

Abstract
Four and a half LIM domain protein 1 (FHL1) is the founding member of the FHL family of proteins characterized by the presence of four and a half highly conserved LIM domains. The LIM domain is a protein-interaction motif and is involved in linking proteins with both the actin cytoskeleton and transcriptional machinery. To date, more than 25 different protein interactions have been identified for full length FHL1 and its spliced variants, and these interactions can be mapped to a variety of functional classes. Because FHL1 is expressed predominantly in skeletal muscle, all of these proteins interactions translate into a multifunctional and integral role for FHL1 in muscle development, structural maintenance, and signalling. Importantly, 27 FHL1 genetic mutations have been identified that result in at least six different X-linked myopathies, with patients often presenting with cardiovascular disease. FHL1 expression is also significantly up-regulated in a variety of cardiac disorders, even at the earliest stages of disease onset. Alternatively, FHL1 expression is suppressed in a variety of cancers, and ectopic FHL1 expression offers potential for some phenotype rescue. This review focuses on recent studies of FHL1 in muscular dystrophies and cardiovascular disease, and provides a comprehensive review of FHL1s multifunctional roles in skeletal muscle.
Keywords: skeletal muscle LIM, muscle, myopathies, Emery–Dreifuss muscular dystrophy, X-linked reducing body myopathy, X-linked myopathy with postural muscle atrophy
Overview
Fifteen years ago, the initial characterization of an cDNA fragment abundantly expressed in skeletal muscle predicted the existence of a multi-LIM domain containing protein subsequently named skeletal muscle LIM protein, or SLIM [1]. With the discovery of several highly homologous proteins and elucidation of the complete protein sequence, the homodomain SLIM protein was aptly renamed four and a half LIM domain protein 1 (FHL1) [2, 3]. In humans, the FHL LIM-only protein family consists of four members, designated FHL1, FHL2, FHL3 and activator of CREM in testis (ACT), also referred to as FHL5 [3–6]. The FHL protein family is defined by a particular secondary structural arrangement of LIM domains (Fig. 1A). All proteins are composed of four complete LIM domains arranged in tandem and separated by eight amino acid residues. There is an N-terminal single zinc finger domain with a consensus sequence equivalent to the C-terminal half of a LIM motif [6]. Comparison between amino acids for FHL1, 2, 3 and 5 revealed ∼44–59% conservation across these entire proteins (Fig. 1B).
Fig 1.
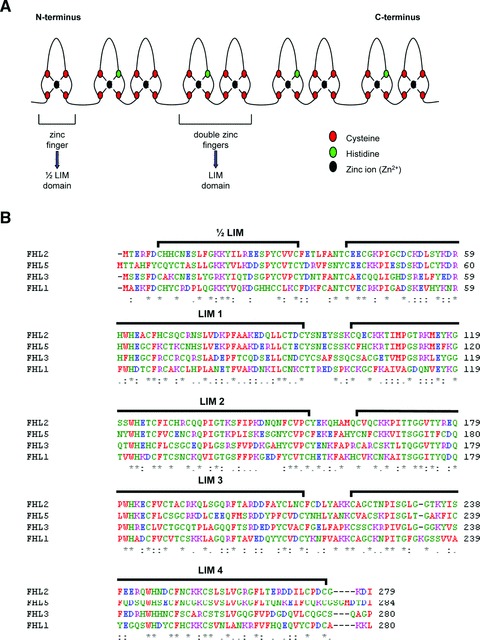
Secondary structural analysis and homology of FHL Proteins. (A) Schematic of the secondary structural features common to all members of the FHL family of proteins. (B) Amino acid sequence alignment of all human FHL proteins was performed using ClustalW. Conserved residues among all FHL proteins are identified by (*), whereas conserved substitutions are indicated with (:), semi-conserved substitutions are denoted with (.).
FHL proteins are expressed primarily in striated muscle, with the exception of ACT. Until recently, FHL1 was the least characterized of the FHL proteins. In the last 5 years, however, scientific findings have grown exponentially, particularly fuelled by discoveries of novel associations of FHL1 mutations and various myopathies. For example, to date 27 mutations have been identified in the human FHL1 gene that result in at least six different muscular dystrophies. In most cases, the skeletal muscle disorders are also coupled with cardiovascular diseases. Our current understanding of FHL1 is suggestive of important muscular functions, from development to force transmission, and a role in cell migration, although the precise mechanisms have yet to be elucidated. In addition to muscle diseases, FHL1 expression has been found to be consistently down-regulated in an even greater number of cancers types, and often more profoundly in metastatic disease. Thus, considering its involvement in some of the most leading causes of human morbidity and mortality, further studies surrounding FHL1 are warranted. FHL2 is most abundant in cardiac muscle whereas FHL3 is almost exclusively expressed in skeletal muscle. Although predominantly a skeletal muscle protein, FHL1 transcripts have also been detected at high levels in the heart, kidney, and lung [7–11] and lower levels in other organs such as the testis and pituitary gland [3, 5, 6, 9, 12–14]. ACT, however, is a testis specific protein expressed in the spermatids of adult testis [6, 14]. In mouse, another FHL family member, FHL4, has been shown to be expressed exclusively in the testis, although not as abundantly as ACT [14, 15]. To date, however, the human equivalent of murine FHL4 has not been identified, nor has translation of the murine FHL4 mRNA been validated [15]. In this review, we discuss the multifunctional roles of FHL1 and highlight its role in specific disease.
FHL1: structure, organization and function
The LIM domain
The acronym LIM was derived from the first letter of three homeodomain transcription factors from which the domain was initially discovered: (1) Lin-11 promotes asymmetric cell divisions in Caenorhabditis elegans during vulval development and regulates vulval morphogenesis [16, 17]; (2) Isl-1 participates in murine motor neuron generation and development [18] and (3) Mec-3 regulates the differentiation of mechanosensory neurons in C. elegans [19]. The LIM domain is a highly conserved module found in all eukaryotes examined thus far, from ascidians to man, but absent in prokaryotes [20, 21]. Within the human genome, there are 135 identifiable LIM domain-encoding sequences located within 58 different genes [20]. It is of particular interest that this motif is as common as other established protein interaction domains, such as Sh2 and Sh3 (Src-homology-2 and -3) domains with 115 and 253 occurrences, respectively [20].
The cysteine-rich motif is present in a number of proteins involved in a diverse array of cellular functions [20]. It has been proposed to function as a modular protein–protein binding interface upon which the coordinated assembly of multimeric protein complexes occurs [20, 22]. Structurally, it consists of a double zinc finger motif separated by two amino acids, and is composed of ∼55 amino acids with eight highly conserved residues. The consensus amino acid sequence of LIM domains has been defined as Cys-X2-Cys-X16–23-His-X2-Cys-X2-Cys-X2-Cys-X16–21-Cys-X2-Cys/His/Asp, where X represents any amino acid [1, 2, 20]. The cysteine and histidine residues coordinate the binding of two Zn21 for every LIM domain, contributing to the stabilization of the secondary and tertiary structure of the protein [3, 20].
Considering its protein interaction properties, LIM domain containing scaffold proteins are capable of interacting with other LIM domain proteins, forming homo- or heterodimers [9]. Furthermore, LIM domains can also associate with tyrosine-containing motifs, PDZ domains, ankyrin repeats and helix-loop-helix domains [9]. Nonetheless, multiple efforts to identify conserved preferences for discrete binding sequences have not been successful [20]. Multidimensional NMR spectroscopy and X-ray crystallography structural analysis studies have revealed a resemblance between the C-terminal zinc finger of LIM domains and the DNA-binding zinc finger of the GATA and steroid-hormone-receptor classes of transcription factors [2, 20]. However, there is no evidence to date that LIM domains directly bind DNA [20, 22]. Furthermore, LIM domains have usually been observed to exert negative effects on the DNA binding of LIM homeodomain proteins [22].
The presence of a LIM domain was recently recognized as a potential hallmark of proteins associating with both the actin cytoskeleton and transcriptional machinery [20, 23]. For instance, the cysteine-rich proteins 1 and 2, each containing two LIM domains, interact with zyxin and alpha-actinin in the cytoplasm and participate in cytoskeletal remodelling [24, 25]. In addition, they translocate to the nucleus and act as bridging molecules interacting with both serum response factors and GATA proteins. In smooth muscle cells, this tetrameric complex activates gene targets and facilitates differentiation [26]. However, the physiological processes responsible for regulating the shuttling of LIM domain proteins between the cytoplasm and nucleus have not been elucidated [22].
The presence of multiple LIM domains, each with a capacity for mediating protein interactions through diverse heterodimeric domain combinations, suggests considerable interaction possibilities for FHL1. In fact, 19 protein interactors have been identified thus far for full length FHL1 (Table 1). In addition, several additional ‘putative’ FHL1 interactions have been identified in large-scale studies and have yet to be validated (Table 2) [27]. These FHL1 interactors can be mapped to a variety of different functional categories, including structural proteins, signal transducers, transcription regulators, receptors and a channel. Characterization of these interactions has so far provided much of the insight into the multi-functional roles of FHL1 and the majority of the interactions were identified from and/or validated in skeletal muscle or cancer cells. Thus, alterations in protein interactions, resulting from gene mutations and aberrant expression levels, could have significant implications in various disease conditions.
Table 1.
FHL1 interacting proteins
| Gene | Protein | Interaction domain | Interaction detection | Reference |
|---|---|---|---|---|
| MYBPC1 | Myosin binding protein C, slow type | C10 domain of MyBP-C | Y2H screen of human skeletal muscle library (FHL1 = bait); in vivo FHL1 co-immunoprecipitation (co-IP) from murine Sol8 skeletal myotubes; immunofluorescence co-localization in murine soleus muscle | [32] |
| MYBPC3 | Myosin binding protein C, cardiac | C10 domain of MyBP-C | In vitro GST-pull down (cardiac MyBP-C = bait); in vivo tag co-IP from COS-1 cells | [32] |
| SRF | Serum response factor | via LIM domains | In vitro GST-pull down | [86] |
| FHL2 | Four and a half LIM domains protein 2 | n.d. | Immunofluorescence co-localization in co-transfected rat cardiomyocytes; in vitro GST-pull down | [87] |
| KCNA5 | Voltage-gated potassium channel subunit Kv1.5 | n.d. | GST-pull down (KCNA5 = bait) of human atrial lysate, followed by MS; in vivo KCNA5 co-IP from human atrium, co-transfected CHO cells; immunofluorescence co-localization in co-transfected CHO cells | [62] |
| NFATC1 | Nuclear factor of activated T cells, cytosolic component 1 | n.d. | GST-tagged FHL1 purification from co-transformed (with His-NFATc1) E. coli; GST-pull down (FHL1 = bait) of murine skeletal muscle lysate; in vivo tag co-IP from co-transfected C2C12 cells; immunofluorescence co-localization in co-transfected C2C12 cells, and co-localization with reducing body myopathic FHL1 mutant in C2C12 and patient samples | [52] |
| TLN1 | Talin 1 | n.d. | In vivo FHL1-myc co-IP from mouse embryonic fibroblast cells (NIH 3T3), followed by in-gel digestion and MS; FHL1-myc co-IP from human PASMCs; immunofluorescence co-localization in human PASMCs and human lung tissue | [64] |
| RAF1 | Raf proto-oncogene serine/ threonine protein kinase | LIMS 1, 2 essential | Y2H assay; Raf1 co-IP from murine cardiac muscle; immunofluorescence co-localization in adult cardiomyocytes | [51] |
| MAP2K2 (MEK2) | Mitogen-activated protein kinase kinase 2 | LIMS 1, 2 essential | Y2H assay; MEK1/2 co-IP from murine cardiac muscle; immunofluorescence co-localization in adult cardiomyocytes | [51] |
| ERK2 | Extracellular signal-regulated kinase 2 | LIMS 1, 2 essential | Y2H assay; ERK2 co-IP from murine cardiac muscle; immunofluorescence co-localization in adult cardiomyocytes | [51] |
| ERK2 (TYDD) | Constitutively phosphorylated ERK2 mutant | LIMS 1, 2 essential | Y2H assay; co-IP from murine cardiac muscle | [51] |
| TTN | Human cardiac Titin | N2B element of titin | In vivo co-IP of HA-FHL1 from COS cells (co-transfected with GFP-titin N2B); immunofluorescence co-localization in adult cardiomyocytes | [51] |
| SMAD2 | Mothers against decapentaplegic homologue 2 | n.d. | In vitro GST-pull down; in vivo co-IP from co-transfected HEK-293 cells; co-IP from human hepatoma HepG2 cells (endogenous) | [77] |
| SMAD3 | Mothers against decapentaplegic homologue 3 | n.d. | In vitro GST-pull down; in vivo co-IP from co-transfected HEK-293 cells; co-IP from human hepatoma HepG2 cells (endogenous) | [77] |
| SMAD4 | Mothers against decapentaplegic homologue 4 | n.d. | In vitro GST-pull down; in vivo co-IP from co-transfected HEK-293 cells; co-IP from human hepatoma HepG2 cells (endogenous) | [77] |
| CSNK1D | Casein kinase 1, delta | n.d. | In vitro GST-pull down; in vivo co-IP from co-transfected HEK-293 cells; co-IP from human hepatoma HepG2 and SMMC7721 cells (endogenous) | [77] |
| RIP140 | Receptor interacting protein of 140 kDa | All domains required (deletion of N-terminal 1/2 LIM or LIM4 abolished interaction) | Y2H screen of human mammary library (FHL1 = bait); direct two-hybrid binding assay; in vitro GST-pull down; in vivo co-IP from co-transfected HEK-293T cells; co-IP from human breast cancer MCF7 cells (endogenous) | [72] |
| ERα | Estrogen receptor α | LIMS 1, 2, 3 necessary; ERα (1-185) containing N-terminal estrogen independent activation function domain | In vitro GST-pull down; co-IP from co-transfected HEK-293T cells; co-IP from human breast cancer MCF7 cells (endogenous) | [78] |
| ERβ | Estrogen receptor β | ERB (1-145) containing N-terminal estrogen independent activation function domain | Y2H screen of human mammary gland library (ERβ 5 bait); direct two-hybrid binding assay; in vitro GST-pull down; co-IP from co-transfected HEK-293T cells | [78] |
Shown are all the known full length FHL1 protein interactions to date, with a description of the identification and validation methods used and, when known, the interaction domains involved.
