

Abstract
To evaluate the impact of hypoxia on the angiogenic capability of endothelial cells (ECs), and further investigate whether the cyclooxygenase-2 (COX-2)/prostaglandin E2 (PGE2) signalling is involved in the angiogenic response of ECs to hypoxia. We explored the impact of various periods (1, 3, 6, 12, 24 hrs) of hypoxia (2% O2) on human umbilical vein endothelial cells (HUVECs) in vitro. We observed cell viability, migration, tube formation, analysed COX-2, vascular endothelial growth factor (VEGF), AQP1 mRNA transcription, protein expression and measured PGE2, VEGF protein concentration in cell supernatants. Then we treated HUVECs with COX-2 selective inhibitor NS398, EP1/2 combined antagonist AH6809 and exogenous PGE2 to investigate the role of COX-2/PGE2 signalling in the angiogenic response of ECs to hypoxia. The results demonstrated that short-term hypoxic treatment enhanced HUVECs proliferation, migration, tube formation, significantly up-regulated COX-2, VEGF, AQP1 mRNA level, protein expression and promoted PGE2, VEGF release. The pharmacological inhibition study revealed that exposure of HUVEC to NS398 and AH6809 under hypoxia impaired the biological responses of ECs to hypoxia. Exogenous PGE2 augments the effects of hypoxia on HUVECs, and partially reversed the inhibitory effects of NS398 on HUVECs proliferation and angiogenic capability. Short-term hypoxic treatment enhanced angiogenic capability of ECs, and COX-2/PGE2 signalling may play a critical role in the biological response of ECs to hypoxia.
Keywords: hypoxia, endothelial cells, angiogenesis, COX-2/PGE2 signalling, VEGF, AQP1
Introduction
Angiogenesis, formation of new capillaries which enables delivery of oxygen and nutrients, is essential for tumour growth and metastasis [1], ischaemic disorder recovering [2] and periodontal tissue remodelling [3]. It is a highly organized process and involves a complex sequence of steps that include endothelial cells (ECs) proliferation, maturation and assembly [4]. As the main cell type involved in angiogenesis, ECs migrate from capillaries toward the pathological zone, undergoing various stress conditions including hypoxia and acidic pH [5]. Moreover, the endothelium plays a predominant role in modulating many aspects of vascular homeostasis. Dysfunction of ECs structure and function may contribute to the overall endothelial dysfunction in a range of clinic settings, including ischaemic diseases [6]. Understanding specific mechanisms underlying angiogenesis from ECs point of view offers the best approach to develop therapies for cancer, ischaemic diseases and modulate the periodontal remodelling process, where neovascularization is either impaired or activated [2].
Hypoxia is a characteristic feature of many physiological or pathological processes in vivo: driving angiogenesis in tumours [7], taking part in ischaemic diseases [8] and participating in bony or soft tissue injury [9]. It has been shown to be an important regulator of blood vessel tone, vessel structure and a potent stimulus of angiogenesis [10]. Hypoxic environment primarily affect the fundamental characteristics of ECs, which situates at the interface between blood and tissue and the first line to sense hypoxia [11]. Vascular ECs cope with hypoxia to maintain vascular homeostasis by expressing a number of genes, which are mediated by a variety of oxygen-sensitive signalling cascades [12]. Interestingly, the presence of hypoxia is always associated with and accompanied by inflammation [13]. For instance, the poorly vascularized regions of tumours with low oxygen tension is closely linked with inflammation in the form of tumour-associated macrophages accumulation [14]; injury in acute myocardial infarction is characterized by inflammatory event of neutrophils infiltration after a period of ischaemia [15]; the inflammatory mediators interleukin-1β and β-glucuronidase have been shown to present in the gingival crevicular fluid of children undergoing orthodontic treatment [16]. Reciprocity of inflammation, oxidative stress and neovascularization is emerging as an important mechanism underlying numerous processes from tissue healing and remodelling to cancer progression [5]. However, the mechanism of hypoxia-driven angiogenesis and the role of inflammatory mediators in the biological response of ECs to hypoxia remains unclear.
Previous study [17] revealed that short-term hypoxia can directly activate ECs towards a pro-inflammatory phenotype through the synthesis of lipid mediators. ECs produce several PGs, such as PGE2, PGI2 and thromboxane A2 in response to various stimuli [18, 19] and these prostanoids, particularly PGE2, have been implicated in angiogenesis [20]. These pro-inflammatory eicosanoids directly stimulate the synthesis of angiogenic factors, promote vascular sprouting, migration and tube formation, and also enhance endothelial survival [20, 21]. Specifically, it has been reported that PGE2 stimulates vascular endothelial growth factor (VEGF) expression in osteoblasts [22], fibroblasts [23], as well as in rat ECs [24] and that COX-2–dependent VEGF induction enhances angiogenesis in vivo[25].
Cyclooxygenase (COX) is a rate-limiting enzyme in the prostaglandin (PG) biosynthetic pathway. Cyclooxygenase-1 is found constitutively expressed in a wide range of tissues, while COX-2 is an inducible enzyme that produces prostaglandins during inflammatory and tumorigenic settings [26]. Furthermore, it is one of the genes induced by hypoxia and acts as a mediator of both inflammation and angiogenesis. EC-derived COX-2 is suggested important in angiogenesis [27]. Although there have been many reports regarding the mechanism of angiogenesis, few studies have explored the direct effect of COX-2/PGE2 signalling on angiogenesis.
Vascular endothelial growth factor (VEGF) is a major regulator of endothelial proliferation and migration [28], and is the most potent inducer of angiogenesis and capillary permeability [29]. It has been reported the key role of oxygen tension in regulating the expression of a variety of genes, and VEGF mRNA is induced by exposure to low oxygen tension under various pathophysiological circumstances [30]. Moreover, the hypoxia-induced COX-2 activation may augment PGE2 release, resulting in either an autocrine or paracrine action that enhances expression of VEGF through the generation of hypoxia inducible factor (HIF)-1α[31, 32].
The aquaporins (AQP) are a family of proteins which mediate water resorption in mammalian renal tubules and several other tissues [33]. Aquaporin-1 is one family member of AQP with a molecular weight of 28 kDa. Previous studies validated that AQP1 is responsible for the high vascular permeability and interstitial fluid pressure. AQP1 is reported to be required for hypoxia-inducible angiogenesis in human retinal vascular ECs [34]. However, the role of COX-2/PGE2 signalling in the VEGF and AQP1 expression of ECs are rarely reported. Therefore, the mechanisms involved in the impact of hypoxia on VEGF and AQP1 expression of ECs remains to be further elucidated from the perspective of inflammation.
In order to better understand how ECs adapt to hypoxic environment, we explored the impact of hypoxia on the human umbilical vein endothelial cells (HUVECs) by observing cell viability, migration, tube formation, analysing COX-2, VEGF and AQP1 expression and measuring PGE2, VEGF concentration in supernatants of HUVECs with different periods of hypoxic treatment. Then to further investigate into the potential role of COX-2/PGE2 signalling in the biological response of ECs to hypoxia, we used COX-2 selective inhibitor NS398, E-prostanoid receptor 1/2 (EP1/2) combined antagonist AH6809 and exogenous PGE2 to treat HUVEC under hypoxia. The findings of present study may elucidate the relationship between inflammation and angiogenesis in human ECs response to hypoxia, and provide theoretical basis for further understanding of the mechanisms involved in cancer, ischaemic disorder and periodontal tissue remodelling.
Materials and methods
Culture and characterization of HUVECs
Human umbilical vein endothelial cells were purchased from American Typical Culture Collection (ATCC code: CRL-1730). Cells were cultured in minimal essential α medium (α-MEM; Gibco BRL, Grand Island, NY, USA) supplemented with 10% (v/v) foetal bovine serum (FBS; CS, Hyclone, Auckland, New Zealand), 100 units/ml penicillin and 100 mg/ml streptomycin. All cells were cultured in 25 cm2 flasks (Corning Glass Works, Corning, NY, USA) and maintained in an incubator supplied with 95% air, 5% CO2 and 100% humidity at 37°C. Every three days the media was exchanged with fresh α-MEM (plus 10% FBS, 100 U/ml penicillin, 100 mg/ml streptomycin). Cells were washed twice with phosphate buffered solution (PBS) and detached with 0.25% trypsin plus 0.05% ethylenediaminetetraacetic acid (EDTA) (Sigma-Aldrich, St. Louis, MO, USA) for passage when they reached a confluence of 80% approximately [35].
