

Abstract
Antigen-presenting cells (APC), like dendritic cells (DC), are essential for T-cell activation, leading to immunity or tolerance. Multiple DC subsets each play a unique role in the immune response. Here, a novel splenic dendritic-like APC has been characterized in mice that has immune function and cell surface phenotype distinct from other, described DC subsets. These were identified as a cell type continuously produced in spleen long-term cultures (LTC) and have an in vivo equivalent cell type in mice, namely ‘L-DC’. This study characterizes LTC-DC in terms of marker phenotype and function, and compares them with L-DC and other known splenic DC and myeloid subsets. L-DC display a myeloid dendritic-like phenotype equivalent to LTC-DC as CD11cloCD11bhiMHC-II−CD8α− cells, distinct by high accessibility and endocytic capacity for blood-borne antigen. Both LTC-DC and L-DC have strong antigen cross-presentation ability leading to strong activation of CD8+ T cells, particularly after exposure to lipopolysaccharide. However, they have weak ability to stimulate CD4+ T cells in antigen-specific responses. Evidence is presented here for a novel DC type produced by in vitro haematopoiesis which has distinct antigen-presenting potential and reflects a DC subset present also in vivo in spleen.
Keywords: dendritic cells, antigen presentation, spleen
Introduction
In human and murine spleen, two main classes of dendritic cells (DC) are present: conventional (c)DC and plasmacytoid (p)DC. These are distinct in terms of cytokine production, cross-presentation capacity and T helper response generated [1, 2]. Conventional DC are steady-state, mature DC with CD11chiMHC-IIhi marker expression [3] and include CD8α− and CD8α+ subsets. The distinct lineage of CD11cloB220+MHC-IIlo plasmacytoid pre-DC (p-preDC) in spleen [4] produces high levels of interferon (IFN)-α upon viral exposure [5]. Further spleen DC subsets in mice derived in vitro loosely fall under the term ‘regulatory’ DC. These include diffDC [6], CD11cloCD11bhiMHC-IIlo DC [7] and IL-10 DC [8]. Under inflammatory or infectious settings, myeloid DC in spleen may also include tumour necrosis factor (TNF)/iNOS-producing DC [9] and monocyte-derived DC (mo-DC) [10, 11]. The latter represent a very different cell type to cDC and pDC, developing under inflammatory conditions which drive cells from blood into lymph nodes for antigen presentation [12]. Although subsets of myeloid cells including monocytes have been described in spleen, this lineage is less well defined in terms of subsets than is the DC lineage [13].
Most DC subsets present antigen in the context of both MHC-I and MHC-II, and some have a unique ability to cross-present exogenous antigen via MHC-I [14]. Exogenous antigen is normally routed through the MHC-II pathway for presentation to CD4+ T cells [15]. Cytosolic antigens are usually loaded on to MHC-I for CD8+ T-cell activation [16]. However in cross-presentation, exogenous antigens are directed into the MHC-I pathway for recognition by CD8+ T cells. CD8α+ cDC have been described as the major cross-presenting antigen-presenting cells (APC) subset in spleen [2, 17]. Although some DC are recognized as very potent cross-presenting cells, they are not unique in this role. Reports claim that macrophages/monocytes [18] and neutrophils [19] can cross-present antigen, although their efficiency, and the extent and range of T cells activated are not characterized.
Our laboratory has characterized a novel dendritic-like cell type produced in vitro in stroma-dependent long-term cultures (LTC) of spleen [20]. LTC continuously shed large cells resembling immature myeloid DC that differ from monocytes by large size and expression of CD11c. The LTC-DC phenotype has remained remarkably stable over years of culture as CD11c+CD11b+CD80/86+MHC-I+MHC-II−FcR+DEC-205−/lo B220−CD8α− cells [21]. Their development occurs independently of inflammatory factors like granulocyte macrophage-colony stimulating factor and TNF-α[22]. LTC-DC are highly endocytic but have limited ability to stimulate CD4+ T-cell responses [21], likely due to ignorance because LTC-DC express low surface MHC-II. Following adoptive transfer, LTC-DC can reduce tumour load in mice given leukemic T cells, and can induce anti-tumour cytotoxic T cells [23]. Thus, the question has emerged as to whether an equivalent cell type exists in vivo, and what functional role it plays.
We show here that LTC-DC have very high capacity to cross-present nominal antigen to CD8+ T cells, particularly after exposure to LPS as a danger signal. Furthermore, we show isolation of an in vivo LTC-DC equivalent cell type, bearing LTC-DC cell surface phenotype with high endocytic and cross-presenting function. These cells are referred to as ‘L-DC’; an interim designation due to similarity with LTC-DC but distinctiveness from cDC, pDC, monocyte and macrophage subsets in spleen.
Materials and methods
Animals
Mice were handled according to guidelines of the Animal and Experimental Ethics Committee at the Australian National University (Canberra, Australia). C57BL/6J, CBA/H, C57BL/6.Tg (TcraTcrb)1100Mjb (OT-I), C57BL/6.SJL/J.OT-II.CD45.1 (OT-II) and C57BL/6.SJL-PtprcaPep3b/BoyJ (B6.SJL) mice were bred under specific pathogen-free conditions. They were used at >4 weeks and were sex matched in experiments.
Long-term cultures
LTC were established from spleens of female mice aged 8 days and maintained in supplemented DMEM (sDMEM) as described previously [20]. Non-adherent cells were collected for experimentation from culture flasks at medium change by resuspension of loosely adhered cells. LTC used here were tested clear of mycoplasma contamination using published methodology [24].
Flow cytometry
Flow cytometry was performed on antibody labelled cells using a Fluorescence Activated Cell Sorter (FACS) to assess cell surface marker expression as described previously [25]. Anti-CD16/32 (FcR block) (eBioscience, San Diego, CA, USA) was used to block non-specific binding, except before treatment with goat anti-rat antibody (Southern Biotechnology, Birmingham, AL, USA). Antibody staining was performed in FACS buffer [DMEM/1%FCS/0.1%NaN3]. Biotin- or fluorochrome-conjugated antibodies specific for CD11c (N418; allophycocyanin), CD11b (M1/70; PE-Cy7 (phycoerythrin-Cy7) or fluorescein isothiocyanate (FITC), CD45RB (C363.16A; PE), ckit (2B8; PE or FITC) and CD34 (RAM34; FITC) were purchased from eBioscience. Antibodies specific for CD11c (HL3; allophycocyanin), CD11b (M1/70; biotin), CD8 (53–6.7; PE or FITC), Sca1 (E13–161.7; biotin), Ly6G (Gr-1: 1A8; PE), CD69 (H1–2F3; biotin), MHC-I (AF6–88.5; biotin), MHC-II (25–9-17; biotin), CD80 (16–10A1; FITC) and CD86 (GL1; biotin) were purchased from Becton Dickinson (San Jose, CA, USA). Anti-CD205 (NLDC-145) was purchased from Serotec (Oxford, UK), and anti-33D1 antibody was prepared from hybridoma cells. Isotype control antibodies were purchased from eBioscience and Becton Dickinson. Secondary conjugates included streptavidin (SA)-allophycocyanin-Cy7 (eBioscience), SA-PE (Becton Dickinson) and goat anti-rat IgG-FITC (Southern Biotechnology). For flow cytometric analysis, cells were resuspended at 106–107 cells/ml in FACS buffer and propidium iodide (PI: 1 μg/ml; Sigma-Aldrich, St Louis, MO, USA) was added prior to flow cytometry for discrimination of live cells. A BD LSRII flow cytometer (Becton Dickinson) was used for analysis, and 3 × 104 to 106 events were collected. Data analysis involved post-acquisition gating using FlowJo software (Tree Star, Ashland, OR, USA).
