

Abstract
Hormones and their receptors play an important role in the development and progression of breast carcinoma. Although the primary focus has been on oestrogen and oestrogen receptor (ER), androgen, androgen receptor (AR) and its coactivator(s) have been implicated in tumorigenesis of breast carcinoma and warrant further investigation. AR coactivator p44/Mep50 is identified as a subunit of methylosome complex and lately characterized as an AR coactivator that enhances AR mediated transcription activity in a ligand dependent manner. In prostate cancer, p44 is expressed in the nucleus of benign epithelia and translocated into the cytoplasm in cancer cells. Furthermore, nuclear expression of p44 inhibits prostate cancer growth. In this report, we examined the expression and function of p44 in breast cancer. In addition to being an AR coactivator, p44 also functions as an ER coactivator. In contrast to findings in prostate cancer, the expression of p44 shows strong cytoplasmic expression in morphologically normal terminal ductal lobular units, while nuclear p44 is observed in both ductal carcinoma in situ and invasive carcinoma. Further, overexpression of nuclear-localized p44 stimulates proliferation and invasion in MCF7 breast cancer cells in the presence of oestrogen and the process is ERα dependent. These findings strongly suggest that p44 plays a role in mediating the effects of hormones during tumorigenesis in breast.
Keywords: androgen receptor coactivator, breast cancer, p44/mep50
Introduction
Invasive breast cancer is a major cause of death in women, especially those living in developed countries [1]. The majority of invasive breast carcinoma is of ductal type, comprising up to 80% of all cases. Among the unfavourable prognostic predictors, auxiliary nodal status is the most significant, as positive status of nodes dramatically decrease survival rate [2]. Tumorigenesis is a multi-step process, starting from transformation of benign epithelial cells to dysplastic and neoplastic cells, leading to invasion and metastasis. Prior to invasion and metastasis, malignant epithelial cells undergo a process referred to as epithelial–mesenchymal transition [3–5], and result in loss of cell polarity, acquisition of migratory phenotype and breakdown of the basement membrane. Multiple factors, including hormonal signalling, cytokine signalling, altered cell-matrix interactions and subsequent intracellular signalling, play a role in this process [6, 7].
It has been well known that oestrogen and oestrogen receptor (ER)-associated signalling cascades are the important mediators of the development of carcinoma of breast [8]. ER serves as prognostic and predictive markers of breast cancer, and ER coactivators, including SRC3/AIB1, have been linked to breast cancer development [9]. However, the roles of androgen, its receptor and coactivators on the development of breast cancer is a less studied area, although androgen and AR have been implicated to play a role in breast cancer growth by their presence in cancerous breast tissue [10–12] and by their proliferative and growth inhibitory effects in different breast cancer cell lines [13, 14]. Approximately 70% to 90% of primary breast cancers express AR, suggesting that AR may modulate tumorigenesis [11, 12, 15]. The in vitro studies of androgen effect on breast cancer using established cell lines provided controversial results [16]. Androgens inhibit the growth of T-47D, ZR-75–1 and MFM-223 cells at certain concentrations, while promotion of the growth of MCF-7 and EFM-19 cells have also been reported [17–19]. Further, the growth regulatory function of AR is different in the breasts of pre-menopausal and post-menopausal women [13, 20].
Androgens function through binding to androgen receptor (AR) and initiating AR-associated intracellular signalling pathways [21]. Initial binding of androgen to AR and augmentation of AR activity by its coactivator(s) are two critical steps in the AR-mediated pathway [22, 23]. There are a number of tumorigenic mechanisms in breast cancer involving alteration of the AR pathway. Alteration of AR protein structure could be associated with breast cancer [16]. Germ line mutation resulting in androgen insensitivity appears to be associated with an increased risk of male breast cancer development [24, 25]. An exon 3 deletion splice variant has been identified in three human breast cancer cells lines [26]. Analogues to ER, dysregulation of AR coactivator(s) may play a role in the development of breast cancer. It has been suggested that AR coactivator(s), such as ARA70, could function as an ER coactivator, possibly mediating the interplay between AR and ER in breast cancer [27].
p44 is a recently identified AR interacting protein [28]. p44 exhibits a salt-sensitive high affinity binding to AR, and enhances AR driven transcriptional activation. We have previously reported that p44 is expressed as a nuclear protein in benign prostate epithelia and translocates to the cytoplasm in prostate cancer cells [29, 30]. Similar observations are made in benign and malignant testicular cells [31]. Moreover, we show that the nuclear expression of p44 inhibits prostate cancer cell growth both in vitro and in vivo[29, 30]. In this study, we examined the expression of p44 in benign and malignant human ductal epithelia as well as the roles of p44 in growth and invasion in breast cancer cells. Our results indicate distinct patterns of nuclear and cytoplasmic p44 expression and function in breast in contrast to prostate cancer.
Materials and methods
Breast cancer specimen
Thirty-three cases were selected from consecutive surgically removed invasive breast carcinomas at New York University Medical Center. Tissues were formalin-fixed and paraffin-embedded. Sections of tissue (4 μm) were cut and mounted on Superfrost Plus adhesion slides and used for histology and immunohistochemistry.
Immunohistochemistry
The immunohistochemical staining was performed on an automated Ventana machine (Ventana, Tucson, AZ, USA) [30]. Before staining, antigen retrieval was performed by heating the specimens in a microwave oven for 30 min. in citrate buffer (pH 8.0) after dewaxing. An affinity purified rabbit polyclonal anti-p44 antibody was applied to the sections at a 1:100 dilution, and sections were then incubated overnight at 4°C. A streptavidin-biotin peroxidase detection system was used according to the manufacturer’s instruction (DAKO, Carpinteria, CA, USA), with 3,3′-diaminobenzidine as substrate. Pre-immune serum was used as negative control.