Cell characterization was evaluated by their morphology on reaching confluence. Besides, immunostaining was performed in order to detect factor VIII-related antigen and CD31 in HUVECs raised to confluence. Briefly, HUVECs were fixed in 4% (w/v) paraformaldehyde for 20–30 min., washed with PBS three times and air-dried at 4°C. Then treated with 3% hydrogen peroxide for 15 min. at room temperature to block intrinsic peroxidase and washed with PBS three times. Fixed cells were incubated for 1 hr at 37°C with primary antibody anti-factor VIII-related antigen or anti-CD31 (mouse anti-human; Abcam, Cambridge, UK). Subsequently, cells were washed in PBS and incubated with biotin-conjugated rabbit anti-mouse IgG (Santa Cruz, CA, USA) for 1 hr at 37°C. Finally, they were incubated with horseradish peroxidase labelled streptavidin for 20 min. at 37°C. At least five independent experiments were performed and cultures were examined with a microscope (Nikon, Japan).
Hypoxic treatment of HUVECs
At 80% confluence, HUVECs were placed in 6 well plates at 1 χ 104/cm2 and incubated with 5% CO2, 100% humidity at 37°C for two days. The cells were assigned to two groups as hypoxic group (2% O2) and normoxic control group (20% O2), while the former divided into five subgroups (1, 3, 6, 12, 24 hrs) according to different periods of hypoxic exposure. Then, the cells were subjected to hypoxia maintained using a three gas modular hypoxic incubator with CO2/O2 monitoring and CO2/N2 gas sources (Binder, Camarillo, CA, USA). Figure 1 is schematical diagram of the device. Culture medium was pre-equilibrated overnight prior to cell exposure. Cell culture plates were placed in the incubator and saturated with a gas mixture containing 2% oxygen, 5% CO2 and 93% nitrogen for the generation of hypoxia at 37°C for defined time periods (1, 3, 6, 12, 24 hrs), each with three replicates and the experiments were done for at least three times. In some experiments, the combined EP1/2 antagonist AH6809 (Sigma-Aldrich), or exogenous PGE2 (Sigma-Aldrich) and/or NS398 (Sigma-Aldrich), a selective inhibitor of COX-2, were also added into the HUVECs medium 4 hrs before hypoxic treatment.
Fig 1.
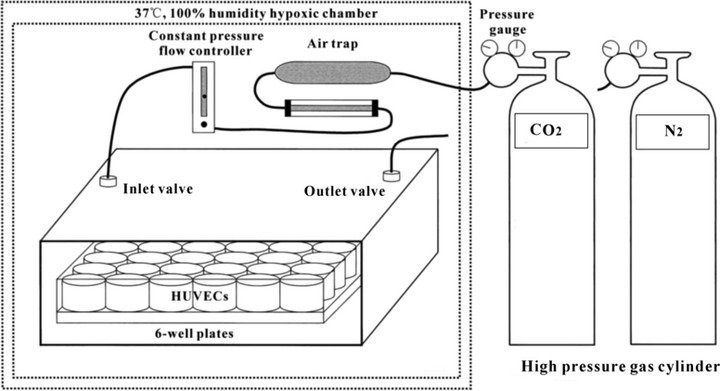
Schematic diagram of the Binder, a three gas modular hypoxic incubator used to simulate hypoxic conditions in vivo. Ambient oxygen concentrations of 2% were maintained using this device with CO2/O2 monitoring and CO2/N2 gas sources. A pure N2 entrance is set for the nitrogen replacement of air in incubator to control the oxygen concentration and achieve the purpose of hypoxia.
3-(4, 5-Dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide assays
The 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay was used to determine HUVECs proliferation and viability. Human umbilical vein endothelial cells were seeded at 1 χ 104/well in the 96 well plates and incubated with normal condition for 48 hrs, then were subjected to hypoxic treatment as mentioned above, each with five replicates. At each set point, the cells were supplemented with MTT (5 mg/ml; Sigma-Aldrich) and incubated for a further 3.5 hrs. The blue formazan thus produced was solubilized with 200 μl/well dimethyl sulfoxide (DMSO) and absorbance was measured at 570 nm by HTS 7000 Plus high efficient analyser (Perkin Elmer, Norwalk, CT, USA).
Transwell migration assay
Human umbilical vein endothelial cell motility was measured using 24-multiwell insert system (Millipore, CA, USA) with 8 μm pore size polycarbonate filter insert that divides the chamber into upper and lower portions. The filters were coated with human recombinant fibronectin. Briefly, HUVECs that were previously starved overnight in 5% foetal calf aerum (FCS; Sigma-Aldrich), were trypsinized and resuspended in 0.1% FCS medium at a density of 4 × 105 cells/ml. Two hundred and fifty microlitres of this suspension were then added to the upper chamber of the insert while 750 μl of 0.1% FCS was added to the lower chamber. After 3 hrs of adhesion, cells were allowed to migrate across the 8 μm pore size polycarbonate membrane under normoxia or hypoxia for 1, 3, 6, 12 and 24 hrs. Cells on the top of the filter were removed by gentle swabbing and the remaining cells on the bottom side of the filter were stained using 0.25% cresyl violet, and counted under a light microscope with an eyepiece grid to visualize set fields. At least four fields in duplicate wells were counted for each condition.
Tube formation assay
Matrigel (BD Biosciences, New Bedford, MA, USA) was added to wells of a cold 96-well plate (45 μl/well), and then incubated at 37°C for 1 hr to allow gelling. Human umbilical vein endothelial cells were removed from confluent cultures by treatment with trypsin and 0.05% EDTA. The cells were washed in serum-containing medium and then resuspended to 105 cells/ml. Into each culture well was added 100 μl cell suspension. Cell culture was carried out at 37°C for 48 hrs in a humidified 5% CO2 atmosphere for complete penetration of cells into the Matrigel. Then after 12 and 24 hrs of normoxic or hypoxic treatment, cells were observed directly and scored on a scale of 0–5 for tube formation using dark field illumination on a light microscope. Based on quality and number of the tubes, they were assigned numeric values: 0, no real tubes; 1, some poorly formed tubes; 2, some formed tubes; 3, network of tubes both formed and poorly formed; 4, network of formed tubes; and 5, network of well formed tubes. Score results from four random fields in duplicate wells were averaged.
Pharmacological COX-2 inhibition
This pharmacological inhibition study was employed to further investigate the biological response of HUVECs to hypoxia. NS398 was added into the HUVECs medium 4 hrs before hypoxic treatment. Minimum effective concentration (MEC) of inhibitor necessary for significant inhibition of COX-2 protein expression was determined by a concentration gradient test, in which NS398 was used at final concentrations of 0, 5, 10 and 20 μM, respectively.
RNA isolation and real-time quantitative PCR
After hypoxic treatment, cells were washed twice with PBS. The total RNA was extracted using TRIzol regent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. Total RNA was quantified, in a spectrophotometer, at an absorbance (A) of 260 nm. The RNA samples had an A260:A280 ratio of 2.0 to guarantee high purity. Two micrograms of total RNA from each sample were subjected to reverse transcription using the SYBR PrimeScript™ RT-PCR Kit (TaKaRa Biotechnology, Dalian, Liaoning, China) according to the manufacturer's protocol. Each real-time PCR was carried out in triplicate in a total of 20 μl reaction mixture (6.8 μl of cDNA, 10 μl of SYBR1 Premix Ex TaqTM, 0.4 μl of ROX Reference Dye II, 0.4 μl of each 10 mM forward and reverse primers and 2 μl of H2O) in an Applied Biosystem (ABI) Prism 7300 Real-time PCR System [Applied Biosystems (ABI), Foster City, CA, USA]. Primers used for real-time PCR analysis are presented in Table 1. The PCR program was initiated by 10 sec. at 95°C for pre-degeneration before 40 thermal cycles, each of 5 s at 95°C (degeneration), 31 s at 60°C (annealing) and 30 s at 72°C (elongation). The starting copy numbers of unknown samples were calculated by the 7300 System SDS Software from the standard curve. The housekeeping gene, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), was concurrently amplified in each sample as control and was used for normalization. The cDNA of control HUVECs untreated of hypoxia normalized to the level of GAPDH mRNA have been ascribed a fold induction of 1. Melting curves for each PCR reaction were generated to ensure the purity of the amplification product. Data analysis was performed using the 2−ΔΔCt method described previously, where GAPDH was used as the reference gene [36].
Table 1.
Primers used for real-time PCR analysis
| Gene | GeneBank | Primers sequences (5′-3′) | Fragment size (bp) |
|---|---|---|---|
| Runx2 | NM_001015051 | R5′-GTGAAGACGGTTATGGTCAAGG-3′; F5′-CAGATGGGACTGTGGTTACTGT-3′ | 169 |
| Osterix | NM_152860 | R5′-CCACTATTTCCCACTGCCTTG-3′; F5′-ACCTACCCATCTGACTTTTGCTC-3′ | 125 |
| COX-2 | NM_000963 | R5′-CTACCAGAAGGGCAGGATACAG-3′; F5′-GCAGGCAGATGAAATACCAGTC-3′ | 165 |
| GAPDH | NM_002046 | R5′-GTAGAGGCAGGGATGATGTTCT-3′; F5′-CTTTGGTATCGTGGAAGGACTC-3′ | 132 |
GAPDH: glyceraldehyde-3-phosphate dehydrogenase.
