

Abstract
Erythropoietin has been shown to promote tissue regeneration after ischaemic injury in various organs. Here, we investigated whether Erythropoietin could ameliorate ischaemic spinal cord injury in the mouse and sought an underlying mechanism. Spinal cord ischaemia was developed by cross-clamping the descending thoracic aorta for 7 or 9 min. in mice. Erythropoietin (5000 IU/kg) or saline was administrated 30 min. before aortic cross-clamping. Neurological function was assessed using the paralysis score for 7 days after the operation. Spinal cords were histologically evaluated 2 and 7 days after the operation. Immunohistochemistry was used to detect CD34+ cells and the expression of brain-derived neurotrophic factor and vascular endothelial growth factor. Each mouse exhibited either mildly impaired function or complete paralysis at day 2. Erythropoietin-treated mice with complete paralysis demonstrated significant improvement of neurological function between day 2 and 7, compared to saline-treated mice with complete paralysis. Motor neurons in erythropoietin-treated mice were more preserved at day 7 than those in saline-treated mice with complete paralysis. CD34+ cells in the lumbar spinal cord of erythropoietin-treated mice were more abundant at day 2 than those of saline-treated mice. Brain-derived neurotrophic factor and vascular endothelial growth factor were markedly expressed in lumbar spinal cords in erythropoietin-treated mice at day 7. Erythropoietin demonstrated neuroprotective effects in the ischaemic spinal cord, improving neurological function and attenuating motor neuron loss. These effects may have been mediated by recruited CD34+ cells, and enhanced expression of brain-derived neurotrophic factor and vascular endothelial growth factor.
Keywords: thoracoabdominal aortic aneurysm (TAAA), spinal cord ischaemia, erythropoietin (EPO), CD34 positive cell, brain-derived neurotrophic factor (BDNF), vascular endothelial growth factor (VEGF)
Introduction
Paraplegia caused by spinal cord ischaemia (SCI) is a devastating complication following thoracoabdominal aortic surgery. Although the incidence of paraplegia is low, ranging between 2.6% and 13.2%, because of the advancement of surgical techniques and the development of various adjuncts [1, 2], patients with paraplegia have poor survival rates [3], and quality of life is significantly impaired. Recently, thoracic endovascular aortic repair (TEVAR) demonstrated favourable results over open surgical repair as a treatment option for descending thoracic aortic aneurysms [4]. However, TEVAR is not a standard treatment option for thoracoabdominal aortic aneurysms. Accordingly, paraplegia remains a critical problem associated with thoracoabdominal aortic surgery.
Erythropoietin (EPO) is known to be a hormone that regulates proliferation and differentiation of erythroid precursor cells. In the last decade, EPO receptors have been shown to be expressed in many other tissues besides the haematopoietic system, and they are considered to be related to the cytoprotective effects of EPO against ischaemia-reperfusion (I/R) injury in various organs including the spinal cord [5, 6]. More interestingly, EPO has been reported to promote tissue regeneration in a variety of disorders via activation of local stem cells or recruitment of bone marrow stem/progenitor cells [7, 8].
There have been some reports in which exogenous EPO has been tested in animal models of SCI [9-11]. These studies demonstrated that EPO improved neurological function and reduced histological injury, conferred by the anti-apoptotic effect of EPO. However, these reports only focussed on the short-term effect of EPO, predominantly within 2 days of treatment. This timeframe is too narrow to evaluate the outcome of EPO treatment after SCI because critical events caused by I/R, such as inflammation, cell death and the repair process, can occur in a delayed manner, sometimes over days and weeks after the initial insult [12]. Indeed, post-operative spinal cord deficits have been reported to occur 12 hrs to some months after surgery, despite the emergence from anaesthesia with intact spinal cord function [3].
Therefore, we investigated the effect of EPO on ischaemic spinal cord injury over a longer term using a mouse model of SCI and sought the underlying mechanism, particularly focusing on the implication of stem cells.
Materials and methods
Mice
All animal experiments were performed in accordance with the guidelines published in the ‘Guide for the Care and Use of Laboratory Animals’ published by the United States National Institutes of Health. Surgical and animal care protocols were reviewed and approved by the local animal care committees of Mecklenburg/Vorpommern in Germany. Adult male C57BL/6 mice (8–12 weeks old; 22–27 g) were purchased from Charles River Laboratories, Sulzfeld, Germany.
Anaesthesia and surgical preparation
Mice were anaesthetized by intraperitoneal injection of ketamin/xylazin (75/25 mg/kg) and placed on a heating table set at 37°C. Rectal temperature was monitored during the surgery. Mice were intubated and mechanically ventilated with room air (300 μl/stroke, 130 strokes/min.). For anti-coagulation, heparin (400 IU/kg) was injected subcutaneously 5 min. before the surgery. Abdominal aortic blood pressure and heart rate were monitored through a catheter inserted into the right femoral artery. The data obtained were automatically recorded every minute with a Millar Pressure-Volume System [Ultra-Miniature Pressure-Volume Catheter (Model SPR-838), Millar Pressure Conductance Unit (Model MPCU-200) and Millar PowerLab data acquisition hardware; Emka Technologies, Paris, France].
Surgical procedure for SCI
A thoracotomy at the second intercostal space on the left side was used to approach the descending thoracic aorta. The left thymus was removed and the left recurrent nerve was carefully separated from the aortic arch. After isolation from the surrounding tissue, the descending thoracic aorta distal to the origin of the left subclavian artery was cross-clamped with a small aneurysm clip (Fine Science Tools GmbH, Heidelberg, Germany) for 7 or 9 min. Complete cessation of blood flow was confirmed by disappearance of blood pressure waves on the monitor screen. Mice that maintained abdominal aortic blood pressure with small waves or above 20 mmHg during the clamping were excluded from this study. After release of the clip, mice were observed for 10 min. until blood pressure returned to stable levels. Afterwards, the chest was closed and the blood pressure catheter was removed. Mice were kept on a heating table until they awoke and were extubated.
Post-operative care
Mice received an intraperitoneal injection of isotonic saline (1 ml) to compensate for fluid loss during surgery before extubation. A subcutaneous injection of 0.9% saline (1.5 ml) was administered daily until animals were killed. Manual bladder expression was performed twice a day for mice with neurological bladder.
