

Abstract
In the context of obesity, perivascular fat produces various adipokines and releases free fatty acids, which may induce inflammation and proliferation in the vascular wall. In this study we investigated how adipokines, oleic acid (OA) and the combined treatment regulate human vascular smooth muscle cell (hVSMC) proliferation and migration and the underlying signalling pathways. Adipocyte-conditioned media (CM) generated from human adipocytes induces a prominent proliferation and migration of hVSMC. Autocrine action of adiponectin totally abolishes CM-induced proliferation. Furthermore, OA but not palmitic acid induces proliferation of hVSMC. CM itself does not contain fatty acids, but CM in combination with OA markedly enhances proliferation of hVSMC in a synergistic way. Both the nuclear factor (NF)-κB and the mammalian target of rapamycin (mTOR) pathway were synergistically activated under these conditions and found to be essential for hVSMC proliferation. Expression of iNOS and production of nitric oxide was only enhanced by combined treatment inducing a marked release of VEGF. Combination of OA and VEGF induces an additive increase of hVSMC proliferation. We could show that the combination of CM and OA led to a synergistic proliferation of hVSMC. Expression of iNOS and production of nitric oxide were only enhanced under these conditions and were paralleled by a marked release of VEGF. These results suggest that the combined elevated release of fatty acids and adipokines by adipose tissue in obesity might be critically related to hVSMC dysfunction, vascular inflammation and the development of atherosclerosis.
Keywords: human smooth muscle cells, adipokines, oleic acid, proliferation, inflammation
Introduction
Obesity is a major determinant of mortality of all causes including cardiovascular disease in industrial countries [1]. Adipocytes in expanded fat are active secretory cells capable of releasing lipid mediators and a variety of cytokines, the so-called adipokines [2]. Many studies in human beings and in various animal models have shown that obesity is strongly related to the development of atherosclerosis [3, 4]. Adipose tissue has a prominent role in the development of a low-grade systemic inflammatory state that contributes to obesity-associated vascular dysfunction and cardiovascular risk [5]. The local secretion of adipokines by perivascular fat may provide a new direct link between obesity and vascular complications [6]. However, the mechanism how perivascular fat increases the risk of metabolic and cardiovascular disease is not yet fully elucidated. Endothelial cells and smooth muscle cells (SMC) represent the major cell types of the artery wall preserving vessel wall homeostasis. The migration of vascular smooth muscle cells (VSMC) from the media to intima and their concomitant proliferation occurring in the synthetic state are critical causes of arterial wall thickening. Adipokines such as leptin and resistin have been shown to affect the vasculature by influencing the proliferation and function of SMC [7, 8]. Adiponectin (AN) is an anti-atherogenic adipokine and hypoadiponectinemia is not only associated with obesity but also with cardiovascular disease and diabetes [9]. VEGF-induced SMC proliferation and migration is inhibited by AN making it a positive regulator of vascular remodelling [10]. It is apparent that expanded adipose tissue, especially by its secretory output, is a strong risk factor for the development of cardiovascular diseases. The crosstalk of adipose tissue with cells in the arterial wall such as SMC is not yet fully understood. Although effects of specific adipokines on SMC function have been studied, effects of the whole secretory output of human adipocytes have not been investigated. Apart from a single study using conditioned media (CM) from mouse cell lines and rat adipose tissue explants [11] showing induction of proliferation in human vascular smooth muscle cell (hVSMC), no other data on a direct interaction of adipocytes and SMC exist. Therefore, the main objective of this study was to provide insight into the complex cellular mechanisms linking obesity and atherosclerosis by assessing the role of protein factors and lipid mediators in the crosstalk between human SMC and subcutaneous and perivascular adipocytes.
Materials and methods
Materials
Reagents for SDS-PAGE were supplied by Amersham Pharmacia Biotech (Braunschweig, Germany) and by Sigma-Aldrich (München, Germany). Polyclonal antibodies anti-phospho-mTOR (Ser2448), anti-mTOR, anti-inter-cellular adhesion molecule (ICAM)-1, anti-phospho-NF-κB (P65) (Ser536), anti-NF-κB (P65), anti-p38 mitogen-activated protein kinase (MAPK), anti-phospho-p38 MAPK (Thr180/Tyr182), anti-p44/42 MAP kinase (ERK1/2) and anti-phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) were supplied by Cell Signalling Technology (Frankfurt, Germany). Anti-actin antibodies and anti-iNOS came from Abcam (Cambridge, UK), anti-tubulin from Calbiochem (Merck Biosciences, Schwalbach, Germany) and anti-VCAM-1 from Acris (Herford, Germany). Horseradish-peroxidase (HRP)-conjugated goat anti-rabbit and goat antimouse IgG antibodies came from Promega (Mannheim, Germany). Collagenase NB4 was obtained from Serva (Heidelberg, Germany). Rapamycin and SNAP was obtained from Calbiochem. The IκB kinase (IKK)-inhibitor I229 was from Sanofi-Aventis (Frankfurt, Germany). I229 has sub-micromolar activity on the isolated IKK complex and is highly specific on IKK. Its general structure is described in PCT/EP00/05340. Troglitazone, cytochalasin B, tumour necrosis factor (TNF)-α, bovine serum albumin (BSA; fraction V, fatty acid free, low endotoxin), sodium palmitate and sodium oleate were obtained from Sigma-Aldrich. AN was purchased from BioVendor GmbH (Heidelberg, Germany). The Cell Proliferation ELISA (BrdU, chemiluminescent) and protease inhibitor cocktail tablets were from Roche (Mannheim, Germany). Foetal calf serum (FCS) was supplied by Gibco (Invitrogen, Carlsbad, CA, USA). VEGF was purchased from Millipore (Schwalbach, Germany). The Transwell Cell migration assay (8-μm-pore-size, colorimetric) was from Cell Biolabs, Inc. (San Diego, CA, USA). 4-amino-5-methylamino-2′, 7′-difluorofluorescein diacetate was obtained from Molecular Probes (Invitrogen GmbH, Karlsruhe, Germany). All other chemicals were of the highest analytical grade commercially available and were purchased from Sigma-Aldrich.
