

Abstract
Growth hormone releasing hormone (GHRH) and its receptors are expressed in a wide variety of human tumours and established cancer cell lines and are involved in carcinogenesis. In addition, GHRH antagonists exert an antitumour activity in experimental cancer models. Recent studies indicate that the mechanisms involved in the mediation of the effects of GHRH include the regulation of the metabolism of the reactive oxygen species. This work demonstrates the expression of GHRH receptors and GHRH in the A549 human lung cancer cell line and shows that the mitogenic effect of GHRH in these cells is dependent on the activation of the extracellular receptor kinase (ERK)1/2 pathway. The action of GHRH can be suppressed by GHRH antagonist MZ-5–156 and mitogen activated protein kinase (MAPK) inhibitor PD 098059. These results are reflected in the effect in the proliferating cell nuclear antigen. In addition, our study shows that GHRH increases the expression of the inducible nitric oxide synthase, an enzyme which is strongly involved in various human diseases, including cancer and augments key intracellular regulators of its expression, such as pNF (nuclear factor)κBp50 and cyclooxygenase 2. GHRH antagonist MZ-5–156 counteracts the effects of GHRH in these studies, indicating that this class of peptide antagonists may be useful for the treatment of diseases related to increased oxidative and nitrosative stress.
Keywords: growth factors, oxidative stress, nitrosative stress
Introduction
Deregulation of cell cycle progression and differentiation contributes to the malignant transformation. Growth factors such as neuropeptide growth hormone releasing hormone (GHRH) are involved in the maintenance of these phenomena, as was recently shown by the knocking down of this hormone in breast, prostate and lung cancer cell lines [1]. When the tumoral mitogenic signals of GHRH were disrupted by specifically designed siRNA, the growth of these cancers was rapidly suppressed. In an endeavour to develop new anticancer agents, one of us (A.V.S.) synthesized GHRH antagonists [2]. These compounds can antagonize the mitogenic effects of GHRH in cancers, and thus possess strong anticancer activity in diverse tumours [3]. The action of GHRH antagonists is mediated through the pituitary GHRH receptor, as well as its splice variant 1 (SV1), which can exert a ligand independent activity [4, 5].
Recent study showed that GHRH and GHRH antagonists can influence the reduction/oxidation (redox) status of LNCaP cancer cells [6]. GHRH increased the production of the reactive oxygen species (ROS) and enhanced the lipid and protein oxidation levels of these cells. In contrast, GHRH antagonists showed anti-oxidative activity and decreased the oxidation levels of these cells [6]. The ability of these compounds to reduce the metabolism of the ROS is of major importance, because these species are strongly involved in the initiation, promotion and progression of cancers [7, 8]. Another oxidant species is the peroxynitrite, which results from the reaction of nitric oxide and superoxide [9]. The chemistry of nitric oxide and the related nitrogen oxygen species as well as their endogenously production adds a new dimension to redox pathology and pathophysiology and increases the complexity of the understanding of free radical biology [10].
Nitric oxide and nitric oxide derived reactive nitrogen species are an essential part of the immunoresponse and physiological signal transduction pathways [11–13]. They are also involved in the pathogenesis of a broad variety of diseases [14–19] through the induction of oxidative and nitrosative stress [10, 20–22]. Nitric oxide is produced by the family of the nitric oxide synthases. Inducible nitric oxide synthase (iNOS) is one of the three isoforms of nitric oxide synthase, which catalyses the oxidative deamination of L-arginine to produce citrulline and nitric oxide [23].
The iNOS expression in tumour cells is associated with cell proliferation and enhancement of cancer migration and invasion [24]. The importance of the nitric oxide expression in the pathogenesis of various diseases including cancer is underlined by the fact that various nitric oxide inhibitors have been developed [20] in order to modulate the intracellular levels of this molecule [25].
The influence of GHRH and its antagonists on the expression of the iNOS, which is strongly involved in tumour progression [16, 24, 26], has not been investigated so far. In the present study, we report the expression of GHRH receptors and GHRH in A549 lung cancer cells and show the activation of ERK1/2 by GHRH and the suppression of this pathway by GHRH antagonist. We also measured the proliferation rate of these cells and demonstrated that it is ERK1/2 dependent. GHRH increased the proliferation rate of A549 cells whereas GHRH antagonist MZ-5–156 and a selective ERK1/2 inhibitor (PD098059) decreased it. These results were also reflected by the expression of the proliferating cell nuclear antigen, a major proliferative marker.
Cells exposed to GHRH expressed lower amounts of p53, whereas those treated with MZ-5–156 increased the expression of this tumour suppressor with anti-oxidative function. We also demonstrated the induction of the expression of the iNOS by GHRH and the inhibition of its expression by GHRH antagonist MZ-5–156. Finally, GHRH was shown to activate the NF-κB transcription factor as well as the cyclooxygenase 2 protein, which are both involved in the regulation of the nitric oxide synthase.
