. 2006;71:236–46. doi: 10.1016/j.cardiores.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 23.Peng T, Lu X, Feng Q. Pivotal role of gp91phox-containing NADH oxidase in lipopolysaccharide-induced tumor necrosis factor-alpha expression and myocardial depression. Circulation. 2005;111:1637–44. doi: 10.1161/01.CIR.0000160366.50210.E9. [DOI] [PubMed] [Google Scholar]
- 24.Glogauer M, Marchal CC, Zhu F, et al. Rac1 deletion in mouse neutrophils has selective effects on neutrophil functions. J Immunol. 2003;170:5652–7. doi: 10.4049/jimmunol.170.11.5652. [DOI] [PubMed] [Google Scholar]
- 25.Rui T, Feng Q, Lei M, et al. Erythropoietin prevents the acute myocardial inflammatory response induced by ischemia/reperfusion via induction of AP-1. Cardiovasc Res. 2005;65:719–27. doi: 10.1016/j.cardiores.2004.11.019. [DOI] [PubMed] [Google Scholar]
- 26.Song W, Lu X, Feng Q. Tumor necrosis factor-alpha induces apoptosis via inducible nitric oxide synthase in neonatal mouse cardiomyocytes. Cardiovasc Res. 2000;45:595–602. doi: 10.1016/s0008-6363(99)00395-8. [DOI] [PubMed] [Google Scholar]
- 27.Bonnans C, Fukunaga K, Keledjian R, et al. Regulation of phosphatidylinositol 3-kinase by polyisoprenyl phosphates in neutrophil-mediated tissue injury. J Exp Med. 2006;203:857–63. doi: 10.1084/jem.20052143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fessler MB, Arndt PG, Just I, et al. Dual role for RhoA in suppression and induction of cytokines in the human neutrophil. Blood. 2007;109:1248–56. doi: 10.1182/blood-2006-03-012898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bosco EE, Mulloy JC, Zheng Y. Rac1 GTPase: a “Rac” of all trades. Cell Mol Life Sci. 2009;66:370–4. doi: 10.1007/s00018-008-8552-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dang PM, Cross AR, Babior BM. Assembly of the neutrophil respiratory burst oxidase: a direct interaction between p67phox and cytochrome b558. Proc Natl Acad Sci USA. 2001;98:3001–5. doi: 10.1073/pnas.061029698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nisimoto Y, Freeman JL, Motalebi SA, et al. Rac binding to p67phox. Structural basis for interactions of the Rac1 effector region and insert region with components of the respiratory burst oxidase. J Biol Chem. 1997;272:18834–41. doi: 10.1074/jbc.272.30.18834. [DOI] [PubMed] [Google Scholar]
- 32.Gorzalczany Y, Alloul N, Sigal N, et al. A prenylated p67phox-Rac1 chimera elicits NADPH-dependent superoxide production by phagocyte membranes in the absence of an activator and of p47phox: conversion of a pagan NADPH oxidase to monotheism. J Biol Chem. 2002;277:18605–10. doi: 10.1074/jbc.M202114200. [DOI] [PubMed] [Google Scholar]
- 33.Sarfstein R, Gorzalczany Y, Mizrahi A, et al. Dual role of Rac in the assembly of NADPH oxidase, tethering to the membrane and activation of p67phox: a study based on mutagenesis of p67phox-Rac1 chimeras. J Biol Chem. 2004;279:16007–16. doi: 10.1074/jbc.M312394200. [DOI] [PubMed] [Google Scholar]
- 34.Bendall JK, Cave AC, Heymes C, et al. Pivotal role of a gp91phox-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation. 2002;105:293–6. doi: 10.1161/hc0302.103712. [DOI] [PubMed] [Google Scholar]
- 35.Rajagopalan S, Kurz S, Munzel T, et al. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation: contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916–23. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Warnholtz A, Nickenig G, Schulz E, et al. Increased NADH-oxidase-mediated superoxide production in the early stages of atherosclerosis: evidence for involvement of the renin-angiotensin system. Circulation. 1999;99:2027–33. doi: 10.1161/01.cir.99.15.2027. [DOI] [PubMed] [Google Scholar]
- 37.Murdoch CE, Zhang M, Cave AC, et al. NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovasc Res. 2006;71:208–15. doi: 10.1016/j.cardiores.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 38.Gao L, Yin H, Smith RJ, et al. Role of kallistatin in prevention of cardiac remodeling after chronic myocardial infarction. Lab Invest. 2008;88:1157–66. doi: 10.1038/labinvest.2008.85. [DOI] [PubMed] [Google Scholar]
- 39.Rosengart MR, Arbabi S, Garcia I, et al. Interactions of calcium/calmodulin-dependent protein kinases (CaMK) and extracellular-regulated kinase (ERK) in monocyte adherence and TNF-alpha production. Shock. 2000;13:183–9. doi: 10.1097/00024382-200003000-00003. [DOI] [PubMed] [Google Scholar]