Cell culture and construction of MCF7 and MDA-MB-231 cell lines and dual luciferase assays
pBabe, pBabeNLSp44 and pBabeNESp44 retroviral constructs were transfected into phoenix A amphotropic packaging cells (American Type Culture Collection) via Lipofectamine–mediated transfection to produce virus. Following transfection, the retroviral packaging cells are maintained at first at 37°C in RPMI 1640 media with the addition of foetal bovine serum and penicillin/streptomycin for 24 hrs and then incubated for another 24 hrs at 328C to increase viral titre. The virus containing supernatant was collected by centrifugation, filtered and transduced with a mixture of viral supernatant and fresh media at a ratio of 1:4 in the presence of 4 mg/ml of polybrene for 24 to 48 hrs in MCF7 and MDA-MB-231 cells. Single colonies, selected by protein expression on Western blot, or pooled cells after retroviral infection were used in this study to examine growth and invasion.
Luciferase assays were performed as described previously [28]. Briefly, 105 cells were plated per well of 24-well plates approximately 24 hrs before transfection. After being washed with phosphate-buffered saline, cells in each well were transfected with 30 ng of a plasmid expressing AR or ERα, 100 ng of the reporter plasmids (either 4 × ARE or 3 × ERE), 2.5 ng of the pR-LUC internal control plasmid, and different amounts of the p44 expression vector. The total amount of DNA was adjusted to 1 μg with pcDNA3.1. Cells were cultured for another 48 hrs and harvested for the dual luciferase assay (Promega, Madison, WI, USA).
Real Time qRT-PCR and Chromatin immunoprecipitation assays
Total RNA was isolated with RNAqueous®-4PCR kit (Ambion, Austin, TX, USA) following the manufacturer’s instructions. A total of 1 mg RNA was used for reverse transcription. Primers, 5′-GCCACGTCTCCACACATCAG-3′ and 5′-TCTTGGCAGCAGGATAGTCCTT-3′, were used to amplify MYC oncogene. 18S RNA was used as internal control [32]. For chromatin immunoprecipitation [33], 2 mg of anti-p44 antibody and IgG were mixed with 25mg of the cross-linked chromatin and incubated overnight at 4°C. After crosslink reversal and recovery of the immunoprecipitated chromatin DNA, PCR was performed with primers 5′- actctgcactgccagacaaa -3′ and 5′- tggaaaccacattttggtca-3′ corresponding to promoter region from –5 to –224.
Proliferation assay
Cell proliferation was performed in 24-well plates and measured by cell number counting under different growth conditions, hormone free (Phenol red free medium and Charcoal stripped serum), androgen (10 nM R1881) or oestrogen (10 nM 17-β-estradiol) media [34]. For cell counting, cells (1 × 104) were plated into 24-well plates and counted with hemocytometer every other day. The assays were done in triplicate.
Matrigel invasion and migration assays
A total of 750 μl media with chemoattractant (10% fetal bovine serum [FBS]) were added to the lower chamber of BD Biocoat Matrigel Invasion Chamber (BD Bioscience, San Jose, CA, USA) [34]. A cell suspension (5 × 104) in 0.5 ml DMEM with 0.1% BSA was placed on the insert of the 24-well chambers. After 24 hrs of incubation, the non-invading cells on the upper surface of the filter member were removed using a cotton swab. Invasive cells on the lower surface of the filter member were stained via Diff Quik stain and counted under light microscope. The average number of cells from three representative high-power fields (400× magnifications) was recorded.
For migration/wound healing assays, cells were seeded evenly in 12-well plates at the density of 4 3 105/ml. Cells were incubated for 12 to 24 hrs until they became fully confluent. A linear wound was introduced with a 10 μl pipette tip, the plate was washed with PBS, and was replaced with fresh oestrogen medium. The distance that cells migrated was measured. The assays were done in triplicate.
Statistical analysis
The statistical analyses of the above results were performed by pairwise Student’s t-test. Differences are considered statistically significant if P < 0.05.
Results
Expression of p44 in benign and malignant breast cells
We examined the expression profile of p44 in benign and malignant breast ductal cells by immunohistochemical studies (IHC). IHC was performed on formalin-fixed paraffin-embedded breast cancer sections and the staining patterns were measured semi quantitatively (0 as negative, 1+ as weak staining, 2+ as moderate staining, 3+ as strong staining) for its intensity in cytoplasm and nuclei. A value 1+ is considered a positive value. Of 33 cases with invasive carcinoma evaluated, 22 had morphologically normal glandular elements and in situ carcinoma (DCIS) on the same section. In these 22 cases with benign terminal ductal lobular units, p44 was expressed as cytoplasmic protein in all cases from weak (n = 2, 9%), to moderate (n = 9, 41%) to strong (n = 11, 50%) cytoplasmic staining (Fig. 1A), but nuclear localization of p44 observed in rare benign cells. In contrast to benign ductal epithelium, p44 was localized in nuclei of DCIS lesions (Fig. 1B) in 18 of 22 (82%) cases and invasive carcinoma in 28 of 31 (90%) cases. Interestingly, nuclear localization of p44 (Fig. 1C) was more frequently seen in invasive rather than in situ components of the ductal carcinoma of the same patient in 17 of 21 (81%) cases. These findings suggest an association of translocation of p44 from the cytoplasm to nucleus in benign to malignant breast epithelia.
Fig 1.