Experimental groups
All mice (n= 148) were divided into three groups: sham (n= 5), 7-min. ischaemia (n= 72), and 9-min. ischaemia (n= 71). Exposure of the descending thoracic aorta was only applied to the sham group. The 7- and 9-min. ischaemia groups were subjected to 7 and 9 min. of cross-clamping, respectively. The 7- and 9-min. ischaemia groups were further divided into two treatment groups, which were control groups (7-min. ischaemia = 7C, n= 37; 9-min. ischaemia = 9C, n= 33) and EPO groups (7-min. ischaemia = 7E, n= 35; 9-min. ischaemia = 9E, n= 38). Groups 7E and 9E received a single dose of 5000 IU/kg of recombinant human EPO (Epoetin-α/Erypo®; Ortho Biotech, Neuss, Germany) intravenously 30 min. before ischaemia. Groups 7C and 9C received an equal volume of saline.
Blood gas analysis
Blood gas analysis was performed with three mice from each group at two different time points, which were 10 min. before ischaemia and 10 min. after the onset of reperfusion. Blood samples (70 μl) were drawn from the right femoral artery and pH, PaO2, PaCO2, base excess and haematocrit of each sample were measured with a blood gas/pH analyser (RAPIDLab 348, Siemens, Germany).
Behavioural test
Mice were subjected to a behavioural test at 3 hrs, 6 hrs, and daily thereafter up to 7 days (days 1–7) after onset of reperfusion. A following paralysis score system [13] was used to grade hind limb neurological functions: (a) walking with hind limbs: 0, normal; 1, toes flat under body when walking but ataxia is present; 2, knuckle-walking; 3, movement in hind limbs but unable to walk; 4, no movement, drags hind limbs; (b) placing/stepping reflex: 0, normal; 1, weak; 2, no stepping. Each grade was obtained by adding the scores of (a) and (b); a grade of 0 was considered normal and 6 was considered complete paralysis. Both hind limbs were assessed separately and a final grade was expressed as an average.
Histological evaluation of ischaemic spinal cord injury
Histological evaluation was performed at day 2 (7C, n= 10; 7E, n= 10; 9C, n= 8; 9E, n= 12) and day 7 (7C, n= 13; 7E, n= 11; 9C, n= 12; 9E, n= 12) after SCI. Mice were killed using an over dose of ketamine/xylazine (150/50 mg/kg), and the thoracic and lumbar part of the spinal cord was harvested en bloc and fixed in formafix 4% (v/v) (Grimm Med. Logistik GmbH, Germany) overnight. The samples were then kept in ethylenediaminetetraacetic acid (EDTA) decalcification solution (pH 7.4) for 2 weeks, embedded in paraffin and sectioned at 4 μm thickness. Twelve cross sections from T6-L5 level of the spinal cord in each mouse were stained with haematoxylin/eosin (HE) and observed under a light microscope (Leica, Hamburg, Germany). Ischaemic injury was quantified by counting the number of normal motor neurons in the bilateral ventral horn of each cross section [14]. The final value of each sample was given as a sum of 12 cross sections.
Immunohistochemistry
Deparaffinized cross sections were incubated in boiling citric buffer (pH 6.0) for 10 min. and allowed to cool down for 20 min. Non-specific binding was blocked by protein blocking reagent (DAKO, Hamburg, Germany). Subsequently, the cross sections were incubated with goat polyclonal anti-CD34 antibody (1:20 dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA) or rabbit polyclonal anti-brain–derived neurotrophic factor (BDNF) antibody (1:50 dilution; Santa Cruz Biotechnology) at 4°C overnight. Cross sections were incubated with donkey anti-goat or goat anti-rabbit Alexa-Fluor 488–conjugated secondary antibody (1:350 dilution; Invitrogen, Darmstadt, Germany) for 2 hrs at room temperature. Nuclei were counterstained with TOPRO3 (5 μM; Invitrogen). The cross sections were mounted in Fluosaver (Merck, Germany). The labelled cross sections were observed with a Leica SP2 confocal microscope (Leica). The number of CD34+ cells in the entire transverse section from the L1-2 level of the spinal cord was counted under a 400χ magnification lens and final values were given as an average of three cross sections. For detection of vascular endothelial growth factor (VEGF) expression, deparaffinized cross sections treated with citric buffer were incubated with hydrogen peroxide for 5 min. and subsequently with 5% (v/v) normal donkey serum for 20 min. The cross sections were then incubated with rabbit polyclonal anti-VEGF antibody (1:50 dilution; Santa Cruz Biotechnology) at 4°C overnight. Immunoreactivity to VEGF was visualized with a linked streptavidin–biotin method using a goat ImmunoCruz™ Staining Kit (Santa Cruz Biotechnology). Cross sections were then counterstained with haematoxylin, dehydrated and mounted in Pertex (Medite, Germany).
Statistical analysis
Values are expressed as the mean ± S.D. After confirmation whether the data were approximated to the normal distribution, statistical analysis for neurological and histological data between control and EPO groups at different time points were performed by Student's t-test with Bonferroni correction. Survival rates were expressed using the Kaplan–Meier survival curve and the significance of these differences was analysed using the Log-rank test. Student's t-test was applied for physiological parameters. Differences between groups were considered significant at P < 0.05.
Results
Mouse model of SCI
The mouse model of SCI was first described in 2000 by Lang-Lazdunski et al. [13]. They used two aneurysm clips, one of which was placed across the aortic arch between the left common carotid artery and the left subclavian artery and the other across the left subclavian artery through a cervical mediastinotomy. In our initial experience of this procedure, it was somewhat difficult to reach both clamping sites through the cervical mediastinotomy. Instead, we performed single clamping across the descending aortic artery through a thoracotomy. We found that this procedure made it easier to reach the descending thoracic aorta and place an aneurysm clip precisely across it, allowing abdominal aortic blood pressure to drop to a sufficient level for developing SCI without additional clamping on the left subclavian artery (Fig. 1). On preliminary examination, 5-min. ischaemia developed no paralysis whereas 12-min. ischaemia resulted in complete paralysis, which is referred to as grade 6 on the paralysis score, in all mice (data not shown). Therefore, 7-min. and 9-min ischaemia were applied in this study.
Fig 1.