Western blotting
To obtain whole-cell extracts, cells that were treated with or without hypoxia were washed twice with ice-cold PBS and then lysed and sonicated in a lysis buffer (Keygen total protein extraction kit; Keygen Biotech., Nanjing, China). The cytosolic fraction was collected as the supernatant after centrifugation at 14,000 ×g at 4°C for 15 min. and assayed it quantitatively with the BCA method. After boiling for 5 min., 20–25 ml of the lysate (50 mg of protein) was applied to SDS–12% PAGE at 80 V for 30 min. and 120 V for 1 hr. The proteins in the gel were then transferred to a PVDF membrane (Millipore). After blocking, the membranes were probed with 1:1000 dilutions of the anti-AQP1 and 1:100 dilutions of the anti-VEGF (Abcam, Hong Kong), followed by the addition of horseradish peroxidase (HRP)–conjugated secondary antibody (diluted 1:5000) at 37°C for 1 hr. Immunoreactive proteins were visualized using a chemiluminescence kit (Immobilon Western Chemiluminescent HRP Substrate; Millipore). Band intensities were determined using the ChemiDoc XRS Gel documentation system and Quantity One software (Bio-Rad, CA, USA).
Enzyme-linked immunosorbent assay assays
The supernatant media of HUVECs, stored in aliquots at −70°C, were thawed on ice before the measurement of VEGF and PGE2 using specific enzyme-linked immunosorbent assay (ELISA) kits obtained from R&D Systems (Minneapolis, MN, USA) according to the manufacturer's instructions. Results were expressed after normalization to total protein contents of the supernatant media and in pg of VEGF or PGE2 per mg of total protein. α-MEM–10% FBS was used as control.
Statistical analysis
All experiments were performed at a minimum of three times. Measurements are expressed as mean ± S.D. Statistical comparisons were made using factorial analysis of variance (ANOVA), for comparing treatments from controls. In MTT, cell migration and tube formation assay, Student's t-test was used to compare between the two groups at same time point. A value of P < 0.05 was statistically considered significant.
Results
Characterization of HUVECs
The cells exhibited typical cobblestone morphology on reaching confluence (Fig. 2A), and immunocytochemical staining showed that they expressed factor VIII-related antigen and CD31 (Fig. 2B and C), which initially suggested that the cells are ECs.
Fig 2.
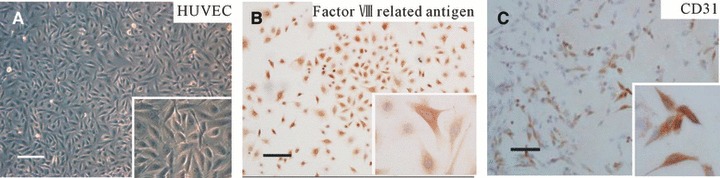
Characterization of HUVECs by morphological observation and immunocytochemical staining. (A) The cells exhibited typical cobblestone morphology on reaching confluence, insert with higher magnification of ×200. (B and C) Immunocytochemical stained with factor VIII-related antigen and CD31 antibody, respectively, the immunoreactive positive cells were stained brownish-yellow, insert with higher magnification of ×400 confirming brownish-yellow stained cytoplasm. Scale bars: 100 μm.
Hypoxia enhances HUVECs proliferation, migration and tube formation
To ensure that hypoxia used in this study (2% oxygen, 5% CO2 and 93% nitrogen in a modular hypoxic incubator) induces typical cell responses to hypoxic stress, we initially verified HUVECs proliferation and viability following hypoxic exposure by MTT assay. As shown in Figure 3A, an up-regulation of HUVECs viability occurs after temporary exposure to hypoxia (1, 3 and 6 hrs), the cells viability was increased gradually with increasing time of hypoxia. At 3 and 6 hrs, it was significantly augmented by hypoxia compared with normoxia control (P < 0.05) and reached maximum at 6 hrs. With increasing time of hypoxia (12 and 24 hrs), it was decreased, less than those of the normoxia control group (P < 0.05).
Fig 3.
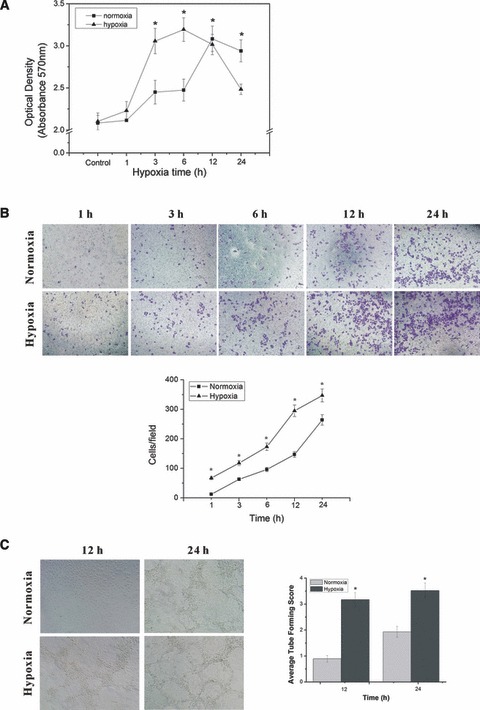
HUVECs proliferation, migration and tube formation under hypoxic exposure. (A) HUVECs proliferation and viability following hypoxic exposure by MTT assay. *P < 0.05 versus control group. (B) HUVECs were assayed for migration under normoxia or hypoxia for indicated periods of time. The cells migrate across the membrane were counted under a light microscope with an eyepiece grid to visualize set fields. At least four fields in duplicate wells were counted for each group to generate the bar chart. (C) The tube formation of HUVECs after 12 and 24 hrs of normoxia or hypoxia was viewed by phase-contrast microscopy at ×100 magnification, tube formation of each group was scored on a scale of 0–5 based on quality and number of the tubes, score results from four random fields in duplicate wells were averaged to generate the bar chart. *P < 0.05 versus normoxia group.
Then, we assessed in vitro migration of HUVECs using fibronectin-coated 8 μm filters under either normoxic or hypoxic conditions for 1, 3, 6, 12 and 24 hrs. We found that hypoxia induced migration of HUVECs in a time-dependent manner. As compared with the migration value under normoxia of 12 ± 0.8, 63 ± 4.9, 96 ± 7.3, 147 ± 9.4 and 264 ± 17.7 cells/field at 1, 3, 6, 12 and 24 hrs, respectively, and the amount of cell migration induced by various periods of hypoxia were 67 ± 4.2, 118 ± 8.3, 173 ± 12.6, 295 ± 19.5, 347 ± 21.6, which were significantly higher than that of normoxia at each time point (P < 0.05) (Fig. 3B).
Finally, to study the effect of hypoxia on capacity of HUVECs to form functional capillaries in vitro, wells containing Matrigel were used in a tubeforming assay. Human umbilical vein endothelial cells were observed after 12, 24 hrs of normoxic or hypoxic treatment and analysed for the quality and number of the tubes formed in the gel. Overall, hypoxia led to a significantly higher level of tube formation than that of normoxia (P < 0.05) (Fig. 3C).
Hypoxia increases angiogenesis-related gene transcription and protein expression after temporary exposure
To find out the angiogenic capability in ECs under hypoxia, we proceeded to demonstrate the VEGF, AQP1 mRNA synthesis, protein expression and VEGF release of HUVECs after hypoxic treatment. As shown in Figure 4A, the VEGF mRNA was significantly increased by hypoxia at all time points (P < 0.05). During cultivation, it transiently increased about 2.3 folds, then peaked at 3 hrs and decreased after 6 hrs of the culture, still higher than those of the control group. Vascular endothelial growth factor protein expression increased significantly at 1 hr after hypoxic exposure and reached summit at 3 hrs (P < 0.05), at 6 hrs, it decended and there was no significant difference between 6 hrs group and control (P > 0.05), then declined to the level lower than control at 12 and 24 hrs (P < 0.05) (Fig. 4B). The accumulation of VEGF protein in ECs supernatants treated by hypoxia was also measured. Data showed that VEGF secretion was significantly increased by hypoxia at all time points. (P < 0.05), and reached the summit at 6 hrs, with the concentration of 255.29 pg/105cells/ml (Fig. 4C).
Fig 4.
