

Abstract
It is evident that the symptoms of Alzheimer's disease (AD) are derived from severe neuronal damage, and especially pyramidal neurons in the hippocampus are affected pathologically. Here, we analysed the proteome of hippocampal neurons, isolated from post-mortem brains by laser capture microdissection. By using 18O labelling and mass spectrometry, the relative expression levels of 150 proteins in AD and controls were estimated. Many of the identified proteins are involved in transcription and nucleotide binding, glycolysis, heat-shock response, microtubule stabilization, axonal transport or inflammation. The proteins showing the most altered expression in AD were selected for immunohistochemical analysis. These analyses confirmed the altered expression levels, and showed in many AD cases a pathological pattern. For comparison, we also analysed hippocampal sections by Western blot. The expression levels found by this method showed poor correlation with the neuron-specific analysis. Hence, we conclude that cell-specific proteome analysis reveals differences in the proteome that cannot be detected by bulk analysis.
Keywords: Alzheimer's disease, 18O labelling, quantitative proteome, laser capture microdissection, pyramidal neuron, immunohistochemistry
Introduction
Alzheimer's disease is characterized by the presence of senile plaques and neurofibrillary tangles, and a loss of synapses and neurons that ultimately lead to brain atrophy [1-3]. The neurofibrillary tangles are detected in the cornu ammonis 1 (CA1) region of hippocampus already at an early stage of AD or mild cognitive impairment [4], and their appearance is followed by neuronal loss with mild Aβ plaque burden compared to other cortex regions. The pathogenesis in AD is not fully understood, but has been proposed to include amyloid β-peptide (Aβ) induced toxicity, tangle formation, excitotoxicity of neurons, oxidative stress and inflammation. We reasoned that investigating alterations in the protein expression in cells associated with AD would give further insight in the mechanisms behind AD.
The gene chip or microarray analysis is now a commonly used technique for transcriptome analysis. However, there is no linear relationship between gene expression and protein levels [5], and analysis of protein levels gives a more direct correlation to phenomena at a cellular or organ level. There are some reports based on proteome analysis using pieces of brain tissue [6, 7]. However, such samples contain a variety of cell types and extracellular proteins, and neuron-specific alterations cannot be observed by this approach. As an alternative to this bulk analysis, the laser capture microdissection (LCM) technique can be used to specifically isolate neurons from brain tissue [8]. For the unbiased identification of proteins, mass spectrometry (MS) is an excellent technique. Because of recent advancements in sensitivity and resolution power of MS, it is now possible to analyse the proteome of selected neurons captured by LCM from post-mortem human brain. However, MS analysis is not inherently quantitative, and to overcome this issue, differential stable isotope labelling methods have been developed. For instance, the 18O labelling technique results in the incorporation of two 18O atoms in the peptides obtained after proteolytic digestion of the sample.
Here, we combine LCM and 18O labelling to identify and compare the expression levels of 150 proteins in CA1 pyramidal neurons from post-mortem AD and control brains. To further study the most differently expressed proteins at a cellular level, we performed immunohistochemistry (IHC) and compared qualitative alterations and pathological changes in hippocampal sections from AD and control brains.
Materials and methods
Brain samples
All brain materials were obtained from the Huddinge Brain Bank at Karolinska Institutet Alzheimer Disease Research Center and stored at −80°C before use. All AD subjects, which were sporadic cases, met the criteria for definitive AD according to the Consortium to Establish a Registry for AD [9, 10]. The control subjects had no known symptoms of neurological or psychiatric disorders. Hippocampal cryosections were used for proteome analysis, and formalin fixed and paraffin embedded sections were used for IHC. We included six sporadic AD and six control cases. F-test showed equal variance, and Student's t-test (two-tailed) was used for a statistical analysis. There were no statistical differences between control and sporadic AD cases in terms of mean age at death and mean post-mortem intervals (Table 1). This study was approved by the Regional Ethical Committee at Karolinska Institutet.
Table 1.
Demographic of Alzheimer's disease and control subjects
| Group | Case label | Age of death (years old) | Gender | Post-mortem time (hour) | Diagnosis* | ApoE allele |
|---|---|---|---|---|---|---|
| Control | 1 | 91 | Female | 24 | Unaffected | n.d. |
| 2 | 78 | Male | 22 | Unaffected | 3/3 | |
| 3 | 89 | Female | 16 | Unaffected | 2/3 | |
| 4 | 82 | Female | 24 | Unaffected | 2/3 | |
| 5 | 80 | Male | 16 | Unaffected | 3/4 | |
| 6 | 84 | Male | 42 | Unaffected | 3/3 | |
| Sporadic AD | 7 | 83 | Female | 32 | Definite AD | 3/4 |
| 8 | 81 | Female | 26 | Definite AD | 2/3 | |
| 9 | 79 | Male | 17 | Definite AD | n.d. | |
| 10 | 74 | Male | 12 | Definite AD | 4/4 | |
| 11 | 72 | Female | 8 | Definite AD | n.d. | |
| 12 | 80 | Male | 12 | Definite AD | n.d. |
Diagnosis by CERAD criteria; n.d.: not determined.
Mean age of death (mean ± S.E.M.): control cases: 84 ± 2.1 yr; sporadic AD cases: 78 ± 1.7 yr.
Mean post-mortem intervals (mean ± S.E.M.): control cases: 24 ± 3.9 hrs; AD: 18 ± 3.8 hrs.
18O labelling efficacy
In a pilot study we investigated the labelling efficacy of 18O using one sample prepared from the CA1 region in hippocampus of case 9 (sporadic AD case) in Table 1. The hippocampal cryosections were mounted on polyethylene naphthalate membrane-covered slide glasses (Carl Zeiss AG, Oberkochen, Germany), fixed in ethanol and Nissl-stained. We dissected the CA1 region (95 μg), together with the polyethylene naphthalate membrane from the slide glass, from the surrounding tissue by using a microscope and a needle. After addition of 20 μl of 360 mM ammonium bicarbonate (Ambic) containing 4 mM CaCl2, 8 μl of 1% RapiGest SF (Waters Corporation, Milford, MA, USA) and 50 μl of Milli-Q water into the tube, the sample was sonicated for 5 min. and incubated at 95°C for 5 min. After cooling, 2 μg of trypsin (Trypsin Gold, Promega Corporation, WI, USA) was added and the sample was digested for 18 hrs. Three microlitres of HCl was added, and the sample was centrifuged at 13,000 r.p.m. for 10 min. to remove the hydrophobic debris from hydrolysed RapiGest SF. The peptides in the clear supernatant were concentrated by using a ZipTipC18 (Millipore, MA, USA) according to the instruction manual. Peptides were eluted from the ZipTipC18 by 80% CH3CN/0.3% formic acid, and the solvent was removed by a spin-vacuum system. After removal of the solvent, the sample was dissolved in 1.7 μl of 0.3 M sodium acetate pH 5.2 (prepared in 90% H218O), 1 μl of 50 mM CaCl2 and 47.3 μl of H218O (99% H218O; Sigma-Aldrich, Inc., St. Louis, MO, USA). To label the peptides with 18O, 1 μl of trypsin (0.5 mg/ml: reconstituted in H218O) was used, and the sample was incubated at 37°C for 48 hrs. To stop the labelling reaction, 8 μl of 5% formic acid (diluted in H218O) was added (pH 2–3). Trypsin was heat inactivated by incubation at 95°C for 90 min. The peptides were stored at −80°C. Only the AD sample was labelled with H218O, the control sample was treated with ordinary water (H216O). For the neuronal proteome analysis, we compared the proteome from controls (non-labelled) with that from AD cases (18O labelled). The 18O labelled peptides were diluted in 2% CH3CN/0.2% formic acid (in H218O) to a concentration of 0.3 μg protein/μl.
For verification of labelling efficacy, an Agilent 6330 Ion Trap liquid chromatography/mass spectrometer (Agilent Technologies, CA, USA) was used. Three microlitres of the sample was injected onto the enrichment column (160 nl) and separated on a 150 mm χ 75 μm analytical column packed with 5 μm Zorbax 300SB-C18. The peptides were eluted at a flow rate of 200 nl/min. by a gradient supplemented with 0.1% formic acid: from 3% to 26% CH3CN in 184 min. and from 26% to 36% CH3CN in 40 min. Mass spectra were recorded from m/z 230 to m/z 1,800. The peptide identification was performed by Mascot software version 2.2 (Matrix Science, Tokyo, Japan) and the Swiss-Prot database (release 55).
