

Abstract
Microtubule integrity is important in cardio-protection, and microtubule disruption has been implicated in the response to ischemia in cardiac myocytes. However, the effects of Taxol, a common microtubule stabilizer, are still unknown in ischemic ventricular arrhythmias. The arrhythmia model was established in isolated rat hearts by regional ischemia, and myocardial infarction model by ischemia/reperfusion. Microtubule structure was immunohistochemically measured. The potential mechanisms were studied by measuring reactive oxygen species (ROS), activities of oxidative enzymes, intracellular calcium concentration ([Ca2+]i) and Ca2+ transients by using fluorometric determination, spectrophotometric assays and Fura-2-AM and Fluo-3-AM, respectively. The expression and activity of sarcoplasmic reticulum Ca2+-ATPase (SERCA2a) was also examined using real-time polymerase chain reaction, Western blot and pyruvate/Nicotinamide adenine dinucleotide-coupled reaction. Our data showed that Taxol (0.1, 0.3 and 1 μM) effectively reduced the number of ventricular premature beats and the incidence and duration of ventricular tachycardia. The infarct size was also significantly reduced by Taxol (1 μM). At the same time, Taxol preserved the microtubule structure, increased the activity of mitochondrial electron transport chain complexes I and III, reduced ROS levels, decreased the rise in [Ca2+]i and preserved the amplitude and decay times of Ca2+ transients during ischemia. In addition, SERCA2a activity was preserved by Taxol during ischemia. In summary, Taxol prevents ischemic ventricular arrhythmias likely through ameliorating abnormal calcium homeostasis and decreasing the level of ROS. This study presents evidence that Taxol may be a potential novel therapy for ischemic ventricular arrhythmias.
Keywords: Taxol, microtubule, ischemic ventricular arrhythmias, calcium overload, reactive oxygen species
Introduction
Ventricular arrhythmias are a major public health problem and are common events leading to sudden death [1]. Most traditional pharmacological therapies for this condition have yielded disappointing results [2–4]. More novel and reliable approaches to drug development are desirable [3, 4].
Microtubules are conserved structures that are primarily composed of tubulin, and a dynamic equilibrium exists between free tubulin and polymerized tubulin [5, 6]. Microtubules support cellular architecture, plasma membranes, myofibrils and other cellular organelles via their filamentous network and provide pathways for transporting mitochondria, endoplasmic reticulum and other membranous organelles throughout the cell [7]. Microtubules play a role in the compartmentalization and anchoring of many proteins, including receptors and ion channels [8].
Microtubule integrity is important in cardio-protection [9]. Microtubule disruption can inhibit the protective effects of ischemic preconditioning and isoflurane-induced myocardial preconditioning [9, 10]. In cardiac cells from newborn rats, disruption of microtubules has been found to stimulate the rate of spontaneous contraction [11]. Tubulin-binding agents that effectively stabilize microtubules have been reported to reverse adriamycin-induced arrhythmias and to affect the probability of eliciting stretch-induced arrhythmia in rabbits [11, 12].
Ischemia is the most common cause of ventricular arrhythmias [13, 14]. Microtubule disruption has been implicated in the response to ischemia in cardiac myocytes [15]. Taxol is a common microtubule stabilizer, and its main mechanism of action is to promote tubulin polymerization and to stabilize existing microtubules [7]. However, the effects of Taxol on ischemic ventricular arrhythmias remain unknown.
Materials and methods
Establishment of the ischemic ventricular arrhythmia model
The ischemic ventricular arrhythmia model was established as previously described [16, 17]. Adult male Sprague-Dawley rats weighing 250–280 g were killed by stunning and cervical dislocation. Hearts were rapidly removed and placed in ice-cold Krebs–Henseleit (K–H) buffer containing the following (mM): NaCl 118.0, KCl 4.7, KH2PO4 1.2, NaHCO3 25, MgSO4 1.2, CaCl2 2.5, glucose 11.0. The hearts were then mounted onto a Langendorff apparatus and perfused retrogradely at a constant pressure of 75 mmHg with K–H buffer gassed with 95% O2/5% CO2 equilibrated at pH 7.35–7.45 and maintained at 37°C for 20 min. The left main coronary artery was ligated within 2 mm of where it emerged adjacent to the left atrium. With this procedure, coronary flow was reduced by at least 25%. The duration of coronary artery ligation was 30 min. No necrosis was found in this model, as determined by 2,3,5-triphenyl tetrazolium chloride (TTC) staining (Fig. 1B). The electrogram and coronary flow rate were monitored before and during coronary artery ligation. Animals used in this study were maintained in accordance with the Guide for Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH publication No. 85–23, revised 1996) and the Policy of the Animal Care and Use Committee of Tongji University.
fig 1.
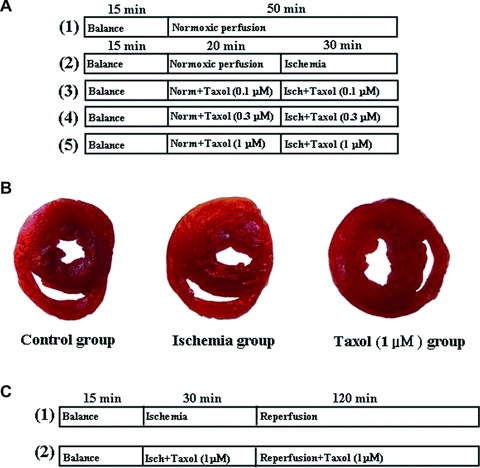
Pharmacological intervention protocols and TTC staining pictures. (A) The pharmacological intervention protocol of the ischemic ventricular arrhythmia study. Norm, normoxic; Isch, ischemia. (B) TTC staining pictures. (C) The pharmacological intervention protocol of the myocardial infarction study. Isch, ischemia.