Table 2.
Putative FHL1 protein interactions
| Gene | Protein | Species | Interaction detection | Database | Reference |
|---|---|---|---|---|---|
| EPB41 | Protein 4.1 (Band 4.1) | Human | Co-IP of Flag-EBP41 from HEK-293 cells, followed by mass spectrometry | IntAct, I2D | [27] |
| MCC | Colorectal mutant cancer protein (Protein MCC) | Human | Co-IP of Flag-MCC from HEK-293 cells, followed by mass spectrometry | IntAct, I2D | [27] |
| HLA-B | HLA class I histocompatibility antigen, B-42 alpha chain | Human | Co-IP of Flag-HLA-B from HEK-293 cells, followed by mass spectrometry | IntAct, I2D | [27] |
| IKBKE | Inhibitor of nuclear factor kappa-B kinase subunit epsilon (I kappa-B kinase epsilon) | Human | Co-IP of Flag-IKBKE from HEK-293 cells, followed by mass spectrometry | IntAct, I2D | [27] |
| PRKAB1 | 5’-AMP-activated protein kinase subunit beta-1 | Human | Co-IP of Flag-PRKAB1 from HEK-293 cells, followed by mass spectrometry | IntAct, I2D | [27] |
| Slc2a4 | Solute carrier family 2, facilitated glucose transporter member 4 | Rat | Co-IP of Myc-GLUT4 from rat L6 myoblast cells, followed by mass spectrometry | IntAct, I2D | [88] |
This table outlines the putative FHL1 interactions, with a description of the identification and validation methods used and the organism the interaction was detected from. The databases searched for identifying the interactions are also listed.
Spliced variants of FHL1
Although FHL1 is classified as a LIM-only protein, spliced variants have been identified containing additional domains resulting in differential localization patterns, protein interactions and functions. Similar to full-length FHL1 (isoform FHL1A), two additional isoforms (FHL1C and FHL1B) were initially identified from murine studies and referred to as KyoT2 and KyoT3, respectively (Fig. 2) [7–11]. FHL1 is also the only FHL member to comprise protein isoforms. Table 3 summarizes the known FHL1 isoform-specific protein interactions.
Fig 2.
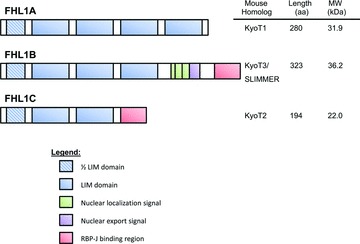
Domain features of FHL1 and spliced variants. Schematic representation of the domain structures present in each of the three human isoforms of FHL1. The chart on the left indicates the murine homologue, protein length and molecular weight of each isoform.
Table 3.
FHL1 isoform specific protein interactions
| Gene | Protein | Isoform specificity | Interaction domain | Interaction detection | Reference |
|---|---|---|---|---|---|
| HIVEP3 | Human immunodeficiency virus type I enhancer binding protein 3 | KyoT2 | In vivo tag co-IP, in vivo mammalian two hybrid assay (luciferase reporter) | [29] | |
| RBPJ | J kappa-recombination signal binding protein | KyoT2, KyoT3, *KyoT1 (weak) | RBP-J-binding motif | KyoT2: Y2H screen of mouse 9.5-dpc embryos and HeLa cells (RBP-J = bait), in vitro GST-pull down, EMSA, in vivo tag co-IP from COS-7 cells; KyoT3: in vivo tag co-IP from HeLa cells; KyoT1: weak Y2H and in vitro GST pull down | [7] |
| RING1 | Ring finger protein 1 | KyoT1, KyoT2 | LIM domains of KyoT1/2; C-terminal fragment of RING1 | KyoT2: Y2H screen of human lymph node cDNA library (KyoT2 5 bait), Y2H assay, in vitro GST-pull down, in vivo tag co-IP from HEK-293 cells, in vivo mammalian two hybrid assay (luciferase reporter) from HEK-293 cells; KyoT1: Y2H assay | [11] |
| CBX4 (HPC2) | Chromobox protein homologue 4 (Polycomb 2 homologue) | KyoT1, KyoT2 | LIM domains of KyoT1/2; C-terminal fragment of HPC2 | KyoT2: Y2H screen of human lymph node cDNA library (KyoT2 = bait), Y2H assay, in vitro GST-pull down, in vivo tag co-IP from HEK-293 cells, in vivo mammalian two hybrid assay (luciferase reporter) from HEK-293 cells; KyoT1: Y2H assay | [30] |
| PIAS1 | Protein inhibitor of activated STAT-1 (Signal transducer and activator of transcription-1) | KyoT2 | Y2H screen of human lymph node cDNA library (KyoT2 = bait); in vitro GST-pull down; in vivo tag co-IP from HEK-293 cells; in vivo mammalian two hybrid assay (luciferase reporter) from HEK-293 cells | [31] | |
| SIVA-1 | KyoT3/SLIMMER | SLIMMER unique sequences, likely nuclear localization and/or nuclear export signals | Y2H screen of human skeletal muscle library (SLIMMER = bait), Y2H assay, GST-tagged Siva-1 purification from co-transformed (with truncated His-SLIMMER) E. coli, in vitro GST-pull down (Siva-1 = bait), in vivo tag co-IP from COS-1 cells, endogenous co-IP from murine skeletal muscle, immunofluorescence co-localization in co-transfected C2C12 myoblasts and myotubes and in mature murine gastrocnemius skeletal muscle | [89] |
Shown are all known protein interactions for the alternatively spliced variants of FHL1, with a description of the interaction identification and validation methods used.
FHL1C (KyoT2) is the shorter isoform of FHL1, encoding for a 22.0 kDa protein sharing the N-terminal two and a half LIM domains with FHL1. However, alternative splicing of exon 5 results in a frameshift in translation, producing a 27 amino acid putative RBP-J binding region at the C-terminus [7, 10]. In addition, similar to FHL1, within the cell the isoform is distributed diffusely in the cytoplasm and nucleus, as determined from GFP-fusion protein expression from C2C12 myoblasts and HepG2 hepatocells [10]. Because FHL1C lacks any typical nuclear localization signal, its translocation to the nucleus may be mediated by particular protein modifications and/or protein interactions [10].
Northern blot and RT-PCR tissue distribution analysis revealed FHL1C is expressed specifically in testis, skeletal muscle and the heart, albeit at lower levels than FHL1 [10]. In the human heart, FHL1C transcript expression was more precisely localized in the left and right ventricles, with lower expression detected in the aorta and left atrium [10]. In contrast, murine KyoT2 exhibits a broader distribution, with transcripts expressed at higher levels in skeletal muscle, brain, lung, kidney and genital organs with lower detection from the thymus, lymph nodes and liver [7, 10]. The disparities in tissue distribution between FHL1C and KyoT2 could be attributable to functional differences in human and mouse.
Although FHL1B (KyoT3) is the larger isoform, encoding for a 34 kDa protein, it contains only the first three and a half LIM domains found in FHL1 [9]. However, it was originally referred to as SLIMMER for SLIM1 with extra regions [9]. The occurrence of a 200 bp insertion at position 741 results in the generation of three tandem putative bipartite nuclear localization signal motifs, followed by a nuclear export sequence and the identical putative RBP-J binding region found in FHL1C [8, 9]. Unlike FHL1C however, FHL1B is predominantly distributed in the nucleus of C2C12 myoblasts and HepG2 hepatocells, mainly attributable to the first bipartite nuclear localization signal [8, 9]. Interestingly, in differentiated myotubes, it is localized exclusively in the cytosol, similar to FHL1 [9]. Northern blot and RT-PCR tissue distribution analysis revealed greater abundance of FHL1B in skeletal muscle compared to heart, colon, prostate and small intestine. In addition to these tissues, murine KyoT3 mRNA was also detected in spleen, thymus, testis, ovary, brain, placenta, lung, liver, kidney and pancreatic tissue [8, 9].
Notch signalling and KYOT2/3 proteins
The Notch signalling pathway is an evolutionarily conserved pathway participating in the control of a broad range of developmental processes, including cell fate determination, differentiation, proliferation and apoptosis through local cell–cell interaction [28]. Mammals express four members of the Notch family of receptors, all designated a type 1 transmembrane receptor with a total of five ligands. The ligands too are single-pass transmembrane proteins, categorized into two families (delta-like 1, 3, 4 and jagged 1, 2), that allow for Notch signalling between neighbouring cells [28, 29]. Direct interaction of the ligand with the Notch receptor triggers proteolytic cleavage of the Notch receptor by γ-secretase-like protease, releasing the Notch intracellular domain (NIC) into the cytoplasm. NIC translocates into the nucleus and serves as a transcriptional activator of the DNA-binding protein RBP-J, in combination with other co-activators [28, 29]. In the absence of transcriptional activators, RBP-J is capable of suppressing transcription of Notch target genes by binding several co-repressor proteins (Fig. 3) [28, 29]. Numerous Notch transcriptional targets have been described, but the Hairy enhancer of split (Hes) and Hes-related families of transcriptional regulators are some of the best defined. These targets function as transcriptional repressors mediating downstream responses of Notch signalling [28, 29].
Fig 3.