Isolation of CA1 pyramidal neurons and sample preparation
The protocol for LCM was described previously [8]. Briefly, after fixation in ethanol, the sections (10 μm in thickness) were mounted on polyethylene naphthalate membrane-covered slide glasses (Carl Zeiss AG) and Nissl stained. The cryosections were stained in pale blue, because intense staining absorbs too much of the laser energy. Glial cells were morphologically distinguished from neurons and carefully excluded from dissection. By using the Laser Microbeam System (Carl Zeiss AG), 2000 of CA1 pyramidal neurons per case (six AD and six controls) were catapulted into LPC-Microfuge tube caps (PALM tube, Carl Zeiss AG), which contained Milli-Q water. Thus, we captured 12,000 neurons from each group. The captured neurons were recovered at the bottom of a PALM tube after centrifugation. Following the addition of 1 μl of 0.5% RapiGest SF, endogenous proteases in samples were heat-inactivated by incubation at 95°C for 90 min. After rapid cooling on ice, water was removed by using a spin-vacuum system. Two microlitres of 360 mM Ambic containing 4 mM CaCl2, 1 μl of 1% RapiGest SF and 5 μl of Milli-Q water were added, and the samples were then sonicated (bath-type) for 5 min. The AD samples were pooled in one tube and the controls in another, and the total amount of protein was determined by FluoroProfile Protein Quantification Kit (Sigma-Aldrich, Inc.). Each tube contained 96 μl of the material derived from six AD and six control cases, respectively (12,000 neurons/tube). To cleave the proteins, 3 μl of 0.1 mg/ml trypsin was added to each tube, and the samples were incubated at 37°C for 24 hrs. The trypsin cleavage was confirmed by SDS-PAGE (4–12% Bis-Tris gels; Invitrogen, Carlsbad, CA, USA) and silver staining (GE Healthcare UK Ltd., NA, UK). RapiGest SF was removed and the tryptic peptides were recovered as described earlier. For comparison of the proteome from AD with control, the AD sample was labelled with H218O as described earlier, while the control sample was treated with ordinary water (H216O).
Proteome analysis of microdissected neurons
For protein identification and to obtain the ratios of the proteins identified in AD and control samples, an LTQ-Orbitrap mass spectrometer (ThermoFisher, CA, USA) was used. Equal amounts of AD and control samples were mixed, and 20 μl of the mixture was injected into a Paradigm MS4 (Michrom Bioresource, CA, USA) with an analytical column (Zorbax 300SB, 0.1 χ 150 mm Agilent Technologies). In total, the sample was injected five times. The mobile phases consisted of 0.1% acetic acid in water (mobile phase A) and 0.1% acetic acid in methanol (mobile phase B). After sample injection, the column was washed for 5 min. with mobile phase A, and peptides were eluted at 500 nl/min using a linear gradient from 5% to 75% mobile phase B in 90 min., and then to 95% B in 10 min. The capillary high-performance liquid chromatography system was coupled to the LTQ-Orbitrap mass spectrometer using a nano-electrospray ionization source (Protana, Odense, Denmark). The spray voltage was 2.0 kV and the temperature of the heated capillary was 200°C. Eluting peptides were analysed using the data-dependent MS/MS mode over a 400–2000 m/z range. For data processing, the identification of peptides was performed with Mascot software version 2.2 and the Swiss-Prot database (release 55). The quantitative analysis was carried out by Xome software (Mitsui Knowledge Industry, Tokyo, Japan). The parameters used for the database search were: monoisotopic mass, peptide mass tolerance of 10 ppm, fragment ion MS/MS tolerance of 0.8 Da, tryptic peptides only, maximum of one missed cleavages, and double 18O labelling (C-term) and oxidation of methionine as variable modifications. All results were combined together using Xome software, and filtered by the peptide score (>25) and the significance threshold P < 0.05. Then, the MS peak heights from 18O labelled peptide were divided by those from unlabelled peptides. The relative expression of each protein was calculated as the average of ratios (18O versus16O MS peak height) of the peptides that were identified from the five injections.
Immunohistochemistry
Immunohistochemistry was performed on 7 μm-thick sections cut from formaldehyde fixed and paraffin embedded tissues. After the dewaxing and rehydration process, for detection of proteins except Aβ peptides and phosphorylated tau (p-tau, phosphorylation at Ser 202), the sections were treated in a pressure cooker for 25 min. using an antigen retrieval solution (Diva Decloaker BIOCARE MEDICAL, CA, USA).
For the detection of Aβ (and also C99 and APP) by using 6E10 antibody (anti-Aβ1–16 monoclonal antibody; COVANCE, NJ, USA), the sections were treated with 99% formic acid for 5 min. The background was reduced by using 3% goat serum (Abcam Inc., MA, USA) in Tris-buffered saline, pH 7.6 or Protein Block Serum-Free (Dako Denmark A/S, Glostrup, Denmark). The sections were incubated at 4°C overnight with each antibody shown in Table S1, and washed with Tris-buffered saline. To detect proteins, except LRP1 and NCF4, sections were treated with secondary biotinylated goat anti-rabbit or antimouse IgG (Vector Laboratories, Burlingame, CA, USA) at 1:300 for 1 hr at room temperature. Then, the sections were treated with Vectastain Elite ABC kit (Vector Laboratories) and visualized by addition of 3–3-diaminobenzidine-4 HCl (DAB/H2O2). For the detection of LRP1 and NCF4, we used DAB/H2O2, and MACH 3 Mouse and Rabbit HRP Polymer detection system (BIOCARE MEDICAL), respectively. All sections were counterstained with haematoxylin. The antibodies and staining conditions are summarized in Table S1.
Western blot analysis of hippocampal CA1 region
The CA1 region of hippocampal sections was isolated as described earlier (see the preparation of CA1 regions from hippocampus under ‘18O labelling efficacy’), dissolved in RIPA buffer and sonicated. SDS sample buffer (LDS sample buffer; Invitrogen Corporation, CA, USA) was added to the samples, and they were loaded onto an SDS-PAGE gel (4–12%; Invitrogen). In each lane, 29 μg of protein was loaded. MagicMark XP standard was used as a molecular weight marker (Invitrogen). For blotting, we used PVDF membranes (Invitrogen) and a Mini Trans-Blot system (Bio-Rad Laboratories, CA, USA) with 25 mM Tris, 192 mM glycine and 10% (v/v) methanol, pH 8.3 as a transfer buffer. The blocking of membrane was performed by 1 hr incubation in 5% (w/v) non-fat dry milk in PBS containing 0.1% Tween-20 (PBST) at room temperature. Probing for carboxyl-terminal PDZ ligand of neuronal nitric oxide synthase protein (CAPON) with antibody sc-9138 (Santa Cruz Biotechnology, Inc., CA, USA) was performed at a dilution of 1/1000 in PBST containing 5% (w/v) BSA for 20 hrs at 4°C. The membranes were incubated with horseradish peroxidase-linked goat–anti-rabbit IgG (1/50,000; GE Healthcare UK Ltd.) in 5% (w/v) non-fat dry milk in PBST for 1 hr at room temperature. The detection of CAPON was achieved by using SuperSignal® West Dura (Thermo Fisher Scientific, IL, USA).
Results
Evaluation of 18O labelling efficacy
For accurate quantification, efficient and reproducible labelling is necessary. We evaluated the labelling by using a test sample prepared from the CA1 region of hippocampus. The sample was digested and labelled by trypsin in H218O, concentrated and desalted by using a ZipTipC18 and injected into the LC-MS system. There was a large peak from the double-labelled peptide (18O:18O) (TGQAPGFSYTDANK, 730.8 m/z) accompanied with the negligible unlabeled peak (728.8 m/z) (Fig. 1A, lower panel). In total, 54 spectra from 26 proteins were evaluated with respect to the ratio of the monoisotopic peak height of non-labelled and 18O double-labelled peptide (Fig. 1B). The average ratio of non-labelled to 18O double-labelled was 0.047 ± 0.029 (mean ± S.D.) and the ratios were distributed in a narrow range.
Fig 1.
