

Abstract
Multiple sclerosis (MS) is characterized by focal destruction of the white matter of the brain and spinal cord. The exact mechanisms underlying the pathophysiology of the disease are unknown. Many studies have shown that MS is predominantly an autoimmune disease with an inflammatory phase followed by a demyelinating phase. Recent studies alongside current treatment strategies, including glatiramer acetate, have revealed a potential role for brain-derived neurotrophic factor (BDNF) in MS. However, the exact role of BDNF is not fully understood. We used the experimental autoimmune encephalomyelitis (EAE) model of MS in adolescent female Lewis rats to identify the role of BDNF in disease progression. Dorsal root ganglia (DRG) and spinal cords were harvested for protein and gene expression analysis every 3 days post-disease induction (pdi) up to 15 days. We show significant increases in BDNF protein and gene expression in the DRG of EAE animals at 12 dpi, which correlates with peak neurological disability. BDNF protein expression in the spinal cord was significantly increased at 12 dpi, and maintained at 15 dpi. However, there was no significant change in mRNA levels. We show evidence for the anterograde transport of BDNF protein from the DRG to the dorsal horn of the spinal cord via the dorsal roots. Increased levels of BDNF within the DRG and spinal cord in EAE may facilitate myelin repair and neuroprotection in the CNS. The anterograde transport of DRG-derived BDNF to the spinal cord may have potential implications in facilitating central myelin repair and neuroprotection.
Keywords: multiple sclerosis, MS, EAE, BDNF, DRG, anterograde transport
Introduction
Multiple sclerosis (MS) is a chronic, neuroinflammatory disease characterized by immune-cell mediated white matter damage in the central nervous system (CNS) [1]. Myelin reactive Th1-cells transmigrate across the blood brain barrier into the CNS. These Th1-cells promote neuroinflammation by the sustained production of cytokines such as interleukins (IL), tumour necrosis factor-α (TNF-α) and interferon-γ (IFN-γ) [2, 3]. The consequent myelin damage results in a variety of disease-induced symptoms including: fatigue, cognitive dysfunction and sensory abnormalities [1, 4].
Current early MS treatments are directed at inflammation and neuroprotection [1, 5]. However, the exact inflammatory mechanisms that underlie the disease are largely unknown. Studies show that changes in neurotrophins and/or their receptors play a critical role in neuroimmune modulation [6, 7]. One candidate neurotrophin in MS-associated inflammation is brain-derived neurotrophic factor (BDNF) [6-9]. BDNF is a potent mitogen for neurons during CNS development [10-12], and an important regulator of neuronal maturation and protection [8, 13]. Studies have shown an association of BDNF production by immune cells and disease activity, in addition higher BDNF serum levels were observed during relapse in contrast to those seen during the stable phase of the disease [9, 16-19]. However, the exact role for BDNF in MS is still unknown.
We have previously shown that the dorsal root ganglia (DRG) is a pivotal reservoir of the inflammatory cytokine TNF-α in the early inflammatory stage of experimental autoimmune encephalomyelitis (EAE) [20]. We hypothesized that BDNF expression is increased within the DRG in the early stages of inflammation associated with the development of EAE, and that this BDNF is anterogradely transported from the DRG to the dorsal horn of the spinal cord.
This study is the first published study showing evidence that BDNF levels are increased in the DRG and spinal cord of rats induced to a state of EAE in accordance with our model of disease induction [20]. Further, we show that BDNF protein is anterogradely transported from the DRG to the dorsal root entry zone, via kinesin-mediated active transport. Our study provides novel information relating to the mechanisms underlying the efficacy of current immunomodulatory therapies used to ameliorate MS symptoms. In addition, we show a previously unrecognized mechanism of BDNF transport into the spinal cord after immune induction.
Materials and methods
Experimental autoimmune encephalomyelitis model
Experimental autoimmune encephalomyelitis (EAE) was induced using MBP, in adolescent female Lewis rats (Charles River, Montreal, Quebec, Canada) as previously described [20]. Adolescent female Lewis rats were randomly assigned to three experimental groups: naïve control (NC), active control (AC) and active EAE (EAE). For each group there were five time points for killing at 3, 6, 9, 12 and 15 days post-induction (dpi). All animal experiments were conducted according to protocols approved by the University of Manitoba Animal Protocol Management and Review Committee, in full compliance with the Canadian Council on Animal Care.
Tissue harvesting and sectioning
For immunohistochemical (IHC) analysis of protein expression, animals were perfusion fixed with 4% paraformaldehyde as previously described [20]. Spinal columns were dissected free of overlying muscle and connective tissue, and decalcified according to previously described protocols [21]. For gene expression analysis, the DRG and spinal cord were harvested as previously described [20]. Tissue was stored in RNAlater stabilization reagent (cat. no. 76106; Qiagen, Washington, DC, USA) until processed. Total RNA, DNA and proteins were isolated as previously described [22].
Western blot
Protein concentration for each sample was assessed using the Bradford protein assay [23]. For each sample, 30 μg total protein was analysed by Western blot as previously described [20]. Anti-BDNF antibody (1:500; R&D systems Inc., Minneapolis, MN, USA) was used to detect BDNF protein. Following incubation with anti-rabbit secondary antibody (1:10,000; Jackson ImmunoResearch, West Grove, PA, USA), immunoreactivity was detected by chemiluminescence (Alpha Innotech, Santa Clara, CA, USA).