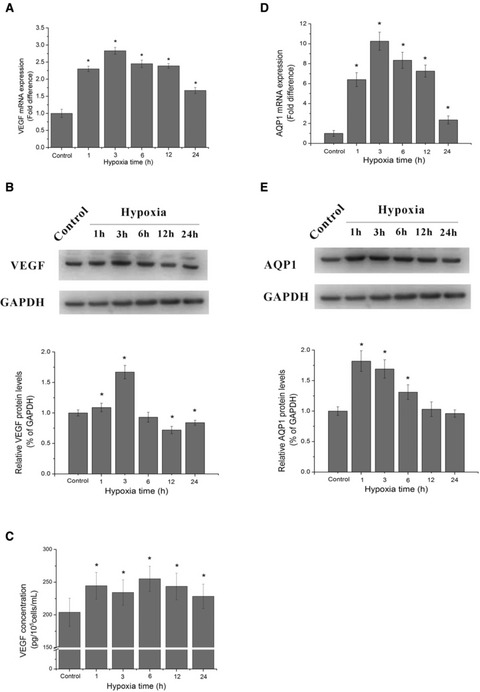
HUVECs proliferation, VEGF, AQP1 mRNA level, protein expression and VEGF accumulation in supernatant under hypoxic exposure. (A) The mRNA levels of VEGF at different hypoxic time points. (B) The protein expression of VEGF at different hypoxic time points by Western blotting analysis, the blots and the bar chart below are in one label. (C) Quantification of VEGF by ELISA assay in supernatant of HUVECs after various periods of hypoxic treatment. (D) The mRNA levels of AQP1 at different hypoxic time points. (E) The protein expression of AQP1 at different hypoxic time points by Western blotting analysis, the blots and the bar chart below are in one label. *P < 0.05 versus control group.
Aquaporin-1 mRNA level was transiently increased about 6.4 folds and peaked at 3 hrs by 10.26 folds. After 6 hrs, it decreased and remained higher than the control (P < 0.05) (Fig. 4D). AQP1 protein expression was significantly up-regulated by hypoxia in short periods (1, 3, 6 hrs, P < 0.05), and declined to the level lower than the control at 12 and 24 hrs, while the differences were not significant (P < 0.05) (Fig. 4E).
Hypoxia up-regulates COX-2 mRNA level, protein expression and promotes PGE2 release in short periods
In order to better understand the effect of hypoxia on the function of HUVECs, we carried out real-time quantitative PCR and Western blot experiments to measure COX-2 mRNA level and protein expression of HUVECs with different periods of hypoxic exposure. The results showed that COX-2 mRNA was at very low levels in unstimulated cells, early minutely induced by 1.35 folds at 1 hr after hypoxic treatment, and reached top at 3 hrs by 2.67 folds, then afterwards declined to level below 2 folds and remained higher than the untreated control group (P < 0.05) (Fig. 5A). Similarly, COX-2 protein expression also increased significantly at 1 hr and maximized by 1.34 folds at 3 hrs, thereafter declined to levels significantly lower than control (P < 0.05) (Fig. 5B).
Fig 5.
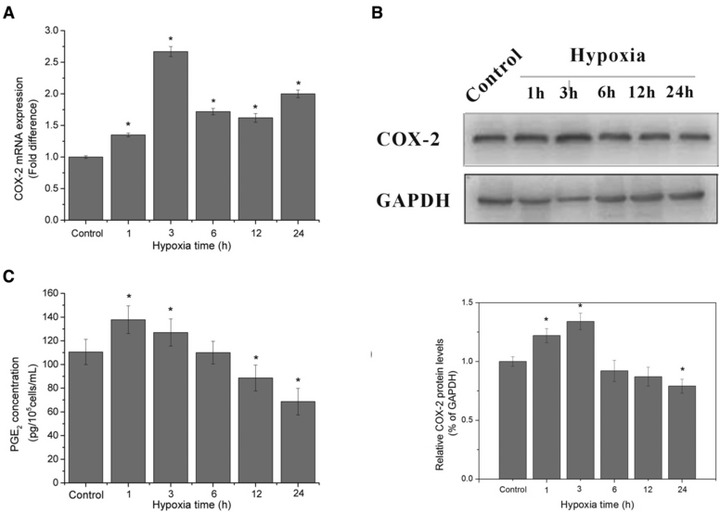
COX-2 mRNA level, protein expression and PGE2 accumulation in supernatant under hypoxic exposure. (A) The mRNA levels of COX-2 at different hypoxic time points. 2−ΔΔCt values were obtained by real-time RT-PCR analysis using GAPDH transcripts for the normalization. (B) The protein expression of COX-2 at different hypoxic time points by Western blotting analysis, the blots and the bar chart below are in one label. (C) Quantification of PGE2 by ELISA assay in supernatant of HUVECs after various periods of hypoxic treatment, the data are expressed in concentration of PGE2. *P < 0.05 versus control group.
Having proved that hypoxia regulates COX-2 at transcription and expression level, we further verified whether it induces COX-2 expression by the production of prostaglandin, determined as PGE2 accumulation in the supernatants of hypoxia treated ECs by ELISA assay. The results indicated that hypoxia stimulated a significant short-term increase in PGE2 release that peaked at 3 hrs, with a concentration of 137.83 ± 11.66 pg/105 cells/ml. Then the amount of PGE2 in supernatants went down, possibly following the pattern of COX-2 expression. Late PGE2 release was decreased, making the quantity of it accumulated in the culture medium significantly lower than the control (P < 0.05), which was consistent with the progression pattern of COX-2 protein expression (Fig. 5C).
Concentration-dependent effect of NS398 on COX-2 expression
The data show that NS398 regulated COX-2 expression in a concentration-dependent manner (Fig. 6). When NS398 was used at 5 μM, COX-2 protein expression was decreased to level about that of control. And when NS398 was used at concentrations of 10 and 20 μM, COX-2 protein expression was decreased to 24.1% and 21.5% of control group. This indicated that 10 μM of NS398 was more effective to inhibit COX-2 activity than 5 μM, but had similar effects to 20 μM under current experimental conditions. To avoid inflicting excessive injury to the cells and unwanted effects, we selected 10 μM as final concentration in the following pharmacological inhibition study.
Fig 6.
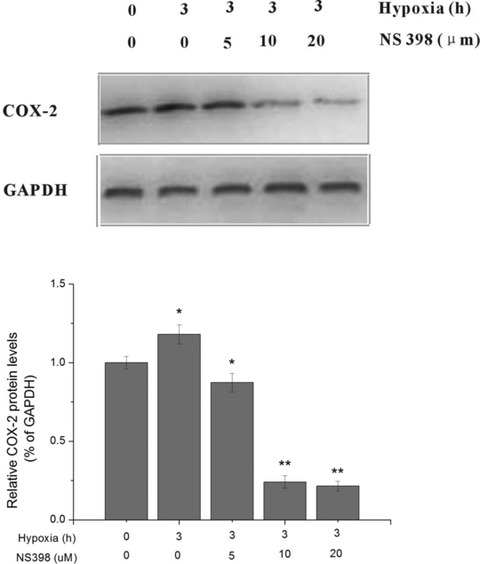
Effect of the selective COX-2 inhibitor NS398 on COX-2 activity by concentration gradient test. HUVECs were exposed to hypoxia and supplemented with NS398 at indicated concentrations of 0, 5, 10 and 20 μM. Cells without hypoxia or NS398 treatment served as control. The COX-2 activity was determined by Western blot analysis. *P < 0.05, **P < 0.01 versus control.
Selectively inhibition of COX-2 activity by NS398 impaires the biological response of HUVECs to hypoxia
In order to verify whether the endogenous COX-2/PGE2 signalling contributes to the biological response of ECs to hypoxia, we pre-treated cell cultures with 10 μM of COX-2-selective inhibitor, NS398 for 4 hrs prior to indicate periods of hypoxic treatment.
First, MTT assay was employed to examine whether NS398 has any effects on HUVECs proliferation. We found that NS398 had an inhibitory effect on HUVECs proliferation under normoxic culture conditions, and the increased proliferation of HUVECs by temporary hypoxia exposure was completely blocked by NS398 (P < 0.05) (Fig. 7A). Moreover, the difference between the cell viability of NS398 + hypoxia-treated group and the hypoxia-treated group was small especially at earlier hypoxic time points of 1 hr (P > 0.05), with prolongation of hypoxic time (3, 6, 12 and 24 hrs), the cell viability of NS398-treated groups significantly decreased compared to the untreated groups.
Fig 7.