Material and methods
Peptides and chemicals
GHRH (1–29)NH2 and GHRH antagonist MZ-5–156[PhAc-Tyr1, D-Arg2, Phe (4-CI)6, Abu15, Nle27 hGHRH (1–28)Agm, where Abu is α-aminobutyric acid, Agm is agmatine, Nle is norleucine, PhAc is phenylacetyl, were synthesized in our laboratory by solid phase methods [2]. GHRH(1–29)NH2 and GHRH antagonist MZ-5–156 were dissolved in dimethyl sulfoxide (DMSO) and diluted with incubation media. The final concentration of DMSO in medium was less than 0.1%. The selective ERK1/2 inhibitor PD098059 was purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture
Prostate cancer cell line LNCaP, mouse fibroblasts 3T3 and lung cancer cells A549 were obtained from American Type Culture Collection (Manassas, VA, USA) and cultured at 37°C in a humidified 95% air/5% CO2 atmosphere. LNCaP cells were cultured in RPMI-1640 medium supplemented with 10% foetal bovine serum and 1% antibiotics/antimycotics. The medium of 3T3, T47D and A549 cells was replaced with DMEM. All the cell culture reagents were purchased from GIBCO (Carlsbad, CA, USA).
Protein isolation and Western blot assay
The proteins were isolated from the cells using CelyticM Lysis Reagent (Sigma-Aldrich) according to manufacturer’s instructions. Protein matched samples were separated by 12% sodium dodecyl sulphate Tris-HCl gels. Wet transfer was used to transfer the proteins onto nitrocellulose membranes (Biorad, Hercules, CA, USA). The membranes were incubated for 1 hr in 5% non-fat dry milk in phosphate-buffered saline – 0.1% (v/v) Tween 20. The blots were then incubated at 4°C overnight with the appropriate antibodies. The signal for the immunoreactive proteins was developed with peroxidase-conjugated secondary antibodies (Santa Cruz, CA, USA; Cell Signaling, Danvers, MA, USA) and visualized by exposure to chemiluminescence substrate (Amersham Biotechnologies, Piscataway, NJ, USA). The β-actin signal was used as a loading control unless otherwise stated. The proliferating cell nuclear antigen (PCNA) (#2586), iNOS (# 2977), p53 (#9282), cyclooxygenase (COX)-2 (#4842) and phospho-MAPK (#9101) antibodies were obtained from Cell Signaling, the GHRHR antibody (ab 28692–100) from Abcam (Cambridge, MA, USA) and the NFK50 (sc-1191), pNFK50 (sc-33022), β actin (sc-47778), ERK2 (sc-81457) and growth hormone-releasing hormone receptor (GHRHR) (sc- 10280) antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Activation of mitogen activated protein kinase pathway by GHRH
A549 human lung carcinoma cells were grown in DMEM with 10% foetal bovine serum. Before this assay, the medium was replaced with DMEM without foetal bovine serum and the cells were treated or not with 1 μM GHRH (1–29)NH2 for 24 hrs in serum free media. The cells were harvested in Cell Lysis Buffer (Sigma-Aldrich) containing proteinase inhibitor cocktail. The cell lysates (100 μl) were separated by electrophoresis according to their molecular weight. The proteins were then transferred to nitrocellulose membranes and probed for phospho-MAPK. The blots were stripped (Restore Plus Western Blot Stripping Buffer, Thermo Scientific, Waltham, MA, USA) and probed for ERK 2 and β-actin.
Cell proliferation rate assay
The rate of cell proliferation was calculated by seeding 10,000 cells in six-well plates and after incubation for 72 hrs counting them under light microscope. Trypan blue assay was used in order to distinguish the alive from the dead cells.
Quantitative analysis of the immunoblot assay
The protein band signals were quantified by Adobe Photoshop and normalized to β-actin signal. In the case of the ERK1/2 and NFκBp50 activation, the bands were normalized to ERK2 and NF-κB50, respectively. The intensity of the bands was equal to their mean value multiplied by their pixel value (absolute intensity). Relative intensity (R.I.) of each band was calculated by dividing its absolute intensity by the absolute intensity of the control band (β-actin, ERK2 or NFκBp50).
Statistical analysis
The data are expressed as the mean ± S.E. Statistical evaluation of the results was performed with a one way ANOVA followed by the least significant difference test. The differences were considered significant at P < 0.05.