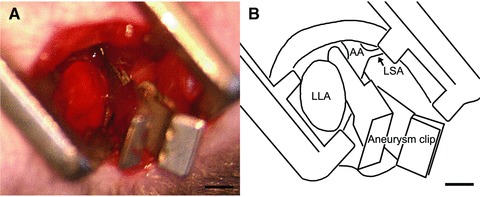
A mouse model of SCI was developed by cross-clamping the descending thoracic aorta through left thoracotomy. A small aneurysm clip was placed across the descending aorta just distal to the left subclavian artery (A). Schematic diagram of A (B). LAA: left atrial appendix; LSA: left subclavian artery; AA: aortic arch; Bar = 1 mm.
There was no significant difference in physiological parameters between the control (7C and 9C) and EPO (7E and 9E) group during SCI surgery except for heart rate at 10 min. after onset of reperfusion, which was higher in the EPO groups than in the control groups (Table 1). These data indicate that SCI surgeries were performed under similar physiological conditions in both groups.
Table 1.
Physiological parameters
| 7-min. ischaemia Control, n= 34(3*) | EPO, n= 32(3*) | P value | 9-min. ischaemia Control, n= 30(3*) | EPO, n= 35(3*) | P value | |
|---|---|---|---|---|---|---|
| HR (beats/min.) | ||||||
| Pre-ischaemia | 265 ± 68 | 286 ± 60 | 0.187 | 265 ± 36 | 270 ± 50 | 0.685 |
| Reperfusion | 269 ± 67 | 315 ± 50 | 0.003† | 266 ± 45 | 298 ± 48 | 0.008† |
| MAABP (mmHg) | ||||||
| Pre-ischaemia | 61.2 ± 18.2 | 62.8 ± 12.9 | 0.671 | 68.1 ± 12.2 | 68.6 ± 14.5 | 0.881 |
| Ischaemia | 11.6 ± 2.0 | 12.0 ± 2.8 | 0.545 | 11.3 ± 4.6 | 10.7 ± 3.7 | 0.622 |
| Reperfusion | 53.5 ± 9.9 | 57.5 ± 10.8 | 0.125 | 59.0 ± 11.6 | 62.3 ± 8.1 | 0.199 |
| RT (°C) | ||||||
| Pre-ischaemia | 36.5 ± 0.7 | 36.5 ± 0.4 | 0.679 | 36.4 ± 0.4 | 36.4 ± 0.4 | 0.962 |
| Ischaemia | 35.8 ± 0.5 | 36.0 ± 0.3 | 0.224 | 35.8 ± 0.3 | 35.8 ± 0.3 | 0.821 |
| Reperfusion | 36.6 ± 0.5 | 36.5 ± 0.4 | 0.325 | 36.6 ± 0.4 | 36.6 ± 0.5 | 0.657 |
| BGA parameters | ||||||
| pH | ||||||
| Pre-ischaemia | 7.36 ± 0.02 | 7.38 ± 0.04 | 0.496 | 7.38 ± 0.05 | 7.35 ± 0.07 | 0.578 |
| Reperfusion | 7.28 ± 0.00 | 7.30 ± 0.03 | 0.368 | 7.22 ± 0.01 | 7.25 ± 0.06 | 0.480 |
| PaO2 (mmHg) | ||||||
| Pre-ischaemia | 76.5 ± 0.1 | 75.4 ± 4.5 | 0.713 | 78.2 ± 3.8 | 80.1 ± 4.2 | 0.592 |
| Reperfusion | 71.0 ± 0.2 | 74.6 ± 3.7 | 0.234 | 79.6 ± 5.2 | 77.7 ± 0.2 | 0.592 |
| PaCO2 (mmHg) | ||||||
| Pre-ischaemia | 44.6 ± 2.1 | 40.3 ± 3.3 | 0.130 | 40.8 ± 3.8 | 40.4 ± 4.3 | 0.910 |
| Reperfusion | 38.1 ± 2.1 | 34.6 ± 2.1 | 0.111 | 36.2 ± 0.4 | 36.5 ± 3.0 | 0.879 |
| BE | ||||||
| Pre-ischaemia | −0.8 ± 0.2 | −0.8 ± 0.9 | 0.935 | −0.2 ± 1.0 | −0.9 ± 1.2 | 0.481 |
| Reperfusion | −9.2 ± 0.7 | −10.0 ± 0.5 | 0.183 | −11.9 ± 1.6 | −11.7 ± 1.8 | 0.893 |
| HCT (%) | ||||||
| Pre-ischaemia | 45.3 ± 1.5 | 43.5 ± 0.7 | 0.164 | 42.7 ± 2.3 | 47.0 ± 1.7 | 0.060 |
| Reperfusion | 46.0 ± 3.5 | 45.5 ± 0.7 | 0.830 | 45.5 ± 4.9 | 48.0 ± 1.4 | 0.476 |
Values are given as mean±S.D. EPO: erythropoietin; HR: heart rate; MAABP: mean abdominal aortic blood pressure; RT: rectal temperature; BE: base excess; HCT: haematocrit; pre-ischaemia, 10 min. before ischaemia; ischaemia, at the end of ischaemia; reperfusion, 10 min. after the onset of reperfusion. *Numbers for BGA analysis. †Significant values.
Survival
Survival rates at day 2 after SCI were 70.6%, 68.8%, 73.3% and 74.3%, and those at day 7 were 65.5%, 63.0%, 62.9% and 63.7% in the 7C, 7E, 9C and 9E groups, respectively. There was no significant difference in the survival rate between the 7C and 7E groups (P= 0.797) or between the 9C and 9E groups (P= 0.971) (Fig. 2). At necropsy, bowel necrosis was detected in three mice from group 7C, three from 9C and one from 9E, indicating that there was no evidence that EPO exacerbated thromboembolism.
Fig 2.
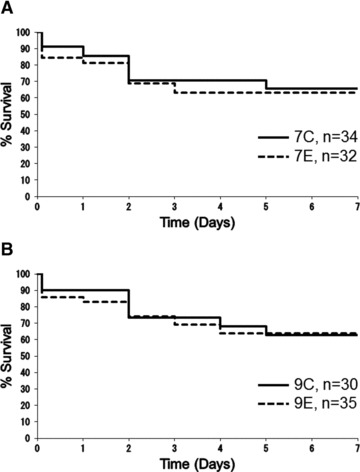
Survival rates of groups subjected to 7-min. ischaemia (A) and 9-min. ischaemia (B) are illustrated using Kaplan–Meier survival curves. 7C: control group subjected to 7-min. ischaemia; 7E: EPO group subjected to 7-min. ischaemia; 9C: control group subjected to 9-min. ischaemia; 9E: EPO group subjected to 9-min. ischaemia. Survival rates at day 2 after SCI were 70.6%, 68.8%, 73.3% and 74.3%, and 65.5%, 63.0%, 62.9% and 63.7% at day 7 in the 7C, 7E, 9C and 9E groups, respectively. There were no significant differences in survival rate between groups 7C and 7E (P= 0.797) or between groups 9C and 9E (P= 0.971), according to the Log-rank test.