Assessment of arrhythmias and pharmacological intervention
Arrhythmias were determined and diagnosed in accordance with the Lambeth Convention criteria [18]. Experiments were terminated or excluded from the final data analysis if any of the following conditions occurred: absence of signs of successful coronary artery occlusion, severe arrhythmias prior to coronary artery occlusion or severe atrioventricular block during the first 5 min. of ischemia. The pharmacological intervention protocol is shown in Figure 1A. Taxol was dissolved in dimethyl sulfoxide (DMSO). The exact concentration of DMSO in the perfusate as well as in the medium used in the later experiments on isolated cells was 1:1000. This concentration does not increase the susceptibility to arrhythmias. The heart rate and coronary flow among all groups in this study were comparable (data not shown).
Measurement of microtubule disruption
Myocardial tissues were perfused with a periodate-lysine-paraformaldehyde fixative at the end of the ischemia protocol. After that, the left ventricles were rinsed successively in phosphate-buffered saline (PBS) solutions containing 10%, 15% and 20% sucrose (2 hrs each). Frozen sections of 2–3 μm in thickness were generated using a cryostat (CM 3050; Leica, Benshiem, Germany) and then mounted on gelatine-coated glass slides, rinsed three times in PBS (30 min. each) and processed for double staining of microtubules and actin filaments. The sections were incubated overnight at 4°C with a monoclonal β-tubulin antibody (1:300; Abcam, Cambridge, UK) and then incubated in fluorescein isothiocyanate-labelled sheep antimouse immunoglobulin (1:300; Abcam) for 1 hr at room temperature. Sections were incubated with rhodamine-phalloidin (1:70; Cytoskeleton Company, Denver, CO, USA) for 20 min. at room temperature and rinsed in PBS. These sections were examined under a fluorescence microscope (BX51; Olympus, Tokyo, Japan). The microtubule disruption scores were analysed as previously reported [19].
Measurement of infarct size
The myocardial infarction model was established as previously described [20]. The pharmacological intervention protocol is shown in Figure 1C. Briefly, the hearts of adult male Sprague-Dawley rats were mounted onto a Langendorff apparatus and perfused with K–H buffer. For hearts subjected to regional ischemia, the left main coronary artery was ligated. Reperfusion was achieved by releasing the ligation. In the present study, the isolated heart was subjected to 30 min. ischemia followed by 120 min. of reperfusion, which caused myocardial infarction. To determine the infarct size, the left main coronary artery was relegated at the end of reperfusion, and 2.5% Evans blue was perfused to delineate the area of risk. The blue staining region was removed and the rest, regarded as the risk area, was frozen and cut into slices, which were then incubated in a sodium phosphate buffer containing 1% (w/v) TTC for 15 min. to visualize the unstained infarcted region. The infarcted region was weighed, and the infarct size was expressed as a percentage of the risk zone.
Measurement of intracellular calcium concentration ([Ca2+]i) and Ca2+ transients
Ventricular myocytes were isolated as previously described [21]. For the [Ca2+]i measurements, myocytes were divided into five groups (n = 50 each) as described in the Langendorff-perfused model. Myocytes were loaded with the fluorescent indicator Fura-2-AM (Fluka, Hauppauge, NY, USA). The concentration of Fura-2-AM used (dissolved in DMSO) was 2.5 μM. Myocytes were shaken gently for 10 min. at room temperature. After loading, myocytes were centrifuged, washed with Tyrode’s solution to remove extracellular Fura-2 and then left for 30 min. to ensure complete hydrolysis of the intracellular ester. Myocytes were then alternately excited at 340/380 nm, and the fluorescence emission was collected through a 510 ± 10 nm band-pass filter (T.I.L.L. Photonic GmbH, Munich, Germany). The ratio of the emitted fluorescence at the two excitation wavelengths (340/380 ratio) was calculated to provide an index of [Ca2+]i.
For the imaging of Ca2+ transients, myocytes were divided into three groups after a 15-min equilibration period as follows: (1) control group: 50 min. normoxic superfusion (n = 50); (2) simulated ischemia group: 20 min. normoxic superfusion, 30 min. superfusion of myocytes with anoxic buffer (n = 50); (3) Taxol group: 20 min. normoxic superfusion, 30 min. superfusion of myocytes with anoxic buffer, with Taxol at a concentration of 1 μM (n = 50) throughout the entire 50 min. superfusion period. Normoxic perfusion was induced by perfusing Tyrode’s solution, while ischemia was simulated with ‘ischemic’ Tyrode’s solution. The composition of the Tyrode’s solution was (mM): NaCl 129.0, KCl 4.0, NaH2PO4 0.9, NaHCO3 20.0, CaCl2 2.5, MgSO4 0.5, and glucose 5.5, pH 7.4, gassed with 95% O2/5% CO2. The ‘ischemic’ Tyrode’s solution simulated conditions of ischemia (e.g. hypoxia, hypercapnia, acidosis, hyperkalaemia, lactate accumulation and no glucose), and its composition was (mM): NaCl 123.0, KCl 8.0, NaH2PO4 0.9, NaHCO3 6.0, CaCl2 2.5, MgSO4 0.5 and sodium lactate 20, pH 6.8, gassed with 90%N2/10%CO2. Myocytes were loaded with the fluorescent membrane-permeant Ca2+ dye Fluo-3–AM (Fluka). Cells were placed on a Nikon microscope fitted with an epifluorescence attachment. Cells were stimulated at a 1.5 excitation threshold through two platinum electrodes at 1 Hz. Myocytes were then excited at 488 nm, and the fluorescence emission was collected through a 520 nm band-pass filter (Nikon A1R/A1, Nikon, Tokyo, Japan). Data obtained were analysed with Image-pro plus 6.0 software (Media Cybernetics, Silver Spring, MD, USA).
Measurement of expression and activities of sarcoplasmic reticulum Ca2+-ATPase (SERCA2a)
To measure the expression and activities of SERCA2a, the left ventricles from hearts subjected to the ischemia protocol were used. SERCA2a mRNA expression was measured by real-time polymerase chain reaction with the green fluorescence dye SYBR Green (Roche, Indianapolis, IN, USA), which was validated with respect to reproducibility and linearity within the measurement range. The assay was performed in quadruplicate using Stratagen 3500 (Stratagen, La Jolla, CA, USA). RNA was extracted and reverse transcribed after complete DNA digestion. RNA yields were comparable in all groups (data not shown). To correct for potential variances between samples in mRNA extraction or in reverse transcribed efficiency, the mRNA content of each gene was normalized to the expression of the stably expressed reference gene GAPDH within the same sample. cDNA sequences were obtained from the public GenBank sequence database of the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov), and primers were designed with Primer3 software (http://fokker.wi.mit.edu/primer3/input.htm). Sequences for all polymerase chain reaction primers were as follows: forward 5′-TCC CTC AAG ATT GTC AGC AA-3′ and reverse 5′-AGA TCC ACA ACG GAT ACA TT-3′ for GAPDH; forward 5′-GGA ATT CCT AGA GAT GTG CAA TGC CCT C-3′ and reverse 5′-CGG GAT CCC GTA ACA ACG CAC ATG CAC GCA-3′ for SERCA2a.