Adipocyte isolation and culture
Subcutaneous adipose tissue was obtained from lean or moderately overweight women (n = 23, body mass index 26.1 ± 1.1, and aged 36.6 ± 2.0 years) undergoing plastic surgery. The procedure was approved by the ethical committee of the Heinrich-Heine-University (Düsseldorf, Germany). All patients were healthy, free of medication and had no evidence of metabolic diseases according to routine laboratory tests. Pre-adipocytes were isolated by collagenase digestion of adipose tissue as previously described by us [12]. Isolated cell pellets were resuspended in Dulbecco’s modified Eagles/Hams F12 (DMEM/F12) medium supplemented with 10% FCS, seeded in 75 cm2 culture flasks and maintained at 37°C with 5% CO2. After overnight incubation, cultures were washed and further incubated in an adipocyte differentiation medium (DMEM/F12, 33 μmol/l biotin, 17 μmol/l d-panthothenic-acid, 66 nM insulin, 1 nM triiodo-L-thyronine, 100 nM cortisol, 10 μg/ml apo-transferrin, 50 μg/μl gentamycin, 15 mmol/l HEPES, 14 nmol/l NaHCO3, pH 7.4) for 15 days with medium change every 2–3 days and addition of 5 μM troglitazone for the first 3 days. The degree of differentiation was determined by oil red staining, induction of AN and repression of pref-1. Differentiated adipocytes were used for the generation of adipocyte-CM, as recently described by us [13]. Briefly, CM was generated by culturing adipocytes for 48 hrs in SMC basal medium (PromoCell) with addition of 50 ng/ml amphotericin b and 50 μg/ml gentamycin. Each CM was tested for its proliferative effect, the content of AN (negatively correlated to proliferation) and interleukin (IL)-6 (not related to proliferation). A more-detailed characterization of CM was described previously by us [13]. The concentration of FFA in CM was measured with a Fatty Acid Assay Kit from Biovision (Biocat, Heidelberg, Germany) and with HPLC [14].
Culture of fat explants and preparation of CM
Human epicardial and subcutaneous fat biopsies were obtained from patients without type 2 diabetes undergoing coronary artery bypass surgery (n = 3, body mass index 27 ± 0.82, and aged 69 ± 2.6 years). Adipose tissue was collected and used to generate CM as described [15]. Briefly, fat explants were cultured in adipocyte tissue medium [DMEM F12 containing 10% fetal calf serum, 33 μmol/l biotin, 17 μmol/l panthothenate and antibiotic-antimycotic (Invitrogen, Carlsbad, USA)]. After 2 days, the medium was replaced with adipocyte tissue medium without serum. After 24 hrs, CM was collected and stored in aliquots at –80°C until further use.
Culture of hVSMC
Primary human coronary artery SMC were obtained from PromoCell (Heidelberg, Germany). hVSMC from four different donors (Caucasian, male, 23, 31, 40 years old; female, 56 years old) were supplied as proliferating cells and kept in culture according to the manufacturer’s protocol. For all experiments, subconfluent cells of passage 3 were used. Cells were characterized as hVSMC by morphologic criteria and by immunostaining with smooth muscle α-actin.
Fatty acid treatment of hVSMC
Sodium salts of fatty acids were dissolved in water as a 6 mM stock solution, and were further diluted in sterile serum-free SMC medium containing 4% (wt/v) BSA. Oleic acid (OA) and palmitic acid (PA) were applied to hVSMC at a final concentration of 100 μmol/l for 18 hrs. All controls of experiments involving fatty acids were treated with BSA alone.
In vitro analysis of growth promoting activity
To monitor DNA synthesis hVSMC were seeded in 96 well culture dishes and allowed to attach for 24 hrs, followed by serum starvation for an additional 24 hrs period. Cells were then stimulated for 24 hrs with the different CM in the presence of BrdU (10 μM). 10.000 hVSMC per 15 mm2 well were incubated with the CM of 35.000 adipocytes. The BrdU ELISA Kit was used to determine proliferation according to the manufacturer’s protocol. Signals were visualized and evaluated on a LUMI Imager work station (Boehringer, Mannheim, Germany). Treatment of three different hVSMC donors (F56, M23, M21) with CM from one adipose tissue donor led to a robust and significant stimulation of proliferation (3–4-fold) (Fig. 1A). Figure 1B shows that the proliferative capacity of CM is adipocyte-donor dependent. 22 CM were tested for their proliferative activity and the majority induced a 2–4-fold proliferation of hVSMC (14 CM showed a proliferation of 2-fold or higher). Only these CM were used for further experiments. Variations in the potency of CM in inducing proliferation may be explained by the differences in AN content (Fig. 1C). This is in accordance with our view of an autocrine function of AN [13].
fig 1.
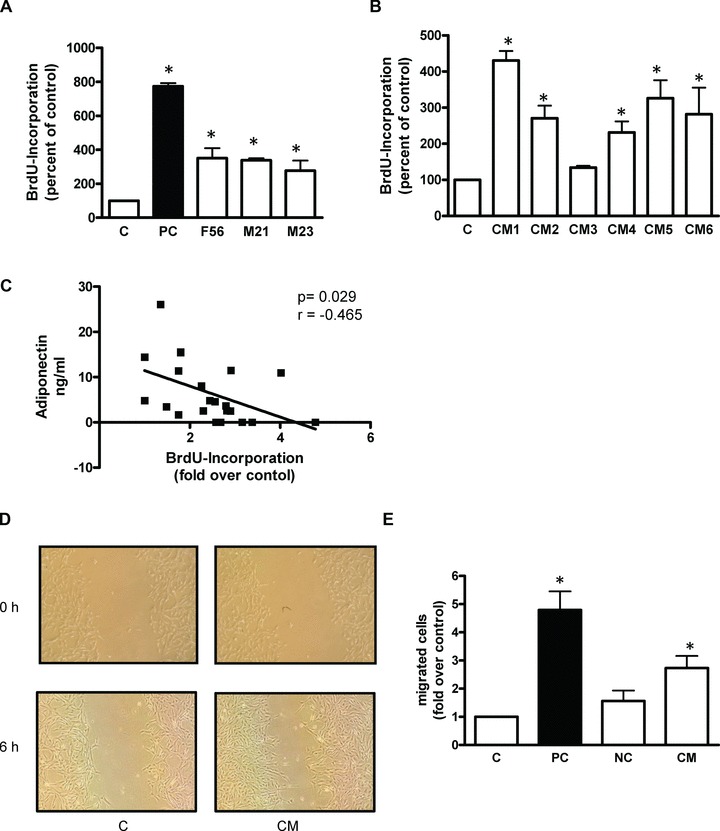
Effect of CM on proliferation (A, B, C) and migration (D, E) of hVSMC. The proliferation was determined by measuring the incorporation of BrdU into DNA. Data are expressed relative to the basal control value, taken as 100%. FCS was used as a positive control (PC). (A) Effect of CM from a single adipose tissue donor on proliferation of three different hVSMC donors. Data are mean values ± S.E.M. of three independent experiments using a specific CM. (B) Proliferative effects of CM from six different adipose tissue donors determined on one hVSMC donor (F56). Data are mean values ± S.E.M. of three independent experiments. (C) Proliferative effect of 22 different CM correlated to their AN content. Representative micrographs are shown. (D) Effects of CM on migration of hVSMC using a in vitro wound scratch assay. (E) Quantitative analysis of hVSMC migration with a Transwell Cell migration assay. 5% FCS was used as PC and in combination with 25 nM cytochalasin B as negative control. Data are presented as mean ± S.E.M. from four independent experiments using four different CM. *P < 0.05 compared to control.