Immunohistochemistry
Qualitative immunofluorescent analysis of cryostat sections was conducted to detect the protein expression of BDNF according to previously described methods [20, 22]. Double-labelled immunofluorescence using monoclonal antibodies against the neuronal markers NF-160 (1:40; Invitrogen, Burlington, Ontario, Canada) or NeuN (1:1000; Chemicon, Billerica, MA, USA) were conducted in conjunction with the polyclonal antibody for BDNF [polyclonal chicken anti-BDNF antibody (1:100; R&D systems Inc.)]. This antibody detects the mature form of BDNF, at 14 kD, that acts via the TrkB receptor to exert its biological effect in the tissue being assessed [24]. Brain-derived neurotrophic factor and NeuN were identified using chicken anti-BDNF antibody, and mouse monoclonal NeuN antibody [22]. Secondary antibodies were biotinylated ∝-chicken IgY (1:100; R&D Systems Inc.) and goat anti-mouse FITC (1:50; Jackson ImmunoResearch), Streptavidin-Alexa Fluor 568 (TRITC/Texas red, 1:500; Molecular Probes/Invitrogen, Burlington, Ontario, Canada). The slides were imaged using an Olympus BX51 configured with FV5000 Confocal laser scanning capability; images were captured in Fluoview Version 4.3. Cell diameter measurements and pseudocoloring were performed using Image Pro Express software (Media Cybernetics, Bethesda, MD, USA). Image sizing, black background balancing and final collation for publication were performed using Adobe Creative Suite 2 v9.0.2 (Adobe Systems Inc., San Jose, CA, USA). No image manipulations were performed other than those described.
Reverse transcription polymerase chain reaction (RT-PCR) and real time PCR
Real time RT-PCR was conducted on tissue samples as previously described [20, 22]. The PCR reaction was performed using a Light-Cycler-DNA master SYBR green 1 kit following manufacturers protocols (Roche, Indianapolis, IN, USA). Brain-derived neurotrophic factor primers were: forward: 5′-TTC TTG TGC AGT GCC AGC CTC GTC-3; reverse; 5′-GCC GTT GAA CTT GCC GTG GGT AGA-3′ and annealing temperature at 54°C. Quantitative reverse transcription polymerase chain reaction (qRT-PCR) results were analysed using one-way analysis of variance (ANOVA) followed by Tukey’s post-hoc comparisons. Statistical significance was confirmed using two-tailed Student’s t-test. The model detected differences between main effect day and group values; and the interaction effect day × group values, which were considered significant at P < 0.05. Normality and homogeneity of error variance of dependent variable were tested by using Kolmogorov–Simirnov and Levene’s test.
Enzyme-linked immunosorbent assay
Total protein was extracted from the samples as described above, and total protein concentration assessed using the Bradford assay [23]. The protein concentrations of the samples were adjusted to 30 μg in the sample volume of 100 μl. Sandwich-style ELISA was performed using the BDNF Emax® ImmunoAssay System (Promega, Madison, WI, USA) according to the manufacturer’s instructions [25]. Brain-derived neurotrophic factor content was interpolated from standard curve runs for each plate (linear range of 7.8–500). For a given DRG and spinal cord segment, samples from the groups of AC and EAE and the naïve control rats were determined in a single run. Each sample was assayed in three separate ELISA assays, with three replicates per sample per ELISA.
Statistical analysis
Statistics was performed using GraphPad Prism version 4.03 for Windows, GraphPad Software, San Diego, CA, USA, www.graphpad.com. Statistical analysis was performed using ANOVA with Tukey’s Multiple Comparison post-hoc test. Student’s t-test was used to assess significance of differences between the groups.
Results
Neurological disability scores
All animals in the EAE groups were assessed for neurological disability according to a previously described global neurological disability assessment tool [20]. Prior to day 6 post-EAE induction (6 dpi), none of the animals displayed clinical neurological deficits thereby scoring zero. However, by 9 dpi all animals started to display clinical signs of neurological disability (mean 0.57 ± 0.45). Neurological disability progressively worsened upon daily assessment until 12 dpi (peak disability; mean 6.42 ± 5.35), then subsided by 15 dpi (mean 1.5 ± 1.41) as the animals entered the remission phase of disease induction, well characterized for this animal model [26] (Fig. 1).
Fig 1.
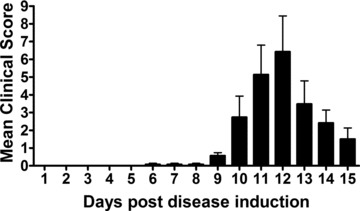
Neurological disability score for EAE animals induced to a state of MS. Disability scores range from a mean clinical disability score of 0 (no disability) to 15 (maximum disability). The bell shaped distribution outlining peak neurological disability in response to EAE induction occurred at day 12 post-EAE induction. Neurological disability is scored according to the following criteria: Tail: 0 = normal; 1 = partially paralysed, weakness; 2 = completely paralysed, limp. Bladder: 0 = normal; 1 = incontinence. Right hind limb: 0 = normal; 1 = weakness; 2 = dragging with partial paralysis; 3 = complete paralysis. Left hind limb: 0 = normal; 1 = weakness; 2 = dragging with partial paralysis; 3 = complete paralysis. Right forelimb: 0 = normal; 1 = weakness; 2 = dragging, not able to support weight; 3 = complete paralysis. Left forelimb: 0 = normal; 1 = weakness; 2 = dragging, not able to support weight; 3 = complete paralysis.
Immunohistochemical analysis of BDNF protein expression in DRG
Comparative IHC analysis of EAE versus AC animals killed at 6, 9, 12 and 15 dpi, revealed markedly increased BDNF immunoreactivity in the EAE animals at 6 dpi through 15 dpi relative to that seen in the AC group at the same experimental time points and/or the NC group (Fig. 2A). Judging by easily detectable sensory neuron morphological criteria, and NeuN double labelling, the increased signal intensity for cytoplasmic BDNF appears to be localized to the sensory neuron population of the DRG. In addition, the satellite cells within the DRG were immunoreactive for BDNF. The overall increase in BDNF from the collaborative effects of satellite cells and neuronal cells appears to reach peak expression around 12 dpi (Fig. 2A). These results suggest that the transient neuronal expression of BDNF in the DRG peaks at day 12 and subsides by day 15 post-induction.
Fig 2.