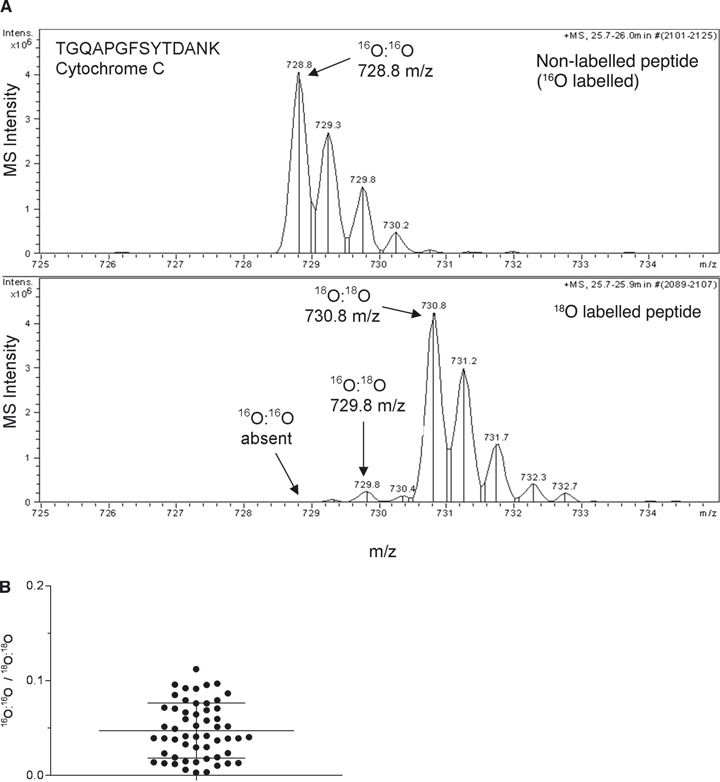
18O labelling of tryptic peptides. (A) Non-labelled (upper) or 18O labelled (lower) MS spectrum from Cytochrome C. A sample from hippocampal CA1 sections was labelled in H216O or H218O. The tryptic peptides were injected into the LC-MS system (Agilent 6330). Peptide identification was performed by MASCOT search. Note the low level of unlabelled peptide in the lower panel. (B) The ratios of unlabelled versus double-labelled peptide in an 18O labelled sample. In the randomly selected 54 MS spectra, the average ratio of non-labelled (16O:16O) and double-labelled peptide (18O:18O) was 0.047 (B). The mean ± S.D. is expressed by lines.
Since, in quantitative proteome analysis, the 18O labelled and the non-labelled samples are mixed before analysis, it is important to control the carboxyl oxygen exchange reaction (back reaction) catalysed by trypsin or low pH [11]. Otherwise, there will be a decrease in labelled peptides over time. We heat-inactivated the sample, followed the back-reaction and found that the percentage of double-labelled peptide within 5 hrs was acceptable for our purpose (Fig. S1A and B).
Quantitative proteome analysis of microdissected pyramidal neurons from human hippocampus
We wanted to compare, between AD cases and controls, the proteome of neurons that were viable before the death of the patients. Therefore, we captured Nissl positive pyramidal neurons by LCM, labelled the AD sample by 18O and analysed the sample by an LTQ-Orbitrap mass spectrometer. The amount of protein from 12,000 neurons was 15 μg in the AD sample as well as in the control tube (59 μl/tube). Equal volumes (10 μl) of the AD and control sample were mixed and injected into the LC-MS system. Hence the amount of total proteins in a single injection was 5 μg. In total, 150 proteins were identified by MS/MS, and the relative expression of those (AD/control) was calculated based on MS peak height (Table S2). Many of these proteins were found to be down-regulated in AD, but there were also those that were up-regulated. The most up-regulated and down-regulated proteins are presented in Table 2 and 3, respectively.
Table 2.
Proteins up-regulated in Alzheimer's disease
| Accession number | Registered record in SWISSPROT | Protein description | Mean ratio (AD/control) |
|---|---|---|---|
| Q93070 | NAR4_HUMAN | Ecto-ADP-ribosyltransferase 4 | 29 |
| 96RP9 | EFG1_HUMAN | Elongation factor G 1, mitochondrial | 22 |
| Q07954 | LRP1_HUMAN | Prolow-density lipoprotein receptor-related protein 1 | 12 |
| Q9Y4Z0 | LSM4_HUMAN | U6 snRNA-associated Sm-like protein LSm4 | 11 |
| Q00872 | MYPC1_HUMAN | Myosin-binding protein C, slow-type | 9.8 |
| Q8IUG5 | MY18B_HUMAN | Myosin-XVIIIb | 9.7 |
| Q9Y222 | DMTF1_HUMAN | Cyclin-D-binding Myb-like transcription factor 1 | 9.6 |
| O75448 | MED24_HUMAN | Mediator of RNA polymerase II transcription subunit 24 | 8.3 |
| Q7RTR0 | NALP9_HUMAN | NACHT, LRR and PYD domains-containing protein 9 | 7.8 |
| Q15080 | NCF4_HUMAN | Neutrophil cytosol factor 4 | 7.0 |
| Q9H4K7 | GTPB5_HUMAN | GTP-binding protein 5 | 6.5 |
| O75052 | CAPON_HUMAN | Carboxyl-terminal PDZ ligand of neuronal nitric oxide synthase protein | 6.5 |
| Q9NY47 | CA2D2_HUMAN | Voltage-dependent calcium channel subunit alpha-2/delta-2 | 5.6 |
| Q9H008 | LHPP_HUMAN | Phospholysine phosphohistidine inorganic pyrophosphate phosphatase | 5.2 |
| P14136 | GFAP_HUMAN | Glial fibrillary acidic protein | 4.9 |
| P07196 | NFL_HUMAN | Neurofilament light polypeptide | 4.7 |
| P35475 | IDUA_HUMAN | Alpha-L-iduronidase | 4.6 |
| P16442 | BGAT_HUMAN | Histo-blood group ABO system transferase | 4.5 |
| P60201 | MYPR_HUMAN | Myelin proteolipid protein | 3.6 |
| Q12888 | TP53B_HUMAN | Tumour suppressor p53-binding protein 1 | 3.2 |
| Q9C0F0 | ASXL3_HUMAN | Putative Polycomb group protein ASXL3 | 2.7 |
| Q92902 | HPS1_HUMAN | Hermansky-Pudlak syndrome 1 protein | 2.5 |
| A2RU48 | CL069_HUMAN | Uncharacterized protein C12orf69 | 2.4 |
| Q8NGA2 | OR7A2_HUMAN | Putative olfactory receptor 7A2 | 2.4 |
| P04271 | S100B_HUMAN | Protein S100-B | 2.2 |
| P21796 | VDAC1_HUMAN | Voltage-dependent anion-selective channel protein 1 | 2.1 |
| P01040 | CYTA_HUMAN | Cystatin-A | 2.0 |
| Q71U36 | TBA1A_HUMAN | Tubulin alpha-1A chain | 1.9 |
| P04406 | G3P_HUMAN | Glyceraldehyde-3-phosphate dehydrogenase | 1.8 |
| P61604 | CH10_HUMAN | 10 kD heat shock protein, mitochondrial | 1.8 |
Table 3.