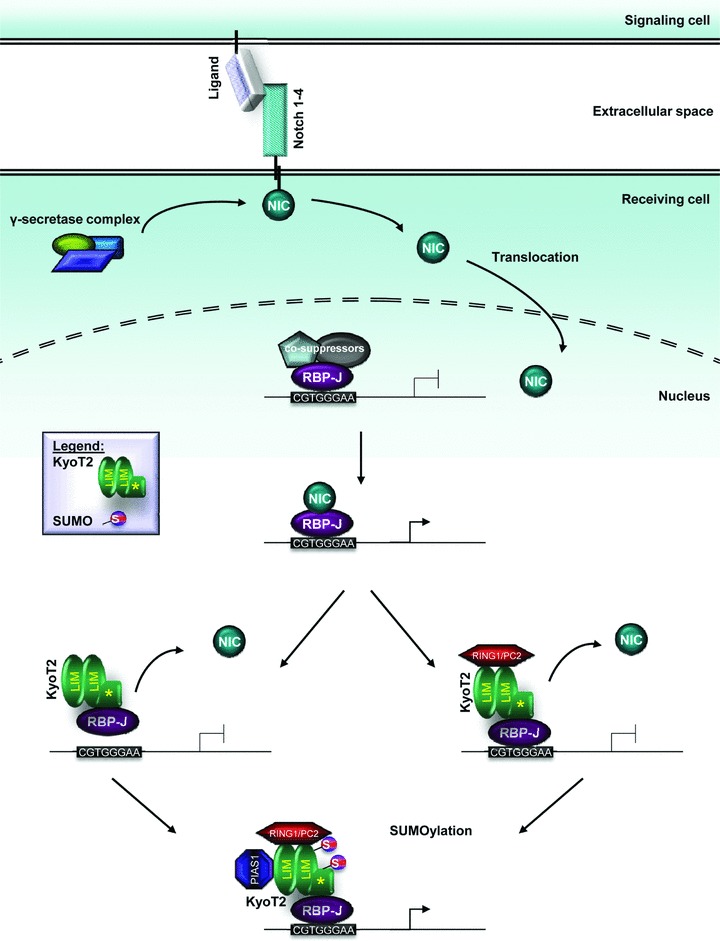
FHL1 isoform KyoT2 and Notch signaling. In mouse, the Notch signaling pathway is activated by proteolytic cleavage of the Notch receptor by γ-secretase-like protease, releasing the Notch intracellular domain (NIC). NIC translocates into the nucleus and transactivates RBP-J. KyoT2 can disrupt this interaction via its RBP-J binding domain and subsequently suppress transcription by displacing RBP-J from DNA and/or recruiting co-suppressors (i.e. RING1 and/or HPC2). Furthermore, sumoylation by PIAS1 antagonizes KyoT2’s repressor activity.
KyoT2 was discovered during yeast two hybrid screenings of mouse embryonic and HeLa cell cDNA libraries using RBP-J as the bait, and the interaction was subsequently verified in mammalian cell systems [7]. Because their binding regions on RBP-J overlap, KyoT2 competes with NIC for binding [7]. In contrast to NIC, KyoT2 interacts with RBP-J to suppress transcription, in a concentration-dependent manner [7, 29]. Furthermore, electrophoretic mobility shift assays revealed that whereas KyoT2 is capable of interacting with the RBP-J-DNA complex, it mostly dis places RBP-J from DNA, thus contributing to its repressional activities [7]. Subsequently, KyoT2 was found to interact with RING1 via its LIM domains [29]. RING1 belongs to the polycomb group proteins which function as transcription suppressors [29]. In co-transfected HEK-293 and COS7 cells, RING1 was shown to form a multimeric complex with KyoT2 and RBP-J, contributing to the repression of RBP-J mediated transactivation. These effects could be abrogated by human immunodeficiency virus type I enhancer binding protein 3, which competes with RING1 to bind KyoT2 at both LIM domains [29]. Similar patterns were also observed for HPC2, another polycomb group protein interactor of KyoT2 (Fig. 3) [30]. These findings suggest there may be two approaches for the KyoT2-mediated suppression of RBP-J. First, KyoT2 could compete with transactivators for binding sites on RBP-J. In addition, it could recruit co-suppressors such as RING1 and/or HPC2. Polycomb group proteins, including HPC2, have been known to form large complexes on promoters to suppress transcription [7, 29, 30].
Similar to KyoT2, KyoT3 was recently demonstrated to compete with NIC for binding RBP-J and repressing transactivation of RBP-J dependent promoters [11]. However, RT-PCR analysis of Hes-1 mRNA levels revealed KyoT3-mediated repression occurred only in the presence of NIC. In the absence of NIC, elevated Hes-1 mRNA was detected in the presence of KyoT3 [11]. It is plausible KyoT3 recruits other molecules to any of its LIM domains to transactivate the Hes-1 promoter or antagonize the repression of RBP-J activity [11].
KyoT2 repression of Notch signalling in cells via interaction with RBP-J can be modulated by PIAS1, promoting transactivation of RBP-J [31]. Considering KyoT2 interacts with two small ubiquitin-related modifier (SUMO) modification E3 ligases (HPC2 and PIAS1), KyoT2 was suggested to be a substrate for SUMOylation [30, 31]. This was verified when the effects of PIAS1 were neutralized in the presence of Sentrin/SUMO-specific protease SENP2, a SUMO hydrolase [31]. PIAS1 promotes SUMOylation of KyoT2 at two sites, K144 in the second LIM domain, and K171 near the RBP-J binding motif, which antagonizes KyoT2’s repressor activity (Fig. 3). In particular, modification of the K171 site most counteracted the repression. Unlike PIAS1, overexpression of HPC2 did not enhance SUMOylation of KyoT2 beyond basal levels [31]. In general, SUMOylation can alter the stability, localization and biological activities of modified proteins [31]. In the case of KyoT2, no effects on its subcellular localization or interaction with RBP-J were detected [31].
Expression patterns of FHL1
Northern blot analyses of a panel of tissue samples obtained from human, rat and sheep subjects identified expression of FHL1 in skeletal muscle across multiple species [1, 5]. In mature skeletal muscle, FHL1 is localized at the I-band, encompassing the Z-line, and transiently at the M-line where it extends partially into the C-zone of the A-band. The resulting transverse banding pattern is of alternating thick and thin bands, corresponding to the I-band and the centre of the A-band, respectively [32].
In embryonic mouse skeletal muscle FHL1 was first detected at E8.75 in the heart and neural tube via in situ hybridisation, using whole mount mouse embryos. At E9.5, FHL1 was prominent in the hindbrain and neural tube, with conspicuous expression in the cardiac outflow tract although the endothelium was not the source of expression in the outflow tract [33]. At E11, however, FHL1 was detected in the somites with a pattern corresponding to the developing myotomes, in addition to a continued expression in the outflow tract and neural tube [33].
Generally consistent with these earlier studies, using whole mount X-gal staining of FHL1 reporter transgenic mouse embryos, a broader FHL1 mRNA distribution has been observed [34]. At E10.5, the heart, somites, limb buds, neural tube, brain and eye stained positive for FHL mRNA. At E14, FHL1 was strongly expressed in the neural tube, and the aorta and pulmonary trunk of the vasculature, and less prominently in the myocardium [34]. Furthermore, strong X-gal staining was also observed in the tongue, limb buds, abdominal muscle, intercostal muscle and thoracic muscle [33, 34].
Enriched FHL1 transcript expression was also detected by in situ hybridization in slow twitch soleus muscle in adult rat, when compared to the fast-twitch gastrocnemius muscle. The specificity of FHL1 expression correlated better with the oxidative properties of muscle fibres rather than the fibre types. Soleus muscle is composed of the highly oxidative type I and type IIa fibres, whereas gastrocnemius is largely composed of the glycolytic type IIb fibres [35].
In the murine C2C12 skeletal muscle cell line, FHL1 expression was detected in myoblasts before the onset of differentiation [5, 36]. During the first 24–48 hrs of differentiation, however, FHL1 expression initially declined by 40% before resuming its inclined expression pattern up to about 6 days [5, 36]. Furthermore, the expression profile of FHL1 was similar to those of the myogenic regulatory factors, myogenin and MRF, and to the growth factor IGF-II. However, FHL1 was present before the onset of myogenin and muscle specific embryonic, neonatal and adult 2b MyHCs expression [5].
FHL1 and disease
FHL1 and skeletal muscle myopathies
Considering the high levels of FHL1 in skeletal muscle, its ontogenic expression pattern, and X-chromosome gene localization, an association of FHLl with muscle disease could almost be anticipated. Indeed, FHL1 was identified recently as the causal gene for six different X-linked myopathies, in accordance with its gene localization on the X-chromosome (Table 4) [3]. One of the first mutations identified in the human FHL1 gene was shown to result in an X-linked dominantly inherited form of scapuloperoneal myopathy (XSPM), identified in a large Italian-American family [37, 38]. XSPM is a neuromuscular disorder characterized by progressive muscular atrophy, initiating in the lower legs and extending to the shoulders and arms with scapular winging, possibly with impaired sensory functions. Serum creatine kinase levels were elevated in all patients, and analysis of two biopsy samples revealed desmin-positive cytoplasmic bodies indicative of a myofibrillary myopathy [37, 38]. These phenotypes were attributed to a Trp-122-Ser substitution, occurring in the second LIM domain at a residue highly conserved across species. Male patients were hemizygous for the mutation with an earlier age of onset (26.1 ± 4.7 years) and more severely affected than the heterozygous affected females (age of onset was 34.4 ± 11.9 years). This suggested that there may be an inverse relationship between the FHL1 protein level and symptoms [37]. Thus, the loss of FHL1 appears to parallel increasing disease severity.
Table 4.
Human FHL1 mutations causing X-linked myopathies and the associated phenotypes
| Myopathy | Gene | Protein | Lim domain | Cytoplasmic bodies | Reducing bodies | Rimmed vacuoles | Scapular winging | Rigid spine | Joint contractures | Scoliosis | Gowers’ sign | Creatine kinase levels | FHL1 expression | Muscle weakness | Muscle athropy | Muscle hypertrophy | Motor development delay | Muscle hypotrophy | Respiratory insufficiency | Cardiac disease | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| X-linked scapuloperoneal myopathy | x | x | x | x | x | ↑ | ↓ | x | x | ND | x | x | [37] | ||||||||
| c.365G>C | p.W122S | 2 | |||||||||||||||||||
| X-linked myopathy with postural muscle atrophy | x | ND | x | x | x | x | x | ↑ | ↓ | x | x | x | x | x | [39, 40] | ||||||
| c.672C>G | p.C224W | 4 | |||||||||||||||||||
| c.381_382insATC | p.D127_T128insIle | 2 | |||||||||||||||||||
| c.838G>A | p.V280M (FHL1B) | – | |||||||||||||||||||
| c.68811:G>A | p.A168GfsX195 | 3 | |||||||||||||||||||
| c.736C>T | p.H246Y | 4 | |||||||||||||||||||
| X-linked reducing body myopathy | xF1 | x | x | x | x | x | x | x | ↑ | ↓/↓ | x | x | x | x | x | [41–43] | |||||
| c.367C>T | p.H123Y | 2 | |||||||||||||||||||
| c.395G>T | p.C132F | 2 | |||||||||||||||||||
| c.457T>C | p.C153R | 2 | |||||||||||||||||||
| c.458G>A | p.C153Y | 2 | |||||||||||||||||||
| c.368A>T | p.H123L | 2 | |||||||||||||||||||
| c.369C>G | p.H123Q | 2 | |||||||||||||||||||
| c.369C>A | p.H123Q | 2 | |||||||||||||||||||
| c.449G>A | p.C150Y | 2 | |||||||||||||||||||
| c.302G>T | p.C101F | 2 | |||||||||||||||||||
| c.304-312delAAGGGGTGC | p.102-104delKFC | 2 | |||||||||||||||||||
| c.310T>C | p.C104R | 2 | |||||||||||||||||||
| n.d. | p.C150R | 2 | [88] | ||||||||||||||||||
| Emery-Dreifuss muscular dystrophy | ND | ND | x | x | x | x | N–↑ | ↓ | x | x | x | x | [44] | ||||||||
| c.841T>G | p.X281E | Stop codon | |||||||||||||||||||
| c.827G>A | p.C276Y | 4 | |||||||||||||||||||
| c.332_688del | p.G111_T229delinsG | 2/3 | |||||||||||||||||||
| c.332_501del | p.D112FfsX51 | 2/3 | |||||||||||||||||||
| c.817dup | p.C273LfsX11 | 4 | |||||||||||||||||||
| c.469_470delAA | p.K157VfsX36 (FHL1A) | 3/4 | |||||||||||||||||||
| c.469_470delAA | p.K157VfsX36 (FHL1B) | 3 | |||||||||||||||||||
| c.469_470delAA | p.K157VfsX62 (FHL1C) | new LIM | |||||||||||||||||||
| c.371_372delAA | p.K124RfsX6 | 2/3/4 | |||||||||||||||||||
| X-linked Emery-Dreifuss-like syndrome | xF+ | ND | ND | x | x | N–↑ | ↓ | ND | ND | x | [89] | ||||||||||
| c.625T>C | p.C209R | 3 | |||||||||||||||||||
| Rigid spine syndrome | x | x | x | x | x | x | x | ↑ | ↓ | x | x | [46] | |||||||||
This table outlines all the human FHL1 myopathies and the causal genetic mutations. The corresponding protein alterations and associated phenotypes are also listed. Legend: x, positive for phenotype; xF+, FHL1 positive aggregates; ND, phenotype not detected; ↑, increased levels; ↓, decreased levels; N-↑, normal to elevated levels.