Results
Expression of GHRH Receptors and GHRH in A549 human lung cancer cell line
The expression of GHRH receptors was examined by Western blot in A549 human lung cancer cells, using 3T3 mouse fibroblast line as a negative [4, 27] and LNCaP as a positive control [1]. The antibody used recognized both type of GHRH receptors (pGHRHR and SV1). Figure 1B also shows the lack of GHRH-R(s) expression in 3T3 cells. T47D cells which express both types of GHRH receptors [1, 28] were used as positive control. In addition, we detected the expression of the GHRH in A549 cells, using LNCaP and T47D cancer cells as positive controls [1]. The results are shown in Figure 1C.
fig 1.
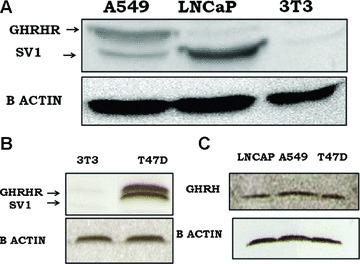
(A) Western blot analysis of the expression of GHRH receptor(s) in A549 lung cancer, LNCaP prostate cancer cell line and 3T3 mouse fibroblast cell line. LNCaP and 3T3 cells were used as positive and negative controls, respectively. (B) Western blot analysis of the expression of GHRH receptor(s) in T47D breast cancer cells and 3T3 mouse fibroblast cell line. T47D cells were used as positive control. (C) Western blot analysis of the expression of GHRH in LNCaP, A549 and T47D cancer cell lines. LNCaP and T47D cells were used as positive controls.
Activation of the ERK1/2 pathway by GHRH in A549 lung cancer cells
We investigated whether GHRH (1–29)NH2 at 0.1 μM and 1 μM concentrations can activate the ERK1/2 pathway in A549 cells. The results show that this hypothalamic hormone activates this pathway at both concentrations, with the R.I. being 0.926 and 1.081, respectively. We also examined the effect of the GHRH antagonist MZ-5–156 on this pathway. GHRH antagonist suppressed the activation of this pathway at 0.1 μM and 1 μM concentrations with the R.I. being 0.379 and 0.339, respectively. The R.I. of the control cells was 0.706. The results are shown in Figure 2.
fig 2.
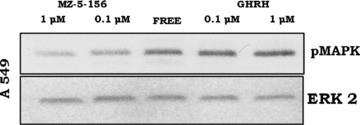
Western blot analysis of the pERK1/2 after incubation of the A549 cells with GHRH antagonist MZ-5–156 and GHRH. The protein levels were normalized to ERK2 signal (loading control). The blot is representative of two independent experiments.
Effect of GHRH(1–29)NH2, GHRH antagonist MZ-5–156 and ERK1/2 inhibitor on the proliferation of A549 cells and 3T3 cells in vitro
A549 lung cancer cell line cultured in vitro was exposed to two concentrations of GHRH(1–29)NH2, GHRH antagonist MZ-5–156 as well as in 10 μM ERK1/2 inhibitor. At the dose of 0.1 and 1 μM, GHRH(1–29)NH2 increased the proliferation rate of the cells by 30.6% and 44.5%, respectively. GHRH antagonist MZ-5–156 at the dose of 0.1 or 1 μM decreased the proliferation rate of A549 cells by 16.1% and 28.4%, respectively. In addition, the ERK1/2 inhibitor in 10 μM final concentration suppressed the proliferation of these cells by 30.6%. The results are shown in Figure 3A. The proliferation of the 3T3 cells, which do not express GHRH receptors, was not influenced by GHRH, MZ-1–156 or the ERK inhibitor. The results are shown in Figure 3B.
fig 3.
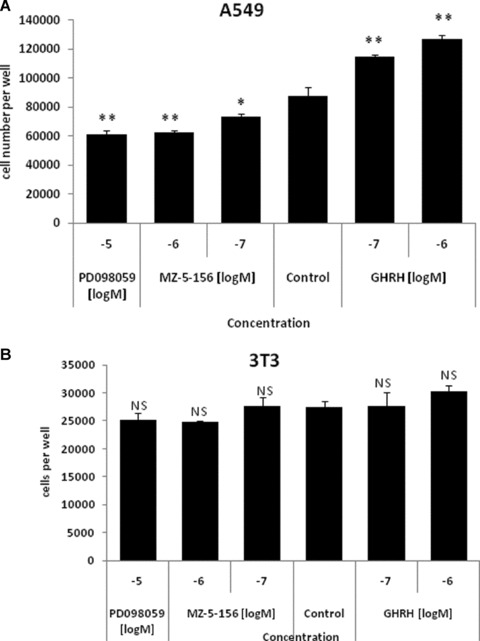
(A) Proliferation rate of the A549 cells exposed to 0.1 μM and 1 μM GHRH (1–29)NH2 and MZ-5–156 as well as 10 μM MAPK inhibitor. *P < 0.01 versus control cells **P < 0.001 versus control cells. NS: non-significant. (B) Proliferation rate of the 3T3 cells exposed to 0.1 μM and 1 μM GHRH (1–29)NH2 and MZ-5–156 as well as 10 μM MAPK inhibitor. NS: non–significant.