Depletion of T and B cells from spleen
Spleen tissue was dissociated and depleted of red blood cells (RBC) using lysis buffer (0.14 NH4Cl, 0.017 M Tris-base [pH 7.5]) for 5 min. at room temperature. Cells were resuspended in FACS buffer (108 cells/ml) and filtered through a 70 μm nylon cell strainer (BD Biosciences). For depletion of T/B cells and RBC, cells were labelled on ice for 15 min. with biotinylated monoclonal antibodies specific for CD19 (eBio1D3), Thy1.2 (30-H12) and TER-119 (eBioscience). Cells were then washed and resuspended at 108 cells/ml in magnetic activated cell sorting (MACS) separation buffer (phosphate-buffered saline [PBS]/0.5%BSA/2 mM ethylenediaminetetraacetic acid /0.1%NaN3]. These cells were then incubated with anti-biotin MACS microbeads (13 μl beads/108 cells; Miltenyi Biotec) for 20 min. on ice. After one wash, splenocytes were resuspended in MACS buffer (500 μl/108 cells) and T/B cells and RBC depleted by running cells through an LS column (Miltenyi Biotec) in a SuperMACS II Separation Unit (Miltenyi Biotec), washing thrice and collecting unbound cells as flow-through.
Isolation and treatment of antigen-presenting cell subsets
To isolate CD11c+ DC, splenocytes were labelled with CD11c+ microbeads (15 μl/108 cells; Miltenyi Biotec) at 108 cells/ml of MACS buffer for 15 min. on ice and washed once. Cells were then resuspended in MACS buffer (500 μl/108) cells, run through a SuperMACS II Separation Unit equipped with an MS column (Miltenyi Biotec), and washed thrice. Flow-through cells were discarded, 1 ml MACS buffer added and CD11c+ cells separated under pressure with no magnet.
To isolate purified APC subsets from T- and B-cell depleted spleen, cells were labelled with antibody specific for the DC markers CD11c, CD11b, CD8α and MHC-II, and cell sorting performed on a BD FACSAria cell sorter (Becton Dickinson). All incubation and washing steps were performed as for antibody staining, with the exception that NaN3-free FACS buffer was used. Following a final wash just prior to sorting, cells were passed through a 70 μm nylon cell strainer (Becton Dickinson).
For in vitro antigen loading of isolated spleen APC subsets or LTC-DC, APC at 107 cells/ml in sDMEM were pulsed for 12 hrs with 10 μg/ml ovalbumin (OVA), or hen egg lysozyme (HEL) as control antigen. For in vitro activation of DC, cells were incubated with LPS (10 μg/ml; Sigma-Aldrich) for 24 hrs at 37°C.
Purification and labelling of T cells
Dissociated lymph node cells were resuspended in anti-IAb/k (TIB120), anti-B220 (RA3–6B2) and anti-CD11b (M1/70) to deplete MHC-II+ APC, B cells and macrophages. For removal of CD4+ T cells or CD8+ T cells, either anti-CD4 (GK1.5) or anti-CD8 (53–6.7) was added. Cells were labelled on ice for 25 min. and washed with MACS buffer. Cells were incubated with Sheep anti-rat Ig Dynabeads (50 μl beads/107 cells; Dynal, Oslo, Norway), and anti-CD11c microbeads (1.5 μl beads/107 cells; Miltenyi Biotec) were added for DC removal. Incubation was performed in MACS buffer (100 μl/107 cells) at 4°C for 25 min., before placing cells in a magnetic particle concentrator (Dynal) for 2 min. Supernatant contained unbound T cells.
To monitor T-cell proliferation, cells were stained with 5- (and 6-) carboxyfluorescein diacetate succinimidyl ester (CFSE; Molecular Probes, Eugene, OR, USA). Proliferation was assessed as dilution of stain with each cell division measured flow cytometrically. T cells were washed and resuspended at <107 cells/100 μl in sDMEM, and CFSE (5 μM) added with vortexing to ensure uniform labelling. Cells were incubated at room temperature for 5 min. before washing twice with sDMEM.
Measurement of endocytosis
To assess endocytosis in vitro, FITC-conjugated OVA (OVA-FITC; Molecular Probes) was added to cells and uptake measured flow cytometrically as described previously [25]. Cells (106) were exposed to OVA-FITC (2 mg/ml) at 37°C for 45 min. Control cells were held on ice over this time. Endocytosis was halted by addition of 500 μl ice-cold FACS buffer. For assessment of endocytosis in vivo, 3 mg OVA-FITC in 200 μl HBSS was injected into recipient mice via the tail vein. After 12–24 hrs, splenocytes were prepared, labelled with antibody, and DC populations gated and analysed for OVA-FITC uptake using flow cytometry.
Mixed lymphocyte reactions
T cells purified from mesenteric lymph nodes of CBA/H mice were labelled with CFSE and co-cultured with LTC-DC, CD11c+ spleen DC, or APC subsets sorted from C57BL/6J mice, or syngeneic control CBA/H mice spleen as stimulators. CFSE-labelled T cells were cultured at 1 × 105 cells/well in a 96-well plate together with diluting concentrations of DC stimulators in a total volume of 200 μl sDMEM. T-cell activation was assessed flow cytometrically among labelled CD4+ and CD8+ T cells in terms of reduction in CFSE intensity and estimation of the number of divided cells.
Antigen-specific T-cell activation by DC
The antigen presenting capacity of DC subsets was assessed using purified CD8+ T cells from OT-I T cell receptor (TCR)-tg mice specific for the Kb restricted peptide OVA257–264[26] and CD4+ T cells from OT-II TCR-tg mice specific for OVA323–339 and IAb[27]. Enriched CFSE-labelled TCR-tg T cells were plated at 104–105 cells/well with diluting concentrations of APC as stimulators in a total volume of 200 μl sDMEM. In some cultures, LPS (10 μg/ml) was added for the first 24 hrs to activate APC, and then washed off. Cells were collected after 1 day to detect up-regulation of the CD69 marker of activation by antibody staining and flow cytometry. After 4 days, T-cell activation was assessed flow cytometrically to analyse cell division in terms of reduction in CFSE intensity as cells divide, as well as blastogenesis indicated by increase in forward scatter (FSC). At this time, supernatant was collected from cultures for assay of IFN-γ production using cytokine bead arrays (Becton Dickinson).
