

Abstract
The major complication of diabetes is accelerated atherosclerosis, the progression of which entails complex interactions between the modified low-density lipoproteins (LDL) and the cells of the arterial wall. Advanced glycation end product-modified-LDL (AGE-LDL) that occurs at high rate in diabetes contributes to diabetic atherosclerosis, but the underlying mechanisms are not fully understood. The aim of this study was to assess the direct effect of AGE-LDL on human vascular smooth muscle cells (hSMC) dysfunction. Cultured hSMC incubated (24 hrs) with human AGE-LDL, native LDL (nLDL) or oxidized LDL (oxLDL) were subjected to: (i) quantification of the expression of the receptors for modified LDL and AGE proteins (LRP1, CD36, RAGE) and estimation of lipid loading, (ii) determination of NADPH oxidase activity and reactive oxygen species (ROS) production and (iii) evaluation of the expression of monocyte chemoattractant protein-1 (MCP-1). The results show that exposure of hSMC to AGE-LDL (compared to nLDL) induced: (a) increased NADPH oxidase activity (30%) and ROS production (28%) by up-regulation of NOX1, NOX4, p22phox and p67phox expression, (b) accumulation of intracellular cholesteryl esters, (c) enhanced gene expression of LRP1 (160%) and CD36 (35%), and protein expression of LRP1, CD36 and RAGE, (d) increased MCP-1 gene expression (160%) and protein secretion (300%) and (e) augmented cell proliferation (30%). In conclusion, AGE-LDL activates hSMC (increasing CD36, LRP1, RAGE), inducing a pro-oxidant state (activation of NADPHox), lipid accumulation and a pro-inflammatory state (expression of MCP-1). These results may partly explain the contribution of AGE-LDL and hSMC to the accelerated atherosclerosis in diabetes.
Keywords: AGE-LDL, CD36, glycated LDL, lipid loading, LRP1, MCP-1, NADPH oxidase, RAGE, SMC proliferation, vascular hSMC
Introduction
In human beings, diabetes mellitus associates with increased incidence of macro- and microvascular complications including coronary artery and peripheral vascular diseases. In diabetic-associated vascular disease, low-density lipoproteins (LDL) are modified and play a key role in the accelerated progression of atherosclerosis [1–3]. Chronic hyperglycaemia enhances glucose-induced LDL oxidation and/or glycation, which increases its pro-atherogenic properties [4] and promotes vascular injury by a mechanism that is scantily defined. Irreversible glycation starts with the non-enzymatic addition of reducing sugars to the native LDL (nLDL) lysine residues, followed by additional reactions leading to the formation of sugar-amino acid adducts, collectively known as advanced glycation end-products (AGE). A correlation between arterial tissue AGE and circulating AGE-ApoB, and the contribution of AGE-specific receptors (RAGE) in atheroma formation was reported [5].
Smooth muscle cells (SMC) are major contributors to the initiation and early progression of the atherosclerotic plaque in human beings and animal models [6]. Within the plaque SMC migrate, proliferate, secrete chemokines that enhance the accrual of monocytes, and ultimately take up transcytosed, modified lipoproteins (Lp) and turn into foam cells. Like all cells of the vessel wall, SMC are exposed to AGE-LDL that accumulates in the atheroma of diabetic patients [7, 8] and animal models of diabetes [9]; the specific effects of AGE-LDL on SMC are not known.
The involvement of the oxidative stress and the NADPH oxidase complex (NADPHox) in the dysfunction of vascular cells is well established [10, 11]. It is known that oxidized LDL (oxLDL) regulates the expression of NADPHox in human endothelial cells and vascular human SMC (hSMC) [12], but there are no data on the effect of AGE-LDL on the modulation of NADPHox in hSMC.