Expression of proliferating cell nuclear antigen in A549 and 3T3 cells after treatment with GHRH or GHRH antagonist
A549 cancer cells cultured in vitro were exposed to two concentrations of GHRH and GHRH antagonist MZ-5–156. GHRH antagonist at 0.1 and 1 μM decreased the expression of PCNA, (R.I.: 0.53, 0.48) compared to the control cells. The results are shown in Figure 4 (upper panel). GHRH at concentrations of 0.1 and 1 μM, increased the expression of this marker (R.I.: 0.75, 0.85). The relative intensity of the control cells was 0.68. The expression of this proliferative marker in 3T3 cells was not influenced by the peptides. The results are shown in Figure 4 (lower panel).
fig 4.
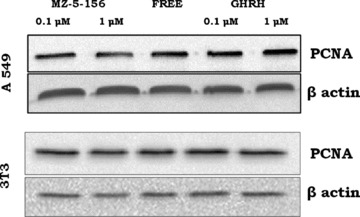
Expression of PCNA by A549 (top) or 3T3 (bottom) cells after exposure to 0.1 μM or 1 μM GHRH (1–29)NH2 and 0.1 μM or 1 μM GHRH antagonist MZ-5–156. Protein levels were normalized to β actin signal (loading control). The blot is representative of two independent experiments.
Expression of wild-type P53 in A549 cells after treatment with GHRH or GHRH antagonist
The expression of the wild-type P53 was measured in the A549 lung cancer cells after exposure to 0.1 μM and 1 μM GHRH or GHRH antagonist MZ-5–156. The expression of this marker was reduced after exposure to GHRH (1–29)NH2 (R.I.: 0.95, 0.94), respectively, and increased after incubation of these cells with 0.1 μM MZ-5–156 (R.I.:1.55) compared to the control cells (R.I.: 1.34). The results are shown in Figure 5.
fig 5.
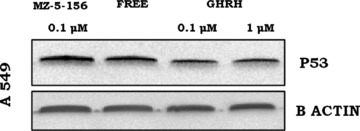
Expression of P53 by A549 cells after exposure to 0.1 μM or 1 μM GHRH (1–29)NH2 and 0.1 μM GHRH antagonist MZ-5–156. Protein levels were normalized to β actin signal (loading control). The blot is representative of two independent experiments.
Effect of GHRH (1–29)NH2 and GHRH antagonist MZ-5–156 on the expression on iNOS in A549 lung cancer cells in vitro
A549 lung cancer cells cultured in vitro were exposed to GHRH and GHRH antagonist MZ-5–156 and the expression levels of iNOS were detected by Western blot. iNOS protein expression was lower in cells exposed to 0.1 μM and 1 μM MZ-5–156 (R.I.: 0.097, 0.086) compared to the control cells (R.I.: 0.149).Treatment of A549 cells with two concentrations of GHRH (0.1 μM and 1 μM) resulted to an increase of the expression of this enzyme, with the relative intensity being (R.I.: 0.164 and 0.196). The results are shown in Figure 6. We also incubated 3T3 cells with 0.1 and 1 μM GHRH and the results indicate that this hormone did not appreciably influence the expression of this enzyme.
fig 6.
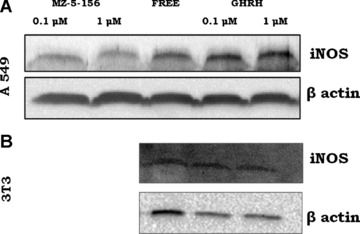
(A) Expression of iNOS by A549 cells after exposure to 0.1 μM or 1 μM GHRH (1–29)NH2 and 0.1 μM GHRH antagonist MZ-5–156. Protein levels were normalized to β actin signal (loading control). The blot is representative of two independent experiments (B) Expression of iNOS by 3T3 cells after exposure to 0.1 μM or 1 μM GHRH (1–29)NH2. Protein levels were normalized to β actin signal (loading control). The blot is representative of two independent experiments,
Effect of GHRH (1–29)NH2 and GHRH antagonist MZ-5–156 on the expression of COX-2 and the activation of the NF-κBp50 in A549 lung cancer cells in vitro
A549 lung cancer cells cultured in vitro were exposed to 1 μM of GHRH (1–29)NH2 and 1 μM antagonist MZ-5–156. The expression levels of COX-2, NFκBp50 and phosphorylated NFκBp50 were detected by Western blot. The results are shown in Figure 7. The expression of COX-2 and phosphorylated NFκBp50 was higher in the cells exposed to the GHRH (R.I.: 1.54, 3.63) and lower to the cells exposed to MZ-5–156 (R.I.: 0.65 and 1.96) as compared to the controls (R.I.: 0.98 and 2.44).
fig 7.