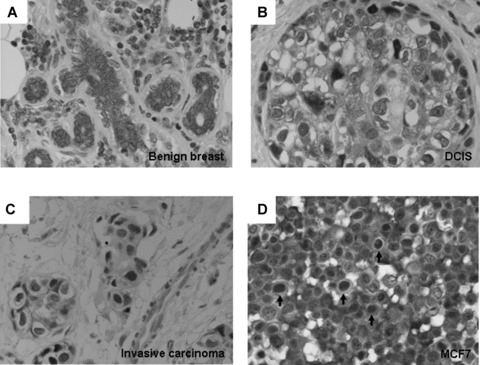
Expression of p44 in benign and malignant breast tissue, Immunohistochemistry showed cytoplasmic expression of p44 in benign breast epithelium (A). Nuclear p44 is observed in DCIS (B) and invasive ductal carcinoma (C). Nuclear p44 is expressed in 25% of MCF7 cells (D). (Magnification: A–D: 400×).
p44 as transcriptional coactivator for oestrogen receptor a
We have shown that p44 increased AR mediated transcriptional activation in a ligand-dependent fashion in prostate cancer PC3 cells. It is of interest to know if p44 can function as an ER coactivator. We previously showed p44 increased ER mediated transcriptional activation only at a minimal level [28]. However, the cell line used in those studies was the prostate cancer cell line, PC3 and not a breast cell line. Thus, we performed dual luciferase assays in breast cancer MCF7 cells to determine AR and ER-mediated transcriptional activation by p44. As shown in Fig. 2A, we were able to consistently achieve at least 2-fold increase in ER mediated transcriptional activation upon transfection with p44 using luciferase reporter genes with three copies of oestrogen responsive element (ERE) in the promoter region [28]. We were also able to increase the levels of AR mediated transcriptional activation by 2.5-fold in MCF7 cells (Fig. 2B) using luciferase reporter genes with four copies of androgen responsive element (ARE) in the promoter region. These results indicate that p44 can function as a transcriptional coactivator for both AR and ER.
Fig 2.
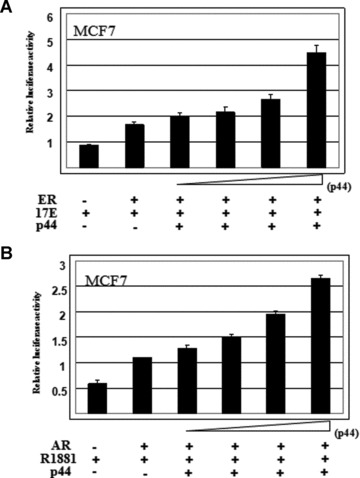
p44 enhances AR and ER mediated transcriptional activation, p44 increased the levels of ER mediated transcriptional activation up to 2-fold (A) and increased the levels of AR mediated transcriptional activation up to 2.5-fold (B) in MCF7 cells in dual luciferase assays. In luciferase assays, the concentration of reagents where appropriate is as follow: androgen (10 nM R1881), oestrogen (10 nM 17-β-estradiol), AR (30 ng), ER (30 ng) and p44 (100 ng, 200 ng, 300 ng and 600 ng).
Establishment of stable clonal MCF7 and MDA-MB-231 cell lines overexpressing nuclear or cytoplasmic p44
To examine the function of p44 in regulating tumour behaviours, stable clonal MCF7 breast cancer cell lines overexpressing nuclear p44 and cytoplasmic p44 were established with p44 fused to a FLAG tag and nuclear localization signal (NLS) resulting pBabeNLSp44 (Fig. 3A, lane 2) or nuclear exporting signal (NES) resulting NESp44 (Fig. 3A, lane 3), respectively. NLSp44 or NESp44, including the FLAG tag, migrate at different rate than endogenous p44 and can be separated. A pooled line and three clonal lines were used in this study. β-actin serves as loading controls (Fig. 3B, lanes 1–3). To confirm correct localization of exogenous p44, western blot analysis was performed with cytoplasmic and nuclear extract from MCF7 cells overexpressing NLSp44 and NESp44. MCF7 overexpressing NESp44 cells showed that both exogenous and endogenous p44 were restricted to the cytoplasm (Fig. 3C, lane 3), while MCF7 cells overexpressing NLSp44 resulted in cytoplasmic and nuclear localization of p44 (Fig. 3C, lanes 5 and 6). The nuclear localization of NLSp44 (Fig. 3D) and cytoplasmic localization of NESp44 (Fig. 3E) was confirmed by immunofluorescent microscopy using EGFP fusion proteins to NLSp44 (EGFP-NLSp44) and NESp44 (EGFP-NESp44) [30]. For MCF7 cells with pBabe control vector, p44 was seen in the cytoplasm but not present in the nuclear fraction since p44 was expressed as nuclear protein in only 25% of MCF7 cells (Fig. 1D, arrows).
Fig 3.
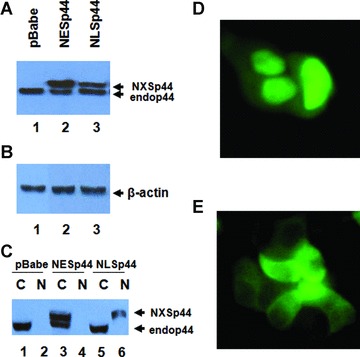
Stable cell lines expressing nuclear and cytoplasmic p44, Clonal and pooled cell lines expressing NLSp44 and NESp44 were established. Western blot analysis of whole cell lysate showed expression of NESp44 (A, lane 2, upper band) and NLSp44 (A, lane 3, upper band). b-actin was used as loading control (B, lanes 1–3). Nuclear and cytoplasmic fraction showed cytoplasmic expression of NESp44 (C, lane 3) and nuclear expression of NLSp44 (C, lane ± versus lane 5). Nuclear expression of EGFP-NLSp44 in MCF7 cells (D). Cytoplasmic expression of EGFP-NESp44 in MCF7 cells (E). NXSp44: either NLS or NES p44. Endop44: endogenous p44. C: cytoplasmic fraction. N: nuclear fraction. The size difference of NXSp44 and Endop44 is due to addition of FLAG tag and localization sequence.