Immunoblotting
hVSMC were treated as indicated and lysed in a buffer containing 50 mM HEPES, pH 7.4, 1% TritonX100, Complete protease inhibitor and PhosStop phosphatase inhibitor cocktail. After incubation for 2 hrs at 4°C, the suspension was centrifuged at 10,000 × g for 15 min. Thereafter, 5 μg protein of lysates were separated by SDS-PAGE using 10% horizontal gels and transferred to polyvenylidene fluorid filters in a semi-dry blotting apparatus [16]. Filters were blocked with Tris-buffered saline containing 0.1% Tween and 5% non-fat dry milk and subsequently incubated overnight with a 1:1000 dilution of the appropriate antibodies. After washing, filters were incubated with secondary HRP-coupled antibody and processed for enhanced chemiluminescence detection using Immobilon HRP substrate (Millipore, Billerica, MA, USA). Signals were visualized and evaluated on a LUMI Imager work station.
ELISA
VEGF secretion by hVSMC and AN release by adipocytes was determined using ELISA kits purchased from BioVendor GmbH. The assays were performed in duplicates according to the manufacturer’s instructions.
hVSMC migration assay
Transwell Cell migration assay was performed with 24-well transwell chambers with 8-μm-pore-size polycarbonat membranes. hVSMC were grown to confluence and serum-starved for 24 hrs. A total of 300 μl cell suspension containing 3 × 104 detached cells was added to the upper compartment. Serum-free medium, CM, FCS or OA were placed in the lower compartment (500 μl/well). In control chambers, 2.5 nM cytochalasin B was added to the upper compartment. The cells were then incubated for 6 hrs. After removal of non-migratory cells, migratory cells were stained and quantified at 570 nm according to the manufacturer’s protocol. We could demonstrate that hVSMC migrated into the scratch already after incubation with CM for 6 hrs (Fig. 1D).
A wound scratch assay was used to visualize the effect of CM on hVSMC migration. hVSMC were seeded (2 × 105 cells/well) into 6-well culture dishes. After 24 hrs starvation, the cell monolayers were scratched using a sterile pipette tip, rinsed repeatedly with PBS to remove residual cell debris and then incubated with 5% FCS (positive control), or with CM for 6 hrs and photographed under a phase-contrast microscope (Olympus, Hamburg, Germany). Treating hVSMC with CM led to a significant 3-fold increase in the migration of hVSMC compared to untreated cells (Fig. 1E).
Measurement of nitric oxide production in hVSMC
Treated hVSMC were washed in PBS, incubated with 10 μM 4-amino-5-methylamino-2′, 7′-difluorofluorescein diacetate for 30 min. As a positive control, cells were treated with 500 μM SNAP. Afterward, cells were lysed in the above mentioned lysis buffer and fluorescence measured using excitation wavelength of 485 nm on an Infinte 200 (Tecan, Männersdorf, Germany).
Presentation of data and statistics
Data are expressed as mean ± S.E.M. Unpaired two-tailed Student’s t-test or one-way anova (post hoc test: Bonferroni’s multiple comparison test) were used to determine statistical significance. All statistical analyses were done using Prism (GraphPad, La Jolla, CA, USA) considering a P-value of less than 0.05 as statistically significant. Corresponding significance levels are indicated in the figures.
Results
CM generated in the presence of AN reduces proliferation of hVSMC
We could show that the treatment with CM led to a robust induction of proliferation (Fig. 1A, B) and migration (Fig. 1D, E) of hVSMC. The proliferative potency of CM is negatively correlated to its AN content (Fig. 1C). To investigate the direct effect of AN on adipocytes we further generated CM in the absence or presence of 10 nM full length AN for 48 hrs (CMAN) similar to earlier work [13] (Fig. 2). The proliferative effect of CMAN was substantially reduced as compared to CM, whereas AN alone had no effect on hVSMC proliferation (Fig. 2A). AN added freshly to CM (CM + AN) just before incubating with hVSMC had no effect on the CM-induced proliferation. These findings are in agreement with our view of an autocrine function of AN [13] and suggest that this protein attenuates the release of growth-mediating factors from adipocytes. As another readout for the pro-atherogenic effects of CM we investigated the expression of the adhesion molecule ICAM-1. We could show that CM led to a significant 2-fold increase in the expression of ICAM-1 (Fig. 2B) that was absent when incubated with CMAN.
fig 2.
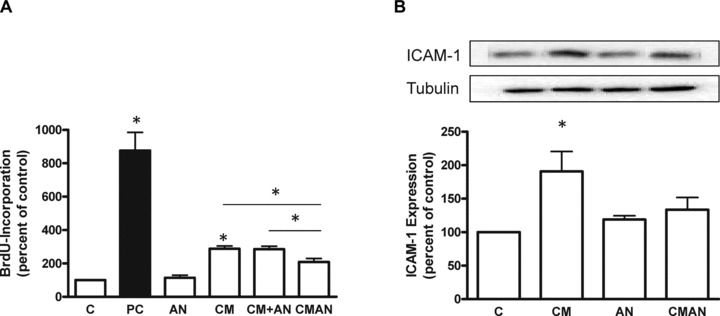
Analysis of CM generated in the absence or presence of AN. CM were generated for 48 hrs with or without 10 nM full length AN (CMAN). Further, AN was added to CM just before the incubation with hVSMC (CM + AN). (A) Effect of AN, CMAN and CM + AN on BrdU incorporation into DNA in hVSMC. Data are expressed relative to the basal control value, which was set as 100%. (B) Analysis of ICAM-1 expression after 24 hrs incubation with CMAN. Total cell lysates were resolved by SDS-PAGE and immunoblotted with a specific ICAM-1 antibody. Data are means ± S.E.M. of three independent experiments and three different adipocyte donors. *P < 0.05 compared to control.