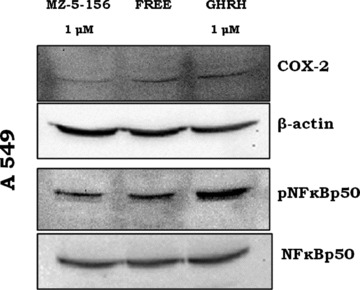
Effect of GHRH and MZ-5–156 on the expression of the COX-2 (upper panel) and the activation of the NF-κBp50 (lower panel) in A549 cells. The blot is representative of two independent experiments.
Discussion
GHRH has been shown to possess a growth factor activity in a variety of experimental cancers and established cancer cell lines. This activity is exerted through the binding to GHRH receptor(s) [29]. However, the signalling pathways which mediate these effects are not completely understood. We have recently demonstrated in transfected cancer cells that the SV1 receptor can activate the ERK1/2 pathway [30], similar to the pituitary type GHRH-R[2], and that the pituitary type receptor can activate the JAK/STAT pathway [31]. GHRH antagonists can counteract these effects.
In addition to the endocrine related pathways, GHRH can also increase the redox status of the LNCaP prostate cancer cell lines through an increase in the metabolism of the ROS [6]. These species have been shown to influence various human diseases and malignancies [32–35]. The reactive nitrogen species can also contribute to protein and lipid modifications as well as deregulate intracellular signalling pathways [36, 37]. The influence of GHRH on the expression of iNOS has not been investigated before.
In the present study, we demonstrated by Western blot the expression of the GHRH-R, SV1 receptor and GHRH in A549 lung cancer cells. When we incubated these cells with GHRH and its antagonists, GHRH activated the ERK1/2 pathway, whereas GHRH antagonist suppressed it. We also measured the in vitro proliferation of these cells after incubation with GHRH, GHRH antagonist and ERK1/2 inhibitor.
We showed that the proliferation of these cells depends on the activation of the ERK1/2 pathway, because a selective MAPK inhibitor and the GHRH antagonist MZ-5–156 suppressed it. In contrast, GHRH, by activating this pathway [38], can increase the proliferation of these cells. These results were also reflected by the expression of the proliferating cell nuclear antigen, a major proliferative marker, with GHRH increasing its expression and the antagonist reducing it. In order to show the specificity of these effects, we treated 3T3 cells, which do not express GHRH receptors, with GHRH and its antagonist. These cells were not influenced by these peptides or by the MAPK inhibitor, as shown not only by their proliferation rate, but also by the expression of the PCNA. Previous studies reported that MAPKs are important regulators of iNOS-nitric oxide expression and that activation of the ERK pathway augments the expression of the iNOS [39, 40]. In vivo studies showed a reduced expression of PCNA and iNOS in animal models after anti-oxidant therapy [41].
We have also detected by Western blots the expression of the wild-type p53 and demonstrated that cells treated with MZ-5–156 show higher expression of this tumour suppressor, whereas cells exposed to GHRH expressed lower amount of p53. These results are consistent with our previous results in LNCaP prostate cell line [6]. It has been previously demonstrated that a balance between p53 and MAPK pathways is critical for preventing or promoting initiation of tumours [42] and that P53 can trigger strong anti-oxidative intracellular mechanisms [43, 44], which involve a down-regulation of inducible nitric oxide and cyclooxygenase 2 [45].
The expression of the iNOS was strongly increased by GHRH in the A549 cells, whereas GHRH antagonist MZ-5–156 reduced it. The activation of ERK1/2, which occurred upon the binding of GHRH to its receptors [30], has been previously shown to increase the expression of the iNOS [46–50] and nitric oxide [12, 21]. GHRH did not influence the expression of iNOS in 3T3 cells, which do not respond to GHRH or its antagonists.
GHRH also stimulated the NF-κB transcription factor, which is not only known to activate angiogenic factors [51] but also the expression of genes involved in encoding enzymes in the prostaglandin-synthesis pathway such as COX-2 and the iNOS [52]. A direct up-regulation of iNOS by COX-2 has also been proposed [53] as well as a model of direct interaction among these proteins [54]. In addition the activation of NF-κB induces the iNOS expression [23, 55–57].
Previous studies have demonstrated the role of nitric oxide in tumour promotion and progression by mediating critical processes, including angiogenesis, tumour cell growth and invasion [21]. Our study shows for the first time that GHRH can activate the iNOS expression and that GHRH antagonists inhibit it; thus GHRH antagonist might be useful for the treatment of diseases related to increased oxidative and nitrosative stress.
