

Abstract
Paradoxically, not only proteinases but also their inhibitors can correlate with bad prognosis of cancer patients, underlining the evolving concept of the protease web as the complex interplay between proteinases, their inhibitors and effector molecules. Elevated levels of tissue inhibitor of metalloproteinases-1 (TIMP-1) render the liver more susceptible to metastasis by triggering urokinase plasminogen activator (uPA) expression as well as hepatocyte growth factor (HGF) signalling, thereby leading to the fatal scattered infiltration of metastasizing tumour cells throughout the parenchyma of the target organ. Here, we investigated whether host uPA is a crucial protagonist for the TIMP-1-induced modulation of a pro-metastatic microenvironment in the liver. Indeed, in livers of uPA-ablated mice elevated TIMP-1 levels did not trigger HGF signalling and did not promote metastasis of a murine T-lymphoma cell line. In contrast, lack of tumour cell-derived uPA induced by gene silencing did not interfere with this pro-metastatic pathway. Furthermore, host uPA was necessary for the recruitment of neutrophilic granulocytes and the associated increase of HGF in livers with elevated TIMP-1 levels. This newly identified co-operation between TIMP-1 and host uPA suggests that therapies, simultaneously interfering with pro- and anti-proteolytic pathways may be beneficial for patients with metastatic disease.
Keywords: TIMP-1, uPA, HGF, scattering, liver metastasis, protease web
Introduction
Metastasis is the decisive prognostic parameter in almost all malignancies. Unfortunately, surgical, radiological or chemo-therapeutic ablations have turned out to be insufficient strategies to deal with advanced metastatic disease, particularly when the secondary site is affected by scattered tumour cell dissemination. It is now an emerging concept that the formation of a ‘pre-metastatic niche’, characterized by a higher susceptibility to metastasizing tumour cells, is one prerequisite for metastasis [1]. Furthermore, it has been suggested that alteration of the organ homeostasis by an imbalance of proteinases and their inhibitors is important for enabling efficient metastatic colonization of distant organs [2]. Therefore, the protease web can be considered as a major determinant of metastatic priming [3].
At first, it was believed that proteinases promoted metastasis due to their path-clearing function, whereas proteinase inhibitors were considered to be anti-metastatic. However, not all proteinases are pro-metastatic or even exhibit anti-metastatic features so that broad-spectrum proteinase inhibition can also induce metastasis [4]. Due to the complexity of proteinase function as well as the dynamics of the protease web, it is not surprising that in many malignancies also high expression of proteinase inhibitors is associated with poor prognosis. In particular, plasminogen activator inhibitor-1 (PAI-1) [5] and tissue inhibitor of metalloproteinases-1 (TIMP-1) [6] have been found to be negative prognostic factors in a large variety of cancers. Under physiological conditions, these natural inhibitors tightly regulate the proteolytic activity of plasminogen activators (PAs) [7] and matrix metalloproteinases (MMPs) [8], respectively. In addition to its inhibitory actions, TIMP-1 exerts strong growth-promoting, anti-apoptotic and pro-angiogeneic functions [9, 10]. Furthermore, we have previously demonstrated that high TIMP-1 levels in the host, although able to abolish the activity of MMPs over the time course of the experiments, lead to an increase of total metastatic burden in the liver [11]. This promotion of metastasis has mainly been a consequence of massive infiltration and scattering of tumour cells throughout the liver parenchyma and has not been due to increased numbers of larger multicellular foci [11]. The observed scattered metastasis phenotype has been based on a particular pro-metastatic gene expression signature involving proteinases of various families such as the serine proteinase urokinase-type plasminogen activator (uPA) [11] and has been triggered by the induction of hepatocyte growth factor (HGF) signalling, as impaired activation of this signalling pathway leads to a subtotal inhibition of scattered micrometastases [11].
Induction of HGF signalling requires binding of proteolytically activated pro-HGF to its receptor c-Met. uPA not only plays a prominent role in the activation of the ligand [12] which is predominantly stored in the extracellular matrix (ECM) [12] and synthesized by activated fibroblasts and endothelial cells [12] as well as immune cells like neutrophils [13] and macrophages [14]. At least in the case of acute liver and lung injury, uPA has also been shown to induce the recruitment of myeloid leucocytes to secondary sites and to mediate their degranulation [15, 16] leading to increase of available HGF in the liver. Consequently, adenoviral gene transfer of the natural uPA-inhibitor PAI-2 leads to a significant reduction of overall metastatic burden as well as of tumour cell scattering in the liver [17]. This raised the question to which extent uPA was involved in TIMP-1-induced liver metastasis.
To elucidate these functional interrelationships, we exploited the well-established L-CI.5s murine T-cell lymphoma model of liver metastasis [18]. Using uPA-ablated mice, we document here that host uPA largely mediates TIMP-1-triggered HGF signalling and subsequent scattering of L-CI.5s murine lymphoma cells throughout the liver parenchyma. Host uPA is crucial for attracting neutrophils to the liver with elevated TIMP-1 levels, thereby increasing the availability of HGF. In contrast to host uPA, uPA derived from tumour cells plays only a minor role in enabling fatal scattered metastasis, as functional genetic down-regulation of uPA in tumour cells did not impair their scattering throughout their target organ. This emphasizes the outstanding importance of modulations of the microenvironment for amplifying the susceptibility to metastasis.
Materials and methods
Cells and viruses
Human embryonic kidney (HEK) 293 and lacZ-tagged L-CI.5s cells were cultured as described previously [18]. Recombinant adenoviruses coding for human TIMP-1 were generated, purified and plaque titred as illustrated [19]. As control, Addl70–3, an adenovirus without transgene, was used [20]. Functionality of adenoviral constructs was tested in vitro as depicted [19]. Adenoviral infection in vivo was performed as described earlier [21]. To achieve a sustained gene-silencing effect in L-CI.5s cells, oligodesoxynucleotides coding for a shRNA against murine uPA mRNA and a corresponding scrambled sequence (both from Eurogentec Deutschland, Cologne, Germany) were cloned into pSiren-RetroQ retroviral vector (Invitrogen, Karlsruhe, Germany). According to standard protocols, HEK 293 cells were co-transfected with these vectors and the helper plasmids pHIT60 [22] and pHCMV-G [23] (both from Eurogentec Deutschland) to generate retroviruses for the transduction of L-CI.5s cells. By means of a limiting dilution and a simultaneous selection with 20 μg/ml puromycin (Sigma-Aldrich Chemie, Deisenhofen, Germany), clones were raised and the amount of their uPA expression was quantified. Therefore, RNA isolation, reverse transcription and qRT-PCR were performed as shown previously [24] using inventoried primers and probes from Applied Biosystems Applera Deutschland, Frankfurt am Main, Germany. Clones of high uPA down-regulation were pooled to generate a cell line (L-CI.5sshuPA). Analogously, a cell line was generated from clones expressing scrambled shRNA (L-CI.5sshscr).