Effect of fatty acids and CM on proliferation, migration and the expression of adhesion molecules of hVSMC
In all experiments using fatty acids these were coupled to fatty acid-free BSA; thus BSA was always present under all conditions. Control experiments indicated that it had no effect on the measured parameters. Subsequently CM was applied at a concentration of 50% (v/v), due to addition of BSA. OA alone induced a significant 3.5-fold increase of hVSMC proliferation. The combination of OA and CM produced a synergistic, substantial stimulation of hVSMC proliferation (7–8-fold) compared to CM and OA alone. By contrast, PA had no proliferative effect on hVSMC. The combination of PA and CM even abrogated the proliferative capacity of CM. Figure 3B shows the capacity of CM, OA and CMOA to induce migration of hVSMC. Although CM induced a robust migration, OA had no effect on hVSMC migration and actually decreased the CM induced migration to the control level. In addition, the increased expression of ICAM-1 in response to CM was prevented by the presence of OA (Fig. 3C). Although CM and OA alone had no effect on the expression of VCAM-1, the combination of both showed a nearly 2-fold increase in the expression of VCAM-1 compared to control (Fig. 3D).
fig 3.

Effect of OA and PA (100 μmol/l) on hVSMC proliferation (A), migration (B) and the expression of adhesion molecules ICAM-1 (C) and VCAM-1 (D). (A) For the proliferation assay, hVSMC were serum starved for 24 hrs and subsequently incubated with BrdU in the absence or presence of CM, OA, PA or the combination of CM with each fatty acid for 18 hrs. Data are expressed relative to the basal control value, which was set as 100%. FCS is used as positive control (PC). Data are means ± S.E.M. of eight independent experiments. (B) Effect of CM, OA and the combined treatment on migration of hVSMC. Data are presented as mean ± S.E.M. from four independent experiments. Analysis of ICAM-1 (C) and VCAM-1 (D) expression after 24 hrs incubation with CM, OA and CMOA. Total cell lysates were resolved by SDS-PAGE and immunoblotted with a specific ICAM-1 or VCAM-1 antibody. Data are mean values ± S.E.M. of three independent experiments. All data were normalized to the level of actin expression and expressed relative to the control. *P < 0.05 compared to control.
CM and OA activate multiple signalling pathways in hVSMC
CMOA produced a substantial phosphorylation of NF-κB after 5 min., which was much more prominent than the effects of OA and CM alone (Fig. 4A). Treatment with CM alone reached a peak value after 30 min., whereas at later time-points NF-κB activation was no longer detectable under all conditions. Activation of p38 MAPK was significantly increased following treatment with CMOA after 10 min., whereas CM alone could activate p38 MAPK more prominently after 60 min. (Fig. 4B). No significant activation of p38 MAPK with OA alone was observed throughout the experiment. Furthermore, CMOA induced mTOR phosphorylation after 5, 10 and 30 min., whereas OA and CM showed no activation of this pathway (Fig. 4C). We did not observe enhanced phosphorylation of ERK after treatment with the combination of CM and OA (data not shown).
fig 4.
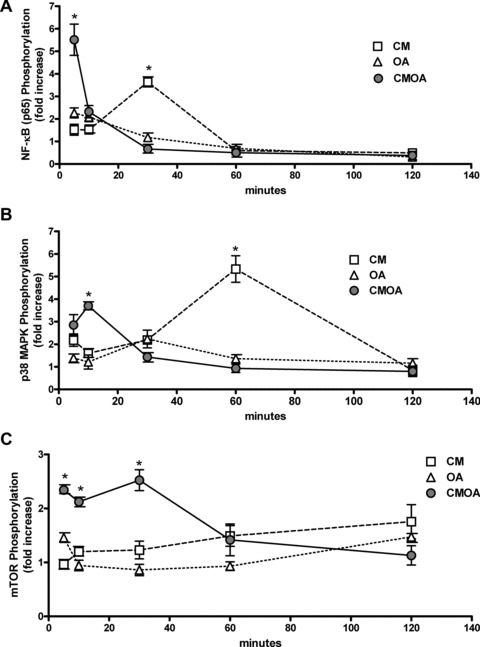
CM, OA and the combination of both acutely activate multiple intracellular signalling pathways. hVSMC were serum starved for 24 hrs and then exposed to CM, 100 μmol/l OA and the combination CMOA for the indicated times. Total cell lysates were resolved by SDS-PAGE and immunoblotted with antibodies to phosphorylated and unphosphorylated forms of NF-κB (A), p38 MAPK (B) and mTOR (C). Data are mean values ± S.E.M. of three independent experiments. All data were normalized to the level of actin expression and are expressed relative to the control. *P < 0.05 compared to control hVSMC (n = 3–4).
mTOR and IKK inhibition abolish CM-, OA- and CMOA-induced proliferation of hVSMC
Inhibition of both mTOR by rapamycin and IKK by compound I229 totally abrogated CM- and OA-induced proliferation of hVSMC. Both inhibitors also abolished the synergistic effect of CM and OA and restored the basal proliferation level (Fig. 5A, B). Compound I229 is a benzimidazole derivative, which showed an IC50 value of 1.9 nM against the IKK complex [17].
fig 5.
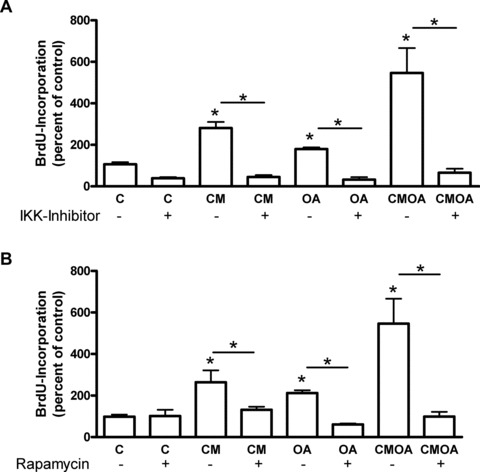
Impact of rapamycin and IKK inhibitor on the proliferative effect of CM, OA and CMOA in hVSMC. Cells were treated with CM, OA and CMOA as described in the legend to Figure 3, without or with 10 nmol/l rapamycin (A) or 10 μmol/l IKK inhibitor (B) for 24 hrs. Proliferation was measured by the incorporation of BrdU into DNA. Data are expressed relative to the basal control value. *P < 0.05 (n = 3–4).
CM and OA induce iNOS expression, VEGF release and nitric oxide production in hVSMC
We determined iNOS expression in hVSMC after incubation with CM, OA and CMOA for 24 hrs (Fig. 6A). CM and OA treatment had no significant effect on iNOS expression. However, the combination of both induced a 2.3-fold increase of iNOS expression in SMC. It is well established that an increased iNOS expression leads to an enhanced VEGF production in different cell types [18–20]. CM alone contains 122 ± 6 pg/ml VEGF (n = 16). Both CM and OA increased VEGF concentration in SMC medium 2.1- and 2.3-fold, respectively, taking into account the endogenous VEGF content of CM (Fig. 6B). In addition, CMOA increased VEGF concentration in a synergistic manner (5.5-fold). Concomitantly, a significant increase in nitric oxide production by 1.5-fold was observed in hVSMC after incubation with the combination CMOA (Fig. 6C). VEGF treatment showed a significant effect (2.5-fold) on proliferation and the combination of VEGF and OA markedly enhanced the proliferation in an additive way (5-fold) (Fig. 6D). Inhibition of NOS by L-nitro-l-arginine methyl ester (NAME) had no effect on the proliferation induced by CM and OA alone, yet it completely abolished the synergistic effect of the two stimuli. Notably, an additive proliferative effect of CM and OA was still observed (Fig. 6E).
fig 6.