Five other FHL1 mutations were discovered to be causal for X-linked myopathy with postural muscle atrophy (XMPMA), clinically characterized by the combined presentation of weakness and atrophy of postural muscles (scapuloperoneal weakness and bent spine) with a pseudoathletic phenotype where alternative muscle groups were hypertrophic [39]. In general, muscle groups composed predominantly of type I fibres were atrophic, whereas those primarily comprised of type II fibres were hypertrophic. Scoliosis and elevated creatine kinase levels were also noted in patients. Furthermore, affected individuals typically died of heart failure, suspected to be caused by hypertrophic cardiomyopathy [39]. Symptoms were typically noticed at about 30 years of age, and follow up studies revealed XMPMA to be a slow but progressive muscular dystrophy. The original mutation, discovered in a large Austrian family, was a Cys-224-Trp substitution located in the fourth LIM domain. In a British family, an isoleucine insertion mutation in the second LIM domain was also found. Three additional XMPMA causing FHL1 mutations were recently identified from five independent German families [40]. Immunoblot and immunostaining analysis revealed almost complete absence of FHL1A protein in both fibres from affected individuals [39, 40]. Granulofilamentous material was observed via electron microscopy, and staining identified the presence of a few cytoplasmimc bodies, although there was no evidence of reducing bodies [40]. Such histopathology suggests XMPMA may share pathophysiological pathways with myofibrillary myopathies [40].
To date however, the greatest number of FHL1 gene mutations have been associated with X-linked reducing body myopathy (XRBM), with all 11 known amino acid substitution mutations and one deletion mutation occurring in the second LIM domain [41–43]. Clinically, XRBM is a rare muscular disorder causing progressive muscular weakness, generally affecting proximal muscles with evidence for early scapuloperoneal involvement. Proximal weakness was also present in the lower extremities, possibly with foot drop or early Achilles tendon contractures. Spinal rigidity was a prominent and presenting feature, whereas scoliosis and elevated creatine kinase levels were also common. The progressive disease course often led to a loss of ambulation and/or respiratory insufficiency, with one documented case of cardiomyopathy [42]. The histochemical criterion for diagnosis however is the presence of intracytoplasmic inclusions, or reducing bodies, in myofibres and identified using the menadione-nitro-blue tetrazolium stain. Muscle fibre degeneration was also evident during muscle biopsies, with the degree of severity associated with an increasing number of reducing bodies [42]. Laser capture microdissection of intracytoplasmic inclusions, followed by nano-flow liquid chromatography-tandem mass spectrometry analysis, identified FHL1 as the most prominent protein in the aggregates. Immunohistochemistry confirmed the enrichment of FHL1 and revealed the primarily juxtanuclear localization of FHL1 positive aggregates. In affected individuals, FHL1 was normally localized to the I-band/Z-disc in myofibres lacking defined aggregates. In contrast, in myofibres heavily afflicted with aggregates, FHL1 expression was diminished at the myofibrils immediate to the aggregates. Patient follow-up exams revealed a time-dependent increase in the number of FHL1 positive aggregates, possibly correlating with the clinical progression of the disease. Taken together, these findings are suggestive of a pathomechanism involving the formation of intracytoplasmic inclusions rather than solely because of a primary dysfunction of FHL1 [42]. Furthermore, reducing bodies have previously been suggested to display morphological and immunohistochemical features reminiscent of aggresomes. In agreement with this, immunolabelling of patient biopsies or C2C12 cells expressing mutant FHL1 revealed the inclusions were positive for γ-tubulin, pericentrin, ubiquitin, GRP78 and desmin. In addition, these aggregates also sequestered wild-type FHL1 and interacting proteins, such as MyBP-C and NFATc1 [41]. As demonstrated in C2C12 cell studies, the presence of mutant FHL1 initiated the formation of inclusion bodies, leading to the incorporation of other proteins.
Another large subset of FHL1 gene mutations was identified recently as causal for Emery-Dreifuss muscular dystrophy (EDMD). EDMD is a rare hereditary disease characterized by the triad of joint contractures, muscular dystrophy and cardiac dysfunctions. Without exception, all index cases examined exhibited joint contractures commonly of the ankles, spine, elbows, knees, neck and hips. Progressive muscle weakness and wasting was also evident, with a predominant scapulo-humeral and pelvic and/or peroneal topography. Finally, arrhythmias and cardiac hypertrophy accounted for most cardiac disease occurrences. Interestingly, this cardiac phenotype contrasts the progression from arrhythmia and/or conduction defects to dilated cardiomyopathy seen in EDMD patients with mutations in either the emerin or lamin A gene, the other causative genes for EDMD. However, combined they account for only 50% of EDMD patients. Genome-wide scans and linkage analysis of six affected families and one isolated case lead to the identification of seven novel causal mutations in the FHL1 gene. Unlike the previously described FHL1 mutations, these mapped to the distal exons (5 to 8) of the gene, affecting LIM domains 2–4 [44]. Also unlike the other myopathies, EDMD biopsies were absent of reducing bodies and myofibrillar protein aggregates. Similarly, however, FHL1 expression was significantly depressed. Immunostaining analysis localized most of the present FHL1 in and/or close to the nucleus.
The reclassification of a single missense mutation (c.625T > C), identified in a large German family, as causal for a X-linked recessive Emery-Dreifuss-like syndrome rather than EDMD further expands the spectrum of FHL1 myopathies [45]. Nine male subjects, ranging from 14 to 60 years of age, presented with joint contractures of the ankles and/or knees with rigid spine syndrome. By contrast to other FHL1 myopathies, muscle weakness, muscular atrophy, athletic habitus, scapular winging and gait problems were absent in all of the patients, except one. Their creatine kinase levels also tended to be normal or only slightly elevated. However, cardiac disease was prominent, as patients also were found to suffer from left ventricular hypertrophy, cardiac fibrosis and hypertension, without evidence of rhythm conduction abnormalities (except for one case of atrial fibrillation) [45]. Histological analysis of muscle biopsy specimens revealed the presence of cytoplasmic bodies, even in asymptomatic female carriers, however reducing bodies were lacking. Furthermore, in a highly progressed case, assessment revealed a pathological variation in fibre size, and the occurrence of necrotic, regenerating and split fibres. A high proportion of fibres also had internalized nuclei, whereas a small proportion was positive for FHL1 containing aggregates. Immunoblot analysis determined FHL1 expression was reduced in both affected males and female carriers of the mutation [45].
Rigid spine was identified as a common clinical feature among patients afflicted with any of the five x-linked myopathies [46]. In accordance with this, an in-frame nine base pair deletion mutation, corresponding to the second LIM domain, was identified in a patient diagnosed with rigid spine syndrome. The patient presented with early scoliosis and prominent muscular weakness and atrophy of the hip and thighs. Winging of the scapula, funnel chest and multiple joint contractures (i.e. neck, spine, hip, and ankle) were also observed. In addition, the patient’s serum creatine kinase level was mildly elevated, whereas his respiratory functions were mildly impaired. Furthermore, intracytoplasmic inclusion bodies were detected from biopsied muscle samples, although only a limited number of fibres were positive for reducing bodies. Immunohistochemistry illustrated an increasingly diffuse FHL1 staining pattern, whereas immunoblotting revealed a reduction in the total FHL1 expression in diseased muscle [46].
In general, all of the described human FHL1 mutations were localized within a LIM domain, usually affecting highly conserved residues. For instance, the Cys-to-Trp mutation causing XMPMA involved a highly conserved cysteine within the fourth LIM domain, and comprises one of four cysteines necessary for the binding of a Zn2+ ion [39]. NMR spectroscopy also predicted complete disruption of the Zn2+ binding sites and collapse of the LIM domain in two severe cases of XRBM, caused by the H123Y and C132F substitution mutations localized to invariant consensus residues in the second LIM domain. Similarly, NMR structural determination identified the disruption of van der Waals contacts, with neighbouring T120 and K102 residues, upon substitution of W122 with a smaller serine causing XSPM [37]. The mutation was predicted to interfere with protein folding or cause conformation instability of the second LIM domain and/or the adjacent second zinc finger binding domain [37]. Metal ions can contribute to the stabilization of a protein’s tertiary structure, as is the case with Zn2+ ions and LIM domains. Unstable or misfolded proteins, such as the mutant FHL1, are likely vulnerable to proteolysis, which could explain the reduced expression observed in most diseased conditions [47–50].
Furthermore, alterations in LIM domains and protein conformation affect the binding interfaces for protein interactions. In XMPMA, insertion of a single isoleucine residue between two amino acids connecting adjacent zinc fingers of the second LIM domain, thereby nonconforming with the consensus C-X2-C spacing, was expected to adversely affect proteins interactions (the protein’s interaction or protein interactions?) [39]. The first two LIM domains, for instance, were deemed essential for mediating interactions with RAF1, MEK2 and ERK2 in cardiac muscle [51]. In C2C12 skeletal muscle cells co-expressing myc-NFATc1 with either HA-FHL1C132F or HA-FHL1H123Y, immunoprecipitation of myc-NFATc1 revealed an ∼80% reduction in interaction with mutant FHL1 [52]. FHL1 positive aggregates in XRBM were also found to sequester FHL1 interacting proteins MyBP-C and NFATc1, possibly contributing to the reduced interaction observed in C2C12 cells [41, 52].
FHL1 expression levels in cardiovascular diseases
Differential FHL1 expression has previously been associated with several cardiovascular diseases. In our group, a transgenic mouse model was generated to recapitulate early onset dilated cardiomyopathy observed in humans and caused by an Arg-9-Cys conversion in phospholamban, an integral membrane phosphoprotein of the sarcoplasmic reticulum [53, 54]. From a large-scale comparative proteomic profiling of ventricular muscle tissue, FHL1 was identified among a subset of 40 top ranked differentially expressed proteins as one of the four most up-regulated, with changes occurring early in disease [54]. Similarly, FHL1 was also detected to be up-regulated during comparative gene expression profiling of transgenic mice overexpressing Gs-alpha, with respect to control littermates [55]. Similar to observations in the transgenic phospholamban mice, a two- to threefold increase in FHL1 transcript level was detected even at the earliest time points tested, although alterations in expression were not detected for either FHL2 or FHL3 [55]. Furthermore, FHL1 overexpression was detected at various time points from three additional mouse models of cardiomyopathy, with chronic stimulation of the β-adrenergic receptor (AR) signalling pathway. In all three mice models, overexpressing β1-AR, β2-AR or PKA, FHL1 was up-regulated by the early stages of disease, and continued to increase with disease progression [55]. Because differential expression could be detected before disease onset, with continued increase throughout disease, it was suggested FHL1 could participate in the transition of phenotype between early and late stages, and thus contributes to the development of cardiomyopathy [55].
Results from FHL1 knockout (Fhl1−/−) mice further support this hypothesis, demonstrating a critical role for FHL1 in pathological cardiac hypertrophy. Fhl1−/− mice were viable, with normal life spans, and demonstrated no differences in cardiac size, dimensions, blood pressure and functions, when compared to age-matched wild-type mice [51]. However, Fhl1−/− mice exhibited a blunted and beneficial response when subjected to transverse aortic constriction, an in vivo model of pressure overload induced cardiac hypertrophy. Following transverse aortic constriction, Fhl1 knockout mice possessed smaller hearts than wild-type mice, resulting in a smaller increase in left ventricular/body-weight ratio, cardiomyocyte cross-sectional area, left ventricular posterior wall thickness and interventricular septal wall thickness. In addition, a lesser reactivation of foetal gene markers occurred in Fhl1−/− mice, such as atrial natriuretic factor, β-MHC and skeletal α-actin, indicative of a blunted hypertrophic response. Echocardiography revealed the percentage of left ventricular fractional shortening in Fhl1−/− mice was comparable to those of sham-operated controls, demonstrating the preservation of cardiac function under pressure overload conditions. Even with prolonged durations, left ventricular end-diastolic pressure and systolic function were preserved, with increased diastolic function. Wild-type mice subjected to transverse aortic constriction, however, exhibited early symptoms of heart failure including reduced left ventricular fractional shortening and increased left ventricular chamber dimensions [51].