SERCA2a protein expression was measured by western blot as previously described [22]. Briefly, liquid nitrogen-frozen left ventricles were homogenized in a buffer containing 50 mM potassium phosphate buffer (pH 7.0), 0.3 M sucrose, 0.5 mM DTT, 1 mM ethylenediaminetetraacetic acid (EDTA; pH 8.0), 0.3 mM phenylmethylsulfonyl fluoride (PMSF), 10 mM NaF, and a phosphate inhibitor cocktail (1:100; Sigma-Aldrich; Saint Louis, MO, USA). Samples were subjected to SDS-PAGE. After electrophoresis, proteins were electrotransferred to a nitrocellulose membrane (Amersham Biosciences, Piscataway, NJ, USA). The blotted membrane was then blocked (5% non-fat dry milk, 10 mM Tris-HCl, pH 7.6, 150 mM NaCl and 0.1% Tween 20) for 2.5 hrs at room temperature and incubated with mouse monoclonal antibody against SERCA2 (1:1000; Abcam) overnight at 4°C. Binding of the primary secondary antibodies (1:1000) was carried out for 1 hr at room temperature, and the membranes were developed using enhanced chemiluminescence (Amersham Biosciences) detected by autoradiography. A quantification analysis of the blots was performed with Quantity-One (Bio-Rad, Hercules, CA, USA).
The SERCA2a activity assay was carried out according to Miyamoto et al. based on pyruvate/NADH-coupled reactions [22]. The sarcoplasmic reticulum membrane was prepared as previously described [22]. Protein content was determined using the BCA Protein Assay Kit (Merck, Darmstadt, Germany). A photometer adjusted to a wavelength of 340 nm was used to assess NADH oxidation (which is coupled to SERCA2a) at 37°C in the membrane preparations using the difference of the total absorbance and basal absorbance. The reaction was carried out in a volume of 1 ml. The SERCA2a activity was calculated as Δ absorbency/ (6.22 × protein × time) in nmol ATP/(mg protein × min.).
Measurement of reactive oxygen species (ROS) and activities of oxidative enzymes
To evaluate ROS qualitatively, the left ventricles from hearts subjected to the ischemia protocol were placed in 20% sucrose for 12 hrs and embedded in optimal cutting temperature compound. Frozen sections of 10 μm in thickness were generated using a cryostat (CM 3050; Leica) and stained with 2′,7′-dichlorofluorescein diacetate (DCFH-DA) from the Tissue Reactive Oxygen Species Assay Kit (Genmed Scientifics, Inc., Arlington, MA, USA). DCFH-DA is membrane permeable and is cleaved intracellularly by non-specific esterases to form DCFH, which is further oxidized by ROS to form the highly fluorescent 2′,7′-dichlorofluorescein (DCF). Fresh-frozen cross sections (10 μm) were stained for 20 min., rinsed, mounted, and observed using fluorescence microscopy with excitation at 490 nm and emission at 520 nm (BX51; Olympus). To study ROS qualitatively, left ventricles from hearts subjected to the ischemia protocol were homogenized in PBS (50 mM sodium phosphate and 150 mM NaCl, pH 7.4) with a PT 10/35 Polytron (Brinkman, Westbury, NY, USA). Some homogenized solutions were used to determine the protein content using the BCA Protein Assay Kit (Merck). Homogenates were stained with DCFH-DA from the Tissue Reactive Oxygen Species Assay Kit (Genmed Scientifics, Inc.). DCFH-DA is oxidized by ROS to form the highly fluorescent DCF. Samples were plated in 96-well plates, and the rate of fluorescence was measured after a 20 min. incubation period at 37°C with an excitation frequency of 488 nm and an emission frequency of 520 nm.
Some homogenized solutions were centrifuged at 5000 × g (4°C) for 10 min., and the supernatant was used for various assays. Xanthine oxidase activity was assayed through H2O2 production using p-hydroxybenzoic acid, 4-aminoantipyrine and horseradish peroxidase according to the manufacturer’s protocol (Genmed Scientifics, Inc.), using a plate reader at 510 nm. Where indicated, the samples were incubated with hypoxanthine for 15 min. at 37°C before reading. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) activity was analysed using a tissue NADPH oxidase assay kit (Genmed Scientifics, Inc.). Briefly, tissue extract (100 μg total protein) was incubated with NADPH. NADPH consumption was monitored by the decrease in absorbance at 340 nm for 5 min. Specific oxidase activity was measured by monitoring the rate of consumption of NADPH inhibited by the addition of DPI. The superoxide dismutase (SOD) assay was performed with an SOD Assay Kit (Genmed Scientifics, Inc.), which is based on the ability of SOD to inhibit the reduction of WST-1 by superoxide. In brief, the reaction mixture contained the cytosolic fraction (100 μg), phosphate buffer, xanthine, xanthine oxidase, EDTA and the water-soluble tetrazolium salt (WST-1). After incubation for 20 min. at 37°C, the optical absorption was measured at 450 nm. One unit was defined as the amount of enzyme that caused one-half of the maximum inhibition of WST reduction.