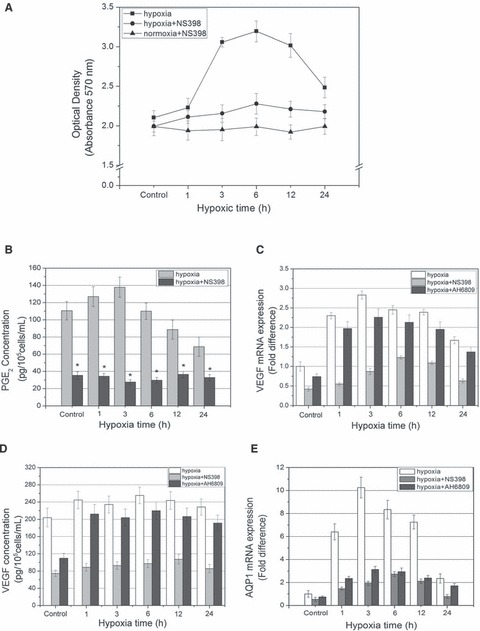
Selectively inhibition of COX-2 activity by NS398 and blockade of EP1/2 by AH6809 impair the biological response of HUVECs to hypoxia. (A) HUVECs proliferation and viability following normoxic, hypoxic or hypoxic + NS398 treatment by MTT assay. (B) Quantification of PGE2 release by ELISA assay in supernatant of HUVECs after various periods of hypoxic treatment with or without 10 μM NS398. (C) VEGF mRNA level assessed by real-time RT-PCR experiment, HUVECs were exposed to hypoxia in the presence or absence of NS398 (10 μM) for indicated periods of time. (D) VEGF concentration in supernatant of HUVECs measured by ELISA assay. (E) AQP1 mRNA level assessed by real-time RT-PCR experiment. *P < 0.05 versus control.
To find out whether NS398 attenuated prostanoid production induced by hypoxia, supernatants were collected and PGE2 concentrations were measured. Results showed that PGE2 synthesis and release were strongly inhibited by NS398 in hypoxia-treated cultures (P < 0.05) (Fig. 7B). NS398 at the concentration of 10 μM completely abolished the inductive effect of PGE2 production by hypoxia, suggesting that the enhancing effect of hypoxia on PGE2 release may be COX-2-dependent.
In order to study the effect of COX-2/PGE2 signalling on angiogenic capability of HUVECs, we examined the effects of NS398 on HUVECs VEGF mRNA transcription, secretion and AQP1 mRNA level. The findings demonstrated that VEGF mRNA transcription was obviously blocked by NS398 (P < 0.05), and its transcription levels gradually rose to a summit (6 hrs) before declination (P < 0.05) (Fig. 7C), and VEGF release was also significantly inhibited (P < 0.05), indicating that NS398 abrogated the inductive effect of hypoxia on VEGF synthesis and secretion (Fig. 7D). Moreover, NS398 significantly down-regulated AQP1 mRNA transcription under normoxic or hypoxic condition (P < 0.05) (Fig. 7E).
Combined blockade of EP1/2 by AH6809 attenuates the biological response of HUVECs to hypoxia
Then, we proceeded to evaluate the effects of AH6809 (combined EP1/2 antogonist) on HUVECs VEGF mRNA transcription, secretion and AQP1 mRNA level to confirm whether PGE2 receptor EP1/2 is involved in pro-angiogenic reaction of ECs to hypoxia. The data demonstrated that VEGF mRNA transcription and secretion were obviously blocked by AH6809 (P < 0.05), but the inhibitory effect was much smaller than that of NS398 (P < 0.05) and hypoxia stimuli could still promote VEGF mRNA transcription and secretion (Fig. 7C and D). In addition, AH6809 significantly decreased AQP1 mRNA transcription to a value which was still higher than that of NS398-treated group under normoxia or hypoxia (P < 0.05) (Fig. 7E).
Exogenous PGE2 enhances and partially reversed the inhibitory effects of NS398 on COX-2 expression, cell viability and VEGF, AQP1 mRNA transcription of HUVECs
From the above mentioned results, we could find that the inhibitory effects of COX-2 inhibitor NS398 on VEGF, AQP1 transcription and VEGF release were stronger than that of combined EP1/2 antagonist AH6809. To better understand the effect of PGE2 as a COX-2 downstream effector, we examined the combinational treatment of NS398 (10 μM) and exogenous PGE2 (10 μM) on COX-2 expression, comparing with that of NS398 or exogenous PGE2 treatment alone. Specifically, HUVECs were treated with 10 μM of NS398 and/or 10 μM of exogenous PGE2 under normoxia for 4 hrs. Results demonstrated that 10 μM of NS398 significantly blocked COX-2 expression, similar to the situation observed in hypoxic condition, while exogenous PGE2 prominently augments the protein expression of COX-2. Interestingly, when treated together with NS398, exogenous PGE2 partially reversed the inhibitory effects of NS398 on COX-2 expression of HUVECs (Fig. 8).
Fig 8.
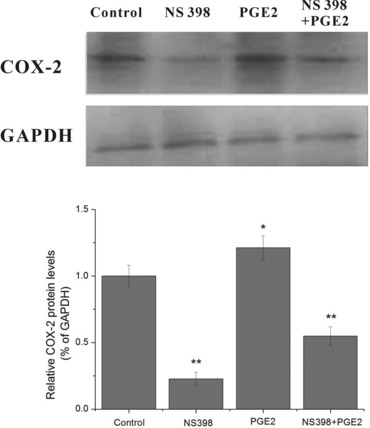
Effect of NS398, exogenous PGE2 and combinational treatment of NS398 and exogenous PGE2 on COX-2 protein expression of HUVECs under normoxia. HUVECs were treated with 10 μM of NS398 and/or 10 μM of exogenous PGE2 under normoxia for 3 hrs. Cells without NS398 or exogenous PGE2 treatment served as control. The COX-2 protein expression was determined by Western blot analysis. *P < 0.05 and **P < 0.01 versus control.
For cell viability, the MTT value of HUVECs under hypoxia was significantly higher than that under normoxia. NS398 exerted a significantly inhibitory effect on HUVECs proliferation both under hypoxia and normoxia, while exogenous PGE2 stimulated them, and combinational treatment of NS398 and PGE2 resulted in a value between the NS398 and PGE2, with hypoxic group significantly higher than the normoxic group (P < 0.05) (Fig. 9A). Similarly, for VEGF and AQP1 mRNA transcription, NS398 alone also significantly blocked while exogenous PGE2 significantly augmented them, and exogenous PGE2 (10 μM) partially reversed the inhibitory effect of NS398 (Fig. 9B and C).
Fig 9.
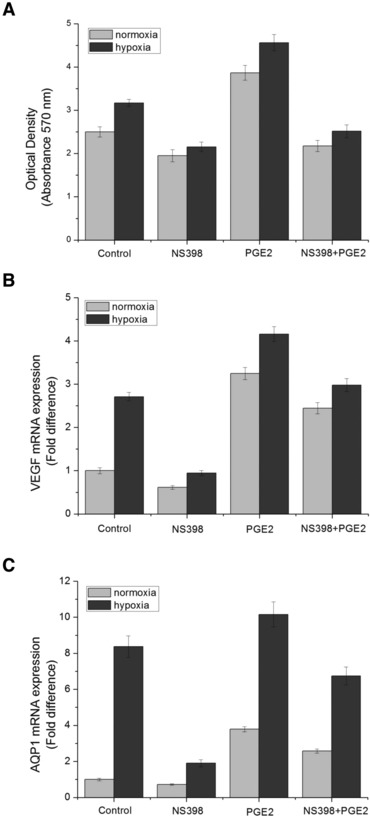
Effect of NS398, exogenous PGE2 and combinational treatment of NS398 and exogenous PGE2 on cell viability and VEGF, AQP1 mRNA transcription of HUVECs under normoxia or hypoxia. HUVECs were treated with 10 μM of NS398 and/or 10 μM of exogenous PGE2 under normoxia or hypoxia for 3 hrs. Cells without NS398 or exogenous PGE2 treatment served as control. (A) HUVECs proliferation and viability was measured by MTT assay. (B) VEGF mRNA level assessed by real-time RT-PCR experiment. (C) AQP1 mRNA level assessed by real-time RT-PCR experiment. *P < 0.05 and **P < 0.01 versus control.
Discussion
The endothelium plays a predominant role in modulating many aspects of vascular homeostasis. When facing a stimulus like hypoxia, this barrier is able to orchestrate a protective cellular response, including angiogenesis, cell proliferation and apoptosis [37, 38]. In the present study, we explored how hypoxia exerts its angiogenic activities on ECs by measuring HUVECs viability, migration, tube formation and angiogenesis related genes/protein (VEGF, AQP1) expression. Vascular endothelial growth factor is a potent angiogenic stimulator with the ability to promote ECs growth [39], enhance ECs survival [40] and induce vasodilatation [41]. Aquaporin-1 is abundantly presented in endothelia of non-fenestrated capillaries [42] and maybe responsible for the vascular hyper-permeability and increases in the hydrostatic interstitial pressure of tissue [43]. Study showed that high degree of AQP1 expression was preferentially associated with enhanced angiogenesis and ECs migration [44, 45]. It has been reported that up-regulation of AQP1 by hypoxia may worked as an O2 transporter in ECs and accelerate intracellular hypoxia directly and induce pro-angiogenic molecules in response to hypoxia, thereby leading to hypoxia inducible angiogenesis [46]. In the present study, we demonstrated specifically that short-term hypoxic treatment: (i) enhanced HUVECs proliferation, motility, tube formation capability; (ii) increased VEGF mRNA level, protein expression and promoted VEGF secretion; (iii) augmented AQP1 mRNA transcription and protein expression. The findings have suggested that temporary hypoxia may promote ECs proliferation and migration, facilitate the angiogenic capability of ECs, and high expression of AQP1 may be associated with the pro-inflammatory phenotype. The adaptive responses of ECs to hypoxia could increase regional blood vessel number, collateral circulation formation and vascular permeability, thus ameliorate local hypoxic microenvironment in ischaemic and remodelling tissue.