Statistical analysis of data
A two-way ANOVA test was used to detect significant experimental effects. The Tukey Honest Significant Differences procedure was used to identify paired cell subsets that were significantly different in prevalence (P ≤ 0.05).
Results
LTC-DC display distinct phenotype and function
Cells produced in LTC reflect myeloid lineage DC in that they express CD11c and CD11b but never CD8α or B220 (Fig. 1) [21]. They express MHC-I but are distinguished by no surface MHC-II expression, defining LTC as unusual by comparison with other culture systems producing immature MHC-II+ DC [28]. LTC-DC co-express the costimulators CD80 and CD86 for T-cell activation and are highly endocytic for FITC-OVA. Cells are characteristically large Forward Scatter (FSC)hi, and negative for markers of regulatory DC and cDC like CD45RB, 33D1 and CD205 [8, 29]. They are also negative for ckit, Sca1 and CD34, markers of haematopoietic precursors, and are distinct from granulocytes as Ly6G−CD11c+ cells (Fig. 1).
fig 1.
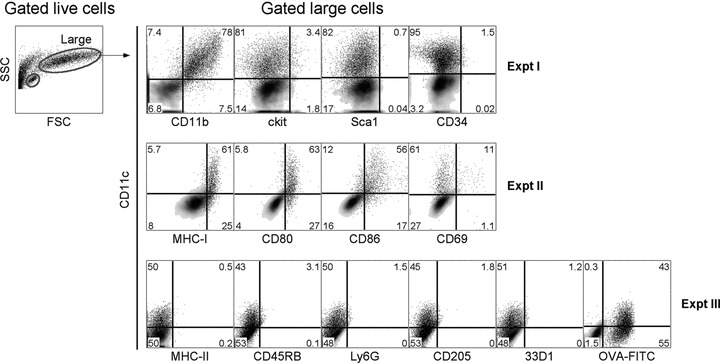
Phenotype of LTC-DC. Non-adherent cells were collected from LTC, stained with antibody (or isotype controls) and PI (1 μg/ml), and analysed flow cytometrically to identify live (PI−) cells. FSC and side scatter (SSC) analysis revealed a clear population of large LTC-DC for analysis of marker expression. To measure endocytic capacity, cells were incubated with 2 mg/ml FITC-OVA for 45 min. at 37°C (or 4°C as control). Density plots reveal background staining with isotype controls (or endocytosis control) used to set gates. Percentage positively staining cells (shown as dot plots) is indicated in quadrants.
LTC-DC were compared with freshly isolated spleen CD11c+ DC for capacity to activate purified CFSE-labelled OVA-specific CD4+ T cells from OT-II TCR-transgenic (tg) mice, and CD8+ T cells from OT-I TCR-tg mice. After 1 day, OVA-pulsed LTC-DC induced activation of CD8+ OT-I T cells indicated by up-regulation of CD69 (Fig. 2A). This occurred for LTC-DC pulsed with OVA ± LPS, but not for LTC-DC pulsed with HEL. In contrast, OVA-pulsed CD11c+ spleen DC weakly activated OT-I CD8+ T cells, but not after LPS treatment. In terms of cell division after 4 days, LTC-DC were superior to CD11c+ spleen DC in activating CD8+ T cells in an antigen-specific manner, but only after LPS activation (Fig. 2A). Fresh splenic CD11c+ DC activated OT-I T cells better in the absence of LPS which may inhibit rather than activate antigen presentation for these DC. Strong activation of CD8+ T cells by LPS-treated LTC-DC was supported by IFN-γ production (Fig. 2A). High dose inhibition of T-cell proliferation induced by OVA + LPS pulsed LTC-DC was likely caused by cell death in cultures, because strong activation of T cells by high numbers of LTC-DC was mirrored in both CD69 up-regulation and IFN-γ production. CD11c+ spleen DC were better activators of CD4+ OT-II T cells than LTC-DC (Fig. 2B). Weak activation of OT-II T cells by LTC-DC was indicated at 4 days, but only with LPS as an activator. This is consistent with minor up-regulation of already negative to low MHC-II expression on LTC-DC following LPS treatment which was reported previously [21]. In that report, LTC-DC gave weak activation of CD4+ T cells from the 3A9 TCR-tg mouse strain but only after LPS activation. LTC-DC represent an APC with restricted ability to activate CD8+ T cells.
fig 2.
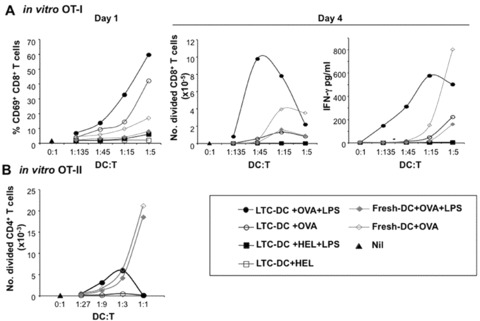
LTC-DC can cross-present antigen. LTC-DC and freshly isolated CD11c+ spleen cells (f-DC) from C57BL/6J mice were pulsed with OVA (10 μg/ml) ± LPS (10 μg/ml). Cells were co-cultured with purified CFSE-labelled CD8+ T cells from OT-I mice, or CD4+ T cells from OT-II mice. T cells alone served as a control (Nil). (A) Activation of CD8+ OT-I T cells was indicated by up-regulation of CD69 after 1 day, and by number of CD8+ T cells undergoing division based on CFSE dilution, or IFN-γ production after 4 days. (B) The number of CD4+ OT-II T cells undergoing division was assessed on the basis of CFSE dilution after 4 days.
LTC-DC also induced proliferation in CD8+ and not CD4+ T cells in an alloreactive mixed lymphocyte reaction (MLR), whereas freshly isolated CD11c+ cDC (f-DC) were effective activators of both CD4+ and CD8+ T cells (Fig. 3A). To determine whether LTC-DC reflect previously described regulatory DC, LTC-DC were tested for ability to inhibit an alloreactive T-cell response induced by CD11c+ spleen DC. LTC-DC from C57BL/6J mice were titrated into both CBA/H anti-C57BL/6J, and C57BL/6J anti-CBA/H responses using spleen CD11c+ DC as APC. However, when LTC-DC were mixed with f-DC, enhanced T-cell proliferation rather than inhibition was detected in both responses (Fig. 3B). This enhanced response suggests more effective antigen presentation due to LTC-DC participation, and argues against LTC-DC as regulatory DC [6].
fig 3.