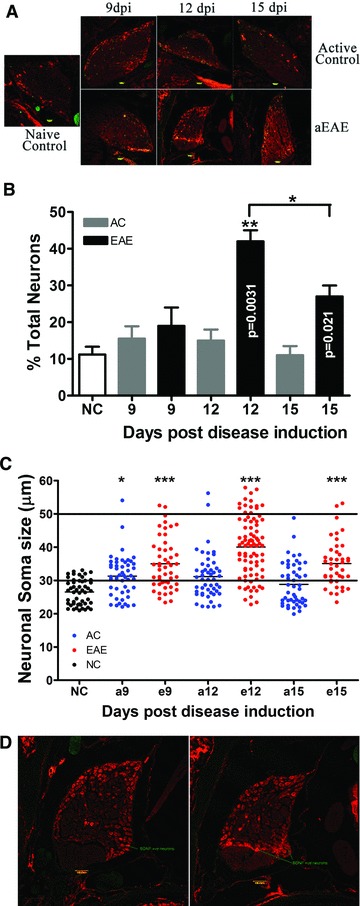
(A) BDNF immunoreactivity in rat DRG. Images shown depict BDNF label (red) co-localized with NeuN (green) in active EAE, active control and naïve animals. 10 μm sections of DRG were stained with BDNF (R&D Systems Inc.). Marked elevations in BDNF labelling were noted in the active EAE animal group at day 12 relative to all other treatment groups. Images were taken at a total magnification of 10× and were exposed for 810 msec. Bar = 50 μm; Dpi = days post-induction of EAE. (B) Percentage of BDNF positive (+) sensory DRG neurons compared to total neurons found within some groups at the various experimental time points. Significantly increased proportions of BDNF+ sensory neurons were identified in the active EAE group which peaks at 12 dpi. (**P < 0.005; *P < 0.05, ANOVA followed by Student’s t-test). (C) Analysis of size of the BDNF positive neurons in DRG. Analysis of the size of BDNF+ neurons obtained from the active EAE group reveals a change in the cell size distribution from small to predominantly medium diameter (<30 μm) sensory neurons which corresponds to C and Aγ fibres, respectively. (*P < 0.05, ***P < 0.001, ANOVA with Tukey’s post test, Student’s t-test was used to assess differences between the means). NC: naïve control; AC: active control; 9, 12 and 15 stand for rat killed date. (D) Comparison of BDNF positive neurons in different area of DRG. Analysis of the size and location of BDNF+ neurons obtained from the active EAE group reveals there are more BDNF+ neurons with lager size in different area from NC group.
A double blind analysis of the EAE group relative to the two control groups showed that the percentage of DRG neurons identified as BDNF positive significantly increased from 9 dpi (19.0% ± 5.0%) to a relative peak 12 dpi (42% ± 3.0%; P = 0.0169), and decreased again at 15 dpi (27.0% ± 3.0%) (Fig. 2B). However, the same increase in neuronal expression of BDNF was not seen in either of the control groups. In NC DRG only 11.2% ± 2.1% of neurons were identified as BDNF positive. In AC controls the percentage of BDNF positive cells did not significantly change between days 9, 12 and 15 post-induction (15.5 ± 3.4, 15.0 ± 3.0, 11.0 ± 2.5, respectively). EAE DRG had significantly more BDNF positive neurons compared to AC DRG at both 12 and 15 dpi (P = 0.0031 and 0.0149, respectively).
A sub-analysis of the BDNF positive neurons identified significant changes in cell sizes across the three groups (Fig. 2C). The mean cell size of BDNF positive neurons in AC9 DRG is significantly larger than in NC DRG (31.93 ± 5.50 μm compared to 26.55 ± 3.82 μm; P < 0.0001). There is no significant difference in mean cell sizes between any of the AC DRG at 9, 12 or 15 dpi (31.93 ± 5.50, 31.46 ± 6.65 and 28.83 ± 6.47 μm, respectively). The cell sizes are significantly larger in EAE DRG compared to AC DRG at all time points assayed (34.97 ± 7.79, 40.05 ± 8.96 and 35.09 ± 7.01 μm at 9, 12 and 15 dpi, respectively). Thus, by 9 dpi, the mean BDNF positive cell size is significantly increased in EAE animals compared to AC animals (P = 0.0091). At 12 dpi, the mean cell sizes are highly significantly different between EAE and AC animals (P = 0.0001). At 15 dpi, the mean BDNF positive cell sizes are also highly significantly different (P = 0.0001). In order to assess potential changes in cell type expressing BDNF, we compared the location of BDNF positive cells in NC DRG to those in the DRG of EAE 12. We found in EAE 12 DRG that BDNF was expressed by small to medium DRG neurons in similar stereological locations to the BDNF positive cells in the NC DRG. In addition, we also saw BDNF expression in medium to large size neurons located in different regions from those seen in the NC DRG (Fig. 2D). Our results show that there are more BDNF expressing medium size neurons in the EAE DRG than in the NC group.
Thus, these data reveal a change in the cell type expressing BDNF, from predominantly small neurons (<30 μm), to predominantly medium diameter sensory neurons (30–50 μm). This corresponds to C and Aγ fibres, respectively.
Enzyme-linked immunosorbent assay analysis of BDNF protein expression in the lumbar DRG
The values for the BDNF expression in the lumbar DRG are presented as the absolute quantity of BDNF per μg of total protein. Levels in NC DRG were 0.599 ± 0.044 pg/μg total protein. AC DRG levels of BDNF were significantly higher (P = 0.0004) at 0.915 ± 0.012, 0.951 ± 0.089 and 1.014 ± 0.023 pg/μg total protein for 9, 12 and 15 dpi, respectively. BDNF levels in day 9 EAE DRG were not significantly different from AC 9 dpi at 1.007 ± 0.053 pg/μg total protein, nor were BDNF levels in day 15 EAE DRG different from AC 15 dpi (1.073 ± 0.022 pg/μg total protein). However, at 12 dpi in the EAE DRG, BDNF levels were significantly increased compared to AC day 12 DRG, at 1.371 ± 0.039 pg/μg total protein compared to 0.951 ± 0.089 pg/μg total protein (P = 0.0049), see Figure 3A.