Mitochondria from left ventricles subjected to the ischemia protocol were isolated as previously described [21]. Two independent methods were used to evaluate the purity of the mitochondrial preparation. First, electron microscopic observations showed very little contamination from broken mitochondria or lysosomes. Second, western blotting indicated that the mitochondrial marker cytochrome c oxidase was enriched at least eight times in the mitochondria preparations, while the endoplasmic reticulum marker calreticulin was markedly reduced to insignificant levels in the purified mitochondria (data not shown). The enzyme activity assay of NADH CoQ reductase (mitochondrial complex I) and ubiquinol ferricytochrome c oxidoreductase (mitochondrial complex III) was carried out according to the instruction manual of the Tissue Mitochondrial Complex I and Complex III Assay Kit (Genmed Scientifics, Inc.). In brief, the reaction mixture of complex I contained mitochondrial extract, Tris/HCl buffer (pH 7.5), EDTA, potassium cyanide, antimycin A and substrates (NADH and decylubiquinone) in the presence or absence of rotenone in a 1 ml total volume. After incubation for 3 min. at 30°C, the activity was read and calculated by following the decrease in absorbance of NADH at 340 nm minus 380 nm using a molar absorption coefficient of 5.5 mM/l/cm. The assay mixture of complex III contained mitochondrial protein (10 ug), EDTA, potassium phosphate buffer (pH 7.5), EDTA, bovine serum albumin, potassium cyanide and substrates (decylubiquinol and oxidized ferricytochrome c) in the presence or absence of antimycin A. After incubation for 5 min. at 30°C, the activity was measured by the increase in absorbance at 550 nm using a molar absorption coefficient of 21.84 mM/l/cm for the reduction of ferricytochrome c.
Statistical analysis
Data are expressed as the mean ± S.E. The time-dependent data were subjected to two-way anova to determine whether there were any significant differences among the groups. In addition, a one sample t-test, chi-squared test or one-way ANOVA was conducted to evaluate the one-way layout data when appropriate. If a significant difference was found, Bonferroni’s post hoc test was conducted to determine which groups differed significantly. P-values <0.05 were considered as statistically significant.
Results
Taxol prevents ischemic ventricular arrhythmias
Taxol (0.1, 0.3 and 1 μM), a microtubule stabilizer, effectively prevented ischemic ventricular arrhythmias, which could be identified from the reduced number of ventricular premature beats and the incidence and duration of ventricular tachycardia (P < 0.05) (Table 1). However, Taxol at a dose of 3 μM affected heart function and did not have a protective effect, which could be considered as an overdose (data not shown).
Table 1.
Effects of Taxol on ischemic ventricular arrhythmias (n = 5)
| Group | VPB | VT | VF | ||
|---|---|---|---|---|---|
| Incidence | Number | Incidence | Duration (sec.) | Incidence | |
| Ischemia | 15/15 | 393 ± 69 | 12/15 | 128 ± 22 | 1/15 |
| Taxol (0.1 μM) | 15/15 | 215 ± 32* | 6/15* | 52 ± 10* | 0/15 |
| Taxol (0.3 μM) | 15/15 | 168 ± 28* | 4/15* | 41 ± 12* | 0/15 |
| Taxol (1 μM) | 9/15* | 100 ± 22* | 2/15* | 12 ± 8* | 0/15 |
P < 0.05, versus Ischemia group.
VPB, ventricular premature beat; VT, ventricular tachycardia; VF, ventricular fibrillation.
Taxol preserves microtubule structure during ischemia
The microtubule disruption score was low (3.2 ± 0.9%) in the control group, whereas it increased significantly to 28.8 ± 6.7% in the ischemia group (P < 0.05). After Taxol treatment (0.1 μM), the microtubule disruption score was reduced to 9.8 ± 1.9%. However, the actin filaments were intact even in cells with microtubule disruption (Fig. 2).
fig 2.
Taxol preserves the microtubule structure in ischemia. (A) Representative pictures of microtubule structure in the control group. Double labelling of the myocardium with mouse monoclonal antibodies against β-tubulin (green) and rhodamine-phalloidin (red) (×400). Bar, 40 μm. (B) Representative pictures of microtubule structure in the ischemia group. Double labelling of the myocardium with mouse monoclonal antibodies against β-tubulin (green) and rhodamine-phalloidin (red) (×400). Bar, 40 μm. (C) Representative pictures of the microtubule structure in the Taxol (0.1 μM) group. Double labelling of the myocardium with mouse monoclonal antibodies against β-tubulin (green) and rhodamine-phalloidin (red) (×400). Bar, 40 μm.
Taxol decreases infarct size
Whether Taxol could increase cardiac repair in a myocardial infarction is a key factor for its potential clinical use in the treatment of ischemic ventricular arrhythmias. Therefore, the effects of Taxol on myocardial infarction were examined. Typical pictures of the size of the myocardial infarctions are shown in Figure 3A. Taxol (1 μM) significantly reduced the infarct size in the isolated perfused rat heart subjected to 30 min. ischemia and 120 min. reperfusion compared with untreated ischemia/reperfusion hearts (Fig. 3B).
fig 3.
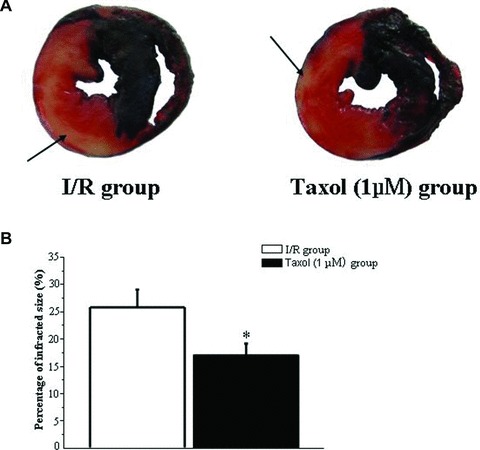
Taxol decreases infarct size. (A) Representative pictures of the infarct size. Myocardial TTC staining was performed immediately after the ischemia/reperfusion (I/R) protocol. 2.5% Evans blue was perfused to delineate the area of risk. Heart slices were incubated with 1% TTC for 15 min. at 37°C to visualize the unstained infarcted region (white), and then digitally photographed. The infarcted region was weighed, and the infarct size was expressed as a percentage of the risk zone. Arrow, white infracted region. (B) Taxol (1 μM) reduces the infarct size. Values represent the mean ± S.E. (n = 8 per group). *P < 0.05 versus I/R group.