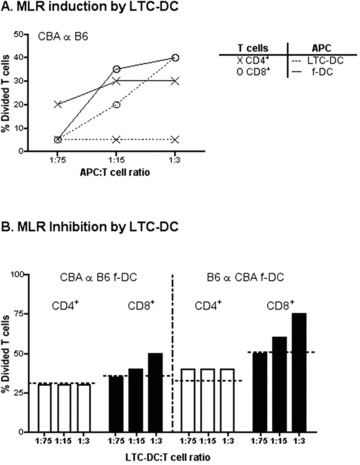
Ability of LTC-DC to induce and inhibit an MLR. T cells were purified from CBA/H or C57BL/6J mouse spleen and CFSE labelled. APC included freshly isolated CD11c+ spleen DC (f-DC) from CBA/H or C57BL/6J mice, or LTC-DC (B6.SJL origin). (A) Diluting numbers of APC were incubated with T cells (105) for 4 days. Cells were collected stained with antibody, and analysed flow cytometrically to determine %CD4+ or %CD8+ T cell division based on decrease in CFSE labelling. T cells cultured with no APC gave 0% division (not shown). (B) MLR co-cultures were established as in (A) using a T cell : f-DC ratio of 15:1. Varying numbers of LTC-DC were added into co-cultures to test their ability to inhibit the MLR reaction. Percentage T-cell division was calculated as in (A), and the level achieved in the absence of added LTC-DC is shown as a dashed line.
An in vivo LTC-DC counterpart in spleen
Spleen was analysed for the presence of an in vivo cell equivalent to LTC-DC, based on the LTC-DC phenotype as highly endocytic CD11c+CD11b+CD8α−MHC-II−FSChi cells. The in vivo LTC-DC counterpart has been named ‘L-DC’. To detect this subset, splenocytes were enriched for DC and myeloid cells by T cell depletion ahead of antibody staining and flow cytometry. Flow cytometric analysis of CD11c and CD8α expression revealed four subsets, three of which expressed CD11c (Fig. 4A). CD11c+CD8α+ cells expressed MHC-II but not CD11b, reflecting a CD8α+ cDC subset [3, 30]. The two remaining CD11c+CD8α− cell populations were CD11clo and CD11chi, representing 21% and 7.5% of live T cell depleted spleen (Fig. 4A). The CD11chiCD8α− population comprised almost exclusively CD11b+MHC-II+ cells (74%), reflecting CD8α− cDC. The CD11cloCD8α− cells comprised three subsets lacking MHC-II expression, only two of which were CD11b+. Only CD11bhiMHC-II− cells displayed large size reflective of LTC-DC (FSChi; 70%). This CD11cloCD8α−CD11bhiMHC-II−FSChi‘L-DC’ population therefore resembled in vitro generated LTC-DC. The majority of CD11bloMHC-II− cells were small, reflecting DC precursors. The CD11b−MHC-II−FSClo subset among CD11cloCD8α− cells reflects p-preDC in line with previous studies [5]. Myeloid cells were identified as CD11c−CD8α−CD11bhiMHC-II− cells with FSClo distribution (Fig. 4A). Flow cytometric analysis of T cell depleted splenocytes led to identification of an L-DC population not previously identified.
fig 4.
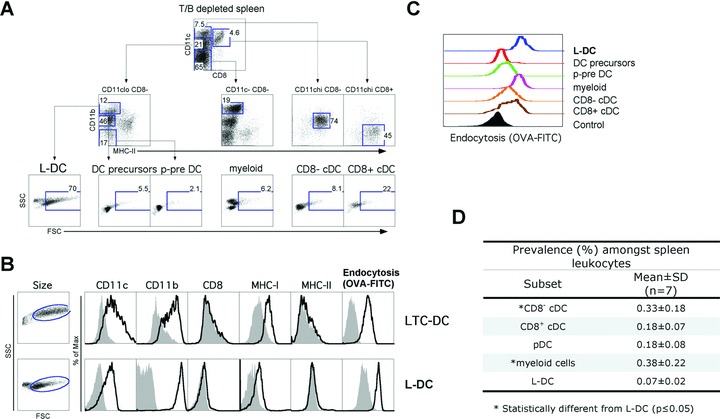
Isolation of a spleen in vivo equivalent of LTC-DC (L-DC). Dissociated, RBC-lysed and T cell depleted spleen cells were stained with specific antibodies and PI (1 μg/ml) prior to flow cytometric analysis. Endocytic capacity was measured in vitro by uptake of FITC-OVA as described in the Figure 1 legend. (A) Live (PI−) cells were gated. Subsets of interest were identified after setting gates to delineate cells on the basis of internal negative and positive cell populations. Forward scatter (FSC) and side scatter (SSC) are shown for each gated population to compare size distribution. Numbers represent percentage cells within gates. Data are representative of a highly consistent pattern of subset distribution across many mice of several strains and age >4 weeks. (B) Phenotypic comparison of size (FSC), marker expression and endocytic capacity was made between LTC-DC and L-DC. Shaded histograms indicate isotype control binding (or endocytosis control). (C) In vitro capacity for endocytosis of OVA-FITC was assessed for each gated spleen subset in relation to a non-endocytic control cell population (filled histogram). (D) The prevalence of each of the defined cell subsets was measured in terms of percentage among total spleen leucocytes.
Direct comparison of marker expression on L-DC and LTC-DC is shown in Figure 4. Both subsets were large in size and highly endocytic for FITC-OVA in vitro (Fig. 4B and C). L-DC, like myeloid cells, showed the highest uptake of FITC-OVA. The cDC subsets (CD8α−, CD8α+) were moderately endocytic, whereas the DC precursor and p-preDC subsets were non-endocytic (Fig. 4C). The prevalence of each subset was estimated after similar cell isolations on seven female C57BL/6J mice. A two-way anova showed no significant (P ≤ 0.05) experimental effect, but a significant T-cell subset effect for prevalence of L-DC compared with myeloid cells and CD8− cDC (Fig. 4D). The prevalence of L-DC among splenic leucocytes was significantly lower than CD8α− cDC and myeloid cells, but not significantly different from pDC and CD8+ cDC. The number of myeloid cells was significantly higher than CD8α+ cDC and pDC. The low frequency of L-DC and their distinct phenotypic characteristics could explain why this subset has not been delineated previously. It is also possible that positive selection procedures that isolate DC as CD11chi or MHC-IIhi cells may not recover L-DC. Also, with their large size, they may be inadvertently eliminated in procedures dependent on buoyant density.