Acknowledgments
This work was supported by the Medical Research Service of the Veterans Affairs Department, University of Miami Miller School of Medicine, Departments of Pathology and Medicine, Division of Hematology/ Oncology, the South Florida Veterans Affairs Foundation for Research and Education (A.V.S.). N.B. would like to thank Dr. Harry Ischiropoulos (University of Pennsylvania, Philadelphia, PA, USA) for inspiration and useful suggestions.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Barabutis N, Schally AV. Knocking down gene expression for growth hormone-releasing hormone inhibits proliferation of human cancer cell lines. Br J Cancer. 2008;98:1790–6. doi: 10.1038/sj.bjc.6604386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schally AV, Varga JL, Engel JB. Antagonists of growth-hormone-releasing hormone: an emerging new therapy for cancer. Nat Clin Pract Endocrinol Metab. 2008;4:33–43. doi: 10.1038/ncpendmet0677. [DOI] [PubMed] [Google Scholar]
- 3.Schally AV. New approaches to the therapy of various tumors based on peptide analogues. Horm Metab Res. 2008;40:315–22. doi: 10.1055/s-2008-1073142. [DOI] [PubMed] [Google Scholar]
- 4.Kiaris H, Chatzistamou I, Schally AV, et al. Ligand-dependent and -independent effects of splice variant 1 of growth hormone-releasing hormone receptor. Proc Natl Acad Sci USA. 2003;100:9512–7. doi: 10.1073/pnas.1533185100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barabutis N, Tsellou E, Schally AV, et al. Stimulation of proliferation of MCF-7 breast cancer cells by a transfected splice variant of growth hormone-releasing hormone receptor. Proc Natl Acad Sci USA. 2007;104:5575–9. doi: 10.1073/pnas.0700407104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barabutis N, Schally AV. Antioxidant activity of growth hormone-releasing hormone antagonists in LNCaP human prostate cancer line. Proc Natl Acad Sci USA. 2008;105:20470–5. doi: 10.1073/pnas.0811209106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Finkel T, Serrano M, Blasco MA. The common biology of cancer and ageing. Nature. 2007;448:767–74. doi: 10.1038/nature05985. [DOI] [PubMed] [Google Scholar]
- 8.Schumacker PT. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell. 2006;10:175–6. doi: 10.1016/j.ccr.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 9.Szabo C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6:662–80. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 10.Wink DA, Mitchell JB. Nitric oxide and cancer: an introduction. Free Radic Biol Med. 2003;34:951–4. doi: 10.1016/s0891-5849(02)01362-x. [DOI] [PubMed] [Google Scholar]
- 11.Gow AJ, Ischiropoulos H. Nitric oxide chemistry and cellular signaling. Journal of Cellular Physiology. 2001;187:277–82. doi: 10.1002/jcp.1085. [DOI] [PubMed] [Google Scholar]
- 12.Thomas DD, Ridnour LA, Isenberg JS, et al. The chemical biology of nitric oxide: implications in cellular signaling. Free Radic Biol Med. 2008;45:18–31. doi: 10.1016/j.freeradbiomed.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xie K, Huang S. Regulation of cancer metastasis by stress pathways. Clin Exp Metastasis. 2003;20:31–43. doi: 10.1023/a:1022590402748. [DOI] [PubMed] [Google Scholar]
- 14.Hofseth LJ, Saito S, Hussain SP, et al. Nitric oxide-induced cellular stress and p53 activation in chronic inflammation. Proc Natl Acad Sci USA. 2003;100:143–8. doi: 10.1073/pnas.0237083100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ischiropoulos H, Beckman JS. Oxidative stress and nitration in neurodegeneration: cause, effect, or association. J Clin Invest. 2003;111:163–9. doi: 10.1172/JCI17638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hofseth LJ, Hussain SP, Wogan GN, et al. Nitric oxide in cancer and chemoprevention. Free Radic Biol Med. 2003;34:955–68. doi: 10.1016/s0891-5849(02)01363-1. [DOI] [PubMed] [Google Scholar]
- 17.Grunewald T, Beal MF. NOS knockouts and neuroprotection. Nat Med. 1999;5:1354–5. doi: 10.1038/70918. [DOI] [PubMed] [Google Scholar]
- 18.Tatsuta T, Langer T. Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J. 2008;27:306–14. doi: 10.1038/sj.emboj.7601972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calabrese V, Mancuso C, Calvani M, et al. Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci. 2007;8:766–75. doi: 10.1038/nrn2214. [DOI] [PubMed] [Google Scholar]