Taxol decreases the rise in [Ca2+]i and preserves the amplitude and decay time of Ca2+ transients during the condition of simulated ischemia
Microtubules are important regulators of Ca2+ handling in the heart, and abnormal calcium homeostasis plays a causal role in the genesis of arrhythmias [5, 23, 24]. Therefore, the effects of Taxol on calcium homeostasis in the condition of simulated ischemia were studied. During the condition of simulated ischemia, [Ca2+]i was increased by 62% (P < 0.05) (Fig. 4A). With 0.1, 0.3 and 1 μM Taxol, the rise in [Ca2+]i was reduced by 23%, 25% and 30%, respectively (P < 0.05) (Fig. 4A). Typical pictures of Ca2+ transients are shown in Figure 4B. During the condition of simulated ischemia, the amplitude of transient decreased by 42%, whereas the decrease was only 13% with Taxol treatment (P < 0.05) (Fig. 4C). During the condition of simulated ischemia, the decay time of the Ca2+ transients was 2.52 times that of the control, while Taxol increased the decay time by 0.59 times (P < 0.05) (Fig. 4D).
fig 4.
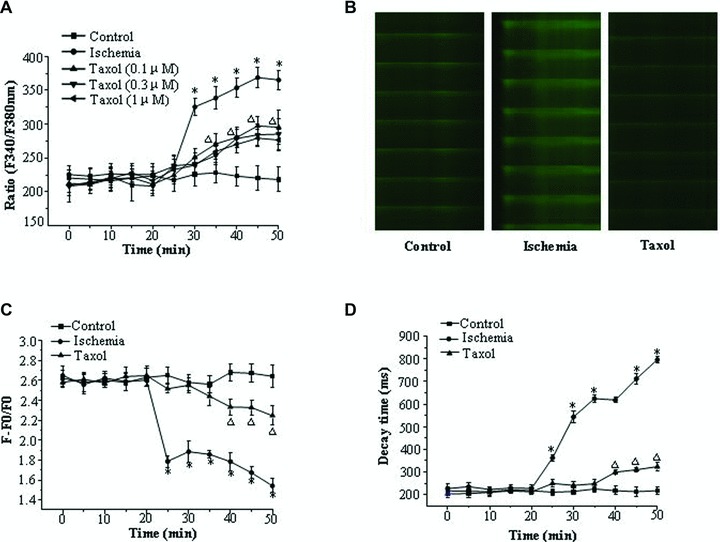
Taxol decreases the rise in [Ca2+]i and preserves the amplitude and decay time of Ca2+ transients in ischemia. (A) [Ca2+]i changes over time. Values represent the mean ± S.E. (n = 50 per group). *P < 0.05 versus control group, □P < 0.05 versus control group and ischemia group. (B) Typical pictures of Ca2+ transients at the end of the experiment. (C) The amplitude of the Ca2+ transients changes over time. Values represent the mean ± S.E. (n = 50 per group). *P < 0.05 versus control group, □P < 0.05 versus control group and ischemia group. (D) The decay time of the Ca2+ transients changes over time. Values represent the mean ± S.E. (n = 50 per group). *P < 0.05 versus control group, □P < 0.05 versus control group and ischemia group.
Taxol preserves SERCA2a activity during ischemia
SERCA2a mRNA and protein expression remained unchanged among the five groups studied (Fig. 5A–C). Compared to the control group, SERCA2a activity was significantly decreased in the ischemia group. Taxol preserved the activity of SERCA2a during ischemia (Fig. 5D).
fig 5.
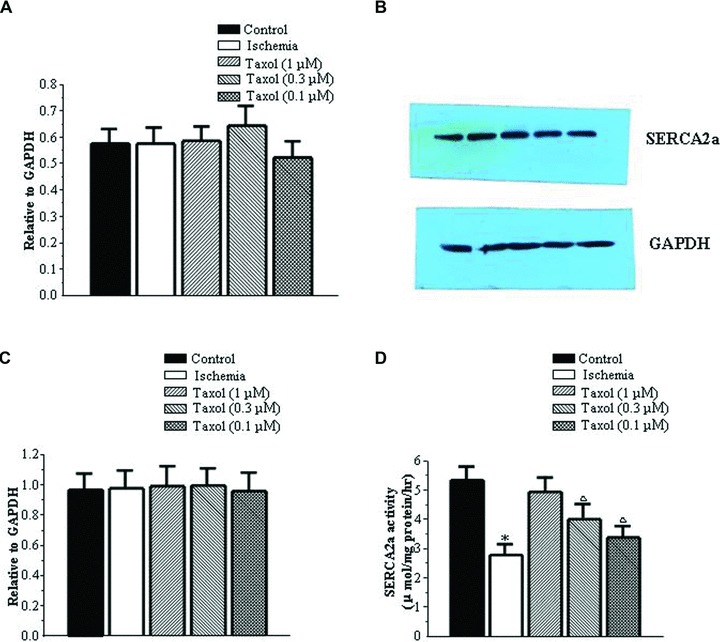
Taxol does not affect the expression of SERCA2a but preserves its activities in ischemia. (A) SERCA2a mRNA expression. Values represent the mean ± S.E. (n = 5 per group). (B) Representative pictures of SERCA2a protein expression. (C) SERCA2a protein expression. Values represent the mean ± S.E. (n = 5 per group). SERCA2a expression, from left to right: control group, ischemia group, Taxol (1 μM) group, Taxol (0.3 μM) group and Taxol (0.1 μM) group. GAPDH expression, from left to right: control group, ischemia group, Taxol (1 μM) group, Taxol (0.3 μM) group and Taxol (0.1 μM) group. (D) SERCA2a activities. Values represent the mean ± S.E. (n = 5 per group). *P < 0.05 versus control group; □P < 0.05 versus control group and ischemia group.
Taxol reduces ROS levels during ischemia
Microtubule disruption can be induced by oxidative stress, and Taxol is able to protect cultured adult mouse cardiac myocytes against oxidative stress induced by H2O2[25, 26]. Oxidative stress plays a pivotal role in ventricular arrhythmias [17, 19, 27–30]. Therefore, the effects of Taxol on ROS generation in ischemia were studied. ROS levels were found to be increased during ischemia, and Taxol reduced this increase in the qualitative evaluation (Fig. 6A). Further study revealed that 0.1, 0.3 and 1 μM Taxol reduced the level of ROS by 33%, 46% and 51%, respectively (P < 0.05) (Fig. 6 B).
fig 6.