Furthermore, ablation of Fhl1 expression was sufficient to prevent cardiomyopathy in transgenic mice overexpressing constitutively active Gq [51]. The Gq signalling pathway is known for its participation in cardiac hypertrophy, and can be stimulated in vivo by transverse aortic constriction. Induction of the pathway was shown to result in elevated FHL1 protein levels in primary rat cardiomyocytes incubated with G protein coupled receptor (GPCR) agonists, in cardiomyocytes overexpressing constitutively active Gq, and in cardiac tissue from Gq transgenic mice. Double Fhl1−/−/Gq transgenic mice, when compared to Gq transgenic mice, displayed both a reduction in left ventricular dimensions and expression of foetal gene markers. In fact, the cardiac chamber size, wall thickness and functions of Fhl12/2/Gq mice were comparable to wild-type and Fhl1−/− mice [51].
In contrast, the opposite expression profile was detected for FHL1 from human dilated cardiomyopathic hearts. Using high-density oligonucleotide arrays, Yang et al. quantified the expression levels of approximately 7000 genes in non-failing and failing human hearts with a diagnosis of end-stage ischaemic and dilated cardiomyopathy [56]. In diseased hearts, FHL1 was found to be down-regulated at both the mRNA and protein level [56]. Thus, the contradictory FHL1 expression profiles from dilated cardiomyopathy in mice and humans might be a consequence of the inherent physiological differences between the species. This has always posed a concern when findings from one species are adapted to another. Alternatively, the discrepancies could also be explained by differences in experimental technicalities (such as probe sets, methodologies, etc.), specificities of detection or the conditions of the hearts obtained from patients of end-stage disease. However, FHL1 has consistently been identified as a strong candidate for overexpression in human cardiac hypertrophy, from three different large-scale gene expression profiling studies [57–59]. Thus, continued study of FHL1 with respect to heart failure is warranted, especially because hypertrophic and dilated cardiomyopathy do share common pathways in disease development [60].
Low-density cDNA array analysis of atrial fibrillation induced by rapid atrial pacing in porcine identified FHL1 transcripts to be significantly up-regulated (3.17 fold), which translated to a 2.9-fold increase at the protein level [61]. Its elevated expression occurred in both the right and left atria at similar levels [61]. A capacity for influencing electrophysiology was demonstrated for FHL1, as described later [62]. Furthermore, although FHL1 was abundant and present at muscle striations, immunostaining revealed a more disorganized and diffuse cytoplasmic staining with slight nuclear localization in the fibrillating atria [61]. Translocation to the nucleus could possibly invoke a transcriptional regulation function for FHL1 in differentiated cardiac cells during arrhythmia. Furthermore, in cultured cardiomyocytes treated with the proarrhythmogenic substrate isoproteronol, FHL1 expression was up-regulated 3.1-fold [61]. The rapid response may have been mediated via activation of the β-AR signal transduction pathway, a relationship that was already implicated above [55, 61]. Isoproterenol is a β-adrenergic agonist and β2-AR was also up-regulated in atrial fibrillation [61]. An involvement of the adrenergic signalling pathway in atrial remodelling during atrial fibrillation has been postulated [61, 63]. Altogether, these results are consistent with the observation that atrial fibrillation was common among patients of FHL1 mutation-induced myopathies.
Elevated FHL1 expression has also been detected in pulmonary hypertension [64]. Proteomic analysis initially identified FHL1 to be up-regulated in lung tissues of a mouse model of hypoxia-induced pulmonary hypertension, and also verified in two rat models. More importantly, the pattern was mirrored in idiopathic pulmonary arterial hypertension human patient samples, and interestingly FHL2 and FHL3 levels were not altered [64]. Furthermore, in mice subjected to prolonged hypoxia a time dependent increase in FHL1 expression was observed [64]. Its expression was revealed to be strongest in vascular smooth muscle cells, with positive staining also present in neomuscularized resistance vessels. However, the hypoxia-dependent up-regulation of FHL1 was restricted to the pulmonary vasculature, whereas time-dependent reduction in FHL1 occurred in the systemic vascula ture [64]. It appears the hypoxia-driven expression of FHL1 was controlled by hypoxia-inducible transcription factors 1 and 2, two major gene regulators active during low oxygen tension. In the absence of either transcription factor, FHL1 levels were significantly lower in hypoxic conditions. In addition, several putative hypoxia-inducible transcription factor-binding sites were detected in the FHL1 gene promoter region, with binding activity by hypoxia-inducible transcription factor proteins occurring only under hypoxia [64].
FHL1 and muscle-associated functions
The pathological consequences of mutations occurring in the FHL1 gene are suggestive of important functional roles for FHL1 in muscle. As such, FHL1 has been associated with numerous functions in muscle cells, although the precise mechanisms have yet to be elucidated. Furthermore, the capacity of FHL1 to function as a transcriptional regulator, similar to other members of the FHL family, has also been demonstrated.
For instance, recombinant expression studies in the murine Sol8 skeletal muscle cell line demonstrated that FHL1 regulates integrin-mediated cytoskeletal rearrangement. Activation of cell-surface integrins results in reorganization of the actin cytoskeleton, with the formation of focal adhesion complexes and stress fibres. Furthermore, integrin-mediated focal adhesion assembly and cytoskeletal signalling regulates cell adhesion, spreading and migration. However, overexpression of GFP-FHL1 in myoblasts plated on fibronectin inhibited myoblast adhesion and enhanced cell spreading and migration, mediated specifically by α5β1-integrin [65]. The use of a modified Transwell migration assay, coated with fibronectin, demonstrated the proportion of GFP-FHL1 expressing myoblasts in the migrating population was twofold greater than in the non-migrating cells. In contrast, migration ceased to occur in the presence of poly-L-lysine, a non-specific inhibitor of integrin activation. In addition, in integrin-activated myoblasts dual localization of FHL1 was observed in the nucleus and the cytoplasm, specifically at focal adhesions and along stress fibres [65]. In myoblasts plated on poly-L-lysine, however, FHL1 was diffusely localized in the cytosol with decreased nuclear expression. Cytochalasin D and nocodazole treatments, respectively, eliminated an intact actin cytoskeleton and microtubules as necessary for the nuclear localization of FHL1 in integrin-activated myoblasts [65].
Similarly, ectopic expression of FHL1 in Sol8 or C2C12 myoblasts, plated on fibronectin and subjected to differentiation conditions, induced two distinct phenotypes, either hyper-elongated or highly branched [36]. Hyper-elongated cells were mononuclear and spindle shaped, with extremely long bipolar extensions, and often thickened in appearance. Alternatively, a significant proportion of myoblasts adopted a branched phenotype, with multiple major cytoplasmic protrusions from the cell body. Both phenotypes are indicative of cytoskeletal remodelling, induced by FHL1 overexpression and integrin activation. Integrin–matrix interactions were sufficient for induction of the branched phenotype, whereas hyper-elongation was dependent specifically on ligand binding to the α5β1-integrin. Furthermore, co-staining for MyoD and myogenin revealed hyper-elongation occurred in differentiating myoblasts, whereas the lack of co-stain identified branched cells as undifferentiated satellite cells [36].
A role for FHL1 has also emerged in pulmonary vascular remodelling following evidence for involvement in the migration and proliferation of primary human pulmonary artery smooth muscle cells (PASMC) [64]. SiRNA mediated silencing of FHL1 significantly decreased human and murine PASMC migration and proliferation activities, whereas overexpresison enhanced these activities [64]. Parallel results were obtained with the silencing of Talin1, an FHL1 interacting protein. Immunofluorescence analysis in human PASMC revealed partial co-localization of FHL1 and Talin1 with the focal adhesion kinase. Thus, FHL1 could possibly induce conformational changes in cytoskeletal proteins and play a role in Talin-mediated regulation of integrin signalling and cytoskeletal organization [64]. In addition, FHL1 could mediate the connection between Talin1 and actin assembly. Interestingly, FHL1 knockdown in PASMC also resulted in reduced cyclinD1 expression, thereby inhibiting G1 phase progression and consequently reduced proliferation [64]. Integrin-mediated cell adhesion has also been shown to regulate cyclinD1 protein levels [64].
Characterization of the FHL1 interaction with MyBP-C revealed a role in myosin filament formation and sarcomere assembly [32]. FHL1 competes with myosin for binding MyBP-C, because they share a common binding region, thus impairing MyBP-C from binding to myosin filaments. When FHL1 was overexpressed in differentiating skeletal muscle cells, disruptions in the formation of the Z-line of the sarcomere and assembly of the myosin thick filament were evident [32]. Similarly, myosin thick filament formation was also inhibited by RNAi-mediated knockdown of FHL1. The knockdown also impeded the incorporation of MyBP-C into the sarcomere, with the formation of dense MyBP-C aggregates instead. Thus, FHL1 regulates myosin filament and sarcomere formation, with consequences for altering the MyBP-C-to-myosin ratio [32].
FHL1 also mediates hypertrophic biomechanical stress responses in mice, upon detection of stretch or agonists induced by GPCR signalling [51]. FHL1 was identified as a component of the stress sensor complex at the sarcomeric I-band, where it interacts with the MAPK cascade components, Raf1, MEK2 and ERK2, at the N2B region of titin in adult cardiomyocytes. Although the precise mechanisms are not known, FHL1 mediates communication between the stretch sensor complex and downstream responses by titin and MAPK components [51]. For instance, in papillary muscles isolated from FHL1 knockout mice and subjected to stretch (Lmax), there was a loss of stretch-induced hypertrophic signalling responses. This was measured by quantitative PCR of ELK1 and atrial natriuretic factor, a transcriptional target of ERK1/2 and marker of hypertrophy, respectively [51]. A specific role for FHL1 in diastolic tension was also suggested when muscle displayed reduced diastolic stress and increased compliance [51]. The N2B element of titin is a component of the extensible region of titin, which contributes to the myofibrillar passive tension generated upon stretch [51].
Furthermore, FHL1 may function as a positive regulator of Raf1/MEK/ERK-mediated signalling, possibly downstream of Gq signalling. For instance, overexpression of FLAG-FHL1 in neonatal cardiomyocytes resulted in increased activation of ERK1/2 by phosphorylation, whereas Akt phosphorylation levels were constant. In addition, FHL1 interactions with Raf1, MEK1/2 and ERK2 were increased in wild-type mouse hearts following transverse aortic constriction. In contrast, a significant reduction in ERK1/2 activation was observed in Fhl1−/− and Fhl1−/−/Gq mice subjected to transverse aortic constriction, but not in Gq transgenic mice with elevated FHL1 expression. An association between FHL1 and Gq-MAPK signalling was demonstrated when inhibition of MAPK/ERK1 activity completely obstructed phenylephrine and angiotensin II, two GPCR agonists, mediated up-regulation of FHL1 expression in cardiomyocytes [51]. The Gq signalling pathway has previously been shown to regulate ERK activation in pressure overload conditions [66].
Overexpression studies also implicated FHL1 as a positive regulator of muscle hypertrophy and myoblast fusion. In HA-FHL1 transfected C2C12 cells induced to differentiate, large ‘sac-like’ myotubes with densely clustered nuclei, and large diameters, were observed. The expression levels of muscle regulatory factors MyoD, Myf5 or myogenin were not affected by FHL1 overexpression, however, a fivefold increase occurred for MHC early in differentiation (48 hrs). In addition, these cells experienced a twofold increase in protein synthesis relative to DNA. According to the myoblast fusion index, scored as the mean number of nuclei per MHC-positive cells, enhanced cell fusion was evident by 96 hrs in differentiation conditions. Taken together, these phenotypes are indicative of myotube hypertrophy with enhanced myoblast fusion [52]. Similarly, skeletal muscle hypertrophy was observed in vivo in FHL1 transgenic mice, where FHL1 functioned to increase whole body skeletal muscle mass and strength, with reduced susceptibility to fatigue. The enhanced muscle strength was a consequence of hypertrophy and not an increased myosin power stroke. Examination of the gastrocnemius muscle demonstrated an increase in the frequency of larger fibres, total fibre cross-sectional area and myofibril width [52]. The hypertrophy was caused by increased muscle fibre dimensions rather than hyperplasia or an increased frequency of naturally occurring large (type 2B) fibres. Furthermore, FHL1-induced hypertrophy was associated with a conversion to oxidative fibre-type expression [52].