The expression of native LDL receptors on arterial cells is tightly controlled and feedback regulated, in contrast to the uncontrolled uptake of modified LDL and subsequent foam cell formation occurring via alternative receptors (CD36, LRP1, scavenger receptors A and B1, and RAGE) [13]; there are no data on the effect of AGE-LDL on these receptors.
MCP-1 is a potent pro-inflammatory chemokine, whose expression in hSMC is up-regulated by high glucose, leading to increased monocyte-SMC adhesive interactions [14]. The effects of AGE-LDL on MCP-1 expression are also not known.
In this study, we provide evidence that AGE-LDL activates hSMC (inducing LRP1, CD36, RAGE), triggering a pro-oxidant state (activation of NADPHox) and pro-inflammatory state (expression of MCP-1) and lipid accumulation. To the best of our knowledge, this is the first report on the effects of AGE-LDL on vascular hSMC, providing a mechanism that may explain accelerated atherosclerosis in diabetic patients.
Materials and methods
Preparation of AGE-LDL
LDL was isolated from the plasma of healthy donors from the Blood Transfusion Center Bucharest by density gradient ultracentrifugation as previously described [15]. The LDL fraction was collected, dialysed against phosphate buffer saline (PBS), pH 7.4, 4°C in the dark, stored in sterile conditions in the presence of antioxidants (1 mg/ml EDTA and 10 μM BHT) at 4°C and used within 7 days. AGE-LDL was prepared by incubation of nLDL (2 mg protein/ml) with D(×)glucose (0.2M final concentration), for 4 weeks, at 37°C, under sterile conditions with antioxidants (1 mg/ml EDTA and 10 mM BHT). Prior to the experiments, AGE-LDL was extensively dialysed against PBS, pH 7.4, 4°C. OxLDL was prepared by incubating nLDL under sterile conditions with 10 μM copper chloride (24 hrs, at 37°C), in the absence of antioxidant protection. The oxidative reaction was stopped by addition of 1 mg/ml EDTA and after extensive dialysis against PBS, pH 7.4, 48C, oxLDL was stored at 4°C, under sterile conditions and used within 7 days.
Characterization of AGE-LDL
Quantification of the free, non-glycated amino groups was done using the trinitrobenzene sulphonic acid assay (TNBSA) [16]. The extent of LDL protein glycation was determined by thiobarbituric assay (TBA) and expressed as nmoles of 5-hydroxymethylfurfuraldehyde (HMF) per mg LDL protein [17]. Protein concentration was measured by the Lowry method as modified for lipoproteins [18] employing bovine serum albumin as a standard. The formation of AGE products, pentosidine and furoyl furanyl imidazole (FFI), was monitored by spectrofluorimetry (excitation/emission 335/385 nm and 370/450 nm) [19]. The lipid peroxide level was determined as thiobarbituric acid reactive substances (TBARS) and expressed as nmol malondialdehyde (MDA)/mg protein [20].
The electric charge of AGE-LDL, oxLDL and nLDL was determined by agarose gel electrophoresis (0.6%) and expressed as the relative electrophoretic mobility (Rf). SDS-PAGE was employed to assess the degradation of LDL’s apoB.
Cell culture and experimental design
Aortic hSMC were obtained by explantation from the media of foetal thoracic aorta and grown in Dulbecco’s modified Eagle’s medium (DMEM) with 10% foetal calf serum (FCS) (v/v) and supplemented with non-essential amino acids and antibiotics [21]. At confluence, hSMC were starved for 24 hrs by incubation with DMEM in the presence of 0.2% FCS and then exposed to nLDL, AGE-LDL or oxLDL (100 μg protein/ml medium) for 24 hrs. As control, hSMC were cultured in the same conditions without LDL exposure. Cultured cells were used between the ninth and twelfth passages.