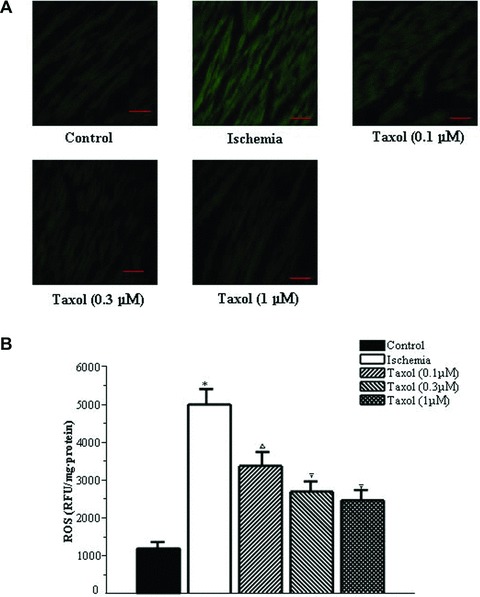
Taxol reduces the levels of ROS during ischemia. (A) Representative qualitative assessment pictures of ROS (×400). Bar, 40 μm. (B) The level of ROS. Values represent the mean ± S.E. (n = 15 per group). *P < 0.05 versus control group; □P < 0.05 versus control group and ischemia group; +P < 0.05 versus control group, ischemia group and Taxol (0.1 and 0.3 μM) groups.
Taxol increases the activity of mitochondrial electron transport chain complexes I and III during ischemia
The enzyme activities of mitochondrial electron transport chain complexes I and III were decreased during ischemia when compared with the control group (P < 0.05) (Table 2). However, 0.3 and 1 μM Taxol increased the activity of mitochondrial electron transport chain complex I, whereas 0.1, 0.3 and 1 μM Taxol increased that of mitochondrial electron transport chain complex III (Table 2).
Table 2.
Effects of Taxol on activities of oxidative enzymes (n = 15)
| Group | Complex I (nM NADH/min./mg) | Complex III (nM ubiquinone/min./mg) | Xanthine oxidase (μM xanthine/min./mg) | NADPH oxidase (μM NADPH/min./mg) | SOD (U/mg) |
|---|---|---|---|---|---|
| Control group | 111.34 ± 2.37 | 971.5 ± 19.95 | 0.16 ± 0.02 | 0.09 ± 0.01 | 0.60 ± 0.09 |
| Ischemia group | 83.99 ± 1.98* | 786.51 ± 18.16* | 0.13 ± 0.01 | 0.10 ± 0.01 | 0.76 ± 0.06 |
| Taxol (0.1 μM) group | 89.47 ± 3.34* | 877.82 ± 12.08*Δ | 0.10 ± 0.02 | 0.07 ± 0.01 | 0.67 ± 0.06 |
| Taxol (0.3 μM) group | 99.11 ± 2.59*Δ | 907.42 ± 16.21Δ | 0.10 ± 0.03 | 0.09 ± 0.02 | 0.77 ± 0.05 |
| Taxol 1(μM) group | 103.49 ± 3.89Δ | 914.73 ± 19.39Δ | 0.16 ± 0.03 | 0.10 ± 0.02 | 0.74 ± 0.04 |
Complex I, mitochondrial complex I; complex III, mitochondrial complex III.
P < 0.05, versus Control group; ΔP < 0.05, versus Ischemia group.
Discussion
The major findings of this study are as follows. First, Taxol prevented ischemic ventricular arrhythmias in rats. Second, Taxol decreased the rise in [Ca2+]i and preserved the amplitude and decay time of Ca2+ transients and the activities of SERCA2a in the condition of simulated ischemia. Third, Taxol decreased ROS during ischemia. Finally, Taxol increased the activity of mitochondrial electron transport chain complexes I and III during ischemia.
Calcium overload and calcium transient alterations play causal roles in the genesis of arrhythmias [21, 31–33]. In this study, Taxol was found to reduce intracellular calcium accumulation and preserve calcium transients during the condition of simulated ischemia. SERCA2a is the main factor contributing to Ca2+ transient decay in rat cardiac myocytes, and its function is critical in maintaining cardiac function [34]. SERCA2a overexpression may effectively prevent ventricular arrhythmias [1, 31]. As predicted, the mRNA and protein levels of SERCA2a did not change after short ischemia. However, it was shown that SERCA2a activity was significantly decreased after short ischemia. We speculated that the anti-arrhythmic effect of Taxol may be related to the preservation of SERCA2a activities during ischemia.
Various ROS are generated during cardiac ischemia, and these deleterious species are highly reactive and prone to promoting oxidative changes in membrane lipids and ion pumps, which may further exacerbate the electrophysiological heterogeneity during ischemia and thus lead to the genesis of arrhythmias [17, 28]. In the present study, Taxol was found to be able to reduce the levels of ROS during ischemia. The main cardiac sources of ROS are the mitochondrial electron transport chain complex I, the mitochondrial electron transport chain complex III ominant endogenous antioxidant is SOD [28]. During ischemia, antioxidant defences are overwhelmed by excess ROS, resulting in serious cellular injuries [28]. Our data showed that the enzyme activities of mitochondrial electron transport chain complexes I and III were decreased during ischemia, consistent with previous studies [27, 35]. In our study, Taxol was found to increase the activities of mitochondrial electron transport chain complexes I and III. Microtubules are in close association with the mitochondria [23], which may be a basis for the effects of Taxol on the activities of complexes I and III. It should be mentioned that ROS generation in this study was measured after the addition of DCFH-DA to homogenates prepared from hearts subjected to ischemia. Due to the instability of ROS, it is possible that the above-mentioned ROS was generated during the in vitro assay. However, for the measurement of ROS, the left ventricles from the control group, ischemia group and Taxol groups were dealt with the same procedure. In the present study, 0.1, 0.3 and 1 μM Taxol reduced the level of ROS by 33%, 46% and 51%, respectively. In view of the fact, ROS we measured were produced during preceding myocardial ischemia, but not during the in vitro assay.