In addition to these described functions, FHL1 also has the capacity for a role in electrophysiology aspects in excitation. Yang et al. identified FHL1 as an interacting partner of the Shaker-related KCNA5 in human atrium, and characterized the interaction in CHO cells [62]. As evidenced from patch clamp recordings of K+ current, FHL1 was capable of modulating KCNA5 activity, by increasing K+ current density, altering channel gating and enhancing slow inactivation [62].
FHL1 and its transcriptional regulation functions in cancer
FHL1 is down-regulated in a variety of cancers as evidenced by comparative microarray profiling and immunohistochemical analysis of human clinical samples. Reduced expression has been identified in lung, prostate, breast, ovarian, colon, thyroid, brain, renal, liver, gastric and skin cancers and melanomas [67–76]. In 115 pairs of hepatocellular carcinoma and matched non-tumour liver tissues, for instance, FHL1 expression was detected in 93.0% of normal tissues and only in 30.4% of cancerous tissues [77]. Similarly, from 46 pairs of breast tumour and non-tumour breast tissues, 91.3% of normal tissue and 30.4% of cancerous tissue were positive for FHL1 [78]. In addition, the expression of FHL1 positively correlated with the expression of epidermal growth factor 2, a prognostic biomarker in breast cancer [78]. In primary gastric cancer patients, a significantly shorter survival was observed from patients with lower FHL1 expression levels [71]. Furthermore, FHL1 suppression appeared greatest in widely invasive and metastatic cases [69–71, 79].
Analogous to these findings, FHL1 was identified as a tumour suppressor gene, which acts to inhibit non-anchored cell growth and migration, downstream of Src- and Crk-associated substrate (Cas) signalling (Fig. 4A) [70]. In Src-transformed cells however, the membrane bound Src tyrosine kinase phosphorylated Cas, a focal adhesion adaptor protein, to suppress FHL1 expression and promote anchorage independence and motility, hallmarks of tumour cell growth [70]. The potential for MAPK, a kinase activated by Src, involvement in FHL1 suppression was eliminated when transformed cells incubated with an MEK blocker failed to express FHL1 [80]. In FHL1 overexpressing cells, colonization was inhibited, whereas non-anchored growth and migration was enhanced [70]. In addition, FHL1 production was induced almost twofold in Src-transformed cells following con tact by adjacent non-transformed cells [70, 81]. Expression of FHL1 also induced expression of the Serum deprivation response protein in Src-transformed cells, by approximately 17-fold [80]. The expression of Serum deprivation response protein, a protein associated with cell growth arrest, was also down-regulated in Src-transformed cells and induced upon contact normalization [70, 81]. Furthermore, FHL1 gene silencing was induced by hyper-methylation of its promoter region, a CpG rich region [80]. Thus, diminished FHL1 expression likely attributes to poorer survival via heightened biological aggressiveness of the tumour [71].
Fig 4.
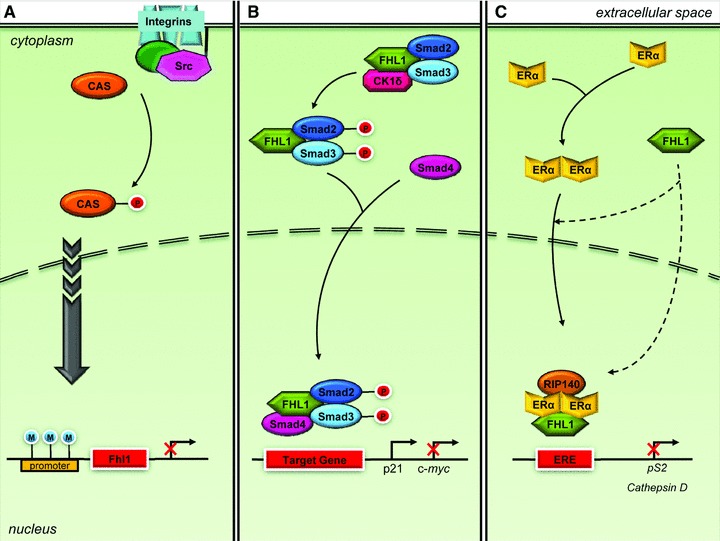
FHL1 and signaling pathways in cancer. (A) Src phosphorylates Cas to suppress FHL1 expression to promote non-anchored tumour cell growth and migration, which would otherwise be inhibited by FHL1. Hypermethylation of the promoter region can induce FHL1 gene silencing. (B) FHL1 inhibits tumour cell growth by transcriptional regulation of TGF-β-responsive genes, although independent of TGF-β and TGF-β receptor. FHL1 phosphorylates cytoplasmic Smad2 and Smad3 through interaction with CK1δ, and facilitates interaction with Smad4. Following nuclear translocation, the FHL1/Smad2/3/4 complex regulates TGF-β responsive gene transcription. (C) FHL1 also inhibits tumour cell growth by co-repressing ER transcriptional activity. FHL1 interacts with Erα, either before or after nuclear translocation, and interacts with RIP140 for synergistic transcription inhibition.
However, in human hepatocellular liver carcinoma cells cultured in vitro or injected into nude mice, expression of FHL1 decreased cancer cell growth. Increased FHL expression also inhibited anchorage-independent growth of hepatoma cells in vitro. The opposite effects were observed with siRNA-mediated knockdown of FHL1. The suppression of tumour growth was mediated via interaction with SMAD proteins for a transforming growth factor (TGF) b-like response (Fig. 4B). FHL1 interacted with SMAD2 and SMAD3 in the cytoplasm, and enhanced phosphorylation of the SMAD proteins through CK1β. Furthermore, FHL1 promoted interaction with SMAD4 and translocation into the nucleus, where it stimulated expression of growth inhibitor genes, such as the CDK inhibitor p21, and suppression of the growth promoting gene c-myc. An identical gene expression pattern was observed in tumours obtained from inoculated nude mice. In patient samples, FHL1 expression was down-regulated in liver tumours, which correlated positively with Smad2/3 phosphorylation, nuclear accumulation of Smad2–4, and p21 expression and correlated negatively with c-myc levels. Although FHL1 enhanced TGF-β responsive transcription, it actually functions independent of TGF-β and the TGF-β receptor. The interaction with CK1β and Smad4, however, was required for FHL1 mediated TGF-β-responsive gene expression and decreased cancer cell growth. Because CK1β mediated differential phosphorylation of the Smad3 that occurs in the TGF-β pathway, FHL1 may regulate additional targets not affected by TGF-β[77].
Similarly, transfection of FHL1 in breast cancer cells also inhibited cell growth, both anchorage-dependent and -independent. Conversely, siRNA mediated silencing of FHL1 significantly increased cell growth, and 17β-estradiol exposure only amplified these effects [78]. These results could be mediated by the interaction between FHL1 and estrogen receptors α or β in breast cancer cells, which occurs independent of 17β-estradiol (Fig. 4C) [78]. FHL1 acts as a negative regulator of estrogen receptor mediated transcription, subsequently repressing expression of estrogen-responsive genes, such as pS2 and cathepsin D [78]. In human breast cancer SKBR3 cells, which lack endogenous estrogen receptor α, FHL1 failed to inhibit target protein expression [78]. Thus, FHL1 acts through the estrogen receptors to repress transcription. Furthermore, FHL1 likely mediates its co-repressor effects by inhibiting the binding of estrogen receptor α to estrogen responsive elements present in the promoter of estrogen responsive genes [78]. Abrogation of FHL1 expression enhanced estrogen signalling, as evidenced from estrogen responsive element driven luciferase assays [82]. When FHL1 is co-expressed with receptor interacting protein of 140 kDa, a cofactor also capable of inhibiting estrogen receptor α, a synergistic inhibition of estrogen-responsive target gene transcription occurs [82].
In contrast to the suppression of FHL1 in many cancers and the phenotype rescue by ectopic FHL1 expression in tumour cells, FHL1 is aberrantly expressed in most T cell acute lymphoblastic leukaemia cell lines, particularly those exhibiting deregulated TLX1/HOX11 expression [83]. TLX1/HOX11 is a homeodomain protein whose abnormal expression, induced by translocations affecting chromosome 10q24, contributes to the leukaemogenesis of T cell tumours [83, 84]. In murine NIH3T3 fibroblasts, expression of TLX1/HOX11 resulted in abnormal growth patterns in vitro and formed tumours in athymic nude mice [84]. Ectopic expression also transcriptionally activated FHL1, functioning as a concentration-dependent regulator [83, 85]. The need for high TLX1/HOX11 levels to stimulate FHL1 expression could explain the lack of consistent FHL1 expression in TLX1/HOX11 positive samples [85]. Regulation of FHL1 expression also required an intact N-terminal transactivation domain and DNA binding capabilities, and was mediated through specific elements located in its proximal promoter [85]. Thus, FHL1 is possibly an important mediator of TLX1/HOX11 function in tumourigenesis.
Perspectives
FHL1 is a LIM-only protein, defined by the tandem arrangement of four and a half LIM domains, and is expressed predominantly in skeletal muscle. Multiple functions have been ascribed to FHL1, belonging to a variety of cellular activities, although the precise molecular mechanisms are poorly characterized. FHL1 is involved in, for instance, sarcomere assembly, cytoskeletal remodelling, biomechanical stress response, muscle hypertrophy and transcriptional regulation. In addition, integrin-mediated, MAPK, β-AR and GPCR signalling pathways are representative of some of the pathway associated with FHL1. The diversity of FHL1’s functional properties may be mediated by the diversity of its interacting partners.
Furthermore, the binding partner specificity of the different LIM domains may contribute to the heterogeneity of FHL1 myopathic phenotypes. Isoform specific effects and possible (skewed) X-inactivation further contribute to the complexity. Twenty-six mutations are causal for six different myopathies, each presenting a combination of various protein aggregates, joint contractures, muscle atrophy/hypertrophy and cardiovascular diseases. This presents complications for diagnostic evaluation screening and genetic counselling of patients or carriers. Alternatively, FHL1 might be an excellent candidate to sequence in unexplained myopathies with an X-linked transmission pattern. In addition, considering the prominence of cardiomyopathies in FHL1 myopathic patients, the FHL1 gene might also be a candidate for hereditary and sporadic cardiomyopathy. The up-regulation of FHL1 expression in numerous cardiovascular diseases, at times even before disease presentation, supports this possibility.
In addition, FHL1 expression is suppressed in cancers and correlates strongly with increased metastatic disease and decreased survival. Diminished FHL1 expression may affect the formation of focal adhesion molecules, thereby contributing to the aggressiveness of cancers. When expressed, FHL1 inhibits tumour cell growth via transcriptional regulation of TGF-β and ER responsive genes. Thus, FHL1 appears to function as a tumour suppressor and regulator of anchorage independence and migration, and tumour cell growth.
In conclusion, FHL1 is a multi-functional protein associated with a variety of disease conditions and responsible for others. Understanding its cellular roles and transcriptional regulation will offer insight into the pathomechanism of diseases and possibly be clinically relevant.