Oestrogen-mediated cell proliferation by nuclear p44 in MCF7 breast cancer cells
To investigate the function of p44 in tumour cell growth, proliferation assays were conducted in MCF7 cells overexpressing NLSp44, NESp44 or pBabe control vector. The assays were performed in hormone free (phenol red free and charcoal stripped FBS) media and media with defined levels of androgen (10 nM R1881) and oestrogen (10 nM 17-β-estradiol) to discern the effects of androgen and oestrogen, since p44 can function as either an AR or ER coactivator. In oestrogen media, NLSp44 stimulated cell proliferation compared to cells expressing NESp44 or control vector (Fig. 4A). This enhanced proliferation was only observed in oestrogen media. Interestingly, p44 inhibited MCF7 cell growth in hormone free (Fig. 4B) and androgen media (data not shown). In contrast, NESp44 decreased the cell proliferation in both hormone free (Fig. 4B) and oestrogen (Fig. 4A) media. To confirm the cell proliferation phenotype of MCF7 cells expressing NLSp44 and NESp44, we performed immunohistochemistry for proliferation index Ki67 marker on these cells. Immunohistochemistry showed statistically significantly increased staining for Ki67 from 87% in MCF7 cells with pBabe control vector to 91% in MCF7 cells expressing NLSp44 (P < 0.04) and decreased staining for Ki67 from 87% in MCF7 cells with pBabe control vector to 79% in MCF7 cells expressing NESp44 (P < 0.003) (Table 1). Because the increased cell growth by p44 is only observed in oestrogen media, we determined whether the enhanced cell proliferation by p44 is oestrogen dependent using an ER– breast cancer cell line MDA-MB-231. In MDA-MB-231 cells, nuclear p44 inhibited and cytoplasmic p44 promoted cancer cell growth, in contrast to MCF7 cells either in the presence (Fig. 4C) or absence (Fig. 4D) of oestrogen.
Fig 4.
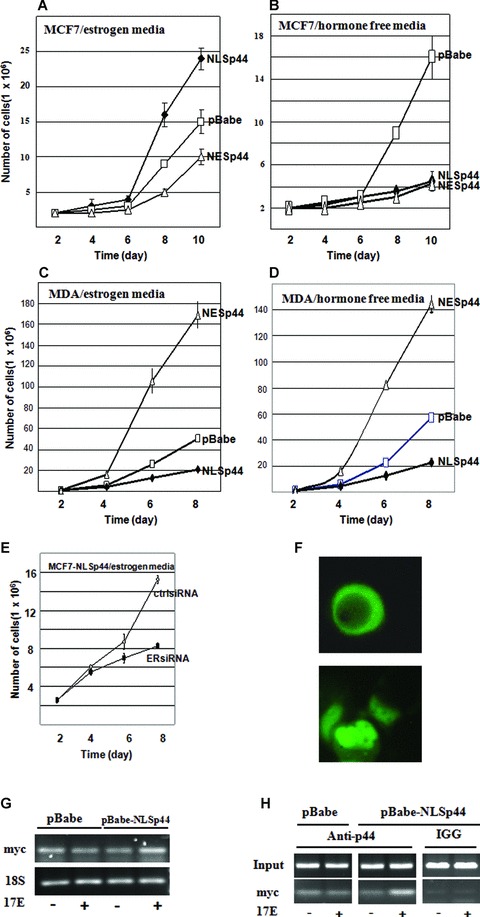
Nuclear p44 stimulates MCF7 breast cancer cell growth through oestrogen pathway, Growth curve of p44 showed increased MCF7 cell proliferation by NLSp44 compared with vector control (A) in oestrogen media. NLSp44 inhibited MCF7 cell growth in hormone free media (B) and NESp44 inhibited MCF7 cell growth in both hormone free (B) and oestrogen (A) media. In MDA-MB-231 cell line, the nuclear p44 inhibits and cytolasmic p44 promotes cancer cell growth both in oestrogen (C) and hormone free (D) media. Nuclear p44 mediated growth promotion is reversed by ERα knockdown in MCF7-NLSp44 cells compared to MCF7-NLSp44 treated with control siRNA (E) in oestrogen media. Cytoplasmic p44 expression in hormone free media (phenol red free and charcoal stripped FBS) (F, upper panel) and nuclear p44 expression in oestrogen media (F, lower panel). Increased ER target gene MYC expression by RT-PCR (G) and enhanced p44 recruitment to MYC promoter by ChIP (H) in the presence of oestrogen in MCF7-NLSp44 cells.
Table 1.
Proliferation index Ki67 in MCF7 cells with NLSp44, NESp44 and pBabe control
| Cells | pBabe | NLSp44 | NESp44 |
|---|---|---|---|
| Ki 67 (%) | 87 ± 2 | 91 ± 1 | 79 ± 1 |
pBabe versus NLSP44, P < 0.04, pBabe versus NESp44, P < 0.003.
To further determine the effects of ERα on the oestrogen-dependent nuclear p44 mediated cell proliferation, we performed ERβ knockdown with siRNA followed by growth kinetic experiments. There is a reduced rate of cell growth with ERα knockdown even in the presence of oestrogen (Fig. 4E). Together with the data that the nuclear p44 only stimulated cell growth in MCF7 (ER+), but not MDA-MB-231 (ER–) cells, there is a strong indication that the nuclear p44 promoted cell proliferation is ERα dependent. We also examined cell cycle gene expression to determine affected pathways involved in nuclear p44 mediated growth promotion by Western blot analysis including p21, p27, cyclin A2, cyclin B1, cyclin D1 and cyclin E. The results revealed increased expression of cyclinB1 (Fig. S1A) when nuclear p44 was overexpressed in MCF7 cells in the presence of oestrogen while the levels of other cyclins remained unchanged (Fig. S1E). The levels of cyclin B1 did not change in MCF7-NLSp44 cells (Fig. S1B). The level of cyclin B1 also did not change in MDA-MB-231 cells in both hormone free and androgen media (Fig. S1C and D), indicating the growth inhibition by nuclear p44 in MDA-MB-231 cells is not through decrease in cyclin B1. The levels of p21 and p27 were not changed (data not shown).