Further flow cytometric analysis of cell surface markers was used to compare L-DC with other known APC. For this study, T/B depleted spleen cells were gated as in Figure 4 to delineate L-DC, CD8α+ cDC, CD8α− cDC and myeloid subsets. Neither LTC-DC nor L-DC expressed 33D1 or CD205 (Fig. 5), two markers which identify CD8α− and CD8α+ cDC [3, 29]. L-DC were also clearly distinct from CD8α− and CD8α+ cDC as ckit− cells. In line with myeloid cells, L-DC were CD80+, but ckit− and CD86−. Both L-DC and myeloid cells were highly endocytic in situ, with L-DC showing highest uptake of FITC-OVA from blood amongst all subsets analysed (Fig. 5). However, L-DC differed from myeloid cells by higher expression of MHC-I, large size (FSChi) and higher endocytic capacity. In common with LTC-DC, L-DC expressed MHC-I, and CD80 but not 33D1, CD205 or ckit. However, LTC-DC differed from L-DC as CD86− cells. They are also distinct as NK1.1– cells, distinguishing them from natural killer (NK) DC [31] (data not shown). Collectively, L-DC reflect a myeloid-lineage cell in that they are CD11b+ cells, although distinct from cDC and monocyte/macrophages on the basis of cell surface marker expression and endocytic capacity.
fig 5.
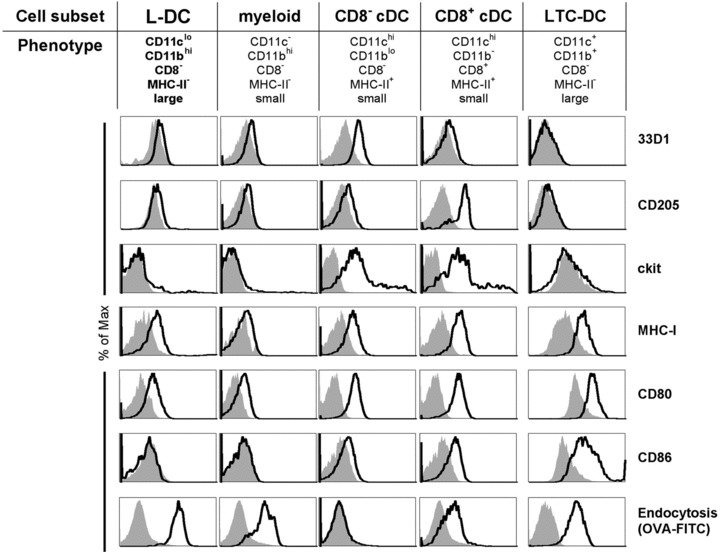
Phenotypic characterization of L-DC. APC subsets including L-DC were identified flow cytometrically according to the gating procedure in Figure 4 using CD11c, CD11b, CD8α, MHC-II and FSC (size) and further assessed for marker expression. In vitro-derived LTC-DC were stained for comparison. Prior to flow cytometry, cells were incubated with PI (1 μg/ml) for gating of live (PI−) cells. In order to measure the in situ capacity of cells for endocytosis, mice were given FITC-OVA (3 mg) i.v. 24 hrs prior to isolation of spleen cells for antibody staining and analysis. Endocytosis was assessed for each gated subset in relation to a non-endocytic control spleen lymphocyte population (filled histogram). For LTC-DC, endocytosis was measured after in vitro treatment with OVA-FITC for 45 min. at 37°C or at 4°C as a control.
L-DC cross-present antigen for T-cell activation
CD8α+ cDC are highly cross-presenting cells both in vitro [32] and in vivo [2, 17]. We therefore compared L-DC with CD8α+ cDC for cross-presenting ability in vitro. APC subsets were isolated and pulsed with OVA or HEL and co-cultured with purified OT-I CD8+ T cells in the presence and absence of LPS as an activator. L-DC were strong activators of OT-I CD8+ T cells but only after LPS activation. The cross-presenting capacity of CD8α+ cDC was very strong, and increased after activation with LPS (Fig. 6A). The main phenotypic effect of LPS activation on L-DC after culture in vitro maps to up-regulation of CD86, which is not expressed on freshly isolated cells (Fig 5; all data not shown).
fig 6.
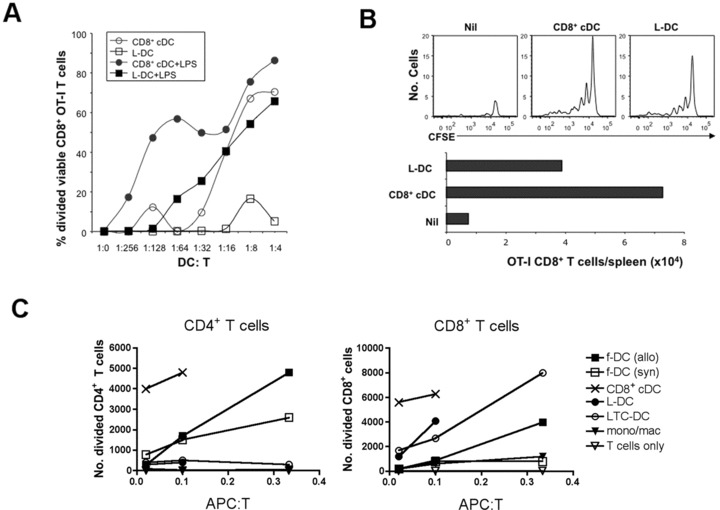
Antigen-presenting capacity of L-DC. CD8α+ cDC, myeloid cells (mono/mac) and L-DC subsets were sorted from C57BL/6J spleen based on gating protocols described in Figure 4. LTC-DC were collected as non-adherent cells from LTC established from spleens of B6.SJL mice. f-DC were enriched from spleens of C57BL/6J and CBA/H mice using CD11c+ microbead positive selection (MACS). (A) To measure in vitro cross-presenting ability, L-DC and CD8α+ cDC were pulsed with OVA (10 μg/ml) and co-cultured with purified CFSE-labelled CD8+ T cells (105) from OT-I mice ±LPS (1 μg/ml). Proliferation of cells was measured by dilution of CFSE after 4 days for calculation of percentage divided cells. (B) Isolated L-DC and CD8α+ cDC were pulsed with OVA (as in A), and 104 cells injected i.v. into B6.SJL (CD45.1) mice previously injected with 105 CFSE-labelled OT-I lymphocytes. Spleens were harvested at 34 days following rechallenge of mice at 25 days with OVA (10 μg, i.v.), and assessed flow cytometrically for CFSE and total number of OT-I CD8+ T cells in spleen. (C) L-DC and LTC-DC were compared with APC subsets isolated from spleen for capacity to induce a MLR. Responder T cells were prepared from CBA/H mice by depletion of B cells, myeloid cells and DC using magnetic beads, labelled with CFSE, and co-cultured with APC, using T cells only as a control. Responding T cells were analysed flow cytometrically at 4 days following antibody staining to gate live (PI−), CD11c−, CD4+ or CD8+ populations. Number of divided cells reflects cells showing a reduction in CFSE intensity.