Acknowledgments
Work in our group was supported by the Heart and Stroke Foundation of Ontario (#T-6281), Canadian Institutes of Health Research (MOP-84267), Genome Canada through the Ontario Genomics Institute, Canadian Foundation for Innovation and the Heart and Stroke/Richard Lewar Centre of Cardiovascular Excellence. AOG and TK are Canada Research Chairs and AOG is a New Investigator of the Heart and Stroke Foundation of Canada.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Morgan MJ, Madgwick AJ, Charleston B, et al. The developmental regulation of a novel muscle LIM-protein. Biochem Biophys Res Commun. 1995;212:840–6. doi: 10.1006/bbrc.1995.2045. [DOI] [PubMed] [Google Scholar]
- 2.Morgan MJ, Madgwick AJ. Slim defines a novel family of LIM-proteins expressed in skeletal muscle. Biochem Biophys Res Commun. 1996;225:632–8. doi: 10.1006/bbrc.1996.1222. [DOI] [PubMed] [Google Scholar]
- 3.Lee SM, Tsui SK, Chan KK, et al. Chromosomal mapping, tissue distribution and cDNA sequence of four-and-a-half LIM domain protein 1 (FHL1) Gene. 1998;216:163–70. doi: 10.1016/s0378-1119(98)00302-3. [DOI] [PubMed] [Google Scholar]
- 4.Chan KK, Tsui SK, Lee SM, et al. Molecular cloning and characterization of FHL2, a novel LIM domain protein preferentially expressed in human heart. Gene. 1998;210:345–50. doi: 10.1016/s0378-1119(97)00644-6. [DOI] [PubMed] [Google Scholar]
- 5.Morgan MJ, Madgwick AJ. The LIM proteins FHL1 and FHL3 are expressed differently in skeletal muscle. Biochem Biophys Res Commun. 1999;255:245–50. doi: 10.1006/bbrc.1999.0179. [DOI] [PubMed] [Google Scholar]
- 6.Morgan MJ, Whawell SA. The structure of the human LIM protein ACT gene and its expression in tumor cell lines. Biochem Biophys Res Commun. 2000;273:776–83. doi: 10.1006/bbrc.2000.3006. [DOI] [PubMed] [Google Scholar]
- 7.Taniguchi Y, Furukawa T, Tun T, et al. LIM protein KyoT2 negatively regulates transcription by association with the RBP-J DNA-binding protein. Mol Cell Biol. 1998;18:644–54. doi: 10.1128/mcb.18.1.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee SM, Li HY, Ng EK, et al. Characterization of a brain-specific nuclear LIM domain protein (FHL1B) which is an alternatively spliced variant of FHL1. Gene. 1999;237:253–63. doi: 10.1016/s0378-1119(99)00251-6. [DOI] [PubMed] [Google Scholar]
- 9.Brown S, McGrath MJ, Ooms LM, et al. Characterization of two isoforms of the skeletal muscle LIM protein 1, SLIM1. Localization of SLIM1 at focal adhesions and the isoform slimmer in the nucleus of myoblasts and cytoplasm of myotubes suggests distinct roles in the cytoskeleton and in nuclear-cytoplasmic communication. J Biol Chem. 1999;274:27083–91. doi: 10.1074/jbc.274.38.27083. [DOI] [PubMed] [Google Scholar]
- 10.Ng EK, Lee SM, Li HY, et al. Characterization of tissue-specific LIM domain protein (FHL1C) which is an alternatively spliced isoform of a human LIM-only protein (FHL1) J Cell Biochem. 2001;82:1–10. doi: 10.1002/jcb.1110. [DOI] [PubMed] [Google Scholar]
- 11.Liang L, Zhang HW, Liang J, et al. KyoT3, an isoform of murine FHL1, associates with the transcription factor RBP-J and represses the RBP-J-mediated transactivation. Biochim Biophys Acta. 2008;1779:805–10. doi: 10.1016/j.bbagrm.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 12.Johannessen M, Moller S, Hansen T, et al. The multifunctional roles of the four-and-a-half-LIM only protein FHL2. Cell Mol Life Sci. 2006;63:268–84. doi: 10.1007/s00018-005-5438-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greene WK, Baker E, Rabbitts TH, et al. Genomic structure, tissue expression and chromosomal location of the LIM-only gene, SLIM1. Gene. 1999;232:203–7. doi: 10.1016/s0378-1119(99)00125-0. [DOI] [PubMed] [Google Scholar]
- 14.Fimia GM, De Cesare D, Sassone-Corsi P. A family of LIM-only transcriptional coactivators: tissue-specific expression and selective activation of CREB and CREM. Mol Cell Biol. 2000;20:8613–22. doi: 10.1128/mcb.20.22.8613-8622.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morgan MJ, Madgwick AJ. The fourth member of the FHL family of LIM proteins is expressed exclusively in the testis. Biochem Biophys Res Commun. 1999;255:251–5. doi: 10.1006/bbrc.1999.0180. [DOI] [PubMed] [Google Scholar]
- 16.Newman AP, Acton GZ, Hartwieg E, et al. The lin-11 LIM domain transcription factor is necessary for morphogenesis of C elegans uterine cells. Development. 1999;126:5319–26. doi: 10.1242/dev.126.23.5319. [DOI] [PubMed] [Google Scholar]
- 17.Gupta BP, Wang M, Sternberg PW. The C. elegans LIM homeobox gene lin-11 specifies multiple cell fates during vulval development. Development. 2003;130:2589–601. doi: 10.1242/dev.00500. [DOI] [PubMed] [Google Scholar]
- 18.Pfaff SL, Mendelsohn M, Stewart CL, et al. Requirement for LIM homeobox gene Isl1 in motor neuron generation reveals a motor neuron-dependent step in interneuron differentiation. Cell. 1996;84:309–20. doi: 10.1016/s0092-8674(00)80985-x. [DOI] [PubMed] [Google Scholar]
- 19.Way JC, Chalfie M. mec-3, a homeobox-containing gene that specifies differentiation of the touch receptor neurons in C. elegans. Cell. 1988;54:5–16. doi: 10.1016/0092-8674(88)90174-2. [DOI] [PubMed] [Google Scholar]
- 20.Kadrmas JL, Beckerle MC. The LIM domain: from the cytoskeleton to the nucleus. Nat Rev Mol Cell Biol. 2004;5:920–31. doi: 10.1038/nrm1499. [DOI] [PubMed] [Google Scholar]
- 21.Bach I. The LIM domain: regulation by association. Mech Dev. 2000;91:5–17. doi: 10.1016/s0925-4773(99)00314-7. [DOI] [PubMed] [Google Scholar]
- 22.Zheng Q, Zhao Y. The diverse biofunctions of LIM domain proteins: determined by subcellular localization and protein-protein interaction. Biol Cell. 2007;99:489–502. doi: 10.1042/BC20060126. [DOI] [PubMed] [Google Scholar]
- 23.Labouesse M, Georges-Labouesse E. Cell adhesion: parallels between vertebrate and invertebrate focal adhesions. Curr Biol. 2003;13:R528–30. doi: 10.1016/s0960-9822(03)00448-2. [DOI] [PubMed] [Google Scholar]
- 24.Pomies P, Louis HA, Beckerle MC. CRP1, a LIM domain protein implicated in muscle differentiation, interacts with alpha-actinin. J Cell Biol. 1997;139:157–68. doi: 10.1083/jcb.139.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmeichel KL, Beckerle MC. LIM domains of cysteine-rich protein 1 (CRP1) are essential for its zyxin-binding function. Biochem J. 1998;331:885–92. doi: 10.1042/bj3310885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang DF, Belaguli NS, Iyer D, et al. Cysteine-rich LIM-only proteins CRP1 and CRP2 are potent smooth muscle differentiation cofactors. Dev Cell. 2003;4:107–18. doi: 10.1016/s1534-5807(02)00396-9. [DOI] [PubMed] [Google Scholar]
- 27.Ewing RM, Chu P, Elisma F, et al. Large-scale mapping of human protein-protein interactions by mass spectrometry. Mol Syst Biol. 2007;3:89. doi: 10.1038/msb4100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.High FA, Epstein JA. The multifaceted role of Notch in cardiac development and disease. Nat Rev Genet. 2008;9:49–61. doi: 10.1038/nrg2279. [DOI] [PubMed] [Google Scholar]
- 29.Qin H, Wang J, Liang Y, et al. RING1 inhibits transactivation of RBP-J by Notch through interaction with LIM protein KyoT2. Nucleic Acids Res. 2004;32:1492–501. doi: 10.1093/nar/gkh295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qin H, Du D, Zhu Y, et al. The PcG protein HPC2 inhibits RBP-J-mediated transcription by interacting with LIM protein KyoT2. FEBS Lett. 2005;579:1220–6. doi: 10.1016/j.febslet.2005.01.022. [DOI] [PubMed] [Google Scholar]
- 31.Wang J, Qin H, Liang J, et al. The transcriptional repression activity of KyoT2 on the Notch/RBP-J pathway is regulated by PIAS1-catalyzed SUMOylation. J Mol Biol. 2007;370:27–38. doi: 10.1016/j.jmb.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 32.McGrath MJ, Cottle DL, Nguyen MA, et al. Four and a half LIM protein 1 binds myosin-binding protein C and regulates myosin filament formation and sarcomere assembly. J Biol Chem. 2006;281:7666–83. doi: 10.1074/jbc.M512552200. [DOI] [PubMed] [Google Scholar]
- 33.Brown S, Biben C, Ooms LM, et al. The cardiac expression of striated muscle LIM protein 1 (SLIM1) is restricted to the outflow tract of the developing heart. J Mol Cell Cardiol. 1999;31:837–43. doi: 10.1006/jmcc.1998.0922. [DOI] [PubMed] [Google Scholar]
- 34.Chu PH, Ruiz-Lozano P, Zhou Q, et al. Expression patterns of FHL/SLIM family members suggest important functional roles in skeletal muscle and cardiovascular system. Mech Dev. 2000;95:259–65. doi: 10.1016/s0925-4773(00)00341-5. [DOI] [PubMed] [Google Scholar]
- 35.Loughna PT, Mason P, Bayol S, et al. The LIM-domain protein FHL1 (SLIM 1) exhibits functional regulation in skeletal muscle. Mol Cell Biol Res Commun. 2000;3:136–40. doi: 10.1006/mcbr.2000.0206. [DOI] [PubMed] [Google Scholar]
- 36.McGrath MJ, Mitchell CA, Coghill ID, et al. Skeletal muscle LIM protein 1 (SLIM1/FHL1) induces alpha 5 beta 1-integrin-dependent myocyte elongation. Am J Physiol Cell Physiol. 2003;285:C1513–26. doi: 10.1152/ajpcell.00207.2003. [DOI] [PubMed] [Google Scholar]
- 37.Quinzii CM, Vu TH, Min KC, et al. X-linked dominant scapuloperoneal myopathy is due to a mutation in the gene encoding four-and-a-half-LIM protein 1. Am J Hum Genet. 2008;82:208–13. doi: 10.1016/j.ajhg.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilhelmsen KC, Blake DM, Lynch T, et al. Chromosome 12-linked autosomal dominant scapuloperoneal muscular dystrophy. Ann Neurol. 1996;39:507–20. doi: 10.1002/ana.410390413. [DOI] [PubMed] [Google Scholar]
- 39.Windpassinger C, Schoser B, Straub V, et al. An X-linked myopathy with postural muscle atrophy and generalized hypertrophy, termed, XMPMA, is caused by mutations in FHL1. Am J Hum Genet. 2008;82:88–99. doi: 10.1016/j.ajhg.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schoser B, Goebel HH, Janisch I, et al. Consequences of mutations within the C terminus of the FHL1 gene. Neurology. 2009;73:543–51. doi: 10.1212/WNL.0b013e3181b2a4b3. [DOI] [PubMed] [Google Scholar]
- 41.Schessl J, Zou Y, McGrath MJ, et al. Proteomic identification of FHL1 as the protein mutated in human reducing body myopathy. J Clin Invest. 2008;118:904–12. doi: 10.1172/JCI34450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schessl J, Taratuto AL, Sewry C, et al. Clinical, histological and genetic characterization of reducing body myopathy caused by mutations in FHL1. Brain. 2009;132:452–64. doi: 10.1093/brain/awn325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shalaby S, Hayashi YK, Nonaka I, et al. Novel FHL1 mutations in fatal and benign reducing body myopathy. Neurology. 2009;72:375–6. doi: 10.1212/01.wnl.0000341311.84347.a0. [DOI] [PubMed] [Google Scholar]
- 44.Gueneau L, Bertrand AT, Jais JP, et al. Mutations of the FHL1 gene cause Emery-Dreifuss muscular dystrophy. Am J Hum Genet. 2009;85:338–53. doi: 10.1016/j.ajhg.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Knoblauch H, Geier C, Adams S, et al. Contractures and hypertrophic cardiomyopathy in a novel FHL1 mutation. Ann Neurol. 67:136–40. doi: 10.1002/ana.21839. [DOI] [PubMed] [Google Scholar]