- 20.Coulter JA, McCarthy HO, Xiang J, et al. Nitric oxide–a novel therapeutic for cancer. Nitric Oxide. 2008;19:192–8. doi: 10.1016/j.niox.2008.04.023. [DOI] [PubMed] [Google Scholar]
- 21.Wink DA, Ridnour LA, Hussain SP, et al. The reemergence of nitric oxide and cancer. Nitric Oxide. 2008;19:65–7. doi: 10.1016/j.niox.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hussain SP, He P, Subleski J, et al. Nitric oxide is a key component in inflammation-accelerated tumorigenesis. Cancer Res. 2008;68:7130–6. doi: 10.1158/0008-5472.CAN-08-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen J, Yan Y, Li J, et al. Differential requirement of signal pathways for benzo[a]pyrene (B[a]P)-induced nitric oxide synthase (iNOS) in rat esophageal epithelial cells. Carcinogenesis. 2005;26:1035–43. doi: 10.1093/carcin/bgi052. [DOI] [PubMed] [Google Scholar]
- 24.Fukumura D, Kashiwagi S, Jain RK. The role of nitric oxide in tumour progression. Nat Rev Cancer. 2006;6:521–34. doi: 10.1038/nrc1910. [DOI] [PubMed] [Google Scholar]
- 25.Muscara MN, Wallace JL. Nitric Oxide. V. therapeutic potential of nitric oxide donors and inhibitors. Am J Physiol. 1999;276:G1313–6. doi: 10.1152/ajpgi.1999.276.6.G1313. [DOI] [PubMed] [Google Scholar]
- 26.Xu W, Liu LZ, Loizidou M, et al. The role of nitric oxide in cancer. Cell Res. 2002;12:311–20. doi: 10.1038/sj.cr.7290133. [DOI] [PubMed] [Google Scholar]
- 27.Barabutis N, Siejka A, Schally AV. Effects of growth hormone-releasing hormone and its agonistic and antagonistic analogs in cancer and non-cancerous cell lines. Int J Oncol. 2010;36:1285–9. doi: 10.3892/ijo_00000613. [DOI] [PubMed] [Google Scholar]
- 28.Siriwardana G, Bradford A, Coy D, et al. Autocrine/paracrine regulation of breast cancer cell proliferation by growth hormone releasing hormone via Ras, Raf, and mitogen-activated protein kinase. Mol Endocrinol. 2006;20:2010–9. doi: 10.1210/me.2005-0001. [DOI] [PubMed] [Google Scholar]
- 29.Kiaris H. GHRH analogs and cancer. Comb Chem High Throughput Screen. 2006;9:161. doi: 10.2174/138620706776055476. [DOI] [PubMed] [Google Scholar]
- 30.Barabutis N, Siejka A, Schally AV, et al. Activation of mitogen-activated protein kinases by a splice variant of GHRH receptor. J Mol Endocrinol. 2010;44:127–34. doi: 10.1677/JME-09-0121. [DOI] [PubMed] [Google Scholar]
- 31.Siejka A, Schally AV, Barabutis N. Activation of Janus kinase/signal transducer and activator of transcription 3 pathway by growth hormone-releasing hormone. Cell Mol Life Sci. 2010;67:959–64. doi: 10.1007/s00018-009-0224-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–95. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 33.Pelicano H, Lu W, Zhou Y, et al. Mitochondrial dysfunction and reactive oxygen species imbalance promote breast cancer cell motility through a CXCL14-mediated mechanism. Cancer Res. 2009;69:2375–83. doi: 10.1158/0008-5472.CAN-08-3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar B, Koul S, Khandrika L, et al. Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res. 2008;68:1777–85. doi: 10.1158/0008-5472.CAN-07-5259. [DOI] [PubMed] [Google Scholar]
- 35.de Magalhaes JP, Church GM. Cells discover fire: employing reactive oxygen species in development and consequences for aging. Exp Gerontol. 2006;41:1–10. doi: 10.1016/j.exger.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 36.Schetter AJ, Heegaard NH, Harris CC. Inflammation and cancer: interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis. 2010;31:37–49. doi: 10.1093/carcin/bgp272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones LE, Jr, Ying L, Hofseth AB, et al. Differential effects of reactive nitrogen species on DNA base excision repair initiated by the alkyladenine DNA glycosylase. Carcinogenesis. 2009;30:2123–9. doi: 10.1093/carcin/bgp256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mayo KE, Miller T, DeAlmeida V, et al. Regulation of the pituitary somatotroph cell by GHRH and its receptor. Recent Prog Horm Res. 2000;55:237–66. [PubMed] [Google Scholar]