The capacity of isolated L-DC and CD8α+ cDC to activate T cells in vivo was compared after adoptive transfer. APC were isolated as in Figure 4, and pulsed with antigen in vitro before intravenous transfer to mice given purified CFSE-labelled OT-I lymphocytes 2 hrs previously. Control mice received only CFSE-labelled OT-I lymphocytes and no OVA-pulsed APC. Mice were left for 25 days and then challenged by intravenous administration of OVA (10 mg). Spleens were harvested 9 days later to identify CFSE-labelled T cells. Mice given either OVA-pulsed L-DC or CD8α+ cDC, showed evidence of division among surviving OT-I T cells (Fig. 6B). There was low survival of OT-I T cells in mice given only OT-I T cells, even following OVA challenge at day 25. This result reinforces L-DC as capable of antigen presentation leading to an immunogenic response in vivo.
The T-cell stimulatory capacity of splenic L-DC and LTC-DC was also confirmed by activation of allogeneic T cells in a MLR. CD8α+ cDC were the strongest stimulators of alloreactive CD4+ and CD8+ T cells, whereas L-DC and LTC-DC stimulated CD8+ T cells but not CD4+ T cells (Fig. 6C). Monocytes/macrophages (or myeloid cells) did not activate either CD4+ or CD8+ T cells, and control freshly isolated spleen DC (f-DC) gave stronger responses among CD4+ than CD8+ T cells. The inability of L-DC to stimulate a CD4+ T-cell response maps to inexpression of MHC-II on freshly isolated cells (0% of cells; Fig. 4). Culture of L-DC for 24 hrs with LPS (10 ug/ml) resulted in low MHC-II expression on 31% of cells, although culture of cells alone in the absence of LPS gave 23% of cells expressing MHC-II (data not shown). Because L-DC once activated with LPS did not activate CD4+ T cells to levels shown by cDC, they do not reflect precursors of cDC. MHC-II expression levels on L-DC are quite distinct from those of cDC (both CD8α+ and CD8α− subsets) which show 100% expression on isolation, with slight reduction to 80–90% upon culture of cells with or with LPS for 24 hrs (data not shown). L-DC are not precursors of cDC and despite activation and culture in vitro, maintain a constant phenotype and distinct T-cell stimulatory function.
Discussion
This paper characterizes a novel APC subset produced in vitro which has an in vivo counterpart in murine spleen. A full understanding of all APC subsets and their immune response potential is needed to optimize vaccination and immunotherapy protocols. The novel APC subset ‘L-DC’ partially characterized here, resembles the immature myeloid ‘LTC-DC’ produced in long-term spleen cultures. Their equivalence is based on common marker expression and function in endocytosis and antigen presentation. Both LTC-DC and L-DC stimulate CD8+ T cells, particularly after exposure to ‘danger signals’ like LPS, but only weakly stimulate CD4+ T cells. L-DC cross-present antigen, and may function in viral infections or cancer where a CD8+ T-cell response is beneficial. We suggest that L-DC may play a distinct role in immunity to blood-borne antigen, and have high accessibility for intravenously delivered antigen. Here we show phenotypic and functional distinction between L-DC and other known APC subsets in spleen, justifying their further investigation as a novel immune system component.
Previous studies have demonstrated clear differences between mo-DC and cDC in terms of phenotype and cell lineage [11]. L-DC are distinct from both mo-DC and cDC which are MHC-II+ cells. We identify clear phenotypic distinction between myeloid (macrophage/monocyte) cells (FSCloCD11bhiCD11c−MHC-II−CD8α−), CD8α−cDC (FSCloCD11b+CD11chiMHC-II+CD8α−), CD8α+ cDC (FSCloCD11b−CD11chiMHC-II+CD8α+) and L-DC (FSChiCD11bhiCD11cloMHC-II−CD8α−). These cell subsets all differ in size, in vivo and in vitro endocytic capacity, and T cell activation ability. Analysis of uptake of intravenously administered antigen by APC subsets in spleen identified L-DC and myeloid cells as more accessible to blood-borne antigen than cDC (Fig. 5). By comparison with CD8α+ cDC [2], L-DC cross-present antigen particularly well after exposure to LPS, again a property shared with LTC-DC. This cross-presenting function distinguishes them from other myeloid lineage cells (i.e. monocytes/macrophages) that can take up antigen in vivo (Fig. 5), but cannot activate antigen-specific T cells in vitro (Fig. 6B). Because LPS is required to activate L-DC for cross-presentation, L-DC may exist in vivo in a resting or immature state, such that inflammation enhances their cross-presentation function. By contrast, CD8α+ cDC subsets are capable of cross-presentation upon ex vivo isolation, and reduce this capacity after LPS treatment [33]. L-DC could be important targets for vaccination against blood-borne pathogens or tumours, because inflammation would induce an optimal response. A DC subset in blood has been described with high accessibility to blood-borne antigens which functions in T-cell-independent marginal zone B-cell activation in spleen [34]. These were characterized as CD11cloCD11bhiCD8α−MHC-IIloCD80loCD86loB220− cells, resembling L-DC. However, the role of L-DC in spleen in induction of B-cell responses has not yet been investigated. When compared with less common DC subsets in spleen, L-DC are distinct from described marginal zone CD8α+CD103+CD207+ DC [35]. They are also distinct from CD11cloCD11b− pre-cDC [10] and from CD11b−CD8α+ cDC precursors [36].
In terms of their relationship with other subsets, LTC-DC do not have the immunosuppressive properties of regulatory DC (Fig. 3) [6, 7]. They also do not resemble TNF/iNOS-producing DC that are F4/80− and CD205+ cells [9, 37]. LTC-DC do not induce CD4+CD25+ regulatory T (Treg) cells as do pDC (unpublished data). CD86 was very weakly expressed on L-DC but was consistently expressed by LTC-DC. However, in vitro culture is known to cause some level of DC activation with up-regulation of CD86 [38], and reculture of LTC-DC directly on to plastic results in weak up-regulation of CD86 [21].
One important issue, considering the myeloid (CD11b+) and immature (MHC-II−) phenotype of LTC-DC and L-DC, is their relationship with the broader group of monocytes/macrophages. In all sorting experiments to isolate L-DC, a spleen cell population of ‘myeloid’ cells (CD11c−CD11bhiMHC-II−FSClo) was isolated for direct comparison. It should be emphasized that this ‘myeloid’ cell population, although heterogeneous, failed to stimulate either CD4+ or CD8+ T cells in an MLR, whereas L-DC displayed clear but restricted CD8+ T-cell activation capacity. This demonstrated a key characteristic of all DC but not other myeloid cell types, which is the ability to stimulate naïve T cells. Because LPS stimulation was required for acquisition of cross-presentation capacity, L-DC must exist in steady-state spleen as immature, and not activated cells. They contrast with mo-DC that require an inflammatory or disease context for development [10]. Although L-DC do not resemble other myeloid cells, the possibility that they share a common progenitor needs further exploration. The high in vivo capacity of L-DC for blood-borne antigen uptake is reflective of macrophages in the marginal zone of spleen [39]. However, macrophages do not contribute to antigen-specific T-cell immunity, and primarily process antigen for marginal zone B cells. L-DC are also F4/80+ cells (preliminary data), and do not resemble any of the spleen macrophage subsets, including marginal zone macrophages, metallophilic macrophages and red pulp macrophages. F4/80 is not expressed by marginal zone macrophages or metallophilic macrophages, and red pulp macrophages that express F4/80 are CD11blo[40].