Experimental metastasis assays
The generation of META/Bomnu/nu mice of variable uPA genotypes was published earlier [25]. For genotyping, DNA from tail biopsies, PCR and qualitative agarose gel electrophoresis were performed in the customary manner. Only immunodeficient knockout and wild-type littermates at an age of 8 to 12 weeks were included in experiments. Animals were injected i.v. 2 × 109 plaque-forming units of AdTIMP-1 and Addl70–3, respectively. Maintenance of the TIMP-1 overexpression until the mice’s killing has been published [11]. Three days later, 5000 L-CI.5s cells were inoculated in the contralateral tail vein. Mice were killed and their livers were removed 6 days later. Wild-type and uPA-ablated META/Bomnu/nu mice were depleted from neutrophils as shown previously [26]. In short, mice were inoculated with 250 μl of anti-Ly-6G rabbit antibody serum (polyclonal; Accurate Chemical & Scientific Corporation, Westbury, NY, USA) 24 hrs and 2 hrs before as well as 5 days after administration of AdTIMP-1, respectively. Three days after TIMP-1 gene transfer, 5000 L-CI.5s cells were inoculated, and another 6 days later, mice were killed and their livers were removed. In a second assay, pathogen-free, athymic CD1nu/nu mice (Charles River, Sulzfeld, Germany) were treated analogously with the exception that 5000 L-CI.5sshuPA or L-CI.5sshscr cells were inoculated. Mice were killed and their livers were removed 6 days later. Different liver tissue samples were snap-frozen in liquid nitrogen for biochemical analysis, sliced for cryo-based histology or stained with 5-bromo-4-chloro-3-indolyl-b-D-galactopyranoside (X-Gal; Roche Diagnostics, Penzberg, Germany) to detect tumour cells as described previously [18]. Multicellular foci exceeding the cut-off size of 0.2 mm in mean diameter were qualified as ‘macrometastases’. Representative close-up pictures were taken to approximately quantify tumour cell scattering. All animal experiments were done in compliance with the guidelines of the Tierschutzgesetz des Freistaates Bayern.
Immunostaining
To detect invaded macrophages and neutrophils, 5-μm-thick cryo-sections were dried, fixed, blocked and intermittently washed in TBS according to standard protocols. Then incubation was performed for 2 hrs at room temperature with anti-FIRE (clone 6F12; Becton Dickinson, Heidelberg, Germany) and anti-Ly-6G antibodies (clone NIMP-R14; Abcam plc, Cambridge, MA, USA), respectively, both diluted 1:50 in TBS. As secondary system biotinylated rabbit anti-rat immunoglobulin (polyclonal; Dako, Hamburg, Germany) and StreptAB Complex (Dako) was used. Staining was performed with AEC Substrate (Dako). All sections were counterstained with Mayer’s hemalaun (Merck, Darmstadt, Germany) for 1 min. and mounted with Kaiser’s glycerol gelatine (Merck). Immunostaining of phosphorylated c-Met was performed on 5-mm-thick frozen sections with anti-phospho-c-Met antibody (polyclonal; Assay designs, Ann Arbor, MI, USA) as described previously [11]. Simultaneous counterstaining was done with 1 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) (AppliChem, Darmstadt, Germany). Sections were mounted with Moviol (Merck).
Biochemical analysis
RNA isolation from snap-frozen liver samples and reverse transcription, and qRT-PCR quantifying HGF mRNA were performed as depicted previously [24] using inventoried primers and probes from Applied Biosystems Applera Deutschland. Liver protein extraction, purification, assessment of protein concentration and the intermittent washing procedures in TBS containing 0.1% (w/v) Tween-20 (TBST; AppliChem) were performed as shown previously [24]. A total of 75 mg of protein were electrophoresed through 8% (w/v) SDS-PAGE and electroblotted at 250 mA for 2 hrs on nitrocellulose membranes (Amersham Biosciences, Amersham, UK). Blots were blocked with 5% (w/v) skim milk (AppliChem) in TBST overnight at 4°C. Then, for 2 hrs at room temperature, slices were incubated with primary antibodies, diluted in 2.5% (w/v) bovine serum albumin (BSA) in TBST as follows: anti-a-tubulin (mouse; Clone DM1A; Calbiochem Immunochemicals, Darmstadt, Germany) 1:500, anti-β-D-galactosidase (mouse; Clone 200–193; Calbiochem Immunochemicals) 1:500, anti-HGF (goat; polyclonal; R&D Systems, Wiesbaden, Germany) 1:1000 (detecting both complete pro-HGF and the separated HGF α-chain) and anti-phospho-c-Met (rabbit; polyclonal; GenScript, Piscataway, NJ, USA) 1:1000. For 1 hr at room temperature, incubation was performed with HRP-conjugated secondary antibodies, diluted in 2.5% (w/v) BSA in TBST as follows: anti-goat (chicken; polyclonal; R&D Systems) 1:1000, antimouse (donkey; polyclonal; Amersham Biosciences, Amersham, UK) 1:1000 and anti-rabbit (donkey; polyclonal; Amersham Biosciences, Amersham, UK) 1:5000. Visualization was carried out using Lumi-Light Western Blotting Substrate (Roche Diagnostics).
Statistical analysis
Data were analysed using Mann-Whitney rank sum test. P < 0.05 was considered significant.
Results
Decreased TIMP-1-induced tumour cell scattering in uPA knockout mice
To assess whether TIMP-1-induced promotion of metastasis was dependent on uPA expression by the host, AdTIMP-1- and Addl70–3-transduced uPA knockout mice and their wild-type controls were challenged with lacZ-tagged L-CI.5s cells and livers were stained with X-Gal. Lack of host uPA significantly decreased TIMP-1-induced overall metastatic burden (Fig. 1A, B). This effect could be attributed to the inhibition of TIMP-1-induced scattering (Fig. 1C), as the number of macrometastases in mice with elevated TIMP-1 levels was not affected by uPA ablation (Fig. 1D).
Fig 1.