Lipid loading induced by AGE-LDL
The cells plated on coverslips placed in 24 well plates (5×104cells/well) were incubated with AGE-LDL, nLDL or oxLDL (100 μg protein/ml) for 24 hrs. After washing with PBS, the cells were fixed in 2% paraformaldehyde (10 min.), stained (6 min.) with Nile Red (150 ng/ml PBS) to evidence the neutral lipids and examined by fluorescence microscopy with a Microphot FXA Nikon microscope using filter block B-2A (λex 5 420–490 nm, λem > 520 nm).
Quantification of free and esterified cholesterol
Total lipids from cells incubated with nLDL, AGE-LDL or oxLDL were extracted in chloroform:methanol (2:1) and the total cholesterol (TC) and free cholesterol (FC) were determined as described [22]. The fluorescence was measured using a Shimadzu spectrofluorimeter (excitation 325 nm, emission 415 nm). The cholesteryl esters (CE) content of the cells was expressed as CE/TC*100 ratio.
Evaluation of the viability, metabolic competence and proliferation rate of hSMC
Cells viability was measured as a function of the mitochondrial activity using a cell growth determination kit based on tetrazolium salt (MTT, Sigma, St. Louis, MO, USA). Evaluation of the energy metabolism of hSMC incubated with AGE-LDL, nLDL and oxLDL was done by measuring the intracellular ATP level with ViaLight Plus Kit (Lonza, Walkersville, MD, USA). In other experiments, we have measured the hexose uptake in hSMC exposed to AGE-LDL, nLDL or oxLDL. Hexose uptake by hSMC was estimated by measuring the glucose concentration in the culture medium before and after incubation with LDL. The proliferation rate was determined by a fluorimetric assay using Hoechst 33258, which measures cellular DNA content [23].
Determination of NADPH oxidase activity
Quiescent hSMC were exposed for 24 hrs to 100 μg/ml AGE-LDL, nLDL or oxLDL. The NADPH activity was determined in cell homogenates by lucigenin-enhanced chemiluminescence assay [24] using a low concentration of lucigenin (5 μmol/l) to oxLDL minimize artifactual O2− production due to redox cycling [25]. The reaction mixture comprised 50 mM PBS, 1 mM EGTA, pH 7.0, 5 μmol/l lucigenin and 100 μmol/l NADPH. The reaction started by addition of cell homogenate (100–150 μg of protein) to the mixture and the light emission was recorded every second for 15 min. in a luminometer (Berthold, Germany). The activity was expressed as relative light units (RLU)/μg of total protein.
Quantification of reactive oxygen species (ROS)
The generation of ROS in hSMC was measured by 2,7-dichlorofluorescein (DCF) fluorescence technique [26]. Briefly, cultured cells were scrapped and resuspended in HEPES-buffered saline solution, pH 7.4, containing (in mM): NaCl, 145; KCl, 5; CaCl2, 1.8; MgCl2, 1; Na2HPO4, 1; glucose, 5; HEPES, 25. After loading with 5 μmol/l DCF for 30 min. at 37°C, the cells were distributed at 104cells/well into a 96-well microplate reader (Tecan, Austria). DCF fluorescence emission was detected at 535 nm with an excitation wavelength of 485 nm. The ROS production was expressed as relative fluorescence units (RFU)/μg of total protein.
Quantification of the gene expression for LDL receptors, NADPH oxidase subunits and MCP-1
Total RNA was isolated from hSMC using the GenElute Mammalian Total RNA Kit (Sigma). The M-MLV Reverse Transcriptase PCR Kit (Invitrogen, Carlsbad, CA, USA) was used to perform the reverse-transcription of RNA and the amplification of complementary DNA (cDNA). The gene expression for LDL receptor (LDL-R), LDL receptor related protein1 (LRP1), scavenger receptor CD36, NADPH subunits (NOX1, NOX4, p67phox, and p22phox) and monocyte chemoattractant protein-1 (MCP-1) was assessed by Real-time PCR (using an Opticon 2 DNA Engine Real-Time thermocycler -MJ Research, USA) using specific primers (Table 1). GAPDH or 28S rRNA was used as housekeeping gene. The PCR reactions were optimized for each pair of primers in order to obtain a single specific reaction product. Optimized amplification conditions were 0.2 μmol/l of each primer, and 2.5 mmol/l MgCl2. Quantification of the PCR products was done using the ‘Fit Point Method’[27], and expressed as arbitrary units (AU). The efficiency of the amplification reaction of target and internal standard were similar. The cDNA was amplified through 35–40 cycles (30 sec., 94°C; 30 sec., 56°C for CD36, 62°C for LDL-R and 60°C for NOX1, NOX4, p67phox, p22phox, LRP1, RAGE and MCP-1; the SyBr green fluorescent complex continuously read; 1 min., 72°C), followed by a melting curve program (from 55°C to 94°C, with compound read at every 1°C) and finally a cooling step at 4°C.