L-DC and LTC-DC phenotypically, but not functionally, reflect some described regulatory DC subsets. These cells are not well characterized, and represent a range of cell types isolated under different conditions, having a common myeloid CD11cloCD11b+/hiMHC-IIlo phenotype. Unlike regulatory DC, LTC-DC have no suppressive capacity for T cells (Fig. 3). Some regulatory DC derive by further differentiation of DC or DC precursors, such as diffDC which develop in stromal cultures from mature mo-DC [6]. One distinct subset of regulatory ‘CD11cloCD45RB+’ DC which induces Tregs derives from Lin−ckit+ bone marrow progenitors cultured over a splenic stroma of fibroblasts and macrophages [41]. Splenic endothelial cells also support development of CD11cloCD11bhiMHC-IIlo regulatory DC from BM Lin−ckit+ precursors [7]. These were shown to have an in vivo spleen counterpart that is more similar in phenotype to L-DC than other regulatory DC. Functionally, these cells secrete nitric oxide and suppress CD4+ T-cell proliferation, reputed characteristics of regulatory DC [7]. The functional competence of L-DC and LTC-DC in terms of cross-presentation would appear to contradict this classification. Further comparative functional studies will be required to determine the extent of similarity between in vivo regulatory DC subsets and the L-DC subset described here. L-DC also resemble myeloid suppressor cells by expression of CD11b, F4/80 and Ly6C [42]. Myeloid suppressors are found in tumour-bearing mice and human beings and are very heterogeneous, representing a range of early myeloid differentiative states. L-DC are distinct, however, by their expression of CD11c and their existence in normal mice. At this stage, L-DC reflect myeloid suppressors only in so far as they are immature myeloid cells. Further experimentation will be directed at identifying the lineage origin of L-DC and understanding their unique functional capacity in regard to vaccination strategies.
Acknowledgments
This work was supported by funding from the National Health and Medical Research Council of Australia to H.O. Both J.K.H.T. and P.P. were supported by an ANU Graduate School scholarship. B.J.Q.C. was recipient of a Peter Doherty Fellowship from the National Health and Medical Research Council of Australia.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Maldonado-Lopez R, De Smedt T, Pajak B, et al. Role of CD8alpha+ and CD8alpha- dendritic cells in the induction of primary immune responses in vivo. J Leuk Biol. 1999;66:242–6. doi: 10.1002/jlb.66.2.242. [DOI] [PubMed] [Google Scholar]
- 2.Schnorrer P, Behrens GM, Wilson NS, et al. The dominant role of CD8+ dendritic cells in cross-presentation is not dictated by antigen capture. Proc Nat Acad Sci USA. 2006;103:10729–34. doi: 10.1073/pnas.0601956103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vremec D, Pooley J, Hochrein H, et al. CD4 and CD8 expression by dendritic cell subtypes in mouse thymus and spleen. J Immunol. 2000;164:2978–86. doi: 10.4049/jimmunol.164.6.2978. [DOI] [PubMed] [Google Scholar]
- 4.O’Keeffe M, Hochrein H, Vremec D, et al. Mouse plasmacytoid cells: long-lived cells, heterogeneous in surface phenotype and function, that differentiate into CD8(+) dendritic cells only after microbial stimulus. J Exp Med. 2002;196:1307–19. doi: 10.1084/jem.20021031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Asselin-Paturel C, Boonstra A, Dalod M, et al. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat Immunol. 2001;2:1144–50. doi: 10.1038/ni736. [DOI] [PubMed] [Google Scholar]
- 6.Zhang M, Tang H, Guo Z, et al. Splenic stroma drives mature dendritic cells to differentiate into regulatory dendritic cells. Nat Immunol. 2004;5:1124–33. doi: 10.1038/ni1130. [DOI] [PubMed] [Google Scholar]
- 7.Tang H, Guo Z, Zhang M, et al. Endothelial stroma programs hematopoietic stem cells to differentiate into regulatory dendritic cells through IL-10. Blood. 2006;108:1189–97. doi: 10.1182/blood-2006-01-007187. [DOI] [PubMed] [Google Scholar]
- 8.Wakkach A, Fournier N, Brun V, et al. Characterization of dendritic cells that induce tolerance and T regulatory 1 cell differentiation in vivo. Immunity. 2003;18:605–17. doi: 10.1016/s1074-7613(03)00113-4. [DOI] [PubMed] [Google Scholar]
- 9.Serbina NV, Salazar-Mather TP, Biron CA, et al. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. 2003;19:59–70. doi: 10.1016/s1074-7613(03)00171-7. [DOI] [PubMed] [Google Scholar]
- 10.Naik SH, Metcalf D, van Nieuwenhuijze A, et al. Intrasplenic steady-state dendritic cell precursors that are distinct from monocytes. Nat Immunol. 2006;7:663–71. doi: 10.1038/ni1340. [DOI] [PubMed] [Google Scholar]
- 11.Xu Y, Zhan Y, Lew AM, et al. Differential development of murine dendritic cells by GM-CSF versus Flt3 ligand has implications for inflammation and trafficking. J Immunol. 2007;179:7577–84. doi: 10.4049/jimmunol.179.11.7577. [DOI] [PubMed] [Google Scholar]
- 12.Randolph GJ, Inaba K, Robbiani DF, et al. Differentiation of phagocytic monocytes into lymph node dendritic cells in vivo. Immunity. 1999;11:753–61. doi: 10.1016/s1074-7613(00)80149-1. [DOI] [PubMed] [Google Scholar]
- 13.Tacke F, Randolph GJ. Migratory fate and differentiation of blood monocyte subsets. Immunobiol. 2006;211:609–18. doi: 10.1016/j.imbio.2006.05.025. [DOI] [PubMed] [Google Scholar]
- 14.Heath WR, Belz GT, Behrens GM, et al. Cross-presentation, dendritic cell subsets, and the generation of immunity to cellular antigens. Immunol Rev. 2004;199:9–26. doi: 10.1111/j.0105-2896.2004.00142.x. [DOI] [PubMed] [Google Scholar]
- 15.Villadangos JA. Presentation of antigens by MHC class II molecules: getting the most out of them. Mol Immunol. 2001;38:329–46. doi: 10.1016/s0161-5890(01)00069-4. [DOI] [PubMed] [Google Scholar]