Proteins down-regulated in Alzheimer's disease
| Accession number | Registered record in SWISSPROT | Protein description | Mean ratio (AD/control) |
|---|---|---|---|
| P04075 | ALDOA_HUMAN | Fructose-bisphosphate aldolase A | 0.22 |
| P80723 | BASP_HUMAN | Brain acid soluble protein 1 | 0.21 |
| Q2PPJ7 | AS250_HUMAN | 250 kD substrate of Akt | 0.21 |
| Q96FN5 | KIF12_HUMAN | Kinesin-like protein KIF12 | 0.21 |
| P62258 | 1433E_HUMAN | 14-3-3 protein epsilon | 0.21 |
| A6NDB9 | PALM3_HUMAN | Paralemmin-3 | 0.20 |
| Q9P273 | TEN3_HUMAN | Teneurin-3 | 0.20 |
| Q13153 | PAK1_HUMAN | Serine/threonine-protein kinase PAK 1 | 0.18 |
| A6NN79 | PERPL_HUMAN | Putative PERP-like protein | 0.17 |
| Q15262 | PTPRK_HUMAN | Receptor-type tyrosine-protein phosphatase kappa | 0.16 |
| Q2KJY2 | KI26B_HUMAN | Kinesin-like protein KIF26B | 0.16 |
| P27348 | 1433T_HUMAN | 14-3-3 protein theta | 0.16 |
| P51991 | ROA3_HUMAN | Heterogeneous nuclear ribonucleoprotein A3 | 0.16 |
| P09972 | ALDOC_HUMAN | Fructose-bisphosphate aldolase C | 0.16 |
| Q6UXK2 | ISLR2_HUMAN | Immunoglobulin superfamily containing leucine-rich repeat protein 2 | 0.15 |
| Q8TEW8 | PAR3L_HUMAN | Partitioning defective 3 homologue B | 0.14 |
| Q5QHF1 | CT199_HUMAN | Uncharacterized protein C20orf199 | 0.13 |
| P07101 | TY3H_HUMAN | Tyrosine 3-monooxygenase | 0.12 |
| Q6W2J9 | BCOR_HUMAN | BCL-6 co-repressor | 0.11 |
| Q5T215 | TPC3L_HUMAN | Trafficking protein particle complex subunit 3-like protein | 0.10 |
| Q9BXS6 | NUSAP_HUMAN | Nucleolar and spindle-associated protein 1 | 0.10 |
| Q15615 | OR4D1_HUMAN | Olfactory receptor 4D1 | 0.10 |
| Q92614 | MY18A_HUMAN | Myosin-XVIIIa | 0.10 |
| P52735 | VAV2_HUMAN | Guanine nucleotide exchange factor VAV2 | 0.09 |
| P62158 | CALM_HUMAN | Calmodulin | 0.09 |
| Q8TAG9 | EXOC6_HUMAN | Exocyst complex component 6 | 0.09 |
| O15226 | NKRF_HUMAN | NF-kappa-B-repressing factor | 0.09 |
| P25713 | MT3_HUMAN | Metallothionein-3 | 0.09 |
| P10809 | CH60_HUMAN | 60 kDa heat shock protein, mitochondrial | 0.08 |
| Q13023 | AKAP6_HUMAN | A-kinase anchor protein 6 | 0.08 |
| O75586 | MED6_HUMAN | Mediator of RNA polymerase II transcription subunit 6 | 0.07 |
| Q96PX9 | PKH4B_HUMAN | Pleckstrin homology domain-containing family G member 4B | 0.06 |
| P02452 | CO1A1_HUMAN | Collagen alpha-1(I) chain | 0.04 |
| Q13451 | FKBP5_HUMAN | FK506-binding protein 5 | 0.02 |
| P02461 | CO3A1_HUMAN | Collagen alpha-1(III) chain | 0.02 |
| P16152 | CBR1_HUMAN | Carbonyl reductase ADPH] 1 | 0.02 |
| Q9HCH0 | K1602_HUMAN | Uncharacterized protein KIAA1602 | 0.02 |
| Q6ZU80 | CN145_HUMAN | Uncharacterized protein C14orf145 | 0.00 |
Most notably, the expression level of ecto-ADP-ribosyltransferase 4 (NAR4) was increased around 29 times compared to the control (Table 2). Most proteins identified were non-transmembrane proteins, but also some membrane bound proteins were detected. We found many different categories of proteins such as transcription-related proteins (cyclin-D-binding Myb-like transcription factor 1, mediator of RNA polymerase II transcription subunit 24, tumour suppressor p53-binding protein 1 (TP53B) and putative Polycomb group protein ASXL3) to be up-regulated in AD, while BCL-6 co-repressor, NF-κB-repressing factor (NKRF) and mediator of RNA polymerase II transcription subunit 6 were down-regulated in AD (Tables 2 and 3). Many DNA- or RNA-binding proteins were also found to be decreased in AD (putative heterogeneous nuclear ribonucleoprotein A1-like protein 3, nucleolar and spindle-associated protein 1 and heterogeneous nuclear ribonucleoprotein A3; Table S2). There are some reports suggesting that the glycolysis is disrupted in neurons in AD brains [7, 12]. In line with this notion, there were some glycolysis-related proteins among the down-regulated proteins in AD, such as pyruvate kinase isozymes M1/M2, gamma-enolase (ENOG), fructose-bisphosphate aldolase A (ALDOA) and fructose-bisphosphate aldolase C (ALDOC). Interestingly, glyceraldehyde-3-phosphate dehydrogenase (G3P), which is involved in glycolysis and thought to be present at a constant level in cells, was up-regulated in AD (Tables 3 and S2).
There is a high amount of tangles, which consist of p-tau, in the hippocampus of severe AD cases. Because p-tau can accelerate breakdown of microtubules into tubulin dimers, we expected decreased levels of tubulin in AD due to a decreased amount of microtubules. Interestingly, however, the levels of four of five tubulin proteins (tubulin α-1A chain, tubulin β-2A chain, tubulin α chain-like 3, tubulin β-3 chain and tubulin β-2C) were found to be unchanged or slightly increased in AD (Table S2). Kinesin proteins are known to act as motor proteins and are involved in axonal transport. We detected a decrease in three kinesin-like proteins: KIF12, KIF26B and KIF27 (Tables 3 and S2). Not only axonal transport but also intracellular transport as well as the release and uptake at the synapses are disturbed in AD brains. Interestingly, we found exocyst complex component 6 involved in docking of exocytic vesicles, and syntaxin-1B (STX1B) and synaptosomal-associated protein 25 (SNP25), which have a role in synaptic vesicle exocytosis, to be down-regulated in AD (Tables 3 and S2). It has been reported that oxidative and nitrosative damage can cause neuronal death in AD. Here, we identified neutrophil cytosolic factor 4 (NCF4), which may be involved in overproduction of reactive oxygen species, as one of the up-regulated proteins (Table 2). Previous reports suggest that the neuronal form of nitric oxide synthase (nNOS) is up-regulated in hippocampal pyramidal neurons in AD [13,14]. Here, we found the CAPON, which is an adaptor protein to nNOS, to be up-regulated in AD (Table 2).
Immunohistochemical studies of proteins with altered expression
Selection of proteins for IHC studies
Based on the protein expression ratios (AD/control) and availability of antibodies, 23 proteins were selected for further studies by IHC. Fifteen of the selected, commercially available, antibodies were found suitable for IHC. Seven of the selected proteins showed similar expression patterns in AD and controls. The alteration in expression, as analysed by MS, was for all of these proteins less than twice, suggesting that it may be difficult to detect such differences by IHC. Hence, antibodies against the remaining eight proteins were used for the studies below. The background derived from the secondary antibody was in all cases undetectable (data not shown), and the experimental conditions are summarized in Table S1.
Estimation of amyloid load and neurofibrillary tangles (p-tau) in hippocampal sections
We assayed sections from all cases used for LCM, and detected amyloid plaques (6E10 immunoreactivity) as well as p-tau (AT-8 antibody) in the CA1 region of the AD case (Fig. S2B and D). Neither amyloid plaques nor p-tau was detected in CA1 of the control cases (Fig. S2A and C).
Pathologically changed expression pattern of S100-B protein (S100B)
In the control case, S100B was specifically expressed in astrocytes and oligodendrocytes (Fig. 2A). In AD cases, the immunoreactivity of S100B in astrocytes was increased as compared to the control case (Fig. 2A–C). S100B immunoreactivity was observed also in endothelial cells in capillaries. However, in one AD case, we detected immunoreactivity not only in glial cells but also in some pyramidal neurons (Fig. 2B, case number 9). We found another AD case to have a high intraneuronal expression of S100B (Fig. 2C, case number 8). In this case, some neurons lacked nuclear structure, suggesting that S100B could be involved in severe neurodegeneration and tangle formation.
Fig 2.
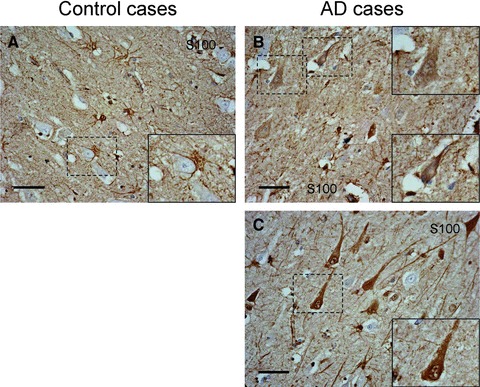
S100B staining in the CA1 region. (A) Control, case number 6. (B, C) Sporadic AD cases, case numbers 9 and 8, respectively (Table 1). Magnification of originals: 40χ. Magnification of insets: 70χ. Scale bar corresponds to 50 μm.