To determine the mechanisms of the p44 enhanced cell growth by oestrogen, we examined p44 nuclear cytoplasmic shuttling by fluorescent microscopy using GFP-p44, as well as promoter occupancy and expression of ER target gene MYC by nuclear p44. P44 was expressed as cytoplasmic protein (25% cells with nuclear p44 expression) under hormone free condition (Fig. 4F upper panel) and as nuclear protein (85% cells with nuclear p44 expression) in the presence of 10 nM 17-β-estradiol (Fig. 4F lower panel). In contrast to MCF7 cells, p44 was localized in both nuclear and cytoplasm of MDA-MD-231 cells. Oestrogen did not affect p44 nuclear localization of MDA-MD-231cells.
We further determined expression of ER target gene MYC using RT-PCR with nuclear p44 overexpression (MCF7-NLSp44 cells) and promoter recruitment of p44 onto MYC promoter using a ChIP analysis. The results of the experiments showed increased MYC expression by RT-PCR (Fig. 4G) and real time qRT-PCR (Fig. S1F) as well as increased promoter occupancy of p44 onto the MYC promoter (Fig. 4H).
Nuclear p44 may promote MCF7 breast cancer cell invasion by in vitro Matrigel assays
To investigate the role of p44 in cell invasion, Matrigel invasion assays were performed with MCF7 cells overexpressing NLSp44, NESp44 or pBabe control. Similarly, we used hormone free, androgen and oestrogen media to define the effects of hormones. The invasiveness of MCF7 cells was significantly enhanced up to 4-fold (P < 0.001) with overexpression of nuclear p44 in the presence of 10 nM oestrogen compared to vector control (Fig. 5B and C). The increased invasion ability was observed in neither hormone free nor androgen media (data not shown). In contrast, NESp44 inhibited invasion up to 2-fold (Fig. 5C) in oestrogen media.
Fig 5.
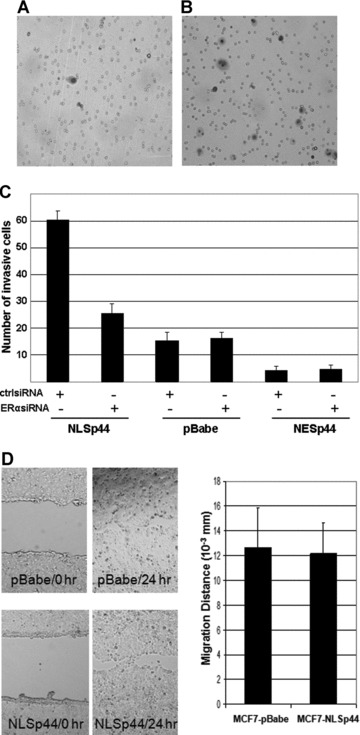
Nuclear p44 promotes MCF7 breast cancer cell invasion through oestrogen pathway, Matrigel invasion assays showed, compared to pBabe control (A), increased invasive cells by NLSp44 (B), up to 4-fold in MCF7 cells. NESp44 moderately decreased invasion ability of MCF7 cells (C). NLSp44 did not alter migration ability of MCF7 cells (D).
To further determine whether increased breast cancer cell invasion ability by nuclear p44 is dependent on ER, we used siRNA to knockdown ERα followed by Matrigel invasion assays. The results showed more than 2-fold decrease in the number of invasive cells for MCF7-NLSp44 cells in the presence of oestrogen (Fig. 5C). The ERα knockdown did not affect the invasion ability of MCF7-pBabe cells (Fig. 5C).
To determine whether the increased invasive ability of MCF7-NLSp44 cells was due to increased cell migration ability, we performed wound healing assays with MCF7-NLSp44 cells in the presence of oestrogen. Cells were seeded evenly and a linear wound was introduced. The results indicated that the distance that MCF7-NLSp44 cells migrated towards the wound area is at a comparable level to the control MCF7-pBabe cells (Fig. 5D). Thus, the increased invasion ability is not due to increased cell migration.
The growth and invasion findings strongly suggest that nuclear p44 may be an important mediator of tumour growth and invasion in ER+ ductal carcinomas.
Discussion
Steroid hormone receptors and their coactivators play an important role in breast cancer oncogenesis and progression. Coactivators modulate the receptor activity through linking hormone receptors, such as ER, with basal transcriptional machinery or by modulating chromatin activity. Several ER coactivators, such as SRC3/AIB1, have also been shown to modulate the development of breast cancer [9]. Androgen, AR and coactivators have been implicated to play a part in breast oncogenesis [15]. A number of coactivators act on both androgen and oestrogen pathways, and possibly mediate the interplay between the two receptors. Coactivator ARA70 can interact with both AR and ER increasing the receptor-mediated transcriptional activities [27]. Dysregulation of ARA70 expression is indicated in cancer development in both breast and prostate cancer [35, 36]. In this study, we show p44 can also function as ER coactivator to activate the ER target gene expression.
p44 is recently characterized as an AR coactivator [28] increasing AR mediated transcriptional activation in an androgen-dependent manner. Up-to-date, most of the studies on p44 are focused on prostate and testicular cancers. We reported that p44 is expressed in the nucleus in benign prostate epithelial and testicular Leydig cells and p44 is expressed as cytoplasmic protein in their malignant counterparts. Further, nuclear p44 inhibits growth in prostate cancer cells. Herein, we report that in addition to functioning as an AR coactivator, p44 also can act as an ER coactivator in MCF7 breast cancer cells. In contrast to prostate and testis, p44 is expressed as a cytoplasmic protein in benign breast epithelia of terminal ductal lobular units while p44 is localized in the nucleus in both DCIS and invasive carcinoma. This phenomenon is very intriguing because this cytoplasmic to nuclear translocation from benign to malignant breast epithelial cells could either be an epiphenomenon in breast cancer or alternatively, p44 may have distinct biological functions in mediating growth stimulation and suppression in different cellular compartments.