In conclusion, the results of this study indicate that Taxol, a microtubule stabilizer, prevents ischemic ventricular arrhythmias likely through ameliorating abnormal calcium homeostasis and decreasing the level of ROS. This study provides evidence that Taxol may be a potential novel therapy for ischemic ventricular arrhythmias.
Of note, profound cardiac disturbances have already been reported in 5% of patients treated with Taxol. Symptomatic bradycardia has also been observed in 29% of ovarian cancer patients treated with Taxol [11]. Moreover, a previous study found that the probability of eliciting a stretch-induced arrhythmia increased in the hearts of rabbits treated with Taxol [12]. However, the present study found that the probability of eliciting ischemic ventricular arrhythmia decreased in rat hearts with Taxol. This difference may be attributed to the following reasons: (1) the Taxol doses used in this study are approximately one-tenth of those observed clinically in serum levels after continuous intravenous infusion, thus preventing the normal cardiac function from being affected [12]; (2) arrhythmias in the present study were elicited by ischemia, whereas those in the previous study were elicited by stretch [12]. The mechanisms of stretch-induced arrhythmia and ischemia-induced arrhythmia are somewhat different. In addition, the species difference also needs to be considered. In the present study, Taxol was found to prevent ischemic ventricular arrhythmias in rats, which is consistent with the fact that microtubule integrity is important for cardio-protection [9, 10].
A potential weakness of the study was that the exact pathways by which Taxol preserves calcium handling in cardiomycytes and reduces oxidative stress remain elusive. Nevertheless, our analysis of the effects of Taxol on ameliorating abnormal calcium homeostasis and decreasing the level of ROS supports its predominant role in preventing ischemic ventricular arrhythmias. Moreover, the time at which Taxol should be given before anticipating ischemia and whether it can be used effectively during ischemia without preliminary treatment should be identified in the future. In addition, Taxol has imperfect specificity, as do almost all available drugs. Whether Taxol targets cardiac ion channels should also be determined in future studies.
Acknowledgments
This work was supported by the National Science Fund of China (30425016, 30330290 and 30470961), the ‘973’ Program Fund of China (2007CB512100), the ‘863’ Program Fund of China (2007AA02Z438), the Program Fund for Shanghai Subject Chief Scientists, the Program Fund for Innovative Research Teams by the Ministry of Education of China (Y.-H.C.), the Shanghai Pujiang Program Fund (07PJ14058, L.P.) and the ‘973’ Program Fund of China (2006CB504100, Z.-N.Z.).
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Prunier F, Kawase Y, Gianni D, et al. Prevention of ventricular arrhythmias with sarcoplasmic reticulum Ca2+ ATPase pump overexpression in a porcine model of ischemia reperfusion. Circulation. 2008;118:614–24. doi: 10.1161/CIRCULATIONAHA.108.770883. [DOI] [PubMed] [Google Scholar]
- 2.Farkas A, Qureshi, Curtis MJ. Inadequate ischaemia-selectivity limits the antiarrhythmic efficacy of mibefradil during regional ischaemia and reperfusion in the rat isolated perfused heart. Br J Pharmacol. 1999;128:41–50. doi: 10.1038/sj.bjp.0702778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Members of the Sicilian G. New approaches to antiarrhythmic therapy: emerging therapeutic applications of the cell biology of cardiac arrhythmias. Circulation. 2001;104:345–60. doi: 10.1161/hc4801.099491. [DOI] [PubMed] [Google Scholar]
- 4.Hall MCS, Todd DM. Modern management of arrhythmias. Postgrad Med J. 2006;82:117–25. doi: 10.1136/pgmj.2005.033654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gomez AM, Kerfant BG, Vassort G. Microtubule disruption modulates Ca2+ signaling in rat cardiac myocytes. Circ Res. 2000;86:30–6. doi: 10.1161/01.res.86.1.30. [DOI] [PubMed] [Google Scholar]
- 6.Bloom K. Microtubule composition: cryptography of dynamic polymers. Proc Natl Acad Sci USA. 2004;101:6839–40. doi: 10.1073/pnas.0401266101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gerdes JM, Katsanis N. Small molecule intervention in microtubule-associated human disease. Hum Mol Genet. 2005;14:R291–300. doi: 10.1093/hmg/ddi269. [DOI] [PubMed] [Google Scholar]
- 8.Unno T, Komori S, Ohashi H. Microtubule cytoskeleton involvement in muscarinic suppression of voltage-gated calcium channel current in guinea-pig ileal smooth muscle. Br J Pharmacol. 1999;127:1703–11. doi: 10.1038/sj.bjp.0702711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakamura Y, Miura T, Nakano A, et al. Role of microtubules in ischemic preconditioning against myocardial infarction. Cardiovasc Res. 2004;64:322–30. doi: 10.1016/j.cardiores.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 10.Ismaeil MS, Tkachenko I, Hickey RF, et al. Colchicine inhibits isoflurane-induced preconditioning. Anesthesiology. 1999;91:1816–22. doi: 10.1097/00000542-199912000-00036. [DOI] [PubMed] [Google Scholar]
- 11.Lampidis TJ, Kolonias D, Savaraj N, et al. Cardiostimulatory and antiarrhythmic activity of tubulin-binding agents. Proc Natl Acad Sci USA. 1992;89:1256–60. doi: 10.1073/pnas.89.4.1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parker KK, Taylor LK, Atkinson JB, et al. The effects of tubulin-binding agents on stretch-induced ventricular arrhythmias. Eur J Pharmacol. 2001;417:131–40. doi: 10.1016/s0014-2999(01)00856-1. [DOI] [PubMed] [Google Scholar]