NKRF, TP53B and the low-density lipoprotein-related protein 1 (LRP1) accumulate in the nucleus
The expression of NKRF was detected only in pyramidal neurons, and was predominantly limited to the nucleolus with weak positivity in the nucleoplasm and perinuclear membrane in the control case (Fig. 3A). Although intense staining was detectable in the nucleolus in the AD case, the perinuclear and nucleoplasm staining was weaker than in the controls (Fig. 3B). The expression level in CA1 pyramidal neurons was clearly higher than in pyramidal neurons in CA3 and cortex (data not shown).
Fig 3.
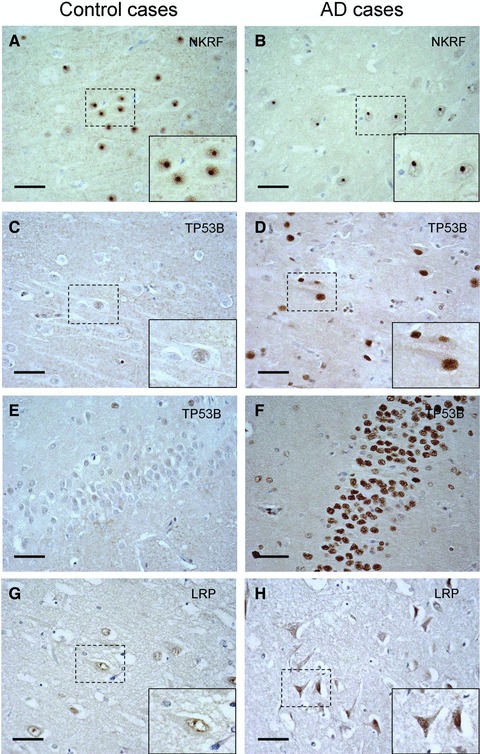
NKRF, TP53B and LRP1 staining in CA1 region. (A, B) NKRF in CA1; (C, D) TP53B in CA1; (E, F) TP53B in gyrus dentatus; (G, H) LRP1 in CA1; (A, C, E, G) Controls, case number 2, 3, 3 and 6, respectively. (B, D, F, H) Sporadic AD, case numbers 9, 10, 10 and 9, respectively. These proteins were detected in nucleolus in AD cases. Magnification of originals: 40χ. Magnification of insets: 70χ. Scale bar corresponds to 50 μm.
The MS analysis indicated that the expression of TP53B, which can bind to the tumour suppressor protein p53, was increased in AD. In the control case, the staining was barely detected in the nuclei of CA1 pyramidal neurons (Fig. 3C). On the other hand, pyramidal neurons and oligodendrocytes from the AD case showed intense nuclear staining, and cells (mostly neurons) in stratum granulosum in gyrus dentatus showed very strong staining in their nucleus (Fig. 3F) compared to the control case (Fig. 3E). Moreover, in the gyrus dentatus in Figure 3F, it was suggested that nuclear staining in astrocytes was weaker than in neurons.
The weak immunoreactivity of LRP1 was diffusely spread throughout the neurons in the controls, and clear staining was detected in the nuclear membrane but rarely at the plasma membrane (Fig. 3G). Interestingly, a condensed signal from the cell body and the nucleus, suggesting neurodegeneration, was detected in neurons from the AD case (Fig. 3H). The most intense signal was detected in neurons having a tangle-like shape and condensed immunoreactivity in the nucleus, suggesting severe insult or neurodegeneration.
Proteins involved in oxidative and nitrosative damage: NCF4 and CAPON
NCF4 was by MS analysis found to be up-regulated in AD. Immunohistochemistry showed high expression in blood cells (possibly monocytes) in the control case, while the neurons were weakly stained (Fig. 4A). In contrast, the pyramidal neurons from the AD case showed more intense, mostly cytoplasmic, staining (Fig. 4B). Interestingly, in this AD case, there was pathognomonic positivity in the extraneuronal space. We found another AD case, in which heavy staining was detected not only in neurons but also in the extraneuronal space (Fig. 4C, case number 10). To investigate whether the immunoreactivity in the area around neurons (Fig. 4B and C) was derived from microglia, an antibody directed to a microglia marker was used (Table S1). Similar morphology as in Figure 4C was observed (Fig. 4E), suggesting that NCF4 is up-regulated also microglia in AD. Furthermore, in case number 10 (Fig. 4C), strong nuclear staining of NCF4 was detected.
Fig 4.

NCF4, microglia marker and CAPON staining in CA1 region. (A–C) NCF4; (D, E) Microglia; (F, G) CAPON; (A, D, F) Controls, case numbers 3, 3 and 6, respectively. (B, C, E, G) Sporadic AD, case numbers 7, 10, 10 and 9, respectively. An antibody against HLA-DP DQ DR was used for the detection of microglia. Arrow heads in (A) and (B) indicate immunoreactivity in blood cells. Magnification of originals: 40χ. Magnification of insets: 70χ. Scale bar corresponds to 50 μm.
CAPON is likely to be ubiquitously expressed, and highly expressed in some parts of the brain [15]. CAPON was weakly expressed in pyramidal neurons and oligodendrocytes in the control case (Fig. 4F), while in the AD case, CAPON was up-regulated not only in neurons but also in oligodendrocytes and in capillaries (Fig. 4G). Interestingly, the expression of CAPON was not increased in all neurons, but dramatically up-regulated in some neurons (Fig. 4G, inset).
Novel proteins: NAR4 and uncharacterized protein C14orf145 (CN145)
By MS analysis, we found some proteins that are not fully investigated by now. NAR4 is predicted to be a member of the ADP ribosyltransferase gene family according to the primary amino acid sequence, and to be expressed at the erythrocyte membrane. In the proteome analysis, NAR4 was found to be extremely up-regulated in hippocampal neurons from AD. Dense immunoreactivity was observed in neurons as well as in the extracellular space of the AD case (Fig. 5B). The increased staining of the extracellular space might suggest an increased cleavage and secretion of NAR4 from neurons in AD. The strongest immunoreactivity was detected in neurons with a tangle-like shape, indicating that up-regulation of NAR4 in CA1 neurons may correlate with tangle formation. Furthermore, we could detect an intense signal in blood cells in the AD case (arrow heads in Fig. 5B).
Fig 5.
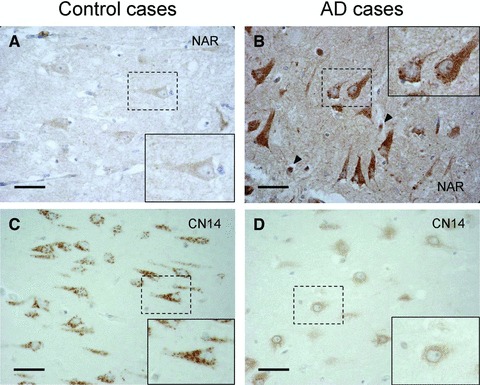
NAR4 and CN145 staining in CA1 region. (A, B) NAR4; (C, D) CN145; (A, C) Controls, cases numbers 6 and 4, respectively. (B, D) Sporadic AD, case number 9 in both. Arrow heads in B indicate immunoreactivity in blood cells. Magnification of originals: 40χ. Magnification of insets: 70χ. Scale bar corresponds to 50 μm.
The intracellular distribution of CN145 has not been fully reported by now. The expression of CN145 was neuron specific (Fig. 5C and D), and in the control case, a more intense and distinct signal was observed in the cell body and possibly in apical dendrites. Interestingly, the staining pattern in the AD case was completely altered into a diffused pattern with weak and perinuclear staining (Fig. 5D, inset), suggesting a shift in compartmentalization. To explore whether the intraneuronal staining was associated with mitochondria or Golgi apparatus, we stained the section with the appropriate markers. However, the immunoreactivity was morphologically quite different (data not shown).
Western blot analysis of CAPON in hippocampal sections
By MS analysis, a tryptic peptide from the long form of CAPON (CAPON-L) was identified and found to be increased in AD (Table 2), and a more intense CAPON staining in the AD case than in the control was observed by IHC (Fig. 5A and B). Because the antibody used in IHC can detect both the long and the short form of CAPON (CAPON-S), we cannot distinguish between them. For comparison, we analysed homogenized hippocampal sections by using Western blot. Both forms of CAPON were detected, the longer form being more abundant. Contrary to LCM-MS analysis, the levels of CAPON were lower in most AD cases compared to controls (Fig. 6). Apparently, the neuron-specific increase in CAPON expression escapes detection due to the presence of extraneuronal tissue. Thus, there is a significant advantage of neuronal analysis by LCM-MS compared to bulk analysis by Western blot.