To discern the function of nuclear and cytoplasmic p44, we devised either NLS or NES N-terminally tagged p44 fusion proteins as described previously [30]. We performed cell proliferation assays in hormone free, androgen (10 nM R1881) and oestrogen (10 nM 17-β-estradiol) media to define the effects of androgen or oestrogen in growth regulation by p44. In our cell proliferation studies by cell counting, we showed that NLSp44 promoted cell growth in MCF7 (ER+) breast cancer cells in the presence of oestrogen, but not in hormone free or androgen media. In hormone free media, NLSp44 actually inhibited MCF7 cell growth. Strikingly, the behaviour of nuclear and cytoplasmic p44 in MDA-MB-231 (ER–) cells is similar to that of prostate cancer. NESp44 increases proliferation in MDA-MB-231 cells. Because MDA-MB-231 cells do not expression ER, it is likely the cytoplasmic process of p44 is not oestrogen or ER dependent. These results indicate that addition of oestrogen can reverse growth inhibition in hormone free media and further stimulate breast cancer cell growth by p44. Related, it would be of interest to determine expression of p44 in ER– human breast cancer cases and correlate its function in ER– cell lines, such as MDA-MD-231, under various conditions (e.g. with and without ER and AR overexpression).
In addition to growth regulation, NLSp44 also enhanced the invasion ability of MCF7 breast cancer cells. Again, the increased invasion is only observed in oestrogen media and ER+ MCF7 cells. The stimulation of growth and invasion by NLSp44 is not observed in ER– MDA-MB-231 cells, regardless of the presence of oestrogen. These results indicate the importance of oestrogen and its receptor for the NLSp44-mediated effect on breast cancer cell growth and invasion. Confirming ERα-dependent nature of nuclear p44 enhanced growth and invasion, ERα knockdown decreased the cell proliferation and invasion of MCF7 cells in the presence of oestrogen. p44 shares identical protein sequence with MEP50, a component of methylosome complex containing PRMT5/JBP1, pICln and Sm proteins, suggesting its role in regulating gene expression by methylation of intranuclear proteins [28, 37, 38]. A recent study addressed the association of p44 with SUZ12, a Polycomb group protein that may bind to histone H2A [39]. In their study, p44 binds to free H2A but not nucleosomal H2A and facilitates the activity of PRMT5 to methylate H2A. Because methylation of H4 by PRMT5 represses transcription [40], it is possible that the methylation of free H2A by PRMT5, mediated by p44, represses the expression of target genes after being incorporated into nucleosome [39]. This converted status of gene transcription may be a critical step in tumorigenesis through transcription regulators such as Polycomb group proteins. Indeed, overexpression of such proteins has been shown to be associated with high proliferation rate and aggressive behaviour in various tumours, including melanoma and carcinomas from breast, endometrium and prostate [41–43]. Overexpression of p44, specifically in nuclei, stimulates proliferation of MCF7 cells in response to oestrogen by targeting ER responsive genes.
Because we show in this study that the expression and function of p44 in breast cancer are distinct from prostate, it is of great interest to determine if the opposite p44 expression pattern in benign and malignant prostate and breast could represent a mechanism in the regulation of p44 nuclear to cytoplasmic translocation by hormone. We also show that p44 colocalizes with PRMT5 in benign and malignant breast tissue (J. Wang and P. Lee, unpublished data). These results indicate that a similar mechanism exists in the regulation of nuclear to cytoplasmic translocation of p44 and PRMT5. This regulation of nuclear and cytoplasmic translocation is at the level of the protein complex rather than single protein level.
In summary, we report that coactivator p44, besides functioning as an AR coactivator, can also function as an ER coactivator. Nuclear p44 promotes breast cancer growth and invasion mediated through oestrogen and its receptor and not by androgen stimulation. Consistently, p44 is expressed as a cytoplasmic protein in benign breast cells and as a nuclear protein in malignant breast cells. These results have great significance in understanding the mechanisms of ER and its cofactors in regulating breast cancer cell proliferation and invasion.
Acknowledgments
MDA-MB-231 cell line was a generous gift from Dr. C. Albarracin at M. D. Anderson Cancer Center. We would like to thank Mr. Junliang Zhou for technical assistant. This work is supported by DOD BRCP and Susan G. Komen Grants to P.L.
Supporting Information
Fig. S1 Cyclin expression in MCF7 and MDA-MD-231 cellsoverexpressing NLSp44 or NESp44. (A) Increased cyclin B1 expression(lane 2) in MCF-NLSp44 cells in the presence of oestrogen. (B)Cyclin B1 expression in MCF-NLSp44 or MCF-NESp44 cells in thehormone free media. (C) Cyclin B1 expression in MDA-MB-231-NLSp44or MDA-MB-231-NESp44 cells in the presence of oestrogen. (D) CyclinB1 expression in MCF-NLSp44 or MCF-NESp44 cells in the hormone freemedia. (E) Cyclin A2, cyclin D1 and cyclin E expression inMCF-NLSp44 or MCF-NESp44 cells in the presence of oestrogen. (F)Increased ER target gene MYC expression by real time qRT-PCR inMCF-NLSp44-LSp44 cells in the presence of 17-b-estradiol.(A)–(E) Lane 1: pBabe, lane 2: NLSp44, lane 3: NESp44.