Outcome of neurological function
In general, average paralysis score in every group improved during the first 24 hrs after SCI. However, scores deteriorated after day 2. Within the first 2 days after SCI, there was no significant difference between groups 7C and 7E or between groups 9C and 9E (Fig. 3A and C). Neurological functions at day 2 were revealed to clearly separate into normal to minor deficit (less than grade 3, defined as ‘mild paralysis’) and complete deficit (grade 6, defined as ‘severe paralysis’) in every group. The percentages of mild paralysis in groups 7C, 7E, 9C and 9E were 47.8%, 47.6%, 20.0% and 25.0%, respectively. There was no significant difference in the percentage of mild paralysis between groups 7C and 7E (P= 0.989) or between groups 9C and 9E (P= 0.974). After day 2 until day 7, mice with mild paralysis and those with severe paralysis were separately analysed in each group (7C-mild, n= 6; 7C-severe, n= 7; 7E-mild, n= 5; 7E-severe, n= 6; 9C-mild n= 2, 9C-severe n= 10; 9E-mild, n= 3; 9E-severe, n= 9; Fig. 3B and D). As a result of the separate analysis, the paralysis score for group 7E-severe significantly improved by day 7, compared to group 7C-severe (4.83 ± 0.98 versus 5.86 ± 0.38 at day 7, respectively, P < 0.05 with Bonferroni correction). A similar improvement in paralysis score was observed for group 9E-severe compared to 9C-severe (5.22 ± 0.67 versus 5.80 ± 0.42 at day 7, respectively, P < 0.05 with Bonferroni correction). Mice with mild paralysis (7C, 7E, 9C and 9E) had maintained relatively good neurological function for the last 5 days and finally recovered to normal.
Fig 3.
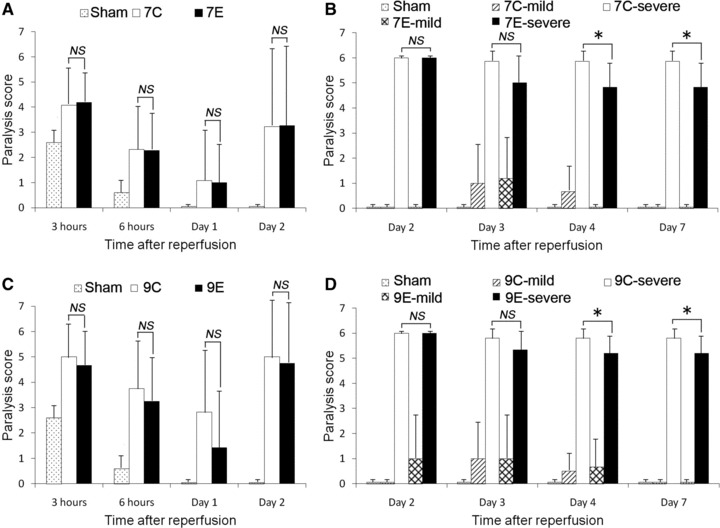
Neurological outcome assessed by paralysis score from 3 hrs to day 2 after SCI with 7-min. ischaemia (A) and 9-min. ischaemia (C) and from day 2 to 7 after SCI with 7-min. ischaemia (B) and 9-min. ischaemia (D). 7C: control group subjected to 7-min. ischaemia; 7E: EPO group subjected to 7-min. ischaemia; 9C: control group subjected to 9-min. ischaemia; 9E: EPO group subjected to 9-min. ischaemia; mild: subgroup including mice with mild paralysis; severe: subgroup including mice with severe paralysis. There were no significant differences in paralysis score between groups 7C and 7E or between groups 9C and 9E until 2 days after SCI (A and C). However, group 7E-severe exhibited significant improvement between day 2 and 7, compared to group 7C-severe (B). A similar tendency was observed in group 9E-severe between day 2 and 7, compared to group 9C-severe (D). Paralysis scores in group 7C-mild, 7E-mild, 9C-mild and 9E-mild all returned to normal (grade 0) at day 7 (B and D). (B and D) *P < 0.05 with Bonferroni correction.
Histological outcome
In cross sections from mice with mild paralysis, typical motor neurons, which contained Nissl substance in the cytoplasm, loose chromatin and prominent nucleoli, were distributed in grey matter. There was no remarkable evidence of ischaemic injury seen in grey matter for groups 7C-mild, 7E-mild, 9C-mild and 9E-mild at day 2 (Fig. 4A, B, E and F), and a small number of inflammatory cells were occasionally located in the grey matter at day 7 (Fig. 4C, D, G and H). The number of motor neurons in these groups was similar at days 2 and 7 and close to that of the sham group (Fig. 4Q and R). In contrast, motor neurons in the ventral horn showed ischaemic changes with shrunken cell bodies, eosinophilic cytoplasm and pyknotic nuclei in cross sections from mice with severe paralysis. The presence of ischaemic motor neurons and vacuolization was pronounced in the grey matter of groups 7C-severe, 7E-severe, 9C-severe and 9E-severe at day 2. A few motor neurons were present only peripherally in grey matter (Fig. 4I, J, M and N). At day 7, affected areas represented by ischaemic motor neurons and vacuolization were replaced with a high density of inflammatory cells including microglia, especially in groups 7C-severe and 9C-severe (Fig. 4K and L). However, such areas were limited in groups 7E-severe and 9E-severe (Fig. 4O and P). There was no significant difference in the number of motor neurons at day 2 between groups 7C-severe and 7E-severe (235 ± 21 versus 240 ± 19, respectively, P= 0.703) or between groups 9C-severe and 9E-severe (190 ± 30 versus 207 ± 28, respectively, P= 0.290). However, there was a significant difference at day 7 between groups 7C-severe and 7E-severe (115 ± 28 versus 195 ± 36, respectively, P < 0.05 with Bonferroni correction), and between groups 9C-severe and 9E-severe (102 ± 21 versus 142 ± 18, respectively, P < 0.05 with Bonferroni correction, Fig. 4Q and R). In summary, in groups 7E-severe and 9E-severe, motor neuron loss from day 2 to 7 was attenuated and the damaged areas in the grey matter were reduced, compared to groups 7C-severe and 9C-severe. Mice with mild paralysis exhibited only minor damage in histology regardless if they were treated with EPO.