- 39.Liu X, Jana M, Dasgupta S, et al. Human immunodeficiency virus type 1 (HIV-1) tat induces nitric-oxide synthase in human astroglia. J Biol Chem. 2002;277:39312–9. doi: 10.1074/jbc.M205107200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chan ED, Riches DW. IFN-gamma + LPS induction of iNOS is modulated by ERK, JNK/SAPK, and p38(mapk) in a mouse macrophage cell line. Am J Physiol Cell Physiol. 2001;280:C441–50. doi: 10.1152/ajpcell.2001.280.3.C441. [DOI] [PubMed] [Google Scholar]
- 41.Chyu KY, Babbidge SM, Zhao X, et al. Differential effects of green tea-derived catechin on developing versus established atherosclerosis in apolipoprotein E-null mice. Circulation. 2004;109:2448–53. doi: 10.1161/01.CIR.0000128034.70732.C2. [DOI] [PubMed] [Google Scholar]
- 42.Ueda K, Arakawa H, Nakamura Y. Dual-specificity phosphatase 5 (DUSP5) as a direct transcriptional target of tumor suppressor p53. Oncogene. 2003;22:5586–91. doi: 10.1038/sj.onc.1206845. [DOI] [PubMed] [Google Scholar]
- 43.Liu B, Chen Y, St Clair DK. ROS and p53: a versatile partnership. Free Radic Biol Med. 2008;44:1529–35. doi: 10.1016/j.freeradbiomed.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sablina AA, Budanov AV, Ilyinskaya GV, et al. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005;11:1306–13. doi: 10.1038/nm1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gallo O, Schiavone N, Papucci L, et al. Down-regulation of nitric oxide synthase-2 and cyclooxygenase-2 pathways by p53 in squamous cell carcinoma. Am J Pathol. 2003;163:723–32. doi: 10.1016/S0002-9440(10)63699-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li QF, Zhu YS, Jiang H. Isoflurane preconditioning activates HIF-1alpha, iNOS and Erk1/2 and protects against oxygen-glucose deprivation neuronal injury. Brain Res. 2008;1245:26–35. doi: 10.1016/j.brainres.2008.09.069. [DOI] [PubMed] [Google Scholar]
- 47.Xu Z, Wang BR, Wang X, et al. ERK1/2 and p38 mitogen-activated protein kinase mediate iNOS-induced spinal neuron degeneration after acute traumatic spinal cord injury. Life Sci. 2006;79:1895–905. doi: 10.1016/j.lfs.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 48.Yuan Z, Feng W, Hong J, et al. p38MAPK and ERK promote nitric oxide production in cultured human retinal pigmented epithelial cells induced by high concentration glucose. Nitric Oxide. 2009;20:9–15. doi: 10.1016/j.niox.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 49.Park GH, Jeon SJ, Ryu JR, et al. Essential role of mitogen-activated protein kinase pathways in protease activated receptor 2-mediated nitric-oxide production from rat primary astrocytes. Nitric Oxide. 2009;21:110–9. doi: 10.1016/j.niox.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 50.Ridnour LA, Thomas DD, Switzer C, et al. Molecular mechanisms for discrete nitric oxide levels in cancer. Nitric Oxide. 2008;19:73–6. doi: 10.1016/j.niox.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–6. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 52.Mantovani A, Allavena P, Sica A, et al. Cancer-related inflammation. Nature. 2008;454:436–44. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 53.Hori M, Kita M, Torihashi S, et al. Upregulation of iNOS by COX-2 in muscularis resident macrophage of rat intestine stimulated with LPS. Am J Physiol Gastrointest Liver Physiol. 2001;280:G930–8. doi: 10.1152/ajpgi.2001.280.5.G930. [DOI] [PubMed] [Google Scholar]
- 54.Xuan YT, Guo Y, Zhu Y, et al. Mechanism of cyclooxygenase-2 upregulation in late preconditioning. J Mol Cell Cardiol. 2003;35:525–37. doi: 10.1016/s0022-2828(03)00076-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adams V, Nehrhoff B, Spate U, et al. Induction of iNOS expression in skeletal muscle by IL-1beta and NFkappaB activation: an in vitro and in vivo study. Cardiovasc Res. 2002;54:95–104. doi: 10.1016/s0008-6363(02)00228-6. [DOI] [PubMed] [Google Scholar]
- 56.Xie QW, Kashiwabara Y, Nathan C. Role of transcription factor NF-kappa B/Rel in induction of nitric oxide synthase. J Biol Chem. 1994;269:4705–8. [PubMed] [Google Scholar]
- 57.Lee JK, Choi SS, Won JS, et al. The regulation of inducible nitric oxide synthase gene expression induced by lipopolysaccharide and tumor necrosis factor-alpha in C6 cells: involvement of AP-1 and NFkappaB. Life Sci. 2003;73:595–609. doi: 10.1016/s0024-3205(03)00317-5. [DOI] [PubMed] [Google Scholar]