- 46.Shalaby S, Hayashi YK, Goto K, et al. Rigid spine syndrome caused by a novel mutation in four-and-a-half LIM domain 1 gene (FHL1) Neuromuscul Disord. 2008;18:959–61. doi: 10.1016/j.nmd.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 47.Molinari M, Galli C, Piccaluga V, et al. Sequential assistance of molecular chaperones and transient formation of covalent complexes during protein degradation from the ER. J Cell Biol. 2002;158:247–57. doi: 10.1083/jcb.200204122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oyadomari S, Yun C, Fisher EA, et al. Cotranslocational degradation protects the stressed endoplasmic reticulum from protein overload. Cell. 2006;126:727–39. doi: 10.1016/j.cell.2006.06.051. [DOI] [PubMed] [Google Scholar]
- 49.Powell SR. The ubiquitin-proteasome system in cardiac physiology and pathology. Am J Physiol Heart Circ Physiol. 2006;291:H1–H19. doi: 10.1152/ajpheart.00062.2006. [DOI] [PubMed] [Google Scholar]
- 50.Schuberth C, Buchberger A. Membrane-bound Ubx2 recruits Cdc48 to ubiquitin ligases and their substrates to ensure efficient ER-associated protein degradation. Nat Cell Biol. 2005;7:999–1006. doi: 10.1038/ncb1299. [DOI] [PubMed] [Google Scholar]
- 51.Sheikh F, Raskin A, Chu PH, et al. An FHL1-containing complex within the cardiomyocyte sarcomere mediates hypertrophic biomechanical stress responses in mice. J Clin Invest. 2008;118:3870–80. doi: 10.1172/JCI34472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cowling BS, McGrath MJ, Nguyen MA, et al. Identification of FHL1 as a regulator of skeletal muscle mass: implications for human myopathy. J Cell Biol. 2008;183:1033–48. doi: 10.1083/jcb.200804077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schmitt JP, Kamisago M, Asahi M, et al. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science. 2003;299:1410–3. doi: 10.1126/science.1081578. [DOI] [PubMed] [Google Scholar]
- 54.Gramolini AO, Kislinger T, Alikhani-Koopaei R, et al. Comparative proteomics profiling of a phospholamban mutant mouse model of dilated cardiomyopathy reveals progressive intracellular stress responses. Mol Cell Proteomics. 2008;7:519–33. doi: 10.1074/mcp.M700245-MCP200. [DOI] [PubMed] [Google Scholar]
- 55.Gaussin V, Tomlinson JE, Depre C, et al. Common genomic response in different mouse models of beta-adrenergic-induced cardiomyopathy. Circulation. 2003;108:2926–33. doi: 10.1161/01.CIR.0000101922.18151.7B. [DOI] [PubMed] [Google Scholar]
- 56.Yang J, Moravec CS, Sussman MA, et al. Decreased SLIM1 expression and increased gelsolin expression in failing human hearts measured by high-density oligonucleotide arrays. Circulation. 2000;102:3046–52. doi: 10.1161/01.cir.102.25.3046. [DOI] [PubMed] [Google Scholar]
- 57.Hwang DM, Dempsey AA, Wang RX, et al. A genome-based resource for molecular cardiovascular medicine: toward a compendium of cardiovascular genes. Circulation. 1997;96:4146–203. doi: 10.1161/01.cir.96.12.4146. [DOI] [PubMed] [Google Scholar]
- 58.Hwang DM, Dempsey AA, Lee CY, et al. Identification of differentially expressed genes in cardiac hypertrophy by analysis of expressed sequence tags. Genomics. 2000;66:1–14. doi: 10.1006/geno.2000.6171. [DOI] [PubMed] [Google Scholar]
- 59.Lim DS, Roberts R, Marian AJ. Expression profiling of cardiac genes in human hypertrophic cardiomyopathy: insight into the pathogenesis of phenotypes. J Am Coll Cardiol. 2001;38:1175–80. doi: 10.1016/s0735-1097(01)01509-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Towbin JA, Bowles NE. The failing heart. Nature. 2002;415:227–33. doi: 10.1038/415227a. [DOI] [PubMed] [Google Scholar]
- 61.Chen CL, Lin JL, Lai LP, et al. Altered expression of FHL1, CARP, TSC-22 and P311 provide insights into complex transcriptional regulation in pacing-induced atrial fibrillation. Biochim Biophys Acta. 2007;1772:317–29. doi: 10.1016/j.bbadis.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 62.Yang Z, Browning CF, Hallaq H, et al. Four and a half LIM protein 1: a partner for KCNA5 in human atrium. Cardiovasc Res. 2008;78:449–57. doi: 10.1093/cvr/cvn038. [DOI] [PubMed] [Google Scholar]
- 63.Goette A, Lendeckel U, Klein HU. Signal transduction systems and atrial fibrillation. Cardiovasc Res. 2002;54:247–58. doi: 10.1016/s0008-6363(01)00521-1. [DOI] [PubMed] [Google Scholar]
- 64.Kwapiszewska G, Wygrecka M, Marsh LM, et al. Fhl-1, a new key protein in pulmonary hypertension. Circulation. 2008;118:1183–94. doi: 10.1161/CIRCULATIONAHA.107.761916. [DOI] [PubMed] [Google Scholar]
- 65.Robinson PA, Brown S, McGrath MJ, et al. Skeletal muscle LIM protein 1 regulates integrin-mediated myoblast adhesion, spreading, and migration. Am J Physiol Cell Physiol. 2003;284:C681–95. doi: 10.1152/ajpcell.00370.2002. [DOI] [PubMed] [Google Scholar]
- 66.Esposito G, Prasad SV, Rapacciuolo A, et al. Cardiac overexpression of a G(q) inhibitor blocks induction of extracellular signal-regulated kinase and c-Jun NH(2)-terminal kinase activity in in vivo pressure overload. Circulation. 2001;103:1453–8. doi: 10.1161/01.cir.103.10.1453. [DOI] [PubMed] [Google Scholar]
- 67.Bhattacharjee A, Richards WG, Staunton J, et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci U S A. 2001;98:13790–5. doi: 10.1073/pnas.191502998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jiang F, Yin Z, Caraway NP, et al. Genomic profiles in stage I primary non small cell lung cancer using comparative genomic hybridization analysis of cDNA microarrays. Neoplasia. 2004;6:623–35. doi: 10.1593/neo.04142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fryknas M, Wickenberg-Bolin U, Goransson H, et al. Molecular markers for discrimination of benign and malignant follicular thyroid tumors. Tumour Biol. 2006;27:211–20. doi: 10.1159/000093056. [DOI] [PubMed] [Google Scholar]
- 70.Shen Y, Jia Z, Nagele RG, et al. SRC uses Cas to suppress Fhl1 in order to promote nonanchored growth and migration of tumor cells. Cancer Res. 2006;66:1543–52. doi: 10.1158/0008-5472.CAN-05-3152. [DOI] [PubMed] [Google Scholar]
- 71.Sakashita K, Mimori K, Tanaka F, et al. Clinical significance of loss of Fhl1 expression in human gastric cancer. Ann Surg Oncol. 2008;15:2293–300. doi: 10.1245/s10434-008-9904-3. [DOI] [PubMed] [Google Scholar]
- 72.Aldred MA, Huang Y, Liyanarachchi S, et al. Papillary and follicular thyroid carcinomas show distinctly different microarray expression profiles and can be distinguished by a minimum of five genes. J Clin Oncol. 2004;22:3531–9. doi: 10.1200/JCO.2004.08.127. [DOI] [PubMed] [Google Scholar]
- 73.Finley DJ, Zhu B, Barden CB, et al. Discrimination of benign and malignant thyroid nodules by molecular profiling. Ann Surg. 2004;240:425–36. doi: 10.1097/01.sla.0000137128.64978.bc. ; discussion 36–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.LaTulippe E, Satagopan J, Smith A, et al. Comprehensive gene expression analysis of prostate cancer reveals distinct transcriptional programs associated with metastatic disease. Cancer Res. 2002;62:4499–506. [PubMed] [Google Scholar]
- 75.Dhanasekaran SM, Barrette TR, Ghosh D, et al. Delineation of prognostic biomarkers in prostate cancer. Nature. 2001;412:822–6. doi: 10.1038/35090585. [DOI] [PubMed] [Google Scholar]
- 76.Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–52. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 77.Ding L, Wang Z, Yan J, et al. Human four-and-a-half LIM family members suppress tumor cell growth through a TGF-beta-like signaling pathway. J Clin Invest. 2009;119:349–61. doi: 10.1172/JCI35930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ding L, Niu C, Zheng Y, et al. FHL1 interacts with estrogen receptors and regulates breast cancer cell growth. J Cell Mol Med. 2009 doi: 10.1111/j.1582-4934.2009.00938.x. Doi: 10.1111/j.1582-4934.2009.00938.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Finley DJ, Arora N, Zhu B, et al. Molecular profiling distinguishes papillary carcinoma from benign thyroid nodules. J Clin Endocrinol Metab. 2004;89:3214–23. doi: 10.1210/jc.2003-031811. [DOI] [PubMed] [Google Scholar]
- 80.Li X, Jia Z, Shen Y, et al. Coordinate suppression of Sdpr and Fhl1 expression in tumors of the breast, kidney, and prostate. Cancer Sci. 2008;99:1326–33. doi: 10.1111/j.1349-7006.2008.00816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Alexander DB, Ichikawa H, Bechberger JF, et al. Normal cells control the growth of neighboring transformed cells independent of gap junctional communication and SRC activity. Cancer Res. 2004;64:1347–58. doi: 10.1158/0008-5472.can-03-2558. [DOI] [PubMed] [Google Scholar]
- 82.Lin J, Ding L, Jin R, et al. Four and a half LIM domains 1 (FHL1) and receptor interacting protein of 140 kDa (RIP140) interact and cooperate in estrogen signaling. Int J Biochem Cell Biol. 2009;41:1613–8. doi: 10.1016/j.biocel.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 83.Greene WK, Bahn S, Masson N, et al. The T-cell oncogenic protein HOX11 activates Aldh1 expression in NIH 3T3 cells but represses its expression in mouse spleen development. Mol Cell Biol. 1998;18:7030–7. doi: 10.1128/mcb.18.12.7030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lichty BD, Ackland-Snow J, Noble L, et al. Dysregulation of HOX11 by chromosome translocations in T-cell acute lymphoblastic leukemia: a paradigm for homeobox gene involvement in human cancer. Leuk Lymphoma. 1995;16:209–15. doi: 10.3109/10428199509049759. [DOI] [PubMed] [Google Scholar]
- 85.Rice KL, Kees UR, Greene WK. Transcriptional regulation of FHL1 by TLX1/HOX11 is dosage, cell-type and promoter context-dependent. Biochem Biophys Res Commun. 2008;367:707–13. doi: 10.1016/j.bbrc.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 86.Foster LJ, Rudich A, Talior I, et al. Insulin-dependent interactions of proteins with GLUT4 revealed through stable isotope labeling by amino acids in cell culture (SILAC) J Proteome Res. 2006;5:64–75. doi: 10.1021/pr0502626. [DOI] [PubMed] [Google Scholar]
- 87.Cottle DL, McGrath MJ, Wilding BR, et al. SLIMMER (FHL1B/KyoT3) interacts with the proapoptotic protein Siva-1 (CD27BP) and delays skeletal myoblast apoptosis. J Biol Chem. 2009;284:26964–77. doi: 10.1074/jbc.M109.036293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schessl J, Columbus A, Hu Y, et al. Familial reducing body myopathy with cytoplasmic bodies and rigid spine revisited: identification of a second LIM domain mutation in FHL1. Neuropediatrics. 2010;41:43–6. doi: 10.1055/s-0030-1254101. [DOI] [PubMed] [Google Scholar]
- 89.Knoblauch H, Geier C, Adams S, et al. Contractures and hypertrophic cardiomyopathy in a novel FHL1 mutation. Ann Neurol. 2010;67:136–40. doi: 10.1002/ana.21839. [DOI] [PubMed] [Google Scholar]