Understanding of the pathways involved in ECs hypoxic response and identifying strategies to activate or inhibit this process in tissue remodelling promotion, ischaemic diseases and cancer therapy would have important clinical implications. The important master regulator of angiogenesis under hypoxic conditions, hypoxia-inducible factor-1 (HIF-1), is a dimeric transcription factor composed of HIF-1α and HIF-1β[47]. It promotes the synthesis of protein that increases the cellular supply with O2 and initiates the defence against hypoxia at different levels [48]. Various traditional studies have focused their efforts on unravelling the importance of HIF-1α pathway in tumour progression and angiogenesis, as well as in the adaptive response of ECs to low oxygen availability [49]. Recent data [50, 51] also suggest that ECs are highly specialized in acquiring a pro-inflammatory phenotype when encountering stressors. They cope with hypoxia by expressing a number of inflammation-related genes, which are mediated by a variety of signalling cascades [52, 53]. However, the reports from the perspective of inflammatory mediators on the relationship between angiogenesis and hypoxia are rare.
Numerous molecules have been implicated in ECs proliferation and angiogenesis. COX-2/PGE2 signalling may be a key regulator of angiogenesis and mediate angiogenesis through multiple mechanisms, including an increase in VEGF transcription and expression [54, 55]. Previous investigations validated that amplification of the COX-2/PGE2 pathway in hypoxic colorectal cancer cells might have important implications for stimulating angiogenesis [56]. There is now ample evidence that COX-2 is involved in controlling cell growth and implicated as a key regulator of angiogenesis. The hypoxia-induced COX-2 activation may augment PGE2 release, resulting in either an autocrine or paracrine action that enhances expression of VEGF [57, 58]. Stimulation of VEGF production by PGE2 has also been demonstrated in previous studies in various cellular systems [59-61]. Our data documented that short-term hypoxia significantly up-regulated COX-2 mRNA transcription, protein expression and promoted PGE2 release (the production of secreted PGE2 is an appropriate measure of COX activity). Accompanied with COX-2 activation and PGE2 secretion, the VEGF and AQP1 expression also increased. Hence, based on the data herein, we demonstrated a potential parallel in the activation way of COX-2 and VEGF to hypoxic stress, and elucidated a positive correlation between COX-2 and VEGF expressions in angiogenesis. The present data also indicated that hypoxia signalling may transfer to COX-2/PGE2 and VEGF signalling, playing cooperative role in the angiogenic action of ECs. Furthermore, we could deduce a possible parallel relationship between inflammation and angiogenesis under hypoxia from the results above, and they might have a positive interaction with each other when faced with hypoxia.
To further study the function of COX-2/PGE2 in the biological response of ECs to hypoxia, we used NS398 (a selective COX-2 inhibitor to inhibit the activity of COX-2) and AH6809 (a combined antogonist of EP1/2) to pre-treat HUVECs prior to indicated periods of hypoxic exposure. Overexpression of COX-2 has been described among various human malignancies [62-64] and blood vessels in pathological conditions [65, 66], suggesting a critical role of COX-2 in tumorigenesis and inflammation. By contrast, selective pharmacological inhibition or genetic depletion of COX-2, which abolishes the eicosanoid synthesis in stimulated ECs, may abrogate an important homeostatic response to hypoxic stress, leading to inhibition of angiogenesis and induction of apoptosis [67]. Previous study has demonstrated that inhibition of COX-2 activity and thus resulting in significantly down regulation of VEGF expression and angiogenesis inhibition in vivo[68]. The present study demonstrated that exposure of HUVECs to NS398: (i) impaired proliferative response to hypoxia; (ii) strongly inhibited PGE2 release; (iii) attenuated VEGF mRNA and AQP1 mRNA transcription, and diminished VEGF secretion. Furthermore, AH6809 modestly abrogated the expression of hypoxia-induced angiogenesis related factors (VEGF and AQP1). Comparing to NS398, the blockade of the PGE2 receptors EP1/2 with a selective antagonist AH6809 received a much smaller effect on angiogenic capability of HUVECs under hypoxia. These results indicate that COX-2 induction ought to be a critical determinant of the angiogenic response of ECs to hypoxia and hypoxia probably exerts its effects in a COX-2-dependent manner. Moreover, the function of EP1/2 is not as crucial as that of COX-2 in the angiogenic capability of HUVECs, but a study has reported that the EP1/2 receptor contributed to growth factor-mediated tubular formation in angiogenesis [69].
To better understand the role of PGE2 in angiogenic response of ECs to hypoxia, we compared NS398, exogenous PGE2 and the combinational treatment of NS398 (10 μM) and exogenous PGE2 (10 μM) on cell viability, COX-2 expression and VEGF, AQP1 mRNA level. The results indicated that PGE2 augmented cell viability and angiogenic capability of HUVECs, and it could partially reversed the inhibitory effect of NS398. From the findings that inhibition of COX-2 activity resulted in attenuated PGE2 secretion, and exogenous PGE2 enhanced COX-2 expression, we described a positive feedback loop of COX-2-PGE2-COX-2. This was consistent with the over-production of PGE2 observed in COX-1 and COX-2 knockout cell lines, and also supported by previous studies using human synovial fibroblasts, prostatic carcinoma cell line (PC-3) and mouse lung fibroblast cells [70-72]. We speculated that PGE2 may lead to increased cell viability and angiogenic capability of HUVECs through its own action and/or the signal amplification of COX-2.
To sum up, the aforementioned results indicated that COX-2 products produced endogenously may serve to facilitate hypoxia mediated cell proliferation and angiogenic capability, and the involvement of endogenously produced prostanoids in promoting ECs proliferation and angiogenic capability occurs dependently of changes in COX-2 activity, and exogenous PGE2 augments the angiogenic effect of hypoxia on HUVECs. Therefore, the hypoxia-induced functional responses and expression of VEGF and AQP1 in ECs is dependent of PGE2 mediated by COX-2 activation. In conclusion, (i) hypoxia could significantly enhance the angiogenic capability of HUVECs; (ii) COX-2 may be a potential therapeutic target for ischaemic disorder and cancer management, and also the critical factor in tissue remodelling as the COX-2/PGE2 pathway influences most, if not all, of the hallmarks of these events; (iii) inflammation and angiogenesis maybe two respects of one response of HUVECs to hypoxic exposure, and they might have a positive interaction with each other.
Acknowledgments
This work was supported by grants from National Nature Science Foundation of China. (Grant nos. 30470436 and 81030034).