References
- 1.Parkin DM, Bray F, Ferlay J, et al. Estimating the world cancer burden: globocan 2000. Int J Cancer. 2001;94:153–6. doi: 10.1002/ijc.1440. [DOI] [PubMed] [Google Scholar]
- 2.Quiet CA, Ferguson DJ, Weichselbaum RR, et al. Natural history of node-positive breast cancer: the curability of small cancers with a limited number of positive nodes. J Clin Oncol. 1996;14:3105–11. doi: 10.1200/JCO.1996.14.12.3105. [DOI] [PubMed] [Google Scholar]
- 3.Boyer B, Valles AM, Edme N. Induction and regulation of epithelial-mesenchymal transitions. Biochem Pharmacol. 2000;60:1091–9. doi: 10.1016/s0006-2952(00)00427-5. [DOI] [PubMed] [Google Scholar]
- 4.Arias AM. Epithelial mesenchymal interactions in cancer and development. Cell. 2001;105:425–31. doi: 10.1016/s0092-8674(01)00365-8. [DOI] [PubMed] [Google Scholar]
- 5.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–54. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 6.Gotzmann J, Mikula M, Eger A, et al. Molecular aspects of epithelial cell plasticity: implications for local tumor invasion and metastasis. Mutat Res. 2004;566:9–20. doi: 10.1016/s1383-5742(03)00033-4. [DOI] [PubMed] [Google Scholar]
- 7.Hood JD, Cheresh DA. Role of integrins in cell invasion and migration. Nat Rev Cancer. 2002;2:91–100. doi: 10.1038/nrc727. [DOI] [PubMed] [Google Scholar]
- 8.Cordera F, Jordan VC. Steroid receptors and their role in the biology and control of breast cancer growth. Semin Oncol. 2006;33:631–41. doi: 10.1053/j.seminoncol.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 9.Singh RR, Kumar R. Steroid hormone receptor signaling in tumorigenesis. J Cell Biochem. 2005;96:490–505. doi: 10.1002/jcb.20566. [DOI] [PubMed] [Google Scholar]
- 10.Agoff SN, Swanson PE, Linden H, et al. Androgen receptor expression in estrogen receptor-negative breast cancer. Immunohistochemical, clinical, and prognostic associations. Am J Clin Pathol. 2003;120:725–31. doi: 10.1309/42F0-0D0D-JD0J-5EDT. [DOI] [PubMed] [Google Scholar]
- 11.Lea OA, Kvinnsland S, Thorsen T. Improved measurement of androgen receptors in human breast cancer. Cancer Res. 1989;49:7162–7. [PubMed] [Google Scholar]
- 12.Soreide JA, Lea OA, Varhaug JE, et al. Androgen receptors in operable breast cancer: relation to other steroid hormone receptors, correlations to prognostic factors and predictive value for effect of adjuvant tamoxifen treatment. Eur J Surg Oncol. 1992;18:112–8. [PubMed] [Google Scholar]
- 13.Ando S, De Amicis F, Rago V, et al. Breast cancer: from estrogen to androgen receptor. Mol Cell Endocrinol. 2002;193:121–8. doi: 10.1016/s0303-7207(02)00105-3. [DOI] [PubMed] [Google Scholar]
- 14.Birrell SN, Bentel JM, Hickey TE, et al. Androgens induce divergent proliferative responses in human breast cancer cell lines. J Steroid Biochem Mol Biol. 1995;52:459–67. doi: 10.1016/0960-0760(95)00005-k. [DOI] [PubMed] [Google Scholar]
- 15.Conzen SD. Minireview: nuclear receptors and breast cancer. Mol Endocrinol. 2008;22:2215–28. doi: 10.1210/me.2007-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Birrell SN, Hall RE, Tilley WD. Role of the androgen receptor in human breast cancer. J Mammary Gland Biol Neoplasia. 1998;3:95–103. doi: 10.1023/a:1018730519839. [DOI] [PubMed] [Google Scholar]
- 17.Hackenberg R, Hofmann J, Holzel F, et al. Stimulatory effects of androgen and antiandrogen on the in vitro proliferation of human mammary carcinoma cells. J Cancer Res Clin Oncol. 1988;114:593–601. doi: 10.1007/BF00398183. [DOI] [PubMed] [Google Scholar]
- 18.Poulin R, Baker D, Labrie F. Androgens inhibit basal and estrogen-induced cell proliferation in the ZR-75–1 human breast cancer cell line. Breast Cancer Res Treat. 1988;12:213–25. doi: 10.1007/BF01805942. [DOI] [PubMed] [Google Scholar]
- 19.Hackenberg R, Luttchens S, Hofmann J, et al. Androgen sensitivity of the new human breast cancer cell line MFM-223. Cancer Res. 1991;51:5722–7. [PubMed] [Google Scholar]
- 20.Honma N, Sakamoto G, Akiyama F, et al. Breast carcinoma in women over the age of 85: distinct histological pattern and androgen, oestrogen, and progesterone receptor status. Histopathology. 2003;42:120–7. doi: 10.1046/j.1365-2559.2003.01542.x. [DOI] [PubMed] [Google Scholar]
- 21.Heinlein CA, Chang C. Androgen receptor (AR) coregulators: an overview. Endocr Rev. 2002;23:175–200. doi: 10.1210/edrv.23.2.0460. [DOI] [PubMed] [Google Scholar]
- 22.Gelmann EP. Molecular biology of the androgen receptor. J Clin Oncol. 2002;20:3001–15. doi: 10.1200/JCO.2002.10.018. [DOI] [PubMed] [Google Scholar]
- 23.Chen Y, Sawyers CL, Scher HI. Targeting the androgen receptor pathway in prostate cancer. Curr Opin Pharmacol. 2008;8:440–8. doi: 10.1016/j.coph.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wooster R, Mangion J, Eeles R, et al. A germline mutation in the androgen receptor gene in two brothers with breast cancer and Reifenstein syndrome. Nat Genet. 1992;2:132–4. doi: 10.1038/ng1092-132. [DOI] [PubMed] [Google Scholar]
- 25.Lobaccaro JM, Lumbroso S, Belon C, et al. Male breast cancer and the androgen receptor gene. Nat Genet. 1993;5:109–10. doi: 10.1038/ng1093-109. [DOI] [PubMed] [Google Scholar]