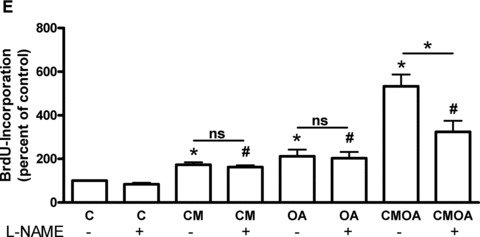
Effects of OA, CM and the combination of both on iNOS expression, VEGF concentration and nitric oxide production and impact of VEGF and NOS inhibitor L-NAME on proliferation. hVSMC were treated as described in the legend to Figure 3. (A) Total cell lysates were resolved by SDS-PAGE and immunoblotted with a specific iNOS antibody. Data are mean values ± S.E.M. of three independent experiments. All data were normalized to the level of actin expression and are expressed relative to the control. (B) After 24 hrs the supernatant were collected and VEGF concentration was measured by ELISA assay. (C) hVSMC were subsequently analysed for their capacity to produce nitric oxide as described in the ‘Materials’ section. As positive control (PC), cells were treated for 30 min. prior the beginning of the experiment with SNAP. (D) Cells were treated with 125 pg VEGF, OA and the combination of VEGF and OA (VEGFOA) for 18 hrs. Proliferation was measured by the incorporation of BrdU into DNA. (E) Cells were treated with CM, OA and CMOA as described in the legend to Figure 3 with or without 1 mM L-NAME for 24 hrs. Data are means ± S.E.M. *P < 0.05 compared to untreated hVSMC (n = 3); #P < 0.05 compared to L-NAME treated hVSMC (n = 3).
Comparison of proliferative capacity between CM from subcutaneous and perivascular adipose tissue
In order to validate our findings obtained with subcutaneous fat, we also assessed the proliferative activity of epicardial fat, which is a perivascular fat depot. CM of both paired subcutaneous and perivascular fat depots were generated from adipose tissue explants from the same patient. Figure 7 shows, that both subcutaneous and epicardial CM induced a significant 1.6-fold proliferation of hVSMC. These results suggest that secreted factors responsible for the induced proliferation are fat depot independent.
fig 7.
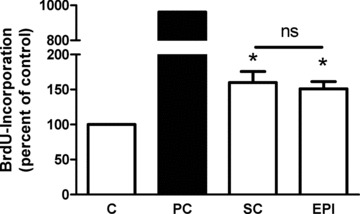
Effect of CM from subcutaneous (sc) and epicardial (epi) fat explants on the proliferation of hVSMC. The proliferation was determined by measuring the incorporation of BrdU into DNA. Data are expressed relative to the basal control value, which was set as 100%. Data are presented as mean ± S.E.M. from three independent experiments using three different CM. Both epicardial and subcutaneous fat were obtained from the same patient. *P < 0.05 compared to control.
Discussion
Obesity is associated with an increased risk for cardiovascular diseases such as atherosclerosis [1]. Inflammation in expanded adipose tissue and a concomitant increased release of adipokines and lipid mediators is linked to obesity and might also be a mechanism underlying the development of atherosclerosis. It has been speculated that perivascular adipose tissue releasing various pro-inflammatory adipokines might directly contribute to the pathogenesis of atherosclerosis [6]. Chemotactic adipokines released by perivascular adipose tissue have already been shown to modulate the function of immune cells infiltrating at the interface of adipose tissue and the adventitia of atherosclerotic aortas [21]. As for effects of adipokines on cells of the vascular wall, it is well known that specific adipocyte-derived factors are involved in regulating vascular functions, including hVSMC proliferation and migration [22, 23]. We could demonstrate in this study that adipokines secreted from in vitro differentiated human adipocytes also induce proliferation and migration of hVSMC. In our system of primary human SMC CM also induces migration, VEGF secretion and increases the expression of adhesion molecules, which might all be critical features in atherosclerosis development. In atherosclerosis, hVSMC increase the expression of adhesion molecules like ICAM-1, VCAM-1, fractalkine (CX3CL1), which allow them to interact with monocytes that differentiate into macrophages [24, 25] suggesting a role of hVSMC in retaining monocytes and macrophages within the atherosclerotic lesion [26]. We could show in the present study that CM-induced expression of ICAM-1 can be completely inhibited by AN. This new finding is in accordance with data demonstrating that AN is able to suppress the expression of adhesion molecules in endothelial cells [27].
CM of human adipocytes contains various growth-promoting and migrative factors such as VEGF, fibroblast growth factor, insulin-like growth factor, plasminogen-derived growth factor (PDGF) and angiotensin II. In our study, the majority of CM induced a 3–4-fold proliferation of hVSMC whereas just a few CM lacked this effect. Measuring AN in all used CM, we could demonstrate that a low AN content is associated with high proliferative action of CM. As earlier work demonstrates that AN exerts an autocrine action on adipocytes decreasing the release of various adipokines [13], we presumed that the AN content in CM might be responsible for the differences in the proliferative potency of CM. Arita and colleagues showed that AN inhibits growth factor-induced proliferation and migration of human aortic SMC by binding these growth factors [10]. Here we can demonstrate that AN added to adipocytes during medium conditioning prevents the proliferative effects of CM. Consistently, AN added to CM after conditioning, and therefore not influencing adipokine release, did not affect CM-induced proliferation. We therefore assume that AN has to be in contact with adipocytes to exert its positive effect on adipokine release rather than binding adipokines and preventing their proliferative effect. The present work emphasizes the importance of the autocrine function of AN and extends this concept to the control of growth promoting factors released by adipocytes.