Fig 3.
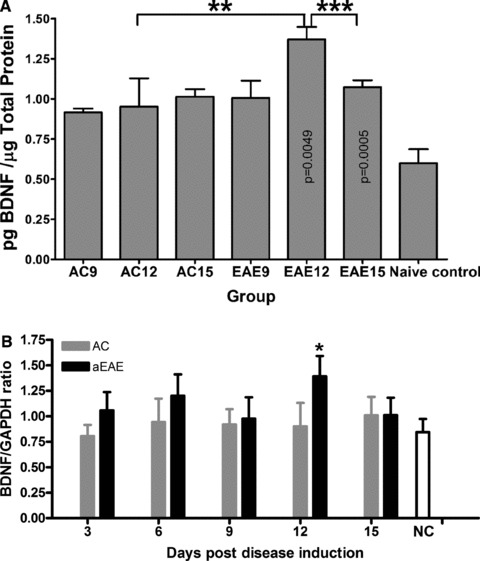
(A) ELISA quantification of BDNF expression in the DRG. BDNF expression in the lumbar DRG was quantified using ELISA. Rats induced to a state of EAE show significantly increased BDNF expression compared to NC animals (**P < 0.005; ***P < 0.001, ANOVA followed by Student’s t-test). Results are shown as pg/ml BDNF/μg total protein. (B) Real-time PCR results of BDNF expression within DRG. The BDNF mRNA expression of DRG for animals of the active EAE group (red bars), killed at day 12 is significantly higher than other groups (naïve control (blue bar) and active control (green bars)) (*P = 0.049) with a one way ANOVA and Tukey’s post test based on two factors (day and group) using software SPSS 16.0.
BDNF gene expression analysis in the DRG
Real time reverse transcription polymerase chain reaction analysis was conducted on DRG from all lumbar, thoracic and cervical regions of the spinal column from the three experimental groups, at the pre-determined experimental time points. The BDNF mRNA expression was assessed in parallel with that of the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The mRNA expression for BDNF within the DRG obtained from the three experimental groups revealed peak expression in the EAE group at 12 dpi compared to all other experimental groups (Fig. 3B). Specifically, mRNA in EAE 12 dpi is significantly higher than other groups (NC and AC) (P = 0.0493).
BDNF protein and gene expression in the spinal cord
Immunohistochemical analysis for BDNF protein expression in the spinal cord reveals marked increases in immunoreactivity in the dorsal horn of the active EAE spinal cord at 12 dpi, compared to AC and NC animals (Fig. 4A). The increased levels of BDNF immunoreactivity in AC animals relative to the NC is expected as there is a significant immune response resulting from the Freund’s adjuvant, MT and PT injections.
Fig 4.
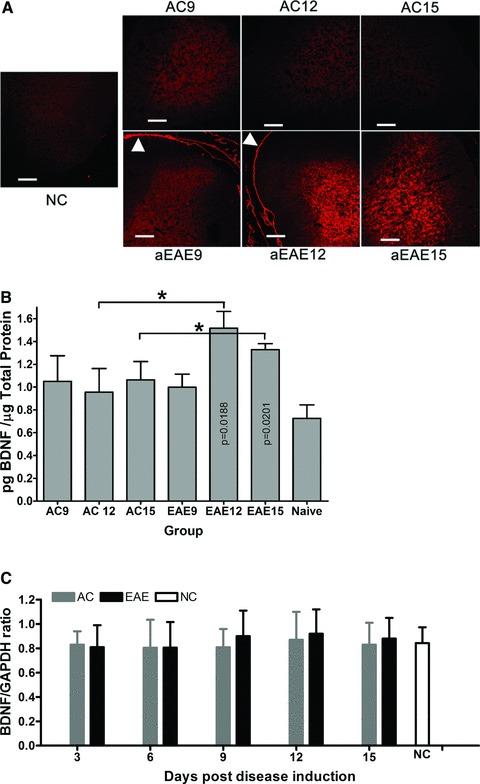
(A) BDNF immunoreactivity in spinal cord. Comparative IHC analysis of 10 μm sections of spinal cord showed markedly increased BDNF immunoreactivity at active EAE 12 dpi relative to active control. Active control spinal cord also shows increased BDNF immunoreactivity at 12 dpi, however, by 15 dpi, immunoreactivity is markedly reduced compared to active EAE spinal cord. Reticulocyte staining shows clearly in the dura of the active EAE spinal cord (arrow head). All images were taken at a total magnification of 100× and were exposed for 764 msec. with BDNF positive cells brightly labelled with red. Bar = 100 μm. (B) ELISA quantification of BDNF expression in the spinal cord. BDNF expression in the lumbar DRG was quantified using ELISA. Rats induced to a state of EAE show significantly increased BDNF expression compared to NC animals (*P < 0.05). Results are shown as pg/ml BDNF/μg total protein. (C) Real-time PCR results of BDNF expression within spinal cord. The BDNF mRNA expression of spinal cord for animals of the active EAE group (grey bars), at 12 dpi, is higher than other groups with an increasing trend (naïve control (white bar) and active control (black bars)) (*P > 0.05) with a one way ANOVA and Tukey’s post test based on two factors (day and group) using software SPSS 16.0.
ELISA analysis of BDNF expression was conducted on spinal cord of animals at 9, 12 and 15 dpi. NC spinal cord contained 0.63 ± 0.06 pg/μg total protein. AC rats at 9, 12 and 15 dpi showed a significantly increased level of BDNF compared to NC at 0.92 ± 0.012, 0.97 ± 0.056, 1.01 ± 0.023 pg/μg total protein, respectively (P > 0.01) (Fig. 4B). Experimental autoimmune encephalomyelitis animals showed a significant increase (P = 0.0195) in BDNF expression in the caudal spinal cord at 12 dpi at 1.41 ± 0.016 pg/μg total protein compared to days 9 and 12 at 0.01 ± 0.05 and 1.07 ± 0.02 pg/μg total protein, respectively (Fig. 4B).