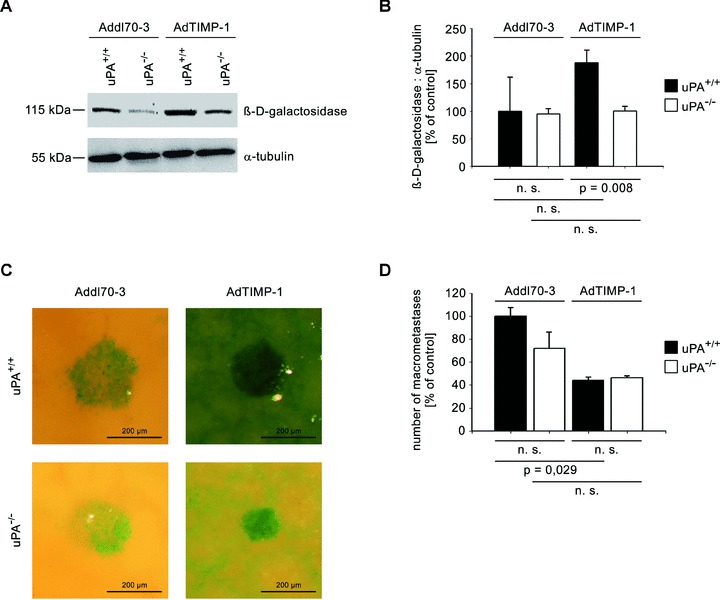
META/Bomnu/nu mice of variable uPA genotype (uPA+/+ and uPA−/−, respectively) were transduced by either AdTIMP-1 or Addl70–3 adenoviruses and 3 days later challenged with lacZ-tagged L-CI.5s cells. Killing and liver removal were performed another 6 days later. (A) Representative Western blot of β-D-galactosidase, the protein encoded by the tumour cell tag lacZ, in liver protein. (B) Densitometry of all performed western blots revealed increased total tumour cell mass in livers with elevated TIMP-1. Loss of host uPA diminished this augmented metastatic burden. Columns: Mean intensities of the β-D-galactosidase bands versusα-tubulin band intensities. The mean of the reference group Addl70–3/uPA+/+ was set as 100%. Bars: S.E. n= 5 mice. Addl70–3/uPA+/+: 100.0% 6 27.7%; Addl70–3/uPA−/−: 95.0%± 4.3%; AdTIMP-1/uPA+/+: 188.1%± 27.7%; AdTIMP-1/uPA−/−: 100.5%± 3.7%. (C) Close-up pictures of surfaces of representative X-Gal-stained metastasis-bearing livers showed increased scattering of tumour cells (indigo-blue signal) in livers with elevated TIMP-1 levels. Loss of host uPA led to a subtotal reduction of this micrometastatic spread. (D) Quantification of macrometastases in X-Gal-stained metastasis-bearing livers revealed a reduction of multicellular foci in livers with elevated TIMP-1 levels. Columns: Mean number of macrometastases. The mean of the reference group Addl70–3/L-CI.5s was set as 100%. Bars: S.E. Addl70–3/uPA+/+: 100.0%± 7.5%, n= 3 mice; Addl70–3/uPA−/−: 71.9%± 14.0%, n= 5 mice; AdTIMP-1/uPA+/+: 44.3%± 2.7%, n= 4 mice; AdTIMP-1/uPA−/−: 46.1%± 1.8%, n= 4 mice.
Reduced TIMP-1-induced HGF signalling in uPA-ablated mice
Next, we investigated whether host uPA was involved in TIMP-1-associated triggering of HGF signalling, which had been shown to account for the increased scattering of tumour cells in the liver (11). The 3-fold TIMP-1-induced increase of phosphorylated c-Met protein (Fig. 2A, B) in uPA wild-type animals was completely abrogated in uPA knockout mice (Fig. 2A, B). When TIMP-1 levels were elevated, prominent phospho-c-Met staining in the liver was detected (Fig. 2C). This induction of HGF signalling in liver compartments affected by metastatic spread was not observed in uPA knockout mice (Fig. 2C).
Fig 2.
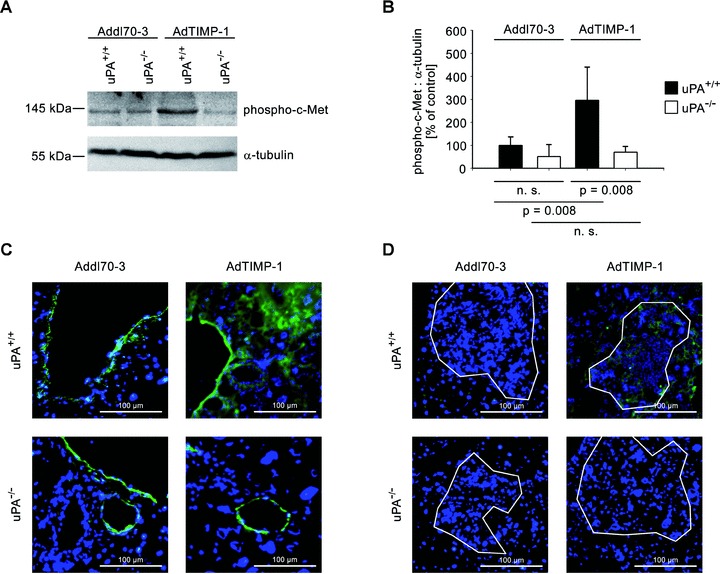
(A) Representative western blot of phosphorylated c-Met in liver protein of META/Bomnu/nu mice (uPA+/+ and uPA−/−, respectively), transduced by either AdTIMP-1 or Addl70–3 adenoviruses. (B) Densitometry of all performed Western blots revealed increased c-Met activation in livers with elevated TIMP-1. Loss of host uPA diminished this induction of HGF signalling. Columns: Mean intensities of the phospho-c-Met bands versus α-tubulin band intensities. The mean of the reference group Addl70–3/uPA+/+ was set as 100%. Bars: S.E. n= 5 mice. Addl70–3/uPA+/+: 100.0%± 16.1%; Addl70–3/uPA−/−: 51.5%± 22.9%; AdTIMP-1/uPA+/+: 296.7%± 63.9%; AdTIMP-1/uPA−/−: 69.9%± 11.2%. (C and D) Representative immunofluorescence analysis of phospho-c-Met (green signal) on cryo-sections (5 μm) of metastasis-bearing bearing livers. Counterstaining was done with DAPI (blue signal). Approximate boundaries of macrometastases were delineated in white. (C) In both Addl70–3- and AdTIMP-1-transduced livers strong staining of phospho-c-Met was found around veins. In livers with elevated TIMP-1, additional c-Met activation in parenchyma was detected. Lack of host uPA did only affect the parenchymatous localization of HGF signalling. (D) Within and around macrometastases, only in livers of uPA wild-type animals with elevated TIMP-1 levels HGF signalling was induced.