It is well established, that OA induces rat VSMC proliferation [28], migration [29] and plays a central role in obesity and fatty acid-induced atherosclerosis [30]. In the current study we could reproduce these effects of OA in hVSMC whereas PA had no effect. This difference between PA and OA are not apoptosis related as both fatty acids do not induce apoptosis at the concentrations used [31]. However, different effects on VSMC proliferation and migration could be explained by differential activation of peroxisome proliferator-activated receptor gamma coactivator (PGC)-1α expression by OA and PA [32, 33]. Zhang and colleagues found that overexpression of PGC-1α blocked OA-induced proliferation, whereas suppression of PGC-1α expression by siRNA amplified these effects. By contrast, PA markedly induced PGC-1α expression [32]. Our study is the first to test a combination of adipokines and fatty acids for their effects on hVSMC demonstrating a markedly enhanced proliferation of primary hVSMC. Previous studies have shown that OA enhances the mitogenic activity of angiotensin II [34] in rat SMC in a synergistic way similar to the synergy between CM and OA in our study. Furthermore, the combination of CM and OA enhanced the expression of VCAM-1 but not of ICAM-1. VCAM-1 is essential for phenotypic modulation of cultured SMC. Interactions of VCAM-1 and its ligand very late antigen (VLA)4 may influence the phenotype and synthetic capacity of SMC [35]. The different expressions of ICAM-1 and VCAM-1 can be explained by the different regulation of these adhesion molecules by mTOR. Minhajuddin et al. showed that mTOR down-regulates thrombin-induced ICAM-1 expression [36], whereas Wood and colleagues showed that inhibition of mTOR decreases VCAM-1 expression [37]. In contrast to proliferation, we could not observe an increase of hVSMC migration after incubation with OA, interestingly OA decreased the CM-induced migration to basal levels. However, other studies in rodents could show that OA induced migration of rat VSMC [32, 33, 38]. Additional work will be needed to elucidate the precise role of OA in CM-induced hVSMC.
Proliferation of SMC is regulated by different pathways including p38 MAPK, NF-κB and mTOR. p38 MAPK can be activated by stress, inflammatory cytokines and growth factors [39]. CM alone and the combination of CM and OA acutely activated p38 MAPK significantly within 1 hr. By contrast, OA alone did not activate p38 MAPK, a finding that confirms similar observations from Lu and colleagues [34]. Proliferation of hVSMC is also regulated by nuclear transcription factors including NF-κB. In SMC cultures, NF-κB is activated by growth stimulants and cytokines [40–42]. In the current study we could show that the combination of CM and OA significantly enhanced NF-κB phosphorylation (5–6-fold) already after 5 min. exposure, in comparison to the moderate effects of CM and OA alone (1–2-fold). The inhibition of the IKK complex upstream from NF-κB with the IKK-inhibitor I229 completely blocked proliferation of hVSMC induced by CM, OA and the combination of both, revealing that NF-κB is an essential pathway for hVSMC proliferation. An initial screening with the Kinex™ Antibody Microarray revealed that the PI3K-Akt-mTOR-P70S6 kinase pathway is activated in hVSMC after incubation with CM for 24 hrs (data not shown). In the present study the combination of CM and OA could acutely activate mTOR significantly within 30 min. Inhibition of mTOR with rapamycin reduced CM, OA and CMOA-induced proliferation of hVSMC to the control level (Fig. 6B). Both the IKK inhibitor and rapamycin completely abrogated the proliferation of hVSMC, indicating that there is a crosstalk between these pathways. A recent study has shown that the down-regulation of PTEN triggered by OA is mediated by a signalling complex made of mTOR and NF-κB in hepatocytes [43]. We therefore suggest that the proliferative potency of the combination of CM and OA could be partly explained by a stronger activation of NF-κB and mTOR.
The expression of iNOS is induced by pro-inflammatory cytokines such as IL-1β, TNF-α, interferon-γ[44, 45] in a number of cell types including SMC. Furthermore Fang et al. have shown that OA induces iNOS expression in human retinal pigment epithelium [46]. In the current study we report that neither CM nor OA alone, but the combination of both significantly enhanced iNOS expression and nitric oxide production in hVSMC. In correlation to this finding, CM and OA only moderately induced VEGF secretion by hVSMC, but the combination of both resulted in a markedly stronger effect. Inhibition of iNOS by L-NAME partly inhibited the CMOA-induced hVSMC proliferation, indicating that the synergistic proliferative effect of CM and OA might be due to an enhanced iNOS expression, nitric oxide production and VEGF release. The potential mechanisms underlying the nitric oxide induced augmentation of VEGF expression in VSMC are not completely understood. Our results demonstrate that cytokine- and OA-mediated iNOS induction enhanced VEGF secretion. Also the combination of human recombinant VEGF and OA leads to an obvious augmentation of hVSMC proliferation, indicating that the enhanced VEGF secretion after CMOA treatment maybe a responsible factor for the markedly increased proliferative effect of CM and OA. Furthermore, VEGF in CM might be an important candidate for the proliferative effect of this complex mixture of adipokines.
To validate our findings obtained with adipocytes from subcutaneous fat, we also assessed the proliferative activity of epicardial fat, which is a perivascular fat depot. For this purpose we generated CM from paired adipose tissue explants from patients undergoing bypass surgery. Although the release of single adipokines may certainly differ between subcutaneous and epicardial fat, these additional data show that the proliferative effect obtained with subcutaneous adipose tissue can be compared to perivascular fat. Despite the fact that arteries are not surrounded by subcutaneous adipose tissue, we consider that this fat depot might have a strong systemic effect on hVSMC within the vessel wall due to its considerable amount. Unfortunately, the amount of perivascular adipose tissue from surgery is technically restricted and its use only for key experiments is a limitation of this study. It should also be noted that CM from explants contain secretory products from all cell types present in adipose tissue and is therefore not completely comparable to CM from in vitro differentiated adipocytes. However, the mechanisms by which CM induces dysfunction of SMC are certainly similar for perivascular and subcutaneous adipose tissue.
In conclusion, we show here for the first time that lipid mediators and adipokines synergistically disturb SMC function inducing augmented proliferation and inflammatory signalling. Enhanced iNOS expression and VEGF release by SMC may be critically involved in this process. We propose that the combined elevated release of fatty acids and adipokines by adipose tissue in obesity might be a link between adipose dysfunction, SMC dysfunction, vascular inflammation and the development of atherosclerosis.