The qRT-PCR analysis of gene expression changes revealed no significant differences in BDNF mRNA in the spinal cord of any of the groups across all time points studied (Fig. 4C).
BDNF protein expression in the dorsal roots
Based on our model of MS induction [20], we hypothesized that BDNF expression is induced in the DRG and translocated to the dorsal horn along the dorsal roots. To test our hypothesis we assessed the presence of BDNF in the dorsal root in the EAE animals. We show markedly increased BDNF immunoreactivity in the dorsal roots of active EAE animals compared to NC (Fig. 4A). Several previous studies have shown that BDNF is anterogradely transported in vesicles, from the neuronal cell body along the axon to the synapse [27, 28]. Vesicles are moved around in the cell along the cytoskeleton, controlled by motor proteins, the kinesin and dynein protein families [29]. Kinesin has been shown to regulate the anterograde transport of BDNF from the neuronal cell body to the synapse [30, 31]. We demonstrate anterograde transport of BDNF along the dorsal roots, using co-localization of BDNF with the motor protein kinesin [30]. Kinesin-BDNF double immunohistochemistry showed co-localization of the two proteins throughout the dorsal root, and dorsal root entry zone which indicates that BDNF is anterogradely transported to dorsal horn from DRG (Fig. 5B).
Fig 5.
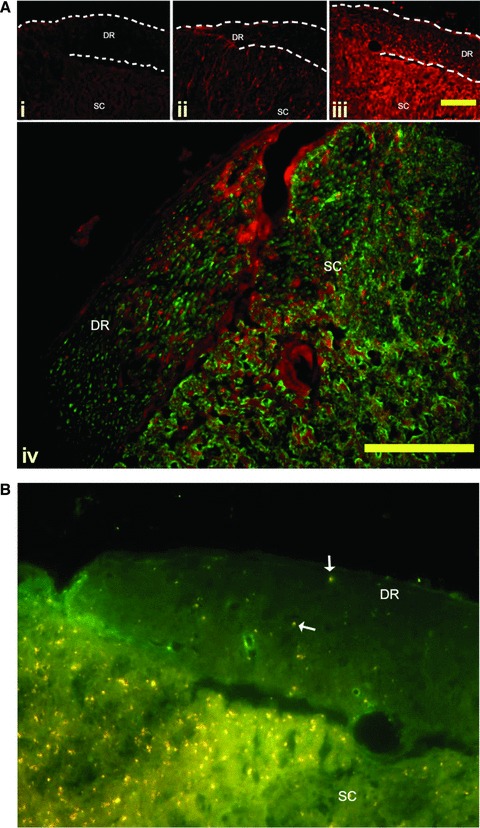
(A) BDNF immunoreactivity in the dorsal roots. Representative sections of dorsal root entry zones from the lumbar spinal cord of Lewis rats induced to a state of EAE. A marked increase in BDNF immunoreactivity (red) is seen in the dorsal roots of active EAE compared to active control animals and NC animals. Panel i—Naïve animals have no BDNF immunoreactivity in the dorsal roots (DR). Panel ii—Active control animals have noticeable BDNF immunoreactivity in the satellite cells of the DR, and in the grey matter of the dorsal horn. Panel iii—Active EAE animals appear to have increased levels of BDNF immunoreactivity in the satellite cells of the DR, as identified as small brightly stained cell surrounding the axons of the root. In addition, there is a marked increase in punctuate staining in the length of the roots. Further, the grey matter of the dorsal horn has a markedly increased immunoreactivity compared to the active control animals. DR = Dorsal root; SC = spinal cord. Bar = 10 μm. Panel iv—BDNF expression (red) is associated with neurons (labelled with Neuron-specific class III beta-tubulin (Tuj1) – green) in both the DR, and the dorsal horn. Bar = 10 μm (B) Anterograde transport of BDNF along the dorsal root is mediated by kinesin. Double immunolabelling of frozen sections of EAE spinal cord shows BDNF (red) co-localized to the motor protein kinesin (green) (arrow). Kinesin is distributed throughout the dorsal root (DR) and spinal cord (SC). BDNF immunoreactivity is markedly higher in the dorsal root entry zone than the root. Co-labelling shows as yellow. Bar = 10 μm.
Discussion
The current study was designed to test our hypothesis that BDNF expression is up-regulated in the DRG and spinal cord in the EAE model of MS. We used MBP to induce EAE in Lewis rat as our research was focussed on the early antigenic induction of inflammation rather than the chronic demyelination aspects of the disease [32]. In this study we show a direct correlation between antigenic immune activation and increased expression of BDNF, at both the gene and protein level within DRG, which peaks at 12 dpi in correlation with peak neurological disability [20]. Further, we show peak BDNF levels in spinal cord at day 12, with maintenance of increased levels at day 15, in contrast to DRG levels which return to normal by day 15. Our results indicate that medium sized sensory neurons of the DRG represent the principle source of BDNF production, and that this BDNF is anterogradely transported from DRG along the roots to the dorsal horn.
The death of oligodendrocytes and subsequent demyelination of the brain and spinal cord, which characterize MS, occur as a direct result of T-cell activation [3]. We have previously shown activation of DRG neurons in the inflammatory stage of EAE, with increased expression of the pro-inflammatory cytokine TNF-α [20]. This lead to the hypothesis that DRG neurons are a primary source of inflammatory mediators in the very early stages of neuroinflammatory disorders such as MS [20]. Although the exact molecular events underlying the pathophysiology of MS remain unclear, immunomodulatory therapies have proven effective for some MS patients [5]. One such therapy is GA [33]. Although the mechanism of action of GA is unknown, studies have shown that GA-active T-cells produce a significantly increased level of BDNF, which is directly neuroprotective and/or neuroregenerative [19, 34]. Although GA-specific Th1, Th2 and Th0 cells are all involved in BDNF production, larger in vitro studies have suggested that Th2 cells play a predominant role in GA modulation of RRMS [19, 35]. Further evidence of a role for BDNF in MS comes from a study showing that BDNF treatment has a beneficial effect on disease progression in EAE [36, 37]. Thus, BDNF may play a significant role in the induction and progression of neuronal and oligodendrocyte damage. Our study provides evidence that BDNF expression is up-regulated in the early inflammatory stage of neuroinflammation. In addition, we show that BDNF levels are maintained in the spinal cord after amelioration of neurological symptoms, suggesting a role for BDNF in the prevention of immediate cell damage resulting from the initial inflammation. Further longitudinal studies are required to clarify the role of BDNF in neuroprotection in the later, demyelinating stages of disease progression.