Decreased TIMP-1-induced levels of latent and activated HGF in uPA knockout mice
We assessed whether changes of protein levels of pro-HGF and its activated form were detectable depending on different levels of TIMP-1 and presence or absence of host uPA. Indeed, gene transfer of TIMP-1 led to increase of pro-HGF protein by about 2.4-fold (Fig. 3A, B). Ablation of host-derived uPA reduced the levels of pro-HGF by 50% (Fig. 3A, B). A similar effect was observed when the amount of activated HGF (HGF-α) was analysed. Transduction with AdTIMP-1 increased HGF-α protein levels even by 3.2-fold, whereas loss of uPA expression by the host again reduced the levels by 50% (Fig. 3A, C). Quantitative RT-PCR of liver tissue revealed no significant change of HGF mRNA transcription after gene transfer of TIMP-1 (Fig. 3D). Thus, the elevated protein levels of HGF had to derive from HGF storing cells invading the liver.
Fig 3.
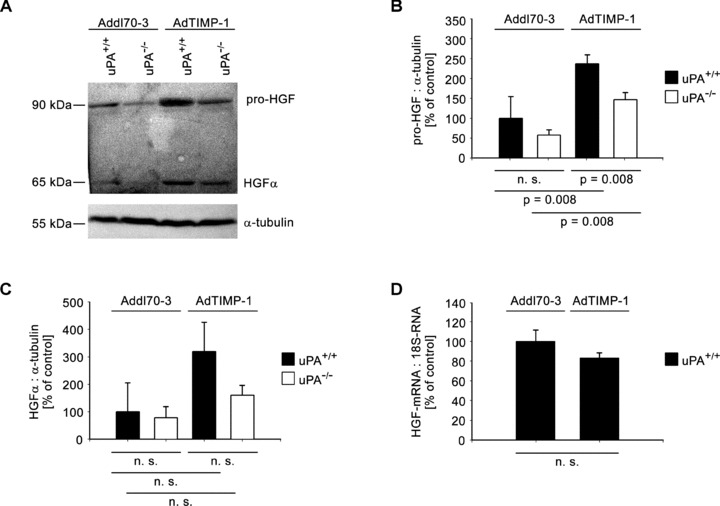
(A) Representative Western blot of pro-HGF and its activated form (HGF-α) in liver protein of META/Bomnu/nu mice (uPA+/+ and uPA−/−, respectively), transduced by either AdTIMP-1 or Addl70–3 adenoviruses. (B and C) Densitometries of all performed Western blots revealed increase of HGF protein levels and its activation in livers with elevated TIMP-1. Loss of host uPA reduced the amounts both of latent and activated HGF. Columns: Mean intensities of either pro-HGF (B) or HGF-a bands (C) versusα-tubulin band intensities. The mean of the reference group Addl70–3/uPA+/+ was set as 100%, respectively. Bars: S.E. n= 5 mice. (B) Addl70–3/uPA+/+: 100.0%± 24.5%; Addl70–3/uPA−/−: 58.0%± 5.8%; AdTIMP-1/uPA+/+: 237.3%± 9.8%; AdTIMP-1/uPA−/−: 146.5%± 8.0%. (C) Addl70–3/uPA+/+: 100.0%± 52.9%; Addl70–3/uPA−/−: 78.6%± 20.1%; AdTIMP-1/uPA+/+: 319.2%± 53.2%; AdTIMP-1/uPA−/−: 159.8%± 18.1%. (D) qRT-PCR of HGF mRNA in livers of META/Bomnu/nu wild-type mice. Columns: Mean amount of the HGF mRNA versus 18S-RNA. The mean of the reference group Addl70–3/uPA+/+ was set as 100%. Bars: S.E. n= 3 mice. Addl70–3/uPA+/+ 100.0%± 11.8%; AdTIMP-1/uPA+/+ 83.0 ± 5.5%.
Requirement of host uPA for TIMP-1-induced neutrophil recruitment to the liver
Next, we addressed whether recruitment to the liver of neutrophils and macrophages as known carriers of HGF [13, 14] is affected by the co-operation of TIMP-1 and host uPA. In wild-type mice with elevated TIMP-1 levels macrophage infiltration was augmented by almost 2-fold (Fig. 4A) and a 6-fold elevation of infiltrated neutrophils even was detected (Fig. 4B) in comparison to the null virus control. When uPA was ablated, TIMP-1-associated infiltration of neutrophils was efficiently inhibited (Fig. 4B), whereas the infiltration of macrophages was not affected (Fig. 4A). In order to test whether the contribution of HGF by neutrophils was crucial for the induction of TIMP-1 induced scattering, we depleted neutrophils from uPA wild-type mice. Comparison with uPA knockout littermates revealed a similarly weak c-Met phosphorylation (Fig. 4C) and no induction of scattered liver metastasis (Fig. 4D) upon neutrophil depletion.
Fig 4.
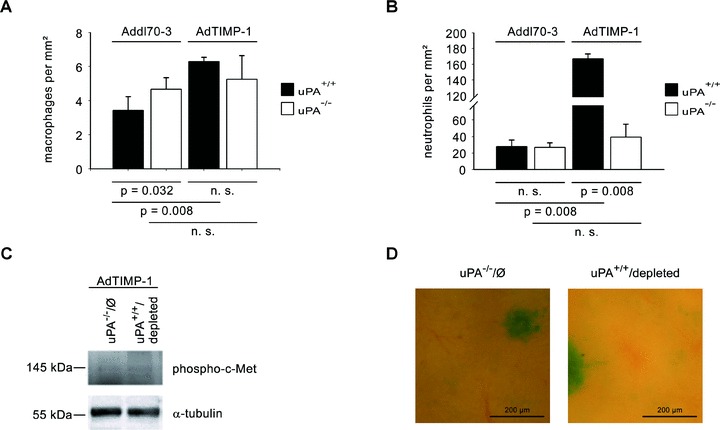
(A and B) Quantification of macrophages (A) and neutrophils (B) in metastasis-bearing bearing livers of META/Bomnu/nu mice (uPA+/+ and uPA−/−, respectively), transduced by either AdTIMP-1 or Addl70–3 adenoviruses, exploiting immunohistochemistry of FIRE (A) and Ly-6G (B) protein, respectively, on liver cryo-sections (5 mm). In livers with elevated TIMP-1 increased infiltration of both macrophages (A) and neutrophils (B) was detected. Loss of host uPA only diminished the number of invaded neutrophils (B), but did not impair macrophage recruitment (A). Columns: Mean number of either invaded macrophages (A) or invaded neutrophils (B) per 1.0 mm2 liver section surface. Bars: S.E. n= 5 mice. (A) Addl70–3/uPA+/+: 3.4 ± 0.4; Addl70–3/uPA−/−: 4.7 ± 0.3; AdTIMP-1/uPA+/+: 6.3 ± 0.1; AdTIMP-1/uPA−/−: 5.3 ± 0.6. (B) Addl70–3/uPA+/+: 27.8 ± 3.5; Addl70–3/uPA−/−: 26.9 ± 2.5; AdTIMP-1/uPA+/+: 167.0 ± 2.7; AdTIMP-1/uPA−/−: 39.1 ± 7.0. (C) Representative Western blot of phosphorylated c-Met in liver protein of META/Bomnu/nu mice (uPA+/+ and uPA−/−, respectively) receiving AdTIMP-1 adenovirus. Analysis revealed similarly weak c-Met phosphorylation in livers of uPA−/−animals without depletion of neutrophils (uPA−/−/Ø) and of neutrophil depleted wild-type littermates (uPA+/+/depleted). (D) Close-up pictures of surfaces of representative X-Gal-stained metastasis-bearing livers of uPA−/−/Ø and uPA+/+/depleted mice, both with elevated TIMP-1 levels. Areas with one large metastasis (indigo-blue) and the immediate environment are shown. Characteristic TIMP-1-induced scattering of tumour cells (indigo-blue signal) in the livers of uPA+/+animals (compare Fig. 1C) is similarly abrogated in conditions without elevated TIMP-1 levels and after depletion of neutrophils.