Fig 4.
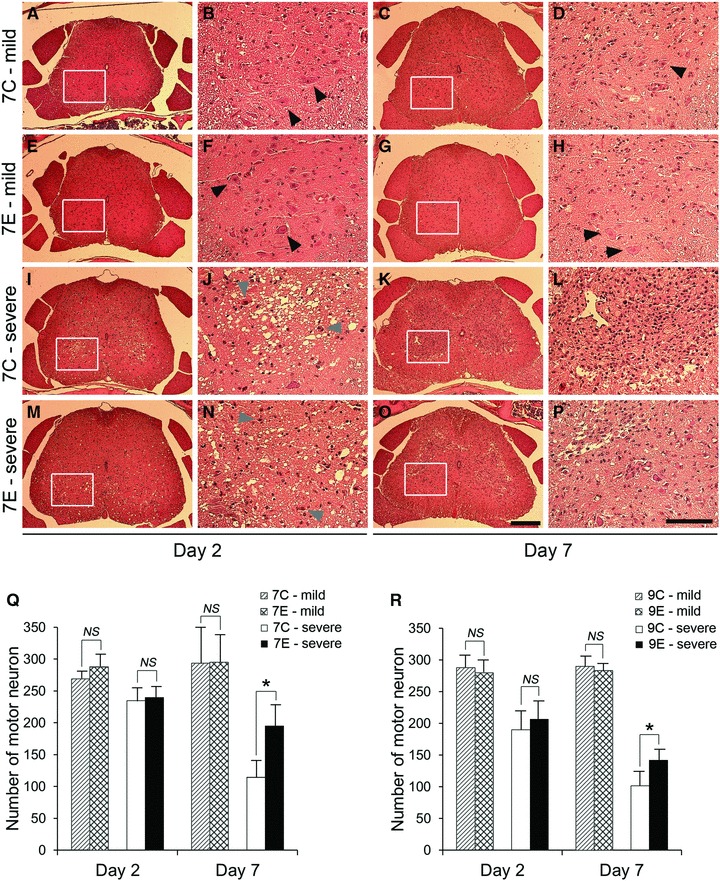
Representative cross sections stained with HE from the L1-2 level of the spinal cord after 7-min. SCI (A–P). An enlargement of the boxed regions is provided on the right side of each cross section. Almost normal morphology was maintained in lumbar spinal cords from group 7C-mild and 7E-mild (A–H). Normal motor neurons are distributed in ventral grey matter (block arrow heads). In contrast, ventral grey matter in the lumbar spinal cord from group 7C-severe and 7E-severe were largely affected at day 2 with ischaemic motor neurons (grey arrow heads) and many vacuolated regions (I, J, M and N). At day 7, affected regions in grey matter were replaced with a high density of inflammatory cells including microglia (K, L, O and P). Note that lesion area was limited in group 7E-severe at day 7 (O and P), compared to group 7C-severe (K and L). Scale bar = 250 μm in low-magnification images and 100 μm in high-power images. Bar graphs illustrating the number of normal motor neurons in the thoracolumbar spinal cord in each group with 7-min. ischaemia (Q) and 9-min. ischaemia (R). Motor neurons were well preserved in groups 7C-mild, 7E-mild, 9C-mild and 9E-mild, whereas the loss of motor neurons was evident in groups 7C-severe, 7E-severe, 9C-severe and 9E-severe. However, higher numbers of motor neurons were preserved in groups 7E-severe and 9E-severe at day 7, compared to groups 7C-severe and 9C-severe. (Q and R) *P < 0.05 with Bonferroni correction.
Immunohistochemical findings
CD34+ cells could be recognized in cross sections, frequently in grey matter of the lumbar spinal cord in every group (Fig. 5A and B). The number of CD34+ cells in group 7E-severe was higher than that of group 7C-severe at day 2 (28.3 ± 9.5 versus 13.8 ± 5.1, respectively, P < 0.05 with Bonferroni correction), and that of group 9E-severe was also higher than that of group 9C-severe at day 2 (44.8 ± 22.4 versus 22.7 ± 11.8, respectively, P < 0.05 with Bonferroni correction, Fig. 5C and D). No significant difference was observed in CD34+ cells from groups 7C-mild, 7E-mild, 9C-mild and 9E-mild. Expression of BDNF was distributed in the ventral grey matter of groups 7E-severe and 9E-severe, whereas expression was rarely detected in groups 7C-severe and 9C-severe (Fig. 5E and F). Similarly, expression of VEGF could be detected only in the ventral or ventrolateral white matter of the spinal cord in groups 7E-severe and 9E-severe, but not in groups 7C-severe and 9C-severe (Fig. 5G and H).
Fig 5.
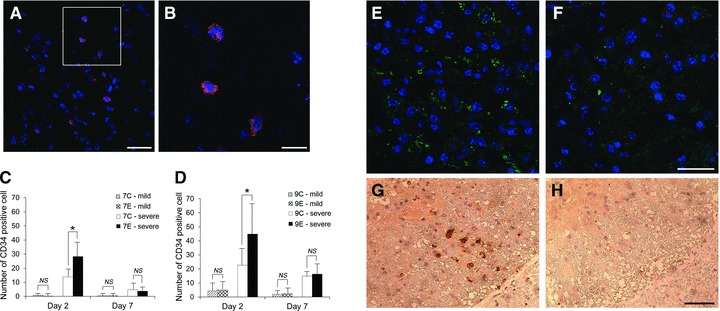
Representative pictures of CD34+ cells in ventral grey matter (A) and enlargement of boxed area (B). Blue: nuclei and red: CD34. (A) Scale bar = 25 μm; (B) Scale bar = 10 μm. The number of CD34+ cells in the lumbar spinal cord after 7-min. (C) and 9-min. SCI (D). CD34+ cells were more abundantly recruited in the spinal cord in groups 7E-severe and 9E-severe than those in groups 7C-severe and 9C-severe at day 2 (C and D, *P < 0.05 with Bonferroni correction). CD34+ cells were not apparent in groups 7C-mild, 7E-mild, 9C-mild and 9E-mild. Representative pictures of immunofluorescence for BDNF in ventral grey matter from groups 9E-severe (E) and 9C-severe (F) at day 7. Blue: nuclei and green: BDNF. BDNF expression in group 9E-severe (E) was more evident than that in group 9C-severe (F). Scale bar = 25 μm. Representative pictures of VEGF expression in the left ventrolateral white matter of the spinal cord from groups 9E-severe (G) and 9C-severe (H) at day 7. VEGF was detectable in the white matter of group 9E-severe and vacuolization in white matter was limited (G). In contrast, no VEGF expression and advanced vacuolization was apparent in the white matter of group 9C-severe (H). Scale bar = 50 μm.