In order to characterize the specific cellular source of the BDNF protein, we used cell soma size to identify the sensory neuron subtypes present in the DRG. Our results show that medium diameter (30–50 μm: Aγ) sensory neurons appear to be the predominant source of BDNF in the EAE DRG. Further, it appears that the specific location of the BDNF positive cells, indicates that the cellular source of BDNF in the EAE DRG is changing from the small C fibres, to the medium Aγ fibres. Based on Aoki’s paper in 2004, the neurons on the outer edge of the DRG are the small NGF dependent (substance P and calcitonin gene related pepride expressing) nociceptive neurons, which are critical for inflammatory hyperalgesia [38]. This cellular source differs from the source of TNF-α in the EAE DRG, which is predominantly produced by small diameter (<30 μm: C) neurons [20]. Interestingly, BDNF enhances the excitability of small diameter neurons, and potentiates their action potential firing, via p75NTR signalling [39]. A study using complete Freund’s adjuvant injection into the rat paw to initiate a pain response showed TrkB, the high affinity BDNF receptor, expression in medium to large sensory neurons of the lumbar DRG, and expression of the low affinity BDNF receptor, p75NTR in the small diameter neurons [39]. Further, microglia activated by peripheral nerve injury secrete high levels of BDNF which subsequently results in the development of neuropathic pain [40]. Further studies are required to identify the BDNF responsive cells in the DRG of the EAE model.
Neurotrophins and cytokines interact to co-regulate their expression in inflammatory states [41, 42]. For example, TNF-α induces the expression of BDNF expression in astrocytes and neurons [41, 42]. Based on our established model [20], we hypothesize that BDNF works in concert with cytokines such as TNF-α and other neurotrophins, such as NGF, to regulate cellular effects on myelin [41, 43]. The current study, in conjunction with our previously published studies [20, 22], provides support for the importance of cytokine–neurotrophin interactions in the induction of neuroinflammatory disorders.
Anterograde and retrograde transport of neurotrophins are known to occur between the DRG and spinal cord [44]. We show evidence for the active transport of BDNF from the DRG to the dorsal root entry zone, via kinesin-mediated anterograde transport. Kinesin is a dimeric molecule that attaches to protein filled vesicles, and walks towards the plus end of a microtubule, transporting the proteins to the synapse. This form of transport is known as anterograde transport. Vesicular transport is the fastest mechanism of transporting proteins at 50–400 mm/day compared to the slower transport of proteins, at less than 8 mm/day [29]. This provides a plausible explanation for the increase in BDNF protein seen in the dorsal horn, even though levels of BDNF mRNA are not increased. This corroborates previous studies showing anterograde transport of BDNF along microtubules via transport vesicles [45]. Our results are also consistent with other studies that have shown transport of TNF-α and BDNF in rodent models of nerve injury [22, 44].
As BDNF is known to affect myelin function [46], it is possible that DRG-derived BDNF contributes to the recovery of the inflammatory damage to myelin that has been shown in previous studies [6]. This potential mechanism of disease induction expands the potential for targeted strategies aimed at attenuating white matter disorders such as MS. One caveat to this therapeutic strategy is the potential for the development of pain, as previous studies have shown that elevated BDNF in the dorsal horn correlates with the development of pain [47]. In addition, we have shown the induction of pain in this model of neuroinflammation, associated with the peak neurological disability (MacNeil, Begum et al., unpublished observations).
Summary
Our study offers new insights into the role of the DRG-derived BDNF in the inflammatory response preceding myelin damage in the early stages of MS. We demonstrate significantly increased BDNF gene and protein levels in the DRG correlating with peak neurological disability. In addition, we show increased BDNF protein levels in the dorsal horn without an increase in mRNA levels, which suggests that BDNF is transported into, rather than synthesized in, the dorsal horn. We show that medium diameter DRG sensory neurons are an important source of BDNF, which is anterogradely transported to the spinal cord during the early stage of autoimmune-induced neuroinflammation. Further, our findings provide support for the immune activation of the DRG as a critical step in the development of myelin disorders of the CNS. This is the first study to identify changes in BDNF expression and transport in the early inflammatory stages of neuroimmune induction.