Fig 6.
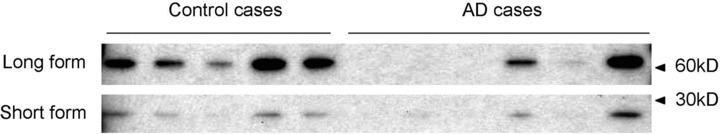
Western blot analysis of CAPON expression in the CA1 region. The samples prepared from the CA1 region were loaded and separated on a 4–12% SDS-PAGE gel. Detection of CAPON was performed by the sc-9138 antibody, and visualized by SuperSignal® West Dura. The bands correspond to the long form (upper bands) and short form (lower bands) of CAPON.
Discussion
To the best of our knowledge, there is today no report on neuron-specific proteome analysis by MS. Here, we employed the 18O labelling technique for finding proteins that are up- or down-regulated in hippocampal pyramidal neurons in AD compared to control cases. In our preliminary experiment using 2000 neurons per tube, we lost almost all proteins during the sample preparation. Therefore, to obtain sufficient material for MS analysis, the amount of neurons captured by LCM was increased to 12,000 neurons/tube, corresponding to 15 μg of protein/tube. To get accurate values from the 18O labelled proteome, it is significant that the efficacy of labelling is high and stable. Hence, we optimized the labelling protocol. The labelling reaction by trypsin was reported to be a double step [16]. Hajkova et al. demonstrated the pH dependency of the labelling reaction, in which the pH above 8 was favourable for proteolytic cleavage of a peptide bond (first reaction), and the optimal pH was round 6 for carboxyl oxygen exchange (second reaction) [17]. Thus, we used Ambic and sodium acetate buffers for the cleavage and the labelling step, respectively. Because the percentage of H218O in the labelling step was 98% and the labelling process by trypsin results in the incorporation of two oxygen atoms, the theoretical maximum percentage of a double-labelled peptide is around 96% (0.982). Therefore, the theoretical ratio of non-labelled/double-labelled peptide is 0.042 (22/96). Because the observed mean ratio, 0.047 (Fig. 1B), was close to the theoretical value, we concluded that the labelling reaction completed adequately. Furthermore, after efficient heat inactivation of the tryptic activity, the labelling was found to be sufficiently stable over time for reliable analysis (Fig. S1A and B). A high sensitivity and a high-resolution power are critical in quantitative proteome analysis. Here, we employed the LTQ-Orbitrap MS system, which has an excellent resolution power, and the Xome software for quantification. Finally, we identified and quantified 150 proteins (Table S2).
Many of the proteins we found are involved in glycolysis (ENOA, ENOG, ALDOA, ALDOC and G3P). There are now several evidences suggesting that glucose metabolism is disrupted in AD brains [18, 19]. A down-regulation of these proteins may suggest an interference of glucose utilization in AD brains. In line with our findings, Brooks et al. reported that the mRNA expression levels of ENOA, ENOG, ALDOA and ALDOC were decreased in AD. On the other hand, we found an increased level of G3P, while Brooks et al. observed the reduced mRNA level [20]. In previous work on hippocampal proteome analysis using brain tissue blocks, fructose bisphosphate aldolase was found to be up-regulated significantly [7], while we found it to be down-regulated. The discrepancies mentioned earlier could be due to the different materials (tissue block or neuronal samples, the stage of AD), analytes (mRNA or proteins) or a combination of these. Intracellular, including axonal, transport is disturbed in AD brains, suggesting an impaired transport by kinesin [21]. Here we detected reduced levels of the kinesin related proteins (KI26B, KIF12 and KIF27), and suggest that these reductions may contribute the transport impairments in AD. Furthermore, we confirmed that SNP25 and STX1B are down-regulated in AD, implying a disruption of synapses.
Great care was taken during the microdissection step to avoid contamination from extraneuronal tissue, and there were several neuron-specific markers, for example β-Tubulin III, neurofilament (light chain and heavy chain), ENOG and tyrosine hydroxylase, among the proteins we identified (Table S2). Still, some of the proteins we found, haemoglobin and albumin, could possibly be due to contamination from blood. Another group of proteins, metallothionein-3 (MT3), glial fibrillary acidic protein (GFAP) and S100B, are not highly expressed in neurons. Although these proteins could come from contaminating fragments of glial cells, we suggest that they are truly expressed in neurons. One general explanation for the identification of ‘non-neuronal’ proteins in our sample is the extremely high sensitivity of MS analysis compared to ordinary immunohistochemical analysis. For instance, we could detect NCF4 by MS analysis but not by IHC using the ABC detection system. Only by using a highly sensitive IHC method (MACH 3 HRP Polymer detection system) it was possible to observe NCF4. Regarding MT-3, there are some studies with widely varying conclusions on the expression levels [22,23]. In our MS analysis, MT-3 was detected both in the control and in the AD sample with a high peptide score, and the expression was found to be decreased in AD. In line with our observation, Yu et al. reported that MT-3 was mainly expressed in neurons and glial cells in temporal cortex from control cases, and that the levels were decreased in AD [24]. Glial fibrillary acidic protein has been considered to be a highly specific marker for astrocytes, but its expression in neurons in hippocampus in AD has been reported [25]. These pathological changes in the expression pattern of GFAP are consistent with our MS data, in which GFAP was identified to be up-regulated in AD. Although S100B is considered to be a mostly astrocytic protein, some previous work also suggest the neuronal expression of S100B [26]. In the control case, the expression of S100B was by IHC found to be limited to astrocytes in the hippocampus (Fig. 2A). However, three out of six AD cases presented intraneuronal S100B positivity. Figure 2B and C are most typical results of neuronal expression. For MS analysis, we pooled all neurons from six AD cases into one tube, and thus, the ratio of AD/control is an average of all cases. Hence, S100B positive neurons such as those represented in Figure 2B and C might contribute to the increased ratio of S100B in AD in the MS analysis. Furthermore, the positivity of S100B in AD cases was seen not only in cytoplasm but also in the nucleus, underscoring the possibility of S100B to have a role as a transcription factor [27]. Thus, the up-regulation of S100B expression may suggest a compensatory mechanism in response to insults in AD.
As expected, NKRF (Fig. 3A and B) and TP53B (Fig. 3D) were detected in the nucleus. Our IHC analysis suggested that the expression of NKRF was highly enriched in nucleoli, and a small fraction of NKRF was present in the nucleoplasm (Fig. 3A). This distribution in the nucleus is completely consistent with a previous report [28]. The IHC analysis, as well as MS data, indicated a decreased level of NKRF in the AD case (Fig. 3B and Table 2), especially in the nucleoplasm. This reduction of NKRF may suggest an activated stage of NF-κB transcription, since NKRF is a transcription factor that interacts with specific negative regulatory elements to mediate transcriptional repression of NF-kappa-B responsive gene such as inducible nitric-oxide synthase [29]. TP53B is proposed to function as a transcriptional co-activator of the p53 tumour suppressor [30]. In the AD case, there was a strong positivity in the nucleus of CA1 neurons and granule cells in gyrus dentatus (Fig. 3D and F), while no staining of neurons was found in the control case (Fig. 3C and D), indicating that the up-regulation of TP53B in AD is a pathological change. Recently it was reported that TP53B is recruited to the sites of DNA damage [31]. Thus, the increase of TP53B may indicate the severe DNA damage of CA1 neurons and granule cells in gyrus dentatus, in which the neurodegeneration is unrelated to tangle formation [32]. Moreover, since p53 is known to be involved in apoptosis, the up-regulation of TP53B may suggest activation of apoptosis in AD [33].
The immunoreactivity raised from the LRP1 antibody was also detected in the nucleus of neurons in AD (Fig. 3H). This antibody was produced by using full length LRP1 as the antigen, and the epitope has not been determined. Possibly, it can detect the intracellular domain of LRP1 (LRP-ICD), which can be cleaved from the full length form by γ-secretase [34], and translocated into nucleus. There is no major evidence for an increased γ-secretase dependent Aβ production in sporadic AD. However, the nuclear accumulation of LRP1-ICD may reflect an increased γ-secretase activity on LRP1 in AD. Recently it was reported that LRP1-ICD attenuates the inflammatory response evoked by LPS [35], suggesting a possible role for LRP1-ICD in response to inflammation in AD.