Unaltered TIMP-1-induced scattering of tumour cells with uPA knockdown
We investigated also whether uPA derived from the tumour cells might play an analogue role in TIMP-1-induced scattered metastasis. For this purpose, we first inoculated AdTIMP-1 or Addl70–3 adenoviruses, respectively, in wild-type mice. Three days later the animals were challenged with tumour cells showing a highly efficient shRNAi-based knockdown of uPA (L-CI.5sshuPA) (Fig. 5A). As control, L-CI.5s cells transduced with a scrambled vector were used (L-CI.5sshscr). Lack of uPA expression by tumour cells did not significantly reduce TIMP-1-associated increase of overall metastatic burden (Fig. 5B, C) and did not inhibit TIMP-1-induced tumour cell scattering throughout the liver parenchyma (Fig. 6A). Immunofluorescence analysis of liver sections revealed that TIMP-1-induced c-Met phoshorylation was not impaired by lack of tumour cell-derived uPA (Fig. 6B). Quantification of multicellular foci on X-Gal-stained liver surfaces showed decreased numbers of macrometastases, independent of elevated TIMP-1 levels (Fig. 5D).
Fig 5.
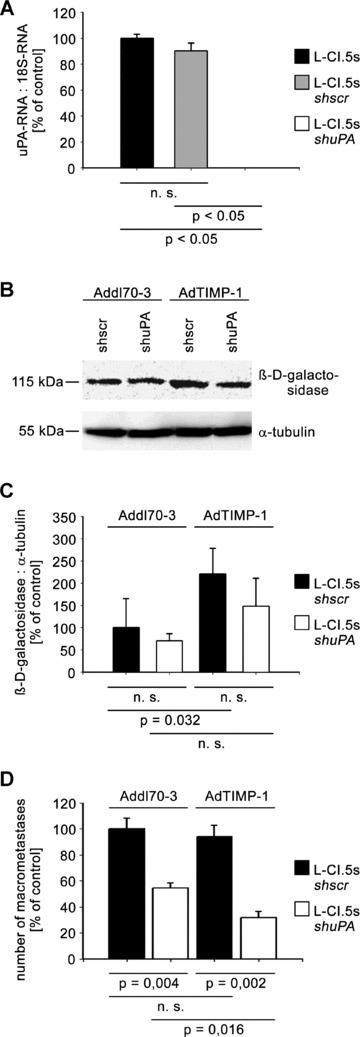
(A) Exploiting shRNAi-based knockdown strategy, lacZ-tagged L-CI.5s cells with a highly efficient uPA-knockdown under the PCR detection limit (L-CI.5sshuPA) were generated as verified by qRT-PCR analysis. Analogue transduction of non-template cDNA resulted in a widely unchanged uPA expression by tumour cells (L-CI.5sshscr). Columns: Mean amount of the uPA mRNA versus 18S-RNA. The mean of the reference group L-CI.5s was set as 100%. Bars: S.E. n= 2 cell pools. L-CI.5s 100.0%± 1.5%; L-CI.5sshscr 90.3 ± 3.0%; L-CI.5sshuPA 0.0 ± 0.0%. (B and C) CD1nu/nu mice were transduced by either AdTIMP-1 or Addl70–3 adenoviruses and 3 days later challenged with L-CI.5s cells either displaying high or low levels of uPA expression. Killing and liver removal were performed another ± days later. (B) Representative Western blot analysis of b-D-galactosidase in liver protein. (C) Densitometry of all performed Western blots revealed an increase of total tumour cell mass in livers with elevated TIMP-1. Lack of tumour cell uPA did not significantly diminish this augmented metastatic burden. Columns: Mean intensities of the β-D-galactosidase bands versusα-tubulin band intensities. The mean of the reference group Addl70–3/uPA+/+ was set as 100%. Bars: S.E. n= 5 mice. Addl70–3/uPA+/+: 100.0%± 29.3%; Addl70–3/uPA−/−: 70.6%± 7.0%; AdTIMP-1/uPA+/+: 220.9%± 5.5%; AdTIMP-1/uPA−/−: 148.3%± 28.1%. (D) Quantification of macrometastases revealed TIMP-1-independent reduction of multicellular foci in livers by lack of tumour cell-derived uPA. Columns: Mean number of macrometastases. The mean of the reference group Addl70–3/L-CI.5sshscr was set as 100%. Bars: S.E. Addl70–3/L-CI.5sshscr: 100.0%± 8.5%, n=± mice; Addl70–3/L-CI.5sshuPA: 54.5%± 3.8%, n= 5 mice; AdTIMP-1/L-CI.5sshscr: 94.3%± 8.4%, n= 8 mice; AdTIMP-1/L-CI.5sshuPA: 31.8%± 4.8%, n= 5 mice.
Fig 6.