Acknowledgments
This work was supported by the Stiftung für Pathobiochemie und Molekulare Diagnostik, the Bundesministerium für Gesundheit, the Commission of the European Communities (Collaborative Project ADAPT, contract number HEALTH-F2–2008-201100), EU COST Action BM0602, the German Science Foundation (Project SE 1922/2-1) and the German-Israel Foundation for Scientific Research and Development (I-750–165.2/2002). We thank Prof. Liebau and her team, Dept. of Plastic Surgery, Florence-Nightingale-Hospital Düsseldorf, for support in obtaining adipose tissue samples. The technical assistance of Andrea Cramer and Angelika Horrighs and the secretarial assistance of Birgit Hurow are gratefully acknowledged.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Calle EE, Thun MJ, Petrelli JM, et al. Body-mass index and mortality in a prospective cohort of U.S. adults. N Engl J Med. 1999;341:1097–105. doi: 10.1056/NEJM199910073411501. [DOI] [PubMed] [Google Scholar]
- 2.Trayhurn P. Endocrine and signalling role of adipose tissue: new perspectives on fat. Acta Physiol Scand. 2005;184:285–93. doi: 10.1111/j.1365-201X.2005.01468.x. [DOI] [PubMed] [Google Scholar]
- 3.Hu FB, Willett WC, Li T, et al. Adiposity as compared with physical activity in predicting mortality among women. N Engl J Med. 2004;351:2694–703. doi: 10.1056/NEJMoa042135. [DOI] [PubMed] [Google Scholar]
- 4.van Dam RM, Willett WC, Manson JE, et al. The relationship between overweight in adolescence and premature death in women. Ann Intern Med. 2006;145:91–7. doi: 10.7326/0003-4819-145-2-200607180-00006. [DOI] [PubMed] [Google Scholar]
- 5.Ruan H, Lodish HF. Insulin resistance in adipose tissue: direct and indirect effects of tumor necrosis factor-alpha. Cytokine Growth Factor Rev. 2003;14:447–55. doi: 10.1016/s1359-6101(03)00052-2. [DOI] [PubMed] [Google Scholar]
- 6.Iacobellis G, Gao YJ, Sharma AM. Do cardiac and perivascular adipose tissue play a role in atherosclerosis. Curr Diab Rep. 2008;8:20–4. doi: 10.1007/s11892-008-0005-2. [DOI] [PubMed] [Google Scholar]
- 7.Calabro P, Samudio I, Willerson JT, et al. Resistin promotes smooth muscle cell proliferation through activation of extracellular signal-regulated kinase 1/2 and phosphatidylinositol 3-kinase pathways. Circulation. 2004;110:3335–40. doi: 10.1161/01.CIR.0000147825.97879.E7. [DOI] [PubMed] [Google Scholar]
- 8.Li L, Mamputu JC, Wiernsperger N, et al. Signaling pathways involved in human vascular smooth muscle cell proliferation and matrix metalloproteinase-2 expression induced by leptin: inhibitory effect of metformin. Diabetes. 2005;54:2227–34. doi: 10.2337/diabetes.54.7.2227. [DOI] [PubMed] [Google Scholar]
- 9.Kadowaki T, Yamauchi T, Kubota N, et al. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest. 2006;116:1784–92. doi: 10.1172/JCI29126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arita Y, Kihara S, Ouchi N, et al. Adipocyte-derived plasma protein adiponectin acts as a platelet-derived growth factor-BB-binding protein and regulates growth factor-induced common postreceptor signal in vascular smooth muscle cell. Circulation. 2002;105:2893–8. doi: 10.1161/01.cir.0000018622.84402.ff. [DOI] [PubMed] [Google Scholar]
- 11.Barandier C, Montani JP, Yang Z. Mature adipocytes and perivascular adipose tissue stimulate vascular smooth muscle cell proliferation: effects of aging and obesity. Am J Physiol Heart Circ Physiol. 2005;289:H1807–13. doi: 10.1152/ajpheart.01259.2004. [DOI] [PubMed] [Google Scholar]
- 12.Hauner H, Petruschke T, Russ M, et al. Effects of tumour necrosis factor alpha (TNF alpha) on glucose transport and lipid metabolism of newly-differentiated human fat cells in cell culture. Diabetologia. 1995;38:764–71. doi: 10.1007/s001250050350. [DOI] [PubMed] [Google Scholar]
- 13.Dietze-Schroeder D, Sell H, Uhlig M, et al. Autocrine action of adiponectin on human fat cells prevents the release of insulin resistance-inducing factors. Diabetes. 2005;54:2003–11. doi: 10.2337/diabetes.54.7.2003. [DOI] [PubMed] [Google Scholar]
- 14.Sell H, Eckardt K, Taube A, et al. Skeletal muscle insulin resistance induced by adipocyte-conditioned medium: underlying mechanisms and reversibility. Am J Physiol Endocrinol Metab. 2008;294:E1070–7. doi: 10.1152/ajpendo.00529.2007. [DOI] [PubMed] [Google Scholar]
- 15.Moro C, Klimcakova E, Lolmede K, et al. Atrial natriuretic peptide inhibits the production of adipokines and cytokines linked to inflammation and insulin resistance in human subcutaneous adipose tissue. Diabetologia. 2007;50:1038–47. doi: 10.1007/s00125-007-0614-3. [DOI] [PubMed] [Google Scholar]
- 16.Wichelhaus A, Russ M, Petersen S, et al. G protein expression and adenylate cyclase regulation in ventricular cardiomyocytes from STZ-diabetic rats. Am J Physiol. 1994;267:H548–55. doi: 10.1152/ajpheart.1994.267.2.H548. [DOI] [PubMed] [Google Scholar]
- 17.Dietze D, Ramrath S, Ritzeler O, et al. Inhibitor kappaB kinase is involved in the paracrine crosstalk between human fat and muscle cells. Int J Obes Relat Metab Disord. 2004;28:985–92. doi: 10.1038/sj.ijo.0802701. [DOI] [PubMed] [Google Scholar]
- 18.Dulak J, Jozkowicz A, Dembinska-Kiec A, et al. Nitric oxide induces the synthesis of vascular endothelial growth factor by rat vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2000;20:659–66. doi: 10.1161/01.atv.20.3.659. [DOI] [PubMed] [Google Scholar]
- 19.Frank S, Stallmeyer B, Kampfer H, et al. Nitric oxide triggers enhanced induction of vascular endothelial growth factor expression in cultured keratinocytes (HaCaT) and during cutaneous wound repair. FASEB J. 1999;13:2002–14. [PubMed] [Google Scholar]
- 20.Xiong M, Elson G, Legarda D, et al. Production of vascular endothelial growth factor by murine macrophages: regulation by hypoxia, lactate, and the inducible nitric oxide synthase pathway. Am J Pathol. 1998;153:587–98. doi: 10.1016/S0002-9440(10)65601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henrichot E, Juge-Aubry CE, Pernin A, et al. Production of chemokines by perivascular adipose tissue: a role in the pathogenesis of atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:2594–9. doi: 10.1161/01.ATV.0000188508.40052.35. [DOI] [PubMed] [Google Scholar]