Discussion
Major findings of this study are as follows. First, our mouse model of SCI was successfully performed and resulted in stable physiological conditions during surgery and standardized neurological and histological outcome following various durations of SCI, which made investigation of EPO treatment reliable. Second, neurological function of EPO-treated mice improved when compared to control mice between day 2 and day 7, and loss of motor neurons was attenuated in EPO-treated mice at day 7 after SCI. Finally, CD34+ cells were densely recruited, and expression of BDNF and VEGF was enhanced in the lumber spinal cord of EPO-treated mice after SCI.
Generally, mouse models are advantageous to study therapeutics, specific genes and molecular signalling pathways because a variety of genetically modified mice are now widely available. Previously, ischaemic models of spinal cord injury have been often described in rabbits and rats. Compared to such animals, mice are less expensive and are equally as easy to operate on. In our model, a skilled surgeon can complete the procedure within 60 min. Rabbit models have been widely used to test the effects of neuroprotective drugs and to examine the pathophysiology of spinal cord injury [15, 16]. However, little is known about the genome of rabbits, and transient occlusion of the infrarenal segment of the aorta causes paraplegia, which implies differences in vascular anatomy because infrarenal aortic cross-clamping rarely causes paraplegia in human beings. In contrast, the vascular anatomy of the spinal cord and distribution of pathology are similar to that in human beings whose spinal cord receives blood flow from one anterior spinal artery and two posterior spinal arteries [13]. In addition, some mice exhibit delayed paralysis between 24 and 48 hrs after an ischaemic insult [17]. This also replicates one aspect of neurological deficit seen after aortic surgery in human beings. However, mice are so small that surgical results are sometimes influenced by only a small change in the surgical procedure, settings or conditions. Therefore, it is crucial to monitor blood pressure down to levels that induce SCI (20 mmHg) during clamping, and to standardize body temperature, bodyweight and age in the mouse SCI model. Another important feature of this mouse SCI model is that neurological outcome is clearly separated into minor deficits and complete paralysis after a 48-hr interval [17]. From this point of view, it is difficult to detect the effect of therapeutics on neurological outcome and it is possible to misjudge the results if both patterns of neurological outcome are analysed together as a uniform result. Accordingly, we looked at results from two perspectives: One compared the percentage of patients who had mild paralysis per total patients between groups. The other, followed the two patterns of neurological outcome separately. Using this method, we revealed that EPO did not decrease the percentage of mice with mild paralysis, but improved neurological function of severely paralysed mice between day 2 and 7.
A possible discrepancy in this study is that neurological function of severely paralysed mice improved between day 2 and 7 whereas the number of motor neurons decreased from day 2 to 7 after SCI even in the EPO-treated groups. However, a similar finding was also reported by Sakamoto et al., which indicated that neurological function gradually recovered despite persistent loss of motor neurons up to 2 weeks after SCI in the rat [18]. These findings indicate that functional outcome depends not only on the number of motor neurons but also on other factors such as functional compensation by viable neuromuscular units or attenuation of inflammation in the injured spinal cord.
Neuroprotection provided by EPO in various pathophysiologies has been well documented, with its action mediated by EPO receptors expressed in many kinds of central nervous system (CNS) cells including neural progenitor cells, neurons, glial cells and endothelial cells [19]. In the rabbit model of SCI, systemic administration of human recombinant EPO reduced apoptosis of motor neurons, leading to favourable neurological outcome compared to saline controls [20]. Dame et al. also described the neuroprotective and neurotrophic properties of EPO observed in the CNS with in vivo models of ischaemic, traumatic or inflammatory brain injury. They also demonstrated that such neuroprotective and neurotrophic effects of EPO were mediated through expression of genes that are related to anti-apoptotic proteins in neuronal cells, such as NF-Ib, IKKa, akt and several Bcl 2 family members [21]. Erythropoietins’ vascular actions have also been demonstrated, one of which is preserving blood–brain barrier (BBB) function, which may contribute to the prevention of brain oedema [22].
Based on the possible anti-apoptotic properties or vascular actions of EPO, we expected to observe the effects of EPO much earlier. However, a 48-hr interval was necessary before the effects of EPO were apparent. This is inconsistent with previous reports that demonstrate the neuroprotective effects of EPO within 2 days after SCI in the mouse, rat and pig [9-11]. However, it is possible that this difference in timing can be attributed to the difference in dosage, timing and route of administration of EPO used. In addition, competitive results in clinical studies have been reported. One such study indicates that EPO improves cardiac function and reduces infarct size after acute myocardial infarction treated with percutaneous coronary intervention [23], whereas EPO failed to do so in another report [24]. Therefore, further investigations would be necessary to clarify this issue.
In our findings, EPO appeared to be more effective at later timepoints, at least 2 days after SCI. This result is supported by literature in which EPO has been shown to have a wide therapeutic window of up to 6 weeks after administration in the case of traumatic spinal cord injury in rats [25]. In addition, EPO is considered to promote neurogenesis and angiogenesis in animal models of acute stroke [26]. This commitment of EPO to tissue regeneration can lead to a wide therapeutic window and delayed neuroprotection. Taguchi et al. have shown that systemic administration of CD34+ cells after stroke accelerated angiogenesis and neurogenesis with functional recovery in mice, suggesting that homing of CD34+ into an ischaemic region was crucial for functional recovery in the CNS [27]. This is consistent with our finding of abundant accumulation of CD34+ cells and improvement of neurological function in EPO-treated groups.