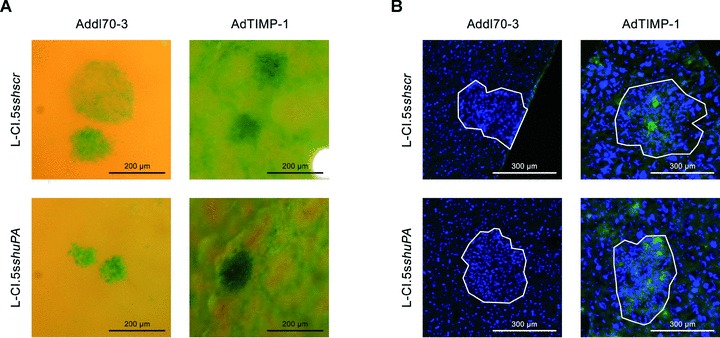
(A) Close-up pictures of surfaces of representative X-Gal-stained metastasis-bearing livers of CD1nu/nu mice, transduced by either AdTIMP-1 or Addl70–3 adenoviruses, showed increased scattering of tumour cells (indigo-blue signal) in livers with elevated TIMP-1 levels. Lack of tumour cell uPA did not reduce micrometastatic spread. (B) Representative immunofluorescence analysis of phospho-c-Met (green signal) on cryo-sections (5 μm) of metastasis-bearing livers. Counterstaining was done with DAPI (blue signal). Approximate boundaries of macrometastases were delineated in white. In both Addl70–3- and AdTIMP-1-transduced livers strong staining for phospho-c-Met was found within and around macrometastases. Lack of tumour cell expression of uPA did not impair this induction of HGF signalling.
Discussion
In this study, we demonstrate that host-derived uPA mediates the recently discovered [11] effect of elevated TIMP-1 levels in inducing HGF signalling-dependent promotion of liver metastasis. Host uPA is here found to be a crucial co-factor for the characteristic accumulation of available HGF in livers with elevated TIMP-1 levels, as it is necessary for the attraction of neutrophils. We further show that host but not tumour cell-derived uPA participates in TIMP-1-induced c-Met phosphorylation in the liver, previously shown to lead to the increased susceptibility of the liver for metastasizing tumour cells [11].
The recognition of the protease web as the complex network regulating tissue homeostasis in physiological and pathophysiological conditions [27] motivated our search for non-canonical interactions among its various components. This, together with the evidence that the initial axiom that proteinases strictly act as pro-metastatic factors whereas their inhibitors exert anti-metastatic features cannot be maintained any more [3], led to our aim to get functional insight into possible pro-metastatic interrelationships between uPA and elevated TIMP-1 levels. Indeed, simultaneous overexpression of TIMP-1 and uPA has been observed in several tumours including breast cancer [28], colorectal cancer [29] and myeloproliferative disorders [30]. Furthermore, decreased TIMP-1-induced liver metastasis after functional-genetic up-regulation of PAI-2 has been the first but indirect evidence for a functional interplay between TIMP-1 and PAs [17]. Here, we determine a direct functional coherence between these two factors elucidating that uPA mediates TIMP-1-triggered c-Met phosphorylation and subsequent tumour cell scattering throughout the liver parenchyma.
In fact, HGF, originally also described as the scatter factor [31], induces a plethora of pro-metastatic genes via the interaction with its receptor c-Met [32]. uPA plays a pivotal role in initiating these intracellular signalling cascades via c-Met as it positively regulates HGF at several levels: uPA is able to induce the expression of HGF [33], to release sequestered pro-HGF from the ECM [34] and to activate it subsequently by limited cleavage [12]. Furthermore, uPA participates in mechanisms attracting to distant organs myeloid leucocytes, cells known to produce and intracellularly store HGF [13, 14], and inducing the release of HGF by degranulation [15, 16]. The present study demonstrates that host uPA is crucial for the infiltration of neutrophils, but not of macrophages, into livers with elevated TIMP-1 levels. This correlated with increased c-Met phosphorylation and subsequent tumour cell scattering throughout the parenchyma.
With regard to tumour progression, a first hint at the overall significance of neutrophils as a main source of HGF has been the recent observation that in non-Hodgkin’s lymphoma the number of neutrophils correlates with the amount of HGF protein [35]. Here, we confirm the importance of infiltrating neutrophils for TIMP-1-triggered progression of metastasis. The fact that elevated TIMP-1 levels do not induce transcription of HGF mRNA in the liver tissue although the amount of HGF protein is increased, indicates that HGF storing cells must infiltrate the liver. Although TIMP-1 has recently been demonstrated to have a certain chemotactic function on neutrophils in myocarditis [36], we provide here for the first time, in the context of metastasis, the evidence that elevated TIMP-1 levels indeed trigger infiltration of HGF storing cells and that uPA mediates this pro-metastatic function of TIMP-1. Neutrophils are found to be the crucial contributors of HGF, as their depletion inhibits TIMP-1-induced c-Met phosphorylation and subsequent tumour cell scattering throughout the liver. The fact that uPA ablation is sufficient to preclude neutrophil attraction to the liver as well as the scattered metastasis phenotype, clearly shows that host uPA is the pivotal mediator of this TIMP-1-triggered effect. This study underlines the eminent role of this proteinase in the TIMP-1-modulated pro-metastatic microenvironment and suggests a link between imbalances of the protease web and the collaborative interactions among metastasizing tumour cells and invading immune cells [37]. Furthermore, these findings stress the importance of the cell signalling function of TIMP-1 [9], whose elevated levels drastically alter the gene expression signature, including the expression of uPA via a yet unknown pathway, in the liver as target organ of metastasis [11].
In several experimental metastasis models, e.g. of breast cancer [38], melanoma [39] and prostate cancer [40], down-regulation of uPA expression in tumour cells has also inhibited metastasis. Therefore, one could suggest that tumour cell-derived uPA was similarly necessary for TIMP-1-induced scattered liver metastasis. However, we document here that neither tumour cell scattering throughout the liver nor c-Met phosphorylation are affected by lack of tumour cell-derived uPA. Nevertheless, we find that lack of tumour cell-derived uPA decreases the number of larger metastatic colonies in the liver. All these findings are in line with ample experimental evidence attributing a predominant role to host-derived uPA in the formation of primary tumours [41] and larger metastatic foci [25, 42]. The observed decrease in the number of macrometastases is independent of modified TIMP-1 levels, reproducing the known effect that uPA expression by tumour cells determines to a certain extent their metastatic potential [39]. Host uPA, however, crucially participates in mediating HGF signalling in the TIMP-1-modulated pro-metastatic liver microenvironment, thereby preparing the soil for the effective scattered infiltration of the target organ.