- 26.Zhu X, Daffada AA, Chan CM, et al. Identification of an exon 3 deletion splice variant androgen receptor mRNA in human breast cancer. Int J Cancer. 1997;72:574–80. doi: 10.1002/(sici)1097-0215(19970807)72:4<574::aid-ijc4>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 27.Lanzino M, De Amicis F, McPhaul MJ, et al. Endogenous coactivator ARA70 interacts with estrogen receptor alpha (ERalpha) and modulates the functional ERalpha/ androgen receptor interplay in MCF-7 cells. J Biol Chem. 2005;280:20421–30. doi: 10.1074/jbc.M413576200. [DOI] [PubMed] [Google Scholar]
- 28.Hosohata K, Li P, Hosohata Y, et al. Purification and identification of a novel complex which is involved in androgen receptor-dependent transcription. Mol Cell Biol. 2003;23:7019–29. doi: 10.1128/MCB.23.19.7019-7029.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou L, Wu H, Lee P, et al. Roles of the androgen receptor cofactor p44 in the growth of prostate epithelial cells. J Mol Endocrinol. 2006;37:283–300. doi: 10.1677/jme.1.02062. [DOI] [PubMed] [Google Scholar]
- 30.Peng Y, Chen F, Melamed J, et al. Distinct nuclear and cytoplasmic functions of androgen receptor cofactor p44 and association with androgen-independent prostate cancer. Proc Natl Acad Sci USA. 2008;105:5236–41. doi: 10.1073/pnas.0712262105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang JJ, Wang Z, Chiriboga L, et al. The expression and function of androgen receptor coactivator p44 and protein arginine methyltransferase 5 in the developing testis and testicular tumors. J Urol. 2007;177:1918–22. doi: 10.1016/j.juro.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 32.Gao S, Lee P, Wang H, et al. The androgen receptor directly targets the cellular Fas/FasL-associated death domain protein-like inhibitory protein gene to promote the androgen-independent growth of prostate cancer cells. Molecular Endocrinol. 2005;19:1792–802. doi: 10.1210/me.2004-0445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Y, Wang L, Zhang M, et al. LEF1 in androgen-independent prostate cancer: regulation of androgen receptor expression, prostate cancer growth, and invasion. Cancer Res. 2009;69:3332–8. doi: 10.1158/0008-5472.CAN-08-3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cai CQ, Peng Y, Buckley MT, et al. Epidermal growth factor receptor activation in prostate cancer by three novel missense mutations. Oncogene. 2008;27:3201–10. doi: 10.1038/sj.onc.1210983. [DOI] [PubMed] [Google Scholar]
- 35.Kollara A, Kahn HJ, Marks A, et al. Loss of androgen receptor associated protein 70 (ARA70) expression in a subset of HER2-positive breast cancers. Breast Cancer Res Treat. 2001;67:245–53. doi: 10.1023/a:1017938608460. [DOI] [PubMed] [Google Scholar]
- 36.Li P, Yu X, Ge K, et al. Heterogeneous expression and functions of androgen receptor co-factors in primary prostate cancer. Am J Pathol. 2002;161:1467–74. doi: 10.1016/S0002-9440(10)64422-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meister G, Eggert C, Buhler D, et al. Methylation of Sm proteins by a complex containing PRMT5 and the putative U snRNP assembly factor pICln. Curr Biol. 2001;11:1990–4. doi: 10.1016/s0960-9822(01)00592-9. [DOI] [PubMed] [Google Scholar]
- 38.Friesen WJ, Wyce A, Paushkin S, et al. A novel WD repeat protein component of the methylosome binds Sm proteins. J Biol Chem. 2002;277:8243–7. doi: 10.1074/jbc.M109984200. [DOI] [PubMed] [Google Scholar]
- 39.Furuno K, Masatsugu T, Sonoda M, et al. Association of Polycomb group SUZ12 with WD-repeat protein MEP50 that binds to histone H2A selectively in vitro. Biochem Biophys Res Commun. 2006;345:1051–8. doi: 10.1016/j.bbrc.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 40.Fabbrizio E, El Messaoudi S, Polanowska J, et al. Negative regulation of transcription by the type II arginine methyltransferase PRMT5. EMBO Rep. 2002;3:641–5. doi: 10.1093/embo-reports/kvf136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Raaphorst FM, Meijer CJ, Fieret E, et al. Poorly differentiated breast carcinoma is associated with increased expression of the human polycomb group EZH2 gene. Neoplasia. 2003;5:481–8. doi: 10.1016/s1476-5586(03)80032-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bachmann IM, Halvorsen OJ, Collett K, et al. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J Clin Oncol. 2006;24:268–73. doi: 10.1200/JCO.2005.01.5180. [DOI] [PubMed] [Google Scholar]
- 43.Collett K, Eide GE, Arnes J, et al. Expression of enhancer of zeste homologue 2 is significantly associated with increased tumor cell proliferation and is a marker of aggressive breast cancer. Clin Cancer Res. 2006;12:1168–74. doi: 10.1158/1078-0432.CCR-05-1533. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Cyclin expression in MCF7 and MDA-MD-231 cellsoverexpressing NLSp44 or NESp44. (A) Increased cyclin B1 expression(lane 2) in MCF-NLSp44 cells in the presence of oestrogen. (B)Cyclin B1 expression in MCF-NLSp44 or MCF-NESp44 cells in thehormone free media. (C) Cyclin B1 expression in MDA-MB-231-NLSp44or MDA-MB-231-NESp44 cells in the presence of oestrogen. (D) CyclinB1 expression in MCF-NLSp44 or MCF-NESp44 cells in the hormone freemedia. (E) Cyclin A2, cyclin D1 and cyclin E expression inMCF-NLSp44 or MCF-NESp44 cells in the presence of oestrogen. (F)Increased ER target gene MYC expression by real time qRT-PCR inMCF-NLSp44-LSp44 cells in the presence of 17-b-estradiol.(A)–(E) Lane 1: pBabe, lane 2: NLSp44, lane 3: NESp44.