The NPXY motif in the cytoplasmic tail of LRP1 can interact with the PTB domain of CAPON [36]. Interestingly, both LRP1 and CAPON, were up-regulated in the AD sample in the MS analysis. Originally, CAPON was identified as an nNOS-binding protein [15]. Recently, several functional roles of CAPON were described, and especially CAPON-S was shown to be functionally involved in cardiac repolarization [37]. Both forms of CAPON affect the number of dendrites, while only CAPON-L is involved in dendrite outgrowth and branching [38]. In our IHC study, the CAPON level was increased in pyramidal neurons from the AD case (Fig. 4G). Since the CAPON antibody detects both CAPON-S and CAPON-L, it is not possible to differentiate the two isoforms by IHC. However, MS analysis showed that a sequence unique for CAPON-L (VEIVAAMR) was up-regulated in AD, and Western blot analysis revealed that CAPON-L is the dominating isoform in the CA1 region. Together these results suggest that CAPON-L was increased in hippocampus CA1 region in AD patients. Furthermore, stimulation of NMDA receptors activates a brain-enriched member of the Ras family of small monomeric G proteins (Dexras1), which induces iron uptake, through the interaction with nNOS–CAPON complex. Hence, the up-regulation of CAPON can lead to iron-induced neurotoxicity via the NMDA receptor signalling cascade [39]. Consequently, increased CAPON levels in AD may contribute to the alteration of the number and/or morphology of dendrites, and enhance neurotoxicity. The intracellular punctiform staining observed in the AD cases indicates that CAPON accumulates in the site of granulovacuolar degeneration in pathologically affected neurons.
NCF4 is one of the components in the NADPH oxidase complex, and contributes to the production of superoxide [40, 41]. In hippocampus from mice, NCF4 was analysed by in situ hybridization and found to be expressed at low levels in CA1 pyramidal neurons and granule cells of the dentate gyrus [42]. In line with these data, we could not detect any signal from NCF4 with the ABC–DAB method. However, by using the MACH3 system (high-sensitive method), we could observe immunoreactivity in CA1 pyramidal neurons (Table S1). NADPH oxidase has a role in host defense, and hence, phagocytes such as neutrophils and macrophages express high levels of NADPH oxidase. Our IHC analysis showed intense staining, possibly from monocytes in vessels (arrow heads in Fig. 4A and A). In addition, a characteristic arboroid structure was observed in the AD case Figure 4B and C. By the M0775 antibody (anti-microglia marker), we identified this structure as microglia (Fig. 4D and E). Both microglia and macrophages originate from haematopoietic stem cells. Thus, it is reasonable that microglia expresses NCF4, and that its expression is induced in the progression of pathological condition caused by the inflammation involved in AD. Indeed, we could detect activated microglia possibly involved in phagocytosis (Fig. 4B). Hence, we conclude that in AD, NCF4 is up-regulated both in pyramidal neurons and in microglia. Moreover, it is interesting to note that in the AD case (Fig. 4C) we detected a strong signal in the nucleolus, suggesting an unexpected role of NCF4 in transcriptional regulation.
There are no detailed reports about the functional roles of NAR4 and CN145 till now. The expression level of NAR4 in the AD sample was increased around 30 times compared to that of control (Table 2). This protein is likely to be expressed in blood cells (arrow head in Fig. 5A and B). The AD case clearly showed an increase of NAR4 expression inside neurons (Fig. 5B).
As described earlier, we found CAPON to be up-regulated in CA1 neuron from AD cases by MS and IHC analysis, while the Western blot analysis using CA1 sections of hippocampus suggested a decrease of CAPON expression in AD (Fig. 6). One possible explanation for the discrepancy between the MS and IHC data and the Western blot analysis is that the neuronal expression of CAPON is up-regulated in AD, while an overall expression of CAPON in this area is decreased due to neuronal loss. These data indicate that our approach for analysis of the neuronal proteome, the combination of LCM and MS, uncovers differences that cannot be detected by bulk analysis. Because the amount of material from LCM is limited, we identify mostly proteins that are abundant in neurons. However, as the development of MS continues, it will be possible in the future to identify and quantify also low-abundant proteins from a limited amount of material.
In summary, the approach to combine LCM and MS analysis made it possible to analyse the neuron-specific proteome. Based on the improved 18O labelling protocol we acquired a list of proteins that are differently regulated in AD, of which several are involved in neurodegeneration. To further examine the differently regulated proteins, IHC was adopted for qualitative analysis. In accordance with MS data, the proteins found to be up-regulated in AD showed intense neuronal positivity in IHC, suggesting some novel pathological events. To truly understand the pathological and quantitative alteration of the identified proteins on events in AD, further studies including functional assays will be necessary. We conclude that this approach revealed neuron-specific alterations in protein expression of importance for AD, and suggest that cell-specific proteome analysis by LCM-MS could be of general use for studies in health and disease.
Acknowledgments
The authors thank Dr Masanori Kusumoto and Dr Diego Iacono for technical assistance on MS and IHC analysis, respectively. We appreciate Dr Yasuhiro Teranishi for giving us useful suggestions on MS analysis. This study was supported by Dainippon Sumitomo Pharma Co., Ltd.
Conflict of interest
The authors confirm that there are no conflicts of interest.
Supporting Information
Summary of experimental conditions for IHC
The expression ratio, AD/Control, of proteins identified by MS
The time-course of the back reaction in the18O labelled peptide.
Amyloid plaque staining by the 6E10 antibody and p-tau staining by the AT-8 antibody.
References
- 1.Wenk GL. Neuropathologic changes in Alzheimer's disease. J Clin Psychiatry. 2003;64:7–10. [PubMed] [Google Scholar]
- 2.Hashimoto M, Rockenstein E, Crews L, et al. Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer's and Parkinson's diseases. Neuromol Med. 2003;4:21–36. doi: 10.1385/NMM:4:1-2:21. [DOI] [PubMed] [Google Scholar]
- 3.Tiraboschi P, Hansen LA, Thal LJ, et al. The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology. 2004;62:1984–9. doi: 10.1212/01.wnl.0000129697.01779.0a. [DOI] [PubMed] [Google Scholar]
- 4.Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 5.Gygi SP, Rochon Y, Franza BR, et al. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999;19:1720–30. doi: 10.1128/mcb.19.3.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muller T, Jung K, Ullrich A, et al. Disease state, age, sex, and post-mortem time-dependent expression of proteins in AD versus control frontal cortex brain samples. Curr Alzheimer Res. 2008;5:562–71. doi: 10.2174/156720508786898488. [DOI] [PubMed] [Google Scholar]
- 7.Sultana R, Boyd-Kimball D, Cai J, et al. Proteomics analysis of the Alzheimer's disease hippocampal proteome. J Alzheimers Dis. 2007;11:153–64. doi: 10.3233/jad-2007-11203. [DOI] [PubMed] [Google Scholar]
- 8.Hashimoto M, Bogdanovic N, Volkmann I, et al. Analysis of microdissected human neurons by a sensitive ELISA reveals a correlation between elevated intracellular concentrations of Aβ42 and Alzheimer's disease neuropathology. Acta Neuropathol. 2010;119:543–54. doi: 10.1007/s00401-010-0661-6. [DOI] [PubMed] [Google Scholar]
- 9.Bogdanovic N, Morris JH. Diagnostic criteria for Alzheimer's disease in multicentre brain banking. In: Cruz-Sanchez FF, Ravid R, Cuzner ML, editors. Neuropathological diagnostic criteria for brain banking. Amsterdam: IOS Press; 1995. pp. 20–9. [Google Scholar]