- 22.Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ Res. 2005;96:939–49. doi: 10.1161/01.RES.0000163635.62927.34. [DOI] [PubMed] [Google Scholar]
- 23.Correia ML, Haynes WG. Leptin, obesity and cardiovascular disease. Curr Opin Nephrol Hypertens. 2004;13:215–23. doi: 10.1097/00041552-200403000-00010. [DOI] [PubMed] [Google Scholar]
- 24.Braun M, Pietsch P, Schror K, et al. Cellular adhesion molecules on vascular smooth muscle cells. Cardiovasc Res. 1999;41:395–401. doi: 10.1016/s0008-6363(98)00302-2. [DOI] [PubMed] [Google Scholar]
- 25.Huo Y, Ley K. Adhesion molecules and atherogenesis. Acta Physiol Scand. 2001;173:35–43. doi: 10.1046/j.1365-201X.2001.00882.x. [DOI] [PubMed] [Google Scholar]
- 26.Cai Q, Lanting L, Natarajan R. Growth factors induce monocyte binding to vascular smooth muscle cells: implications for monocyte retention in atherosclerosis. Am J Physiol Cell Physiol. 2004;287:C707–14. doi: 10.1152/ajpcell.00170.2004. [DOI] [PubMed] [Google Scholar]
- 27.Ouchi N, Kihara S, Arita Y, et al. Novel modulator for endothelial adhesion molecules: adipocyte-derived plasma protein adiponectin. Circulation. 1999;100:2473–6. doi: 10.1161/01.cir.100.25.2473. [DOI] [PubMed] [Google Scholar]
- 28.Lu G, Morinelli TA, Meier KE, et al. Oleic acid-induced mitogenic signaling in vascular smooth muscle cells. A role for protein kinase C. Circ Res. 1996;79:611–8. doi: 10.1161/01.res.79.3.611. [DOI] [PubMed] [Google Scholar]
- 29.Greene EL, Lu G, Zhang D, et al. Signaling events mediating the additive effects of oleic acid and angiotensin II on vascular smooth muscle cell migration. Hypertension. 2001;37:308–12. doi: 10.1161/01.hyp.37.2.308. [DOI] [PubMed] [Google Scholar]
- 30.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–9. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 31.Artwohl M, Lindenmair A, Roden M, et al. Fatty acids induce apoptosis in human smooth muscle cells depending on chain length, saturation, and duration of exposure. Atherosclerosis. 2009;202:351–62. doi: 10.1016/j.atherosclerosis.2008.05.030. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y, Liu C, Zhu L, et al. PGC-1alpha inhibits oleic acid induced proliferation and migration of rat vascular smooth muscle cells. PLoS One. 2007;2:e1137. doi: 10.1371/journal.pone.0001137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu L, Sun G, Zhang H, et al. PGC-1alpha is a key regulator of glucose-induced proliferation and migration in vascular smooth muscle cells. PLoS One. 2009;4:e4182. doi: 10.1371/journal.pone.0004182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu G, Meier KE, Jaffa AA, et al. Oleic acid and angiotensin II induce a synergistic mitogenic response in vascular smooth muscle cells. Hypertension. 1998;31:978–85. doi: 10.1161/01.hyp.31.4.978. [DOI] [PubMed] [Google Scholar]
- 35.Duplaa C, Couffinhal T, Dufourcq P, et al. The integrin very late antigen-4 is expressed in human smooth muscle cell. Involvement of alpha 4 and vascular cell adhesion molecule-1 during smooth muscle cell differentiation. Circ Res. 1997;80:159–69. doi: 10.1161/01.res.80.2.159. [DOI] [PubMed] [Google Scholar]
- 36.Minhajuddin M, Bijli KM, Fazal F, et al. Protein kinase C-delta and phosphatidylinositol 3-kinase/Akt activate mammalian target of rapamycin to modulate NF-kappaB activation and intercellular adhesion molecule-1 (ICAM-1) expression in endothelial cells. J Biol Chem. 2009;284:4052–61. doi: 10.1074/jbc.M805032200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wood SC, Bushar G, Tesfamariam B. Inhibition of mammalian target of rapamycin modulates expression of adhesion molecules in endothelial cells. Toxicol Lett. 2006;165:242–9. doi: 10.1016/j.toxlet.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 38.Yun MR, Lee JY, Park HS, et al. Oleic acid enhances vascular smooth muscle cell proliferation via phosphatidylinositol 3-kinase/Akt signaling pathway. Pharmacol Res. 2006;54:97–102. doi: 10.1016/j.phrs.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 39.Omura T, Yoshiyama M, Izumi Y, et al. Involvement of c-Jun NH2 terminal kinase and p38MAPK in rapamycin-mediated inhibition of neointimal formation in rat carotid arteries. J Cardiovasc Pharmacol. 2005;46:519–25. doi: 10.1097/01.fjc.0000179001.00779.a5. [DOI] [PubMed] [Google Scholar]
- 40.Hishikawa K, Oemar BS, Yang Z, et al. Pulsatile stretch stimulates superoxide production and activates nuclear factor-kappa B in human coronary smooth muscle. Circ Res. 1997;81:797–803. doi: 10.1161/01.res.81.5.797. [DOI] [PubMed] [Google Scholar]
- 41.Lawrence R, Chang LJ, Siebenlist U, et al. Vascular smooth muscle cells express a constitutive NF-kappa B-like activity. J Biol Chem. 1994;269:28913–8. [PubMed] [Google Scholar]
- 42.Obata H, Biro S, Arima N, et al. NF-kappa B is induced in the nuclei of cultured rat aortic smooth muscle cells by stimulation of various growth factors. Biochem Biophys Res Commun. 1996;224:27–32. doi: 10.1006/bbrc.1996.0979. [DOI] [PubMed] [Google Scholar]
- 43.Vinciguerra M, Veyrat-Durebex C, Moukil MA, et al. PTEN down-regulation by unsaturated fatty acids triggers hepatic steatosis via an NF-kappaBp65/mTOR-dependent mechanism. Gastroenterology. 2008;134:268–80. doi: 10.1053/j.gastro.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 44.Aktan F. iNOS-mediated nitric oxide production and its regulation. Life Sci. 2004;75:639–53. doi: 10.1016/j.lfs.2003.10.042. [DOI] [PubMed] [Google Scholar]
- 45.Kleinert H, Schwarz PM, Forstermann U. Regulation of the expression of inducible nitric oxide synthase. Biol Chem. 2003;384:1343–64. doi: 10.1515/BC.2003.152. [DOI] [PubMed] [Google Scholar]
- 46.Fang IM, Yang CH, Yang CM, et al. Comparative effects of fatty acids on proinflammatory gene cyclooxygenase 2 and inducible nitric oxide synthase expression in retinal pigment epithelial cells. Mol Nutr Food Res. 2009;53:739–50. doi: 10.1002/mnfr.200800220. [DOI] [PubMed] [Google Scholar]