Acknowledgments
We would like to thank Elizabeth Tran, Samantha Kendall, Jeremy Paterson, Joumana Mustapha and Kim Madec for technical assistance. We would also like to thank our funding & supporting agencies: Biogen Idec Canada Inc (M.N. and E.E.F.), Manitoba Medical Services Foundation (E.E.F. and M.N.), Manitoba Institute of Child Health (M.N. and E.E.F.), Manitoba Health Research Council (E.E.F.), Pfizer UK Global (M.N. and E.E.F.), and Purdue Pharma (M.N. and E.E.F.). W.Z. was the recipient of a Canada Millennium Scholarship Foundation award. P.V. and F.B. were recipients of MHRC Studentships. W.Z., F.B., B.M. P.P. and P.V. performed the research. W.Z., B.M. and E.E.F. analysed the data. E.E.F. wrote the paper. M.N. and E.E.F. designed the research study.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Namaka MP, Ethans K, Jensen B, et al. Therapeutic choices/e-therapeutic. Ottawa: Canadian Pharmacists Association Publishers, Inc; 2011. Multiple sclerosis. pp. 309–19. [Google Scholar]
- 2.Bar-Or A. The immunology of multiple sclerosis. Semin Neurol. 2008;28:29–45. doi: 10.1055/s-2007-1019124. [DOI] [PubMed] [Google Scholar]
- 3.Codarri L, Fontana A, Becher B. Cytokine networks in multiple sclerosis: lost in translation. Curr Opin Neurol. 2010;23:205–11. doi: 10.1097/WCO.0b013e3283391feb. [DOI] [PubMed] [Google Scholar]
- 4.Zhu W, Le Dorze JA, Prout M, et al. An overview of relapsing remitting multiple sclerosis (RRMS) and current treatment options. Pharmacy Pract. 2010;9:CE1–7. [Google Scholar]
- 5.Namaka M, Leong C, Prout M, et al. Emerging therapies for the management of multiple sclerosis. Clin Med Insights: Therap. 2010;2:1–13. [Google Scholar]
- 6.De Santi L, Annunziata P, Sessa E, et al. Brain-derived neurotrophic factor and TrkB receptor in experimental autoimmune encephalomyelitis and multiple sclerosis. J Neurol Sci. 2009;287:17–26. doi: 10.1016/j.jns.2009.08.057. [DOI] [PubMed] [Google Scholar]
- 7.Linker R, Gold R, Luhder F. Function of neurotrophic factors beyond the nervous system: inflammation and autoimmune demyelination. Crit Rev Immunol. 2009;29:43–68. doi: 10.1615/critrevimmunol.v29.i1.20. [DOI] [PubMed] [Google Scholar]
- 8.Azoulay D, Urshansky N, Karni A. Low and dysregulated BDNF secretion from immune cells of MS patients is related to reduced neuroprotection. J Neuroimmunol. 2008;195:186–93. doi: 10.1016/j.jneuroim.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 9.Azoulay D, Vachapova V, Shihman B, et al. Lower brain-derived neurotrophic factor in serum of relapsing remitting MS: reversal by glatiramer acetate. J Neuroimmunol. 2005;167:215–8. doi: 10.1016/j.jneuroim.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 10.Leibrock J, Lottspeich F, Hohn A, et al. Molecular cloning and expression of brain-derived neurotrophic factor. Nature. 1989;341:149–52. doi: 10.1038/341149a0. [DOI] [PubMed] [Google Scholar]
- 11.Djalali S, Holtje M, Grosse G, et al. Effects of brain-derived neurotrophic factor (BDNF) on glial cells and serotonergic neurones during development. J Neurochem. 2005;92:616–27. doi: 10.1111/j.1471-4159.2004.02911.x. [DOI] [PubMed] [Google Scholar]
- 12.Lykissas MG, Batistatou AK, Charalabopoulos KA, et al. The role of neurotrophins in axonal growth, guidance, and regeneration. Curr Neurovasc Res. 2007;4:143–51. doi: 10.2174/156720207780637216. [DOI] [PubMed] [Google Scholar]
- 13.Cunha C, Brambilla R, Thomas KL. A simple role for BDNF in learning and memory? Front Mol Neurosci. 2010;3:1–14. doi: 10.3389/neuro.02.001.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nishimura K, Nakamura K, Anitha A, et al. Genetic analyses of the brain-derived neurotrophic factor (BDNF) gene in autism. Biochem Biophys Res Commun. 2007;356:200–6. doi: 10.1016/j.bbrc.2007.02.135. [DOI] [PubMed] [Google Scholar]
- 15.Stadelmann C, Kerschenstainer M, Misgeld T, et al. BDNF and gp145trkB in multiple sclerosis brain lesions. Brain. 2002;125:75–85. doi: 10.1093/brain/awf015. [DOI] [PubMed] [Google Scholar]
- 16.Brandes DW. The role of glatiramer acetate in the early treatment of multiple sclerosis. Neuropsychiatr Dis Treat. 2010;6:329–36. doi: 10.2147/ndt.s5898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kala M, Miravalle A, Vollmer T. Recent insights into the mechanism of action of glatiramer acetate. J Neuroimmunol. 2011;235:9–17. doi: 10.1016/j.jneuroim.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 18.Arnon R, Aharoni R. Neuroprotection and neurogeneration in MS and its animal model EAE effected by glatiramer acetate. J Neural Transm. 2009;116:1443–49. doi: 10.1007/s00702-009-0272-3. [DOI] [PubMed] [Google Scholar]
- 19.Ziemssen T, Kumpfel T, Klinkert WE, et al. Glatiramer acetate-specific T-helper 1- and 2-type cell lines produce BDNF: implications for multiple sclerosis therapy. Brain-derived neurotrophic factor. Brain. 2002;125:2381–91. doi: 10.1093/brain/awf252. [DOI] [PubMed] [Google Scholar]
- 20.Melanson M, Miao P, Eisenstat D, et al. Experimental autoimmune encephalomyelitis-induced upregulation of tumour necrosis factor-alpha in the dorsal root ganglia. Mult Scler. 2009;15:1135–45. doi: 10.1177/1352458509106856. [DOI] [PubMed] [Google Scholar]
- 21.Begum F, Zhu W, Namaka MP, et al. A novel decalcification method for adult rodent bone for histological analysis of peripheral-central nervous system connections. J. Neurosci. Methods. 2010;187:59–66. doi: 10.1016/j.jneumeth.2009.12.013. [DOI] [PubMed] [Google Scholar]
- 22.Miao P, Madec K, Gong Y, et al. Axotomy-induced up-regulation of tumour necrosis factor-alpha in the dorsal root ganglia. Neurol Res. 2008;30:623–31. doi: 10.1179/174313208X289606. [DOI] [PubMed] [Google Scholar]