There have been many reports that have addressed the local or systemic delivery of EPO-promoted migration of bone marrow stem cells, such as CD34+ and c-kit+ cells, to damaged tissue [7, 28]. An interaction between the chemokine receptor CXCR-4 and its ligand stromal cell–derived factor-1 (SDF-1) is considered to be a pivotal element in bone marrow stem cell mobilization and homing. The expression of CXCR-4 on the surface of stem cells facilitates their migration along a gradient of SDF-1 [29]. In animal models of myocardial infarction, EPO promotes the recruitment of bone marrow stem cells in the infarct heart with enhancing local SDF-1 expression [7, 28]. A similar mechanism may have been stimulated in the current study to recruit CD34+ cells in the ischaemic spinal cord. In addition to SDF-1, local inflammatory cytokines in response to hypoxic stress are also involved in stem cell migration. Kaminski et al. demonstrated that adhesion of circulating c-kit+ cells onto vascular endothelium, which is the first step of stem cell infiltration into injured tissue, was intensified only when SDF-1 was present together with tumour necrosis factor α (TNF-α) [30]. Therefore, little tissue damage and a subsequent low inflammatory response could account for low numbers of CD34+ cells in mice with mild paralysis, even in the EPO-treated groups.
However, how accumulated CD34+ cells contribute to the improvement of neurological function and preservation of motor neurons is unclear. One possible explanation is that the interaction of CD34+ cells with local parenchymal cells produces trophic factors that support the recovery of neurological function [31]. In addition, BDNF is a growth factor, which is important in the development and maintenance of the nervous system and has a neuroprotective role [32]. Consistent with the present study, Wang et al. demonstrated that expression of BDNF was enhanced after EPO treatment in a rat model of embolic stroke, with functional improvement [26]. Brain-derived neurotrophic factor is considered to be expressed in motor neurons, microglia and endothelial cells in the CNS [26, 32]. Therefore, EPO may induce the expression of BDNF directly or indirectly in these cells through interaction with recruited CD34+ cells. Vascular endothelial growth factor is an endothelial cell-specific growth factor that can be secreted from endothelial progenitor cells (EPCs), which are considered to be CD34+, and mediate angiogenesis together with EPCs [27]. A recent study also revealed that VEGF had a direct neuroprotective effect on motor neurons independently of blood circulation, promoting cell survival and neuronal outgrowth [33]. These reports suggest that enhanced VEGF secretion from recruited CD34+ cells in the spinal cord of the EPO-treated groups may also contribute to functional recovery and neuroprotection.
In conclusion, systemic administration of EPO improved neurological function with preservation of motor neurons in the ischaemic spinal cord after SCI in the mouse. Such beneficial effects may have been provided by recruitment of CD34+ cells and enhanced expression of BDNF and VEGF. This evidence supports EPO as a treatment option that may ameliorate paraplegia after thoracoabdominal aortic aneurysm (TAAA) surgery.
Acknowledgments
This work was supported by the German Research Foundation, Sonderforschungsbereich/Transregio 7, B5, B2 and A4, German Helmholtz Association, Mecklenburg-Vorpommern (Nachwuchsgruppe Regenerative Medizin Regulation der Stammzellmigration 0402710), Bundesministerium für Bildung und Forschung (BMBF), and Referenz- und Translationszentrum für kardiale Stammzelltherapie (RTC). The authors thank Ms. Margit Fritsche, Department of Cardiac Surgery, Ms. Berit Blendow and Ms. Doris Butzlaff, Institute for Experimental Surgery, University of Rostock, for their technical assistance.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Wong DR, Parenti JL, Green SY, et al. Open repair of thoracoabdominal aortic aneurysm in the modern surgical era: contemporary outcomes in 509 patients. J Am Coll Surg. 2011;212:569–79. doi: 10.1016/j.jamcollsurg.2010.12.041. [DOI] [PubMed] [Google Scholar]
- 2.Conrad MF, Crawford RS, Davison JK, et al. Thoracoabdominal aneurysm repair: a 20-year perspective. Ann Thorac Surg. 2007;83:856–61. doi: 10.1016/j.athoracsur.2006.10.096. [DOI] [PubMed] [Google Scholar]
- 3.Wong DR, Coselli JS, Amerman K, et al. Delayed spinal cord deficits after thoracoabdominal aortic aneurysm repair. Ann Thorac Surg. 2007;83:1345–55. doi: 10.1016/j.athoracsur.2006.11.035. [DOI] [PubMed] [Google Scholar]
- 4.Bavaria JE, Appoo JJ, Makaroun MS, et al. Endovascular stent grafting versus open surgical repair of descending thoracic aortic aneurysms in low-risk patients: a multicenter comparative trial. J Thorac Cardiovasc Surg. 2007;133:369–77. doi: 10.1016/j.jtcvs.2006.07.040. [DOI] [PubMed] [Google Scholar]
- 5.Maiese K, Li F, Chong ZZ. New avenues of exploration for erythropoietin. JAMA. 2005;293:90–5. doi: 10.1001/jama.293.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldman SA, Nedergaard M. Erythropoietin strikes a new cord. Nat Med. 2002;8:785–7. doi: 10.1038/nm0802-785. [DOI] [PubMed] [Google Scholar]
- 7.Klopsch C, Furlani D, Gäbel R, et al. Intracardiac injection of erythropoietin induces stem cell recruitment and improves cardiac functions in a rat myocardial infarction model. J Cell Mol Med. 2009;13:664–79. doi: 10.1111/j.1582-4934.2008.00546.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shingo T, Sorokan ST, Shimazaki T, et al. Erythropoietin regulates the in vitro and in vivo production of neuronal progenitors by mammalian forebrain neural stem cells. J Neurosci. 2001;21:9733–43. doi: 10.1523/JNEUROSCI.21-24-09733.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simon F, Scheuerle A, Calzia E, et al. Erythropoietin during porcine aortic balloon-occlusion-induced ischemia/reperfusion injury. Crit Care Med. 2008;36:2143–50. doi: 10.1097/CCM.0b013e31817d7912. [DOI] [PubMed] [Google Scholar]