Table 1.
Real-time PCR primers employed
| Forward | Reverse | |
|---|---|---|
| 28S rRNA | AAACTCTGGTGGAGGTCCGT | CTTACCAAAAGTGGCCCACTA |
| GAPDH | ACCACAGTCCATGCCATCAC | TCCACCACCCTGTTGCGTA |
| LDLR | CAATGTCTCACCAAGCTCTG | TCTGTCTCGAGGGGTAGCTG |
| LRP1 | GAGCTGAACCACGCCTTTG | GGTAGACACTGCCACTCCGATAC |
| CD36 | ATGTAACCCAGGACGCTGAG | GTCGCAGTGACTTTCCCAAT |
| RAGE | TCCAATTGGTGGTGGAGCCAGAA | TCGCCTGGTTCGATGATGCGTAT |
| MCP-1 | AGCATGAAAGTCTCTGCCGCCCTTCTG | ATTACTTAAGGCATAATGTTTCACA |
| NOX1 | CACAAGAAAAATCCTTGGGTCAA | GACAGCAGATTGCCGACACACA |
| NOX4 | TGGCTG CCCATCTGGTGAATG | CAGCAGCCCTCCTGAAACATGC |
| p67phox | CGGTCGTGGCATCTGTGGTGG | GGGATGTCGGAC TGCGGAGAGC |
| p22phox | GTTTGTGTGCCTGCTGGAGT | TGGGCGGCTGCTTGATGGT |
GAPDH: glyceraldehyde 3-phosphate dehydrogenase; LDLR: Low-density lipoprotein receptor; LRP1: LDL receptor-related protein 1; RAGE: receptor for advanced glycation end-products; MCP-1: monocyte chemotactic protein-1.
Quantification of the protein expression by Western blot analysis
Cells were lysed in RadioImmuno Precipitation Assay (RIPA) buffer containing 50 mMTris-HCl, 150 mM NaCl, 1 mM EDTA, 1 mM phenylmethyl sulfonyl fluoride, pH 7.4, 1% Triton X-100, 1% Na-deoxycholate, 0.1% sodium dodecyl sulphate. The proteins (50 μg) were separated on 8% SDS-PAGE, transferred to a nitrocellulose membrane and blocked over night with 5% non-fat milk and 0.05% Tween 20 in Tris-buffered saline pH 7.5. The blots were than exposed to the primary antibodies against human CD36 (rabbit polyclonal), RAGE (mouse monoclonal), MCP-1 (rabbit polyclonal) and β-actin (mouse monoclonal), followed by horseradish peroxidase conjugated goat anti-rabbit or anti-mouse IgG; all antibodies were from Abcam, UK, except for β-actin (Santa Cruz, USA) and the ECL reagents (Pierce, Rockford, IL, USA). The relative protein expression was normalized to β-actin after densitometry with TotalLab100 software. For soluble MCP-1, the medium was collected after 24 hrs incubation of hSMC with LDL, concentrated by ultrafiltration and 10 μl aliquots were applied on 15% SDS-PAGE.