- 16.Jackson MR, Peterson PA. Assembly and intracellular transport of MHC class I molecules. Annu Rev Cell Biol. 1993;9:207–35. doi: 10.1146/annurev.cb.09.110193.001231. [DOI] [PubMed] [Google Scholar]
- 17.den Haan JMM, Lehar SM, Bevan MJ. CD8+ but not CD8- dendritic cells cross-prime cytotoxic T cells in vivo. J Exp Med. 2000;192:1685–96. doi: 10.1084/jem.192.12.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramirez MC, Sigal LJ. Macrophages and dendritic cells use the cytosolic pathway to rapidly cross-present antigen from live, vaccinia-infected cells. J Immunol. 2002;169:6733–42. doi: 10.4049/jimmunol.169.12.6733. [DOI] [PubMed] [Google Scholar]
- 19.Beauvillain C, Delneste Y, Scotet M, et al. Neutrophils efficiently cross-prime naive T cells in vivo. Blood. 2007;110:2965–73. doi: 10.1182/blood-2006-12-063826. [DOI] [PubMed] [Google Scholar]
- 20.O’Neill HC, Wilson HL, Quah B, et al. Dendritic cell development in long-term spleen stromal cultures. Stem Cells. 2004;22:475–86. doi: 10.1634/stemcells.22-4-475. [DOI] [PubMed] [Google Scholar]
- 21.Quah B, Ni K, O’Neill HC. In vitro hematopoiesis produces a distinct class of immature dendritic cells from spleen progenitors with limited T cell stimulation capacity. Int Immunol. 2004;16:567–77. doi: 10.1093/intimm/dxh060. [DOI] [PubMed] [Google Scholar]
- 22.Ni K, O’Neill HC. Development of dendritic cells from GM-CSF-/- mice in vitro: GM-CSF enhances production and survival of cells. Dev Immunol. 2001;8:133–46. doi: 10.1155/2001/68024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Neill HC, Jonas N, Wilson H, et al. Immunotherapeutic potential of dendritic cells generated in long-term stroma-dependent cultures. Cancer Biother & Radiopharmac. 1999;14:263–76. doi: 10.1089/cbr.1999.14.263. [DOI] [PubMed] [Google Scholar]
- 24.Uphoff CC, Drexler HG. Detection of mycoplasma in leukemia-lymphoma cell lines using polymerase chain reaction. Leukemia. 2002;16:289–93. doi: 10.1038/sj.leu.2402365. [DOI] [PubMed] [Google Scholar]
- 25.Periasamy P, Tan JK, Griffiths KL, et al. Splenic stromal niches support hematopoiesis of dendritic-like cells from precursors in bone marrow and spleen. Exp Hematol. 2009;37:1060–71. doi: 10.1016/j.exphem.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 26.Hogquist KA, Jameson SC, Heath WR, et al. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 27.Barnden MJ, Allison J, Heath WR, et al. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol. 1998;76:34–40. doi: 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- 28.Winzler C, Rovere P, Rescigno M, et al. Maturation stages of mouse dendritic cells in growth factor-dependent long-term cultures. J Exp Med. 1997;185:317–28. doi: 10.1084/jem.185.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dudziak D, Kamphorst AO, Heidkamp GF, et al. Differential antigen processing by dendritic cell subsets in vivo. Science. 2007;315:107–11. doi: 10.1126/science.1136080. [DOI] [PubMed] [Google Scholar]
- 30.Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nat Rev Immunol. 2002;2:151–61. doi: 10.1038/nri746. [DOI] [PubMed] [Google Scholar]
- 31.Chen L, Calomeni E, Wen J, et al. Natural killer dendritic cells are an intermediate of developing dendritic cells. J Leukoc Biol. 2007;81:1422–33. doi: 10.1189/jlb.1106674. [DOI] [PubMed] [Google Scholar]
- 32.Naik SH, Proietto AI, Wilson NS, et al. Cutting edge: generation of splenic CD8+ and CD8- dendritic cell equivalents in Fms-like tyrosine kinase 3 ligand bone marrow cultures. J Immunol. 2005;174:6592–7. doi: 10.4049/jimmunol.174.11.6592. [DOI] [PubMed] [Google Scholar]
- 33.Wilson NS, Behrens GM, Lundie RJ, et al. Systemic activation of dendritic cells by Toll-like receptor ligands or malaria infection impairs cross-presentation and antiviral immunity. Nat Immunol. 2006;7:165–72. doi: 10.1038/ni1300. [DOI] [PubMed] [Google Scholar]
- 34.Balazs M, Martin F, Zhou T, et al. Blood dendritic cells interact with splenic marginal zone B cells to initiate T-independent immune responses. Immunity. 2002;17:341–52. doi: 10.1016/s1074-7613(02)00389-8. [DOI] [PubMed] [Google Scholar]
- 35.Qiu CH, Miyake Y, Kaise H, et al. Novel subset of CD8{alpha}+ dendritic cells localized in the marginal zone is responsible for tolerance to cell-associated antigens. J Immunol. 2009;182:4127–36. doi: 10.4049/jimmunol.0803364. [DOI] [PubMed] [Google Scholar]
- 36.Bedoui S, Prato S, Mintern J, et al. Characterization of an immediate splenic precursor of CD8+ dendritic cells capable of inducing antiviral T cell responses. J Immunol. 2009;182:4200–7. doi: 10.4049/jimmunol.0802286. [DOI] [PubMed] [Google Scholar]
- 37.Sunderkotter C, Nikolic T, Dillon, et al. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol. 2004;172:4410–7. doi: 10.4049/jimmunol.172.7.4410. [DOI] [PubMed] [Google Scholar]
- 38.Vremec D, Shortman K. Dendritic cell subtypes in mouse lymphoid organs – cross-correlation of surface markers, changes with incubation, and differences among thymus, spleen, and lymph nodes. J Immunol. 1997;159:565–73. [PubMed] [Google Scholar]
- 39.Geijtenbeek TB, Groot PC, Nolte MA, et al. Marginal zone macrophages express a murine homologue of DC-SIGN that captures blood-borne antigens in vivo. Blood. 2002;100:2908–16. doi: 10.1182/blood-2002-04-1044. [DOI] [PubMed] [Google Scholar]
- 40.Taylor PR, Martinez-Pomares L, Stacey M, et al. Macrophage receptors and immune recognition. Annu Rev Immunol. 2005;23:901–44. doi: 10.1146/annurev.immunol.23.021704.115816. [DOI] [PubMed] [Google Scholar]
- 41.Svensson M, Maroof A, Ato M, et al. Stromal cells direct local differentiation of regulatory dendritic cells. Immunity. 2004;21:805–16. doi: 10.1016/j.immuni.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 42.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]