- 23.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 24.Chadwick W, Magnus T, Martin B, et al. Targeting TNF-alpha receptors for neurotherapeutics. Trends Neurosci. 2008;31:504–11. doi: 10.1016/j.tins.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grimm JW, Lu L, Hayashi T, et al. Time-dependent increases in brain-derived neurotrophic factor protein levels within the mesolimbic dopamine system after withdrawal from cocaine: implications for incubation of cocaine craving. J Neurosci. 2003;23:742–47. doi: 10.1523/JNEUROSCI.23-03-00742.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mix E, Meyer-Rienecker H, Zettl U. Animal models of multiple sclerosis for the development and validation of novel therapies-potential and limitations. J. Neurol. 2008;255:7–14. doi: 10.1007/s00415-008-6003-0. [DOI] [PubMed] [Google Scholar]
- 27.von Bartheld CS, Wang X, Butowt R. Anterograde axonal transport, transcytosis, and recycling of neurotrophic factors: the concept of trophic currencies in neural networks. Mol Neurobiol. 2001;24:1–28. doi: 10.1385/MN:24:1-3:001. [DOI] [PubMed] [Google Scholar]
- 28.Gauthier LR, Charrin BC, Borrell-Pages M, et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–38. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 29.Caviston JP, Holzbaur EL. Microtubule motors at the intersection of trafficking and transport. Trends Cell Biol. 2006;16:530–7. doi: 10.1016/j.tcb.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 30.Miki H, Okada Y, Hirokawa N. Analysis of the kinesin superfamily: insights into structure and function. Trends Cell Biol. 2005;15:467–76. doi: 10.1016/j.tcb.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 31.Park JJ, Cawley NX, Loh YP. A bi-directional carboxypeptidase E-driven transport mechanism controls BDNF vesicle homeostasis in hippocampal neurons. Mol Cell Neurosci. 2008;39:63–73. doi: 10.1016/j.mcn.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mannie M, Swanborg RH, Stepaniak JA. Experimental autoimmune encephalomyelitis in the rat. Curr Protoc Immunol. 2009;15:15.2. doi: 10.1002/0471142735.im1502s85. [DOI] [PubMed] [Google Scholar]
- 33.Skihar V, Silva C, Chojnacki A, et al. Promoting oligodendrogenesis and myelin repair using the multiple sclerosis medication glatiramer acetate. Proc Natl Acad Sci USA. 2009;106:17992–7. doi: 10.1073/pnas.0909607106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lalive PH, Neuhaus O, Benkhoucha M, et al. Glatiramer acetate in the treatment of multiple sclerosis: emerging concepts regarding its mechanism of action. CNS Drugs. 2011;25:401–14. doi: 10.2165/11588120-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.La Mantia L, Munari LM, Lovati R. Glatiramer acetate for multiple sclerosis. Cochrane Database Syst Rev. 2010;5:CD004678. doi: 10.1002/14651858.CD004678.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Makar TK, Bever CT, Singh IS, et al. Brain-derived neurotrophic factor gene delivery in an animal model of multiple sclerosis using bone marrow stem cells as a vehicle. J Neuroimmunol. 2009;210:40–51. doi: 10.1016/j.jneuroim.2009.02.017. [DOI] [PubMed] [Google Scholar]
- 37.Makar TK, Trisler D, Eglitis MA, et al. Brain-derived neurotrophic factor (BDNF) gene delivery into the CNS using bone marrow cells as vehicles in mice. Neurosci Lett. 2004;356:215–9. doi: 10.1016/j.neulet.2003.11.045. [DOI] [PubMed] [Google Scholar]
- 38.Woolf CJ, Allchorne A, Safieh-Garabedian B, et al. Cytokines, nerve growth factor and inflammatory hyperalgesia: the contribution of tumour necrosis factor alpha. Br J Pharmacol. 1997;121:417–24. doi: 10.1038/sj.bjp.0701148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang YH, Chi XX, Nicol GD. Brain-derived neurotrophic factor enhances the excitability of rat sensory neurons through activation of the p75 neurotrophin receptor and the sphingomyelin pathway. J Physiol. 2008;586:3113–27. doi: 10.1113/jphysiol.2008.152439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coull JAM, Beggs S, Boudreau D, et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–21. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- 41.Aloe L, Properzi F, Probert L, et al. Learning abilities, NGF and BDNF brain levels in two lines of TNF-alpha transgenic mice, one characterized by neurological disorders, the other phenotypically normal. Brain Res. 1999;840:125–37. doi: 10.1016/s0006-8993(99)01748-5. [DOI] [PubMed] [Google Scholar]
- 42.Saha R, Liu X, Pahan K. Up-regulation of BDNF in astrocytes by TNF-α: A case for the neuroprotective role of cytokine. J Neuroimmun. Pharmacol. 2006;1:212–22. doi: 10.1007/s11481-006-9020-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takei Y, Laskey R. Tumour necrosis factor alpha regulates responses to nerve growth factor, promoting neural cell survival but suppressing differentiation of neuroblastoma cells. Mol Biol Cell. 2008;19:855–64. doi: 10.1091/mbc.E07-06-0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zweifel LS, Kuruvilla R, Ginty DD. Functions and mechanisms of retrograde neurotrophin signalling. Nat Rev Neurosci. 2005;6:615–25. doi: 10.1038/nrn1727. [DOI] [PubMed] [Google Scholar]
- 45.Altar CA, DiStefano PS. Neurotrophin trafficking by anterograde transport. Trends Neurosci. 1998;21:433–7. doi: 10.1016/s0166-2236(98)01273-9. [DOI] [PubMed] [Google Scholar]
- 46.McTigue DM, Horner PJ, Stokes BT, et al. Neurotrophin-3 and brain-derived neurotrophic factor induce oligodendrocyte proliferation and myelination of regenerating axons in the contused adult rat spinal cord. J Neurosci. 1998;18:5354–65. doi: 10.1523/JNEUROSCI.18-14-05354.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Merighi A, Salio C, Ghirri A. BDNF as a pain modulator. Prog Neurobiol. 2008;85:297–317. doi: 10.1016/j.pneurobio.2008.04.004. [DOI] [PubMed] [Google Scholar]