References
- 1.Folkman J, Shing Y. Angiogenesis. J Biol Chem. 1992;267:10931–4. [PubMed] [Google Scholar]
- 2.Bernas GC. Angiotherapeutics from natural products: from bench to clinics? Clin Hemorheol Microcirc. 2003;29:199–203. [PubMed] [Google Scholar]
- 3.Ku SJ, Chang YI, Chae CH, et al. Static tensional forces increase osteogenic gene expression in three-dimensional periodontal ligament cell culture. BMB Rep. 2009;31:427–32. doi: 10.5483/bmbrep.2009.42.7.427. [DOI] [PubMed] [Google Scholar]
- 4.Grellier M, Ferreira-Tojais N, Bourget C. Role of vascular endothelial growth factor in the communication between human osteoprogenitors and endothelial cells. J Cell Biochem. 2009;106:390–8. doi: 10.1002/jcb.22018. [DOI] [PubMed] [Google Scholar]
- 5.Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9:685–93. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- 6.Scarabelli TM, Stephanou A, Pasini E. Different signaling pathways induce apoptosis in endothelial cells and cardiac myocytes during ischemia/reperfusion injury. Circ Res. 2002;90:745–8. doi: 10.1161/01.res.0000015224.07870.9a. [DOI] [PubMed] [Google Scholar]
- 7.Paris S, Denis H, Delaive E. Up-regulation of 94-kDa glucose-regulated protein by hypoxia-inducible factor-1 in human endothelial cells in response to hypoxia. FEBS Lett. 2005;579:105–14. doi: 10.1016/j.febslet.2004.11.055. [DOI] [PubMed] [Google Scholar]
- 8.Sluimer JC, Gasc JM, vanWanroij JL, et al. Hypoxia, hypoxia-inducible transcription factor, and macrophages in human atherosclerotic plaques are correlated with intraplaque angiogenesis. J Am Coll Cardiol. 2008;51:1258–65. doi: 10.1016/j.jacc.2007.12.025. [DOI] [PubMed] [Google Scholar]
- 9.Salim A, Nacamuli RP, Morgan EF. Transient changes in oxygen tension inhibit osteogenic differentiation and Runx2 expression in osteoblasts. J Biol Chem. 2004;279:40007–16. doi: 10.1074/jbc.M403715200. [DOI] [PubMed] [Google Scholar]
- 10.Tandle AT, Calvani M, Uranchimeg B, et al. Endothelial monocyte activating polypeptide-II modulates endothelial cell responses by degrading hypoxia-inducible factor-1alpha through interaction with PSMA7, a component of the proteasome. Exp Cell Res. 2009;315:1850–9. doi: 10.1016/j.yexcr.2009.03.021. [DOI] [PubMed] [Google Scholar]
- 11.Michiels C. Endothelial cell functions. J Cell Physiol. 2003;196:430–43. doi: 10.1002/jcp.10333. [DOI] [PubMed] [Google Scholar]
- 12.Faller DV. Endothelial cell response to hypoxic stress. Clin Exp Pharmacol Physiol. 1999;26:74–84. doi: 10.1046/j.1440-1681.1999.02992.x. [DOI] [PubMed] [Google Scholar]
- 13.Taylor CT. Interdependent roles for hypoxia inducible factor and nuclear factor-kappaB in hypoxic inflammation. J Physiol. 2008;586:4055–9. doi: 10.1113/jphysiol.2008.157669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumours and other ischaemic tissues. Blood. 2004;104:2224–34. doi: 10.1182/blood-2004-03-1109. [DOI] [PubMed] [Google Scholar]
- 15.Kim YS, Kim JS, Kwon JS, et al. BAY 11-7082, a nuclear factor-κB inhibitor, reduces inflammation and apoptosis in a rat cardiac ischemia-reperfusion injury model. Int Heart J. 2010;51:348–53. doi: 10.1536/ihj.51.348. [DOI] [PubMed] [Google Scholar]
- 16.Tzannetou S, Efstratiadis S, Nicolay O, et al. Interleukin-1beta and beta- glucuronidase in gingival crevicular fluid from molars during rapid palatal expansion. Am J Orthod Dentofacial Orthop. 1999;115:686–96. doi: 10.1016/s0889-5406(99)70295-7. [DOI] [PubMed] [Google Scholar]
- 17.Michiels C, Arnould T, Remacle J. Endothelial cell responses to hypoxia: initiation of a cascade of cellular interactions. Biochim Biophys Acta. 2000;1497:1–10. doi: 10.1016/s0167-4889(00)00041-0. [DOI] [PubMed] [Google Scholar]
- 18.Marcus AJ, Weksler BB, Jaffe EA. Enzymatic conversion of prostaglandin endoperoxide H2 and arachidonic acid to prostacyclin by cultured human endothelial cells. J Biol Chem. 1978;253:7138–41. [PubMed] [Google Scholar]
- 19.Camacho M, Lopez-Belmonte J, Vila L. Rate of vasoconstrictor prostanoids released by endothelial cells depends on cyclooxygenase-2 expression and prostaglandin I synthase activity. Circ Res. 1998;83:353–65. doi: 10.1161/01.res.83.4.353. [DOI] [PubMed] [Google Scholar]
- 20.Leahy KM, Koki AT, Masferrer JL. Role of cyclooxygenases in angiogenesis. Curr Med Chem. 2000;7:1163–70. doi: 10.2174/0929867003374336. [DOI] [PubMed] [Google Scholar]
- 21.Salcedo R, Zhang X, Young HA, et al. Angiogenic effects of prostaglandin E2 are mediated by up-regulation of CXCR4 on human microvascular endothelial cells. Blood. 2003;102:1966–77. doi: 10.1182/blood-2002-11-3400. [DOI] [PubMed] [Google Scholar]
- 22.Harada S, Nagy JA, Sullivan KA, et al. Induction of vascular endothelial growth factor expression by prostaglandin E2 and E1 in osteoblasts. J. Clin. Invest. 1994;93:2490–6. doi: 10.1172/JCI117258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ben-Av P, Crofford LJ, Wilder RL, et al. Induction of vascular endothelial growth factor expression in synovial fibroblasts by prostaglandin E and interleukin-1: A potential mechanism of inflammatory angiogenesis. FEBS Lett. 1995;372:83–7. doi: 10.1016/0014-5793(95)00956-a. [DOI] [PubMed] [Google Scholar]
- 24.Cheng T, Cao W, Wen R, et al. Prostaglandin E2 induces vascular endothelial growth factor and basic fibroblast growth factor mRNA expression in cultured rat Muller cells. Invest. Ophthalmol Vis Sci. 1998;39:581–91. [PubMed] [Google Scholar]
- 25.Vincenti V, Cassano C, Rocchi M, et al. Assignment of the vascular endothelial growth factor gene to human chromosome 6p21.3. Circulation. 1996;93:1493–5. doi: 10.1161/01.cir.93.8.1493. [DOI] [PubMed] [Google Scholar]
- 26.Akarasereenont P, Techatraisak K, Thaworn A, et al. The induction of cyclooxygenase-2 by 17 beta-estradiol in endothelial cells is mediated through protein kinase C. Inflamm Res. 2000;49:460–5. doi: 10.1007/s000110050617. [DOI] [PubMed] [Google Scholar]
- 27.Hull MA, Thomsom JL, Hawkey CJ. Expression of cyclooxygenase 1 and 2 by human gastric endothelial cells. Gut. 1999;45:529–36. doi: 10.1136/gut.45.4.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neufeld G, Cohen T, Gengrinovitch S, et al. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999;13:9–22. [PubMed] [Google Scholar]
- 29.Leung DW, Cachianes G, Kuang WJ, et al. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306–9. doi: 10.1126/science.2479986. [DOI] [PubMed] [Google Scholar]
- 30.Dor Y, Porat R, Keshet E. Vascular endothelial growth factor and vascular adjustments to perturbations in oxygen homeostasis. Am J Physiol. 2001;280:C1367–74. doi: 10.1152/ajpcell.2001.280.6.C1367. [DOI] [PubMed] [Google Scholar]
- 31.Semenza GL, Agani F, Feldser D, et al. Hypoxia, HIF-1, and the pathophysiology of common human diseases. Adv Exp Med Biol. 2000;475:123–30. doi: 10.1007/0-306-46825-5_12. [DOI] [PubMed] [Google Scholar]
- 32.Shweiki D, Itin A, Soffer D, et al. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–5. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- 33.Mobasheri A, Airley R, Hewitt SM, et al. Heterogeneous expression of the aquaporin 1 (AQP1) water channel in tumours of the prostate, breast, ovary, colon and lung: a study using high density multiple human tumour tissue microarrays. Int J Oncol. 2005;26:1149–58. [PubMed] [Google Scholar]
- 34.Kentaro K, Kazuo Y, Asami T, et al. Aquaporin 1 is required for hypoxia-inducible angiogenesis in human retinal vascular endothelial cells. Microvasc Res. 2008;75:297–301. doi: 10.1016/j.mvr.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 35.Seo BM, Miura M, Gronthos S, et al. Investigation of multipotent postnatal stem cells from human periodontal ligament. Lancet. 2004;364:149–55. doi: 10.1016/S0140-6736(04)16627-0. [DOI] [PubMed] [Google Scholar]