- 13.Wyse DG, Friedman PL, Brodsky MA, et al. Life-threatening ventricular arrhythmias due to transient or correctable causes: high risk for death in follow-up. J Am Coll Cardiol. 2001;36:1718–24. doi: 10.1016/s0735-1097(01)01597-2. [DOI] [PubMed] [Google Scholar]
- 14.de Diego C, Pai RK, Chen F, et al. Electrophysiological consequences of acute regional ischemia/reperfusion in neonatal rat ventricular myocyte monolayers. Circulation. 2008;118:2330–7. doi: 10.1161/CIRCULATIONAHA.108.789149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ganote C, Armstrong S. Ischaemia and the myocyte cytoskeleton: review and speculation. Cardiovasc Res. 1993;27:1387–403. doi: 10.1093/cvr/27.8.1387. [DOI] [PubMed] [Google Scholar]
- 16.Thandroyen FT, McCarthy J, Burton KP, et al. Ryanodine and caffeine prevent ventricular arrhythmias during acute myocardial ischemia and reperfusion in rat heart. Circ Res. 1988;62:306–14. doi: 10.1161/01.res.62.2.306. [DOI] [PubMed] [Google Scholar]
- 17.Yang CS, Tsai PJ, Chou ST, et al. The roles of reactive oxygen species and endogenous opioid peptides in ischemia-induced arrhythmia of isolated rat hearts. Free Radic Biol Med. 1995;18:593–8. doi: 10.1016/0891-5849(94)00153-b. [DOI] [PubMed] [Google Scholar]
- 18.Walker MJA, Curtis MJ, Hearse DJ, et al. The Lambeth Conventions: guidelines for the study of arrhythmias in ischaemia, infarction, and reperfusion. Cardiovasc Res. 1988;22:447–55. doi: 10.1093/cvr/22.7.447. [DOI] [PubMed] [Google Scholar]
- 19.Sato H, Hori M, Kitakaze M, et al. Reperfusion after brief ischemia disrupts the microtubule network in canine hearts. Circ Res. 1993;72:361–75. doi: 10.1161/01.res.72.2.361. [DOI] [PubMed] [Google Scholar]
- 20.Cao CM, Xia Q, Gao Q, et al. Calcium-activated potassium channel triggers cardioprotection of ischemic preconditioning. J Pharmacol Exp Ther. 2005;312:644–50. doi: 10.1124/jpet.104.074476. [DOI] [PubMed] [Google Scholar]
- 21.Li J, Xiao J, Liu Y, et al. Mitochondrial benzodiazepine receptors mediate cardioprotection of estrogen against ischemic ventricular fibrillation. Pharmacol Res. 2009;60:61–7. doi: 10.1016/j.phrs.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 22.Miyamoto MI, del Monte F, Schmidt U, et al. Adenoviral gene transfer of SERCA2a improves left-ventricular function in aortic-banded rats in transition to heart failure. Proc Natl Acad Sci USA. 2000;18:793–8. doi: 10.1073/pnas.97.2.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Howarth FC, Calaghan SC, Boyett MR, et al. Effect of the microtubule polymerizing agent taxol on contraction, Ca2+ transient and L-type Ca2+ current in rat ventricular myocytes. J Physiol. 1999;516:409–19. doi: 10.1111/j.1469-7793.1999.0409v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kerfant BG, Vassort G, Gomez AM. Microtubule disruption by colchicine reversibly enhances calcium signaling in intact rat cardiac myocytes. Circ Res. 2001;88:E59–65. doi: 10.1161/hh0701.090462. [DOI] [PubMed] [Google Scholar]
- 25.Skobel E, Kammermeier H. Relation between enzyme release and irreversible cell injury of the heart under the influence of cytoskeleton modulating agents. Biochim Biophys Acta. 1997;1362:128–34. doi: 10.1016/s0925-4439(97)00060-4. [DOI] [PubMed] [Google Scholar]
- 26.Lee CF, Liu CY, Hsieh RH, et al. Oxidative stress-induced depolymerization of microtubules and alteration of mitochondrial mass in human cells. Ann NY Acad Sci. 2005;1042:246–54. doi: 10.1196/annals.1338.027. [DOI] [PubMed] [Google Scholar]
- 27.Petrosillo G, Ruggiero FM, Di Venosa N, et al. Decreased complex III activity in mitochondria isolated from rat heart subjected to ischemia and reperfusion: role of reactive oxygen species and cardiolipin. FASEB J. 2003;17:714–6. doi: 10.1096/fj.02-0729fje. [DOI] [PubMed] [Google Scholar]
- 28.Kevin LG, Novalija E, Stowe DF. Reactive oxygen species as mediators of cardiac injury and protection: the relevance to anesthesia practice. Anesth Analg. 2005;101:1275–87. doi: 10.1213/01.ANE.0000180999.81013.D0. [DOI] [PubMed] [Google Scholar]
- 29.Shah M, Akar FG, Tomaselli GF. Molecular basis of arrhythmias. Circulation. 2005;112:2517–29. doi: 10.1161/CIRCULATIONAHA.104.494476. [DOI] [PubMed] [Google Scholar]
- 30.Ter Keurs HE, Boyden PA. Calcium and arrhythmogenesis. Physiol Rev. 2007;87:457–506. doi: 10.1152/physrev.00011.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.del Monte F, Lebeche D, Guerrero JL, et al. Abrogation of ventricular arrhythmias in a model of ischemia and reperfusion by targeting myocardial calcium cycling. Proc Natl Acad Sci USA. 2004;101:5622–7. doi: 10.1073/pnas.0305778101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bers DM. Calcium and cardiac rhythms: physiological and pathophysiological. Circ Res. 2002;90:14–7. [PubMed] [Google Scholar]
- 33.Clusin WT. Mechanisms of calcium transient and action potential alternans in cardiac cells and tissues. Am J Physiol Heart Circ Physiol. 2008;294:H1–10. doi: 10.1152/ajpheart.00802.2007. [DOI] [PubMed] [Google Scholar]
- 34.Morissette MR, Stricker JC, Rosenberg MA, et al. Effects of myostatin deletion in aging mice. Aging Cell. 2009;8:573–83. doi: 10.1111/j.1474-9726.2009.00508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paradies G, Petrosillo G, Pistolese M, et al. Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: involvement of reactive oxygen species and cardiolipin. Circ Res. 2004;94:53–9. doi: 10.1161/01.RES.0000109416.56608.64. [DOI] [PubMed] [Google Scholar]