Altogether, we elucidate for the first time a functional interplay between an activator (uPA) and an inhibitor (TIMP-1) of two different proteinase families. This interrelation between uPA and TIMP-1, pro- and anti-proteolytic protagonists initially assumed to functionally counteract each other in the overall proteolytic balance, illustrates the enormous intricacy of the protease web. It is important to note that the present study dissects the mechanistic details in a murine model. However, as both molecules were up-regulated in livers of patients with bad prognosis [11] it is possible that they have a similar role in the promotion of metastasis in human disease. This mechanism could represent one of the detrimental side effects that caused the clinical failure of broad-spectrum MMP inhibitors, even more as treatment with batimastat, a pharmaceutical of this group, has been shown to increase host uPA expression [43]. Consequently, simultaneous interference with pro- and anti-proteolytic systems may be a quite unexpected but possibly viable approach in the treatment of malignancies. Moreover, the awareness that uPA mediates TIMP-1-induced HGF signalling suggests that also triggered effector molecule pathways should be included in the therapeutic spectrum. In synopsis, a non-canonical combination of different targeted therapies may enable us to fight cancer more efficiently.
Acknowledgments
We thank Katja Honert, Mareike Lehnhoff and Dr. Susanne Schaten (all from Technische Universität, Munich, Germany) for their expert technical support. Financial support (to A.K.): European Union Framework Programme 6, project LSHC-CT-2003–503297, Cancerdegradome, Framework Programme 7 project HEALTH-2007–201279, Microenvimet and grant KR2047/1–1 of the Deutsche Forschungsgemeinschaft.
Conflict of interest
No conflicts of interest are declared.
References
- 1.Kaplan RN, Rafii S, Lyden D. Preparing the “soil”: the premetastatic niche. Cancer Res. 2006;66:11089–93. doi: 10.1158/0008-5472.CAN-06-2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gupta GP, Massagué J. Cancer metastasis: building a framework. Cell. 2006;127:679–95. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 3.Duffy MJ, McGowan PM, Gallagher WM. Cancer invasion and metastasis: changing views. J Pathol. 2008;214:283–93. doi: 10.1002/path.2282. [DOI] [PubMed] [Google Scholar]
- 4.Folgueras AR, Pendás AM, Sánchez LM, et al. Matrix metalloproteinases in cancer: from new functions to improved inhibition strategies. Int J Dev Biol. 2004;48:411–24. doi: 10.1387/ijdb.041811af. [DOI] [PubMed] [Google Scholar]
- 5.Andreasen PA. PAI-1 – a potential therapeutic target in cancer. Curr Drug Targets. 2007;8:1030–41. doi: 10.2174/138945007781662346. [DOI] [PubMed] [Google Scholar]
- 6.Würtz SØ, Schrohl AS, Mouridsen H, et al. TIMP-1 as a tumour marker in breast cancer – an update. Acta Oncol. 2008;47:580–90. doi: 10.1080/02841860802022976. [DOI] [PubMed] [Google Scholar]
- 7.Myöhänen H, Vaheri A. Regulation and interactions in the activation of cell-asociated plasminogen. Cell Mol Life Sci. 2004;61:2840–58. doi: 10.1007/s00018-004-4230-9. [DOI] [PubMed] [Google Scholar]
- 8.Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases. structure, function, and biochemistry. Circ Res. 2003;92:827–39. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- 9.Chirco R, Liu XW, Jung KK, et al. Novel functions of TIMPs in cell signalling. Cancer Met Rev. 2006;25:99–113. doi: 10.1007/s10555-006-7893-x. [DOI] [PubMed] [Google Scholar]
- 10.Stetler-Stevenson WG. Tissue inhibitors of metalloproteinases in cell signalling: metalloproteinase-independent biological activities. Sci Signal. 2008;1:re6. doi: 10.1126/scisignal.127re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kopitz C, Gerg M, Bandapalli OR, et al. Tissue inhibitor of metalloproteinase-1 promotes liver metastasis by induction of hepatocyte growth factor signalling. Cancer Res. 2007;67:8615–23. doi: 10.1158/0008-5472.CAN-07-0232. [DOI] [PubMed] [Google Scholar]
- 12.Naldini L, Tamagnone L, Vigna E, et al. Extracellular proteolytic cleavage by urokinase is required for activation of hepatocyte growth factor/scatter factor. EMBO J. 1992;11:4825–33. doi: 10.1002/j.1460-2075.1992.tb05588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grenier A, Chollet-Martin S, Crestani B, et al. Presence of a mobilizable intracellular pool of hepatocyte growth factor in human polymorphonuclear neutrophils. Blood. 2002;99:2997–3003. doi: 10.1182/blood.v99.8.2997. [DOI] [PubMed] [Google Scholar]
- 14.Armbrust T, Batusic D, Xia L, et al. Early gene expression of hepatocyte growth factor in mononuclear phagocytes of rat liver after administration of carbon tetrachloride. Liver. 2002;22:486–94. doi: 10.1034/j.1600-0676.2002.01731.x. [DOI] [PubMed] [Google Scholar]
- 15.Abraham E, Gyetko MR, Kuhn K, et al. Urokinase-type plasminogen activator potentiates lipopolysaccharide-induced neutrophil activation. J Immunol. 2003;170:5644–51. doi: 10.4049/jimmunol.170.11.5644. [DOI] [PubMed] [Google Scholar]
- 16.Bezerra JA, Currier AR, Melin-Aldana H, et al. Plasminogen activator direct reorganization of the liver lobule after acute injury. Am J Pathol. 2001;158:921–9. doi: 10.1016/S0002-9440(10)64039-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kopitz C, Gerg M, Gänsbacher B, et al. Plasminogen activator inhibitor-2, but not cystatin C, inhibits the prometastatic activity of tissue inhibitor of metalloproteinases-1 in the liver. Hum Gene Ther. 2008;19:1039–49. doi: 10.1089/hum.2008.078. [DOI] [PubMed] [Google Scholar]
- 18.Krüger A, Schirrmacher V, Von Hoegen P. Scattered micrometastases visualized at the single-cell level: detection and re-isolation of lacZ-labeled metastasized lymphoma cells. Int J Cancer. 1994;58:275–84. doi: 10.1002/ijc.2910580222. [DOI] [PubMed] [Google Scholar]
- 19.Hitt M, Bett AJ, Addison CL. Techniques for human adenovirus vector construction and characterization. In: Adolph KW, et al., editors. Viral gene techniques. San Diego: Academic Press; 1995. pp. 13–30. [Google Scholar]