- 36.Hirakawa S, Hong YK, Harvey N, et al. Identification of vascular lineage specific genes by transcriptional profiling of isolated blood vascular and lymphatic endothelial cells. Am J Pathol. 2003;162:575–86. doi: 10.1016/S0002-9440(10)63851-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gloria1 MA, Cenedeze1 MA, Pacheco-Silva A, et al. The blockade of cyclooxygenases-1 and −2 reduces the effects of hypoxia on endothelial cells. Braz J Med Biol Res. 2006;39:1189–96. doi: 10.1590/s0100-879x2006000900006. [DOI] [PubMed] [Google Scholar]
- 38.Ben-Yosef Y, Miller A, Shapiro S, et al. Hypoxia of endothelial cells leads to MMP-2-dependent survival and death. Am J Physiol Cell Physiol. 2005;289:C1321–31. doi: 10.1152/ajpcell.00079.2005. [DOI] [PubMed] [Google Scholar]
- 39.Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocr. Rev. 1997;18:4–25. doi: 10.1210/edrv.18.1.0287. [DOI] [PubMed] [Google Scholar]
- 40.Gerber HP, Dixit V, Ferrara N. Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J Biol Chem. 1998;273:13313–6. doi: 10.1074/jbc.273.21.13313. [DOI] [PubMed] [Google Scholar]
- 41.Ku DD, Zaleski JK, Liu S, et al. Vascular endothelial growth factor induces EDRF-dependent relaxation in coronary arteries. Am J Physiol. 1993;265:H586–92. doi: 10.1152/ajpheart.1993.265.2.H586. [DOI] [PubMed] [Google Scholar]
- 42.Nielsen S, Smith BL, Christensen EI, et al. Distribution of aquaporin CHIP in secretary and resorptive epithelia and capillary endothelia. Proc Natl Acad Sci USA. 1993;90:7275–29. doi: 10.1073/pnas.90.15.7275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 44.Vacca A, Frigeri A, Ribatti D, et al. Microvessel overexpression of aquaporin 1 parallels bone marrow angiogenesis in patients with active multiple myeloma. Br J Haematol. 2001;113:415–21. doi: 10.1046/j.1365-2141.2001.02738.x. [DOI] [PubMed] [Google Scholar]
- 45.Verkman AS. Aquaporins in endothelia. Kidney Int. 2006;69:1120–3. doi: 10.1038/sj.ki.5000226. [DOI] [PubMed] [Google Scholar]
- 46.Kaneko K, Yagui K, Tanaka A, et al. Aquaporin 1 is required for hypoxia-inducible angiogenesis in human retinal vascular endothelial cells. Microvasc Res. 2008;75:297–301. doi: 10.1016/j.mvr.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 47.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–32. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 48.Bracken CP, Whitelaw ML, Peet DJ. The hypoxia-inducible factors: key transcriptional regulators of hypoxic responses. Cell Mol Life Sci. 2003;60:1376–93. doi: 10.1007/s00018-003-2370-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carroll VA, Ashcroft M. Role of hypoxia-inducible factor (HIF)-1A versus HIF-2A in the regulation of HIF target genes in response to hypoxia, insulin-like growth factor-I, or loss of von Hippel-Lindau function: implications for targeting the HIF pathway. Cancer Res. 2006;66:6264–70. doi: 10.1158/0008-5472.CAN-05-2519. [DOI] [PubMed] [Google Scholar]
- 50.Simon LS. Role and regulation of cyclooxygenase-2 during inflammation. Am J Med. 1999;106:37S–42S. doi: 10.1016/s0002-9343(99)00115-1. [DOI] [PubMed] [Google Scholar]
- 51.Kim SH, Lee SM. Expression of hepatic vascular stress genes following ischemia/reperfusion and subsequent endotoxemia. Arch Pharm Res. 2004;27:769–75. doi: 10.1007/BF02980147. [DOI] [PubMed] [Google Scholar]
- 52.Faller DV. Endothelial cell response to hypoxic stress. Clin Exp Pharmacol Physiol. 1999;26:74–84. doi: 10.1046/j.1440-1681.1999.02992.x. [DOI] [PubMed] [Google Scholar]
- 53.Lo LL, Cheng JJ, Chiu JJ, et al. Endothelial exposure to hypoxia induces Egr-1 expression involving PKC-alpha-mediated Ras/Raf-1/ERK1/2 pathway. J Cell Physiol. 2001;188:304–12. doi: 10.1002/jcp.1124. [DOI] [PubMed] [Google Scholar]
- 54.Chang SH, Liu CH, Conway R, et al. Role of prostaglandin E2-dependent angiogenic switch in cyclooxygenase 2-induced breast cancer progression. Proc Natl Acad Sci USA. 2004;101:591–6. doi: 10.1073/pnas.2535911100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuwano T, Nakao S, Yamamoto H, et al. Cyclooxygenase 2 is a key enzyme for inflammatory cytokine-induced angiogenesis. FASEB J. 2004;18:300–10. doi: 10.1096/fj.03-0473com. [DOI] [PubMed] [Google Scholar]
- 56.Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–64. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 57.Calviello G, Nicuolo DF, Gragnoli S, et al. n-3 PUFAs reduce VEGF expression in human colon cancer cells modulating the COX-2/PGE2 induced ERK-1 and −2 and HIF-1alpha induction pathway. Carcinogenesis. 2004;25:2303–10. doi: 10.1093/carcin/bgh265. [DOI] [PubMed] [Google Scholar]
- 58.Huang SP, Wu MS, Shun CT, et al. Cyclooxygenase-2 increases hypoxia-inducible factor-1 and vascular endothelial growth factor to promote angiogenesis in gastric carcinoma. J Biomed Sci. 2005;12:229–41. doi: 10.1007/s11373-004-8177-5. [DOI] [PubMed] [Google Scholar]
- 59.Casibang MS, Purdom S, Jakowlew L, et al. Prostaglandin E2 and vasoactive intestinal peptide increase vascular endothelial cell growth factor mRNAs in lung cancer cells. Lung Cancer. 2001;31:203–12. doi: 10.1016/s0169-5002(00)00168-9. [DOI] [PubMed] [Google Scholar]
- 60.Pai R, Szabo IL, Soreghan BA, et al. PGE(2) stimulates VEGF expression in endothelial cells via ERK2/JNK1 signaling pathways. Biochem Biophys Res Commun. 2001;286:923–8. doi: 10.1006/bbrc.2001.5494. [DOI] [PubMed] [Google Scholar]
- 61.Hoper MM, Voelkel NF, Bates TO, et al. Prostaglandins induce vascular endothelial growth factor in a human monocytic cell line and rat lungs via cAMP. Am J Respir Cell Mol Biol. 1997;17:748–56. doi: 10.1165/ajrcmb.17.6.2888. [DOI] [PubMed] [Google Scholar]
- 62.Wolff H, Saukkonen K, Anttila S, et al. Expression of cyclooxygenase-2 in human lung carcinoma. Cancer Res. 1998;58:4997–5001. [PubMed] [Google Scholar]
- 63.Eberhart CE, Coffey RJ, Radhika A, et al. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994;107:1183–8. doi: 10.1016/0016-5085(94)90246-1. [DOI] [PubMed] [Google Scholar]
- 64.Tucker ON, Dannenberg AJ, Yang EK, et al. Cyclooxygenase-2 expression is up-regulated in human pancreatic cancer. Cancer Res. 1999;59:987–90. [PubMed] [Google Scholar]
- 65.Alvarez Y, Briones AM, Balfagon G, et al. Hypertension increases the participation of vasoconstrictor prostanoids from cyclooxygenase-2 in phenylephrine responses. J Hypertens. 2005;23:767–77. doi: 10.1097/01.hjh.0000163145.12707.63. [DOI] [PubMed] [Google Scholar]
- 66.Antman EM, DeMets D, Loscalzo J. Cyclooxygenase inhibition and cardiovascular risk. Circulation. 2005;112:759–70. doi: 10.1161/CIRCULATIONAHA.105.568451. [DOI] [PubMed] [Google Scholar]
- 67.Churchman A, Baydoun AR, Hoffman R. Inhibition of angiogenic tubule formation and induction of apoptosis in human endothelial cells by the selective cyclooxygenase-2 inhibitor 5-bromo-2-(4-fluorophenyl)-3-(methylsulfonyl) thiophene (DuP-697) Eur J Pharmacol. 2007;573:176–83. doi: 10.1016/j.ejphar.2007.06.057. [DOI] [PubMed] [Google Scholar]
- 68.Liu XH, Kirschenbaum A, Yao S, et al. Inhibition of cyclooxygenase-2 suppresses angiogenesis and the growth of prostate cancer in vivo. J. Urol. 2000;164:820–5. doi: 10.1097/00005392-200009010-00056. [DOI] [PubMed] [Google Scholar]
- 69.Faour WH, He Y, He QW, et al. Prostaglandin E2 regulates the level and stability of cyclooxygenase-2 mRNA through activation of p38 mitogen-activated protein kinase in interleukin-1b-treated human synovial fibroblasts. J Biol Chem. 2001;276:31720–31. doi: 10.1074/jbc.M104036200. [DOI] [PubMed] [Google Scholar]
- 70.Tjandrawinata RR, Hughes-Fulford M. Up-regulation of cyclooxygenase-2 by product-prostaglandin E2. Adv Exp Med Biol. 1997;407:163–70. doi: 10.1007/978-1-4899-1813-0_25. [DOI] [PubMed] [Google Scholar]
- 71.Kirtikara K, Morham SG, Raghow R, et al. Compensatory prostaglandin E2 biosynthesis in cyclooxygenase 1 or 2 null cells. J Exp Med. 1998;187:517–23. doi: 10.1084/jem.187.4.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Akarasereenont P, Techatrisak K, Chotewuttakorn S, et al. The induction of cyclooxygenase-2 in IL-1beta-treated endothelial cells is inhibited by prostaglandin E2 through cAMP. Mediators Inflamm. 1999;8:287–94. doi: 10.1080/09629359990298. [DOI] [PMC free article] [PubMed] [Google Scholar]