- 10.Smith PD, Puskas F, Fullerton DA, et al. Attenuation of spinal cord ischemia and reperfusion injury by erythropoietin. J Thorac Cardiovasc Surg. 2011;141:256–60. doi: 10.1016/j.jtcvs.2010.09.017. [DOI] [PubMed] [Google Scholar]
- 11.Sönmez A, Kabakçi B, Vardar E, et al. Erythropoietin attenuates neuronal injury and potentiates the expression of pCREB in anterior horn after transient spinal cord ischemia in rats. Surg Neurol. 2007;68:297–303. doi: 10.1016/j.surneu.2006.11.045. [DOI] [PubMed] [Google Scholar]
- 12.Stevens RD, Bhardwaj A. Eryrthropoietin and the promise of ischemic multiorgan protection. Crit Care Med. 2008;36:2446–7. doi: 10.1097/CCM.0b013e3181810513. [DOI] [PubMed] [Google Scholar]
- 13.Lang-Lazdunski L, Matsushita K, Hirt L, et al. Spinal cord ischemia. Development of a model in the mouse. Stroke. 2000;31:208–13. doi: 10.1161/01.str.31.1.208. [DOI] [PubMed] [Google Scholar]
- 14.Kurita N, Kawaguchi M, Horiuchi T, et al. An evaluation of white matter injury after spinal cord ischemia in rats: a comparison with gray matter injury. Anesth Analg. 2005;100:847–54. doi: 10.1213/01.ANE.0000146523.56647.5E. [DOI] [PubMed] [Google Scholar]
- 15.Lafci B, Yasa H, Ilhan G, et al. Protection of the spinal cord from ischemia: comparative effects of levosimendan and iloprost. Eur Surg Res. 2008;41:1–7. doi: 10.1159/000121394. [DOI] [PubMed] [Google Scholar]
- 16.Yamauchi T, Sakurai M, Abe K, et al. Ubiquitin-mediated stress response in the spinal cord after transient ischemia. Stroke. 2008;39:1883–9. doi: 10.1161/STROKEAHA.106.455832. [DOI] [PubMed] [Google Scholar]
- 17.Awad H, Ankeny DP, Guan Z, et al. A mouse model of ischemic spinal cord injury with delayed paralysis caused by aortic cross-clamping. Anesthesiology. 2010;113:880–91. doi: 10.1097/ALN.0b013e3181ec61ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sakamoto T, Kawaguchi M, Kurita N, et al. Long-term assessment of hind limb motor function and neuronal injury following spinal cord ischemia in rats. J Neurosurg Anesthesiol. 2003;15:104–9. doi: 10.1097/00008506-200304000-00007. [DOI] [PubMed] [Google Scholar]
- 19.Noguchi CT, Asavaritikrai P, Teng R, et al. Role of erythropoietin in the brain. Crit Rev Oncol Hematol. 2007;64:159–71. doi: 10.1016/j.critrevonc.2007.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Celik M, Goekmen N, Erbayraktar S, et al. Erythropoietin prevents motor neuron apoptosis and neurologic disability in experimental spinal cord ischemic injury. Proc Natl Acad Sci U S A. 2002;99:2258–63. doi: 10.1073/pnas.042693799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dame C, Juul SE, Christensen RD. The biology of erythropoietin in the central nervous system and its neurotrophic and neuroprotective potential. Biol Neonate. 2001;79:228–35. doi: 10.1159/000047097. [DOI] [PubMed] [Google Scholar]
- 22.Chi OZ, Hunter C, Liu X, et al. Effects of erythropoietin on blood-brain barrier disruption in focal cerebral ischemia. Pharmacology. 2008;82:38–42. doi: 10.1159/000127839. [DOI] [PubMed] [Google Scholar]
- 23.Taniguchi N, Nakamura T, Sawada T, et al. Erythropoietin prevention trial of coronary restenosis and cardiac remodeling after ST-elvated acute myocardial infarction (EPOC-AMI): a pilot, randomized, placebo-controlled study. Circ J. 2010;74:2365–71. doi: 10.1253/circj.cj-10-0267. [DOI] [PubMed] [Google Scholar]
- 24.Ott I, Schulz S, Mehilli J, et al. Erythropoietin in patients with acute ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention: a randomized, double-blind trial. Circ Cardiovasc Interv. 2010;3:408–13. doi: 10.1161/CIRCINTERVENTIONS.109.904425. [DOI] [PubMed] [Google Scholar]
- 25.Coleman T, Brines M. Science review: recombinant human erythropoietin in critical illness: a role beyond anemia? Crit Care. 2004;8:337–41. doi: 10.1186/cc2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L, Zhang Z, Wang Y, et al. Treatment of stroke with erythropoietin enhances neurogenesis and angiogenesis and improves neurological function in rats. Stroke. 2004;35:1732–7. doi: 10.1161/01.STR.0000132196.49028.a4. [DOI] [PubMed] [Google Scholar]
- 27.Taguchi A, Soma T, Tanaka H, et al. Administration of CD34+ cells after stroke enhances neurogenesis via angiogenesis in a mouse model. J Clin Invest. 2004;114:330–8. doi: 10.1172/JCI20622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brunner S, Winogradow J, Huber BC, et al. Erythropoietin administration after myocardial infarction in mice attenuates ischemic cardiomyopathy associated with enhanced homing of bone marrow-derived progenitor cells via the CXCR-4/SDF-1 axis. FASEB J. 2009;23:351–61. doi: 10.1096/fj.08-109462. [DOI] [PubMed] [Google Scholar]
- 29.Petit I, Szyper-Kravitz M, Nagler A, et al. G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat Immunol. 2002;3:687–94. doi: 10.1038/ni813. [DOI] [PubMed] [Google Scholar]
- 30.Kaminski A, Ma N, Donndorf P, et al. Endothelial NOS is required for SDF-1a/CXCR4-mediated peripheral endothelial adhesion of c-kit+ bone marrow stem cells. Lab Invest. 2008;88:58–69. doi: 10.1038/labinvest.3700693. [DOI] [PubMed] [Google Scholar]
- 31.Lu CZ, Xiao BG. G-CSF and neuroprotection: a therapeutic perspective in cerebral ischaemia. Biochem Soc Trans. 2006;34:1327–33. doi: 10.1042/BST0341327. [DOI] [PubMed] [Google Scholar]
- 32.Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–8. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- 33.Rosenstein JM, Krum JM, Ruhrberg C. VEGF in the nervous system. Organogenesis. 2010;6:107–14. doi: 10.4161/org.6.2.11687. [DOI] [PMC free article] [PubMed] [Google Scholar]