- 20.Jones N, Shenk T. Isolation of deletion and substitution mutants of adenovirus type 5. Cell. 1978;13:181–8. doi: 10.1016/0092-8674(78)90148-4. [DOI] [PubMed] [Google Scholar]
- 21.Kopitz C, Anton M, Gänsbacher B, et al. Reduction of experimental human fibrosarcoma lung metastasis in mice by adenovirus-mediated cystatin C overexpression in the host. Cancer Res. 2005;65:8608–12. doi: 10.1158/0008-5472.CAN-05-1572. [DOI] [PubMed] [Google Scholar]
- 22.Soneoka Y, Cannon PM, Ramsdale EE, et al. A transient three-plasmid expression system for the production of high titer retroviral vectors. Nucl Acids Res. 1995;23:628–33. doi: 10.1093/nar/23.4.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yee JK, Miyanohara A, La Porte P, et al. A general method for the generation of high-titer, pantropic retroviral vectors: highly efficient infection of primary hepatocytes. PNAS. 1994;91:9564–8. doi: 10.1073/pnas.91.20.9564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arlt M, Kopitz C, Pennington C, et al. Increase in gelatinase-specificity of matrix metalloproteinase inhibitors correlates with antimetastatic efficacy in a T-cell lymphoma model. Cancer Res. 2002;62:5543–50. [PubMed] [Google Scholar]
- 25.Frandsen TL, Holst-Hansen C, Nielsen BS, et al. Direct evidence of the importance of stromal urokinase plasminogen activator (uPA) in the growth of an experimental human breast cancer using a combined uPA gene-disrupted and immunodeficient xenograft model. Cancer Res. 2001;61:532–7. [PubMed] [Google Scholar]
- 26.Lawson CA, Yan SD, Yan SF, et al. Monocytes and tissue factor promote thrombosis in a murine model of oxygen deprivation. J Clin Invest. 1997;99:1729–38. doi: 10.1172/JCI119337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Auf Dem Keller U, Doucet A, Overall CM. Proteinase research in the era of systems biology. Biol Chem. 2007;388:1159–62. doi: 10.1515/BC.2007.146. [DOI] [PubMed] [Google Scholar]
- 28.Castelló R, Estellés A, Vásquez C, et al. Quantitative real-time reverse transcription-PCR assay for urokinase plasminogen activator, plasminogen activator inhibitor type I, and tissue metalloproteinase inhibitor type i gene expressions in primary breast cancer. Clin Chem. 2002;48:1288–95. [PubMed] [Google Scholar]
- 29.Baker EA, Leaper DJ. The plasminogen activator and matrix metalloproteinase systems in colorectal cancer: relationship to tumour pathology. Eur J Cancer. 2003;39:981–8. doi: 10.1016/s0959-8049(03)00065-0. [DOI] [PubMed] [Google Scholar]
- 30.Jensen MK, Holten-Andersen MN, Riisbro R, et al. Elevated plasma levels of TIMP-1 correlate with plasma suPAR/uPA in patients with chronic myeloproliferative disorders. Eur J Haematol. 2003;71:377–84. doi: 10.1034/j.1600-0609.2003.00096.x. [DOI] [PubMed] [Google Scholar]
- 31.Naldini L, Weidner KM, Vigna E, et al. Scatter factor and hepatocyte growth factor are industinguishable ligands for the MET receptor. EMBO J. 1991;10:2867–78. doi: 10.1002/j.1460-2075.1991.tb07836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benvenuti S, Comoglio PM. The MET receptor tyrosine kinase in invasion and metastasis. J Cell Physiol. 2007;213:316–25. doi: 10.1002/jcp.21183. [DOI] [PubMed] [Google Scholar]
- 33.Bueno M, Salgado S, Beas-Zárate C, et al. Urokinase-type plasminogen activator gene therapy in liver cirrhosis is mediated by collagens gene expression down-regulation and up-regulation of MMPs, HGF and VEGF. J Gene Med. 2006;8:1291–9. doi: 10.1002/jgm.961. [DOI] [PubMed] [Google Scholar]
- 34.Matsuoka H, Sisson TH, Nishiuma T, et al. Plasminogen-mediated activation and release of hepatocyte growth factor from extracellular matrix. Am J Respir Cell Mol Biol. 2006;35:705–13. doi: 10.1165/rcmb.2006-0006OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toyama T, Ido A, Sasak H, et al. Possible involvement of neutrophils in a serum level increase of hepatocyte growth factor in non-Hodgkin’s lymphoma. Oncol Rep. 2005;13:439–44. [PubMed] [Google Scholar]
- 36.Crocker SJ, Frausto RF, Whitmire JK, et al. Amelioration of Coxsackievirus B3-mediated myocarditis by inhibition of tissue inhibitor of metalloproteinase-1. Am J Pathol. 2007;171:1762–73. doi: 10.2353/ajpath.2007.070179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeNardo DG, Johansson M, Coussens LM. Immune cells as mediators of solid tumor metastasis. Cancer Metastasis Rev. 2008;27:11–8. doi: 10.1007/s10555-007-9100-0. [DOI] [PubMed] [Google Scholar]
- 38.Pakneshan P, Szyf M, Farias-Eisner R, et al. Reversal of the hypomethylation status of urokinase (uPA) promoter blocks breast cancer growth and metastasis. J Biol Chem. 2004;279:31735–44. doi: 10.1074/jbc.M401669200. [DOI] [PubMed] [Google Scholar]
- 39.Hearing VJ, Law LW, Corti A, et al. Modulation of metastatic potential by cell surface urokinase of murine melanoma cells. Cancer Res. 1988;48:1270–8. [PubMed] [Google Scholar]
- 40.Pulukuri SMK, Rao JS. Small interfering RNA-directed reversal of urokinase plasminogen activator demethylation inhibits prostate tumour growth and metastasis. Cancer Res. 2007;67:6637–46. doi: 10.1158/0008-5472.CAN-07-0751. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 41.Gutierrez LS, Schulman A, Brito-Robinson T, et al. Tumour development is retarded in mice lacking the gene for urokinase-type plasminogen activator or its inhibitor, plasminogen activator inhibitor-1. Cancer Res. 2000;60:5839–47. [PubMed] [Google Scholar]
- 42.Almholt K, Lund LR, Rygaard J, et al. Reduced metastasis of transgenic mammary cancer in urokinase-deficient mice. Int J Cancer. 2005;113:525–32. doi: 10.1002/ijc.20631. [DOI] [PubMed] [Google Scholar]
- 43.Holst-Hansen C, Low JA, Stephens RW, et al. Increased stromal expression of murine urokinase plasminogen activator in a human breast cancer xenograft model following treatment with the matrix metalloproteinase inhibitor, batimastat. Breast Cancer Res Treat. 2001;68:225–37. doi: 10.1023/a:1012217820507. [DOI] [PubMed] [Google Scholar]