- 10.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–86. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 11.Storms HF, van der Heijden R, Tjaden UR, et al. Considerations for proteolytic labeling-optimization of 18O incorporation and prohibition of back-exchange. Rapid Commun Mass Spectrom. 2006;20:3491–7. doi: 10.1002/rcm.2738. [DOI] [PubMed] [Google Scholar]
- 12.Bigl M, Bruckner MK, Arendt T, et al. Activities of key glycolytic enzymes in the brains of patients with Alzheimer's disease. J Neural Transm. 1999;106:499–511. doi: 10.1007/s007020050174. [DOI] [PubMed] [Google Scholar]
- 13.Luth HJ, Holzer M, Gertz HJ, et al. Aberrant expression of nNOS in pyramidal neurons in Alzheimer's disease is highly co-localized with p21ras and p16INK4a. Brain Res. 2000;852:45–55. doi: 10.1016/s0006-8993(99)02178-2. [DOI] [PubMed] [Google Scholar]
- 14.Simic G, Lucassen PJ, Krsnik Z, et al. nNOS expression in reactive astrocytes correlates with increased cell death related DNA damage in the hippocampus and entorhinal cortex in Alzheimer's disease. Exp Neurol. 2000;165:12–26. doi: 10.1006/exnr.2000.7448. [DOI] [PubMed] [Google Scholar]
- 15.Jaffrey SR, Snowman AM, Eliasson MJ, et al. CAPON: a protein associated with neuronal nitric oxide synthase that regulates its interactions with PSD95. Neuron. 1998;20:115–24. doi: 10.1016/s0896-6273(00)80439-0. [DOI] [PubMed] [Google Scholar]
- 16.Yao X, Freas A, Ramirez J, et al. Proteolytic 18O labeling for comparative proteomics: model studies with two serotypes of adenovirus. Anal Chem. 2001;73:2836–42. doi: 10.1021/ac001404c. [DOI] [PubMed] [Google Scholar]
- 17.Hajkova D, Rao KC, Miyagi M. pH dependency of the carboxyl oxygen exchange reaction catalyzed by lysyl endopeptidase and trypsin. J Proteome Res. 2006;5:1667–73. doi: 10.1021/pr060033z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heiss WD, Szelies B, Kessler J, et al. Abnormalities of energy metabolism in Alzheimer's disease studied with PET. Ann N Y Acad Sci. 1991;640:65–71. doi: 10.1111/j.1749-6632.1991.tb00192.x. [DOI] [PubMed] [Google Scholar]
- 19.Hoyer S, Nitsch R, Oesterreich K. Predominant abnormality in cerebral glucose utilization in late-onset dementia of the Alzheimer type: a cross-sectional comparison against advanced late-onset and incipient early-onset cases. J Neural Transm Park Dis Dement Sect. 1991;3:1–14. doi: 10.1007/BF02251132. [DOI] [PubMed] [Google Scholar]
- 20.Brooks WM, Lynch PJ, Ingle CC, et al. Gene expression profiles of metabolic enzyme transcripts in Alzheimer's disease. Brain Res. 2007;1127:127–35. doi: 10.1016/j.brainres.2006.09.106. [DOI] [PubMed] [Google Scholar]
- 21.Stokin GB, Lillo C, Falzone TL, et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282–8. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- 22.Erickson JC, Sewell AK, Jensen LT, et al. Enhanced neurotrophic activity in Alzheimer's disease cortex is not associated with down-regulation of metallothionein-III (GIF) Brain Res. 1994;649:297–304. doi: 10.1016/0006-8993(94)91076-6. [DOI] [PubMed] [Google Scholar]
- 23.Uchida Y, Takio K, Titani K, et al. The growth inhibitory factor that is deficient in the Alzheimer's disease brain is a 68 amino acid metallothionein-like protein. Neuron. 1991;7:337–47. doi: 10.1016/0896-6273(91)90272-2. [DOI] [PubMed] [Google Scholar]
- 24.Yu WH, Lukiw WJ, Bergeron C, et al. Metallothionein III is reduced in Alzheimer's disease. Brain Res. 2001;894:37–45. doi: 10.1016/s0006-8993(00)03196-6. [DOI] [PubMed] [Google Scholar]
- 25.Hol EM, Roelofs RF, Moraal E, et al. Neuronal expression of GFAP in patients with Alzheimer pathology and identification of novel GFAP splice forms. Mol Psychiatry. 2003;8:786–96. doi: 10.1038/sj.mp.4001379. [DOI] [PubMed] [Google Scholar]
- 26.Donato R, Sorci G, Riuzzi F, et al. S100B's double life: intracellular regulator and extracellular signal. Biochim Biophys Acta. 2009;1793:1008–22. doi: 10.1016/j.bbamcr.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 27.Bernardini C, Lattanzi W, Businaro R, et al. Transcriptional effects of S100B on neuroblastoma cells: perturbation of cholesterol homeostasis and interference on the cell cycle. Gene Expr. 2010;14:345–59. doi: 10.3727/105221610x12718619643013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niedick I, Froese N, Oumard A, et al. Nucleolar localization and mobility analysis of the NF-κB repressing factor NRF. J Cell Sci. 2004;117:3447–58. doi: 10.1242/jcs.01129. [DOI] [PubMed] [Google Scholar]
- 29.Feng X, Guo Z, Nourbakhsh M, et al. Identification of a negative response element in the human inducible nitric-oxide synthase (hiNOS) promoter: The role of NF-κB-repressing factor (NRF) in basal repression of the hiNOS gene. Proc Natl Acad Sci U S A. 2002;99:14212–7. doi: 10.1073/pnas.212306199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iwabuchi K, Li B, Massa HF, et al. Stimulation of p53-mediated transcriptional activation by the p53-binding proteins, 53BP1 and 53BP2. J Biol Chem. 1998;273:26061–8. doi: 10.1074/jbc.273.40.26061. [DOI] [PubMed] [Google Scholar]
- 31.Sordet O, Redon CE, Guirouilh-Barbat J, et al. Ataxia telangiectasia mutated activation by transcription- and topoisomerase I-induced DNA double-strand breaks. EMBO Rep. 2009;10:887–93. doi: 10.1038/embor.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simic G, Kostovic I, Winblad B, et al. Volume and number of neurons of the human hippocampal formation in normal aging and Alzheimer's disease. J Comp Neurol. 1997;379:482–94. doi: 10.1002/(sici)1096-9861(19970324)379:4<482::aid-cne2>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 33.Riley T, Sontag E, Chen P, et al. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9:402–12. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- 34.May P, Reddy YK, Herz J. Proteolytic processing of low density lipoprotein receptor-related protein mediates regulated release of its intracellular domain. J Biol Chem. 2002;277:18736–43. doi: 10.1074/jbc.M201979200. [DOI] [PubMed] [Google Scholar]
- 35.Zurhove K, Nakajima C, Herz J, et al. γ-Secretase limits the inflammatory response through the processing of LRP1. Sci Signal. 2008;1:ra15. doi: 10.1126/scisignal.1164263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gotthardt M, Trommsdorff M, Nevitt MF, et al. Interactions of the low density lipoprotein receptor gene family with cytosolic adaptor and scaffold proteins suggest diverse biological functions in cellular communication and signal transduction. J Biol Chem. 2000;275:25616–24. doi: 10.1074/jbc.M000955200. [DOI] [PubMed] [Google Scholar]
- 37.Chang KC, Barth AS, Sasano T, et al. CAPON modulates cardiac repolarization via neuronal nitric oxide synthase signaling in the heart. Proc Natl Acad Sci U S A. 2008;105:4477–82. doi: 10.1073/pnas.0709118105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carrel D, Du Y, Komlos D, et al. NOS1AP regulates dendrite patterning of hippocampal neurons through a carboxypeptidase E-mediated pathway. J Neurosci. 2009;29:8248–58. doi: 10.1523/JNEUROSCI.5287-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheah JH, Kim SF, Hester LD, et al. NMDA receptor-nitric oxide transmission mediates neuronal iron homeostasis via the GTPase Dexras1. Neuron. 2006;51:431–40. doi: 10.1016/j.neuron.2006.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matute JD, Arias AA, Dinauer MC, et al. p40phox: the last NADPH oxidase subunit. Blood Cells Mol Dis. 2005;35:291–302. doi: 10.1016/j.bcmd.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 41.Wientjes FB, Hsuan JJ, Totty NF, et al. p40phox, a third cytosolic component of the activation complex of the NADPH oxidase to contain src homology 3 domains. Biochem J. 1993;296:557–61. doi: 10.1042/bj2960557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mizuki K, Kadomatsu K, Hata K, et al. Functional modules and expression of mouse p40(phox) and p67(phox), SH3-domain-containing proteins involved in the phagocyte NADPH oxidase complex. Eur J Biochem. 1998;251:573–82. doi: 10.1046/j.1432-1327.1998.2510573.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary of experimental conditions for IHC
The expression ratio, AD/Control, of proteins identified by MS
The time-course of the back reaction in the18O labelled peptide.
Amyloid plaque staining by the 6E10 antibody and p-tau staining by the AT-8 antibody.