Statistical analysis
Data were expressed as means ± SD. Statistical evaluation was done by one-way ANOVA test; P ≤ 0.05 was considered statistically significant. Standard chemicals and reagents were purchased from Sigma (Saint Louis, MO, USA) if not otherwise stated.
Results
Characterization of AGE-LDL
The extent of LDL glycation was estimated using TNBSA, which upon coupling to free, non-glycated amino groups gives the highly chromogenic 2,4,6-trinitrophenyl (TNP) derivatives. Compared to nLDL (0.50 ± 0.015 AU) or oxLDL (0.44 ± 0.055 AU), the assay revealed a 40% reduction of the free amino groups of apoB in AGE-LDL (0.30 ± 0.00 AU, P < 0.05) indicative of the irreversible glycation of the protein (Fig. 1A). In addition, the TBA assay indicated the extensive glycation of apoB: a 4-fold increased glycation of AGE-LDL (6.80 ± 2.73 nmoles HMF/mg protein, P < 0.05) compared to nLDL (1.81 ± 1.34 nmoles HMF/mg protein) (Fig. 1B). A 2-fold increase of the HMF content in oxLDL (4.68 ± 1.40 nmoles HMF/mg protein) was determined. Measurement of the FFI characteristic fluorescence showed a 10-fold increase in AGE-LDL (1.35 ± 0.01 AU, P < 0.01) and oxLDL (2.08 ± 0.12 AU, P < 0.01) compared to nLDL (0.17 ± 0.06 AU) (Fig. 1C). Pentosidine content was increased in AGE-LDL (1.64 ± 0.32 AU, P < 0.05) and oxLDL (1.52 ± 0.28 AU, P < 0.05) compared to nLDL (0.76 ± 0.05 AU) (Fig. 1C).
Fig 1.
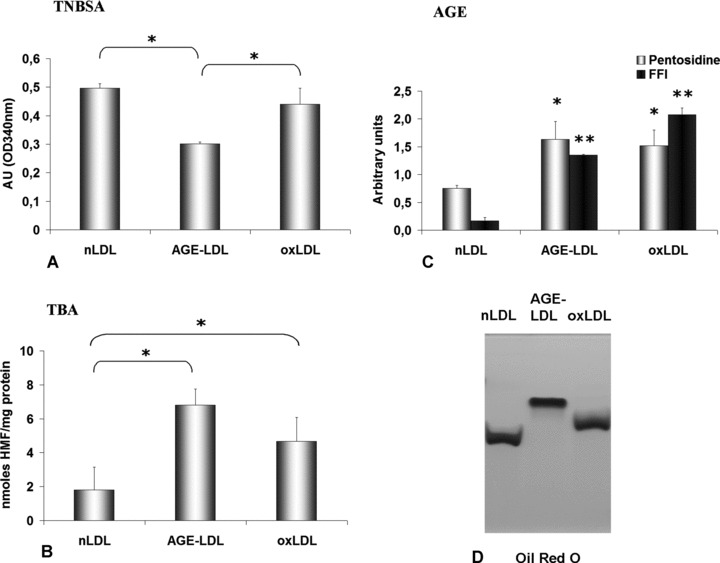
Characterization of AGE-LDL. Comparative evaluation of native LDL (nLDL), copper-oxidized LDL (oxLDL) and AGE-LDL (obtained by incubation of nLDL with 0.2M D(+)glucose for 4 weeks). The glycated epitopes were measured by TNBSA (A) and TBA (B) assays, pentosidine and FFI levels (C), *P < 0.05, **P < 0.01, n= 4, and the electronegative charge in 0.6% agarose gel electrophoresis (Oil Red O staining) (D).
The mean lipid peroxides level was 5-fold higher in AGE-LDL (1.77 ± 0.5 nmoles MDA/mg protein, P < 0.05) and 10-fold higher in oxLDL (4.06 ± 0.02 nmoles MDA/mg protein) than in nLDL (0.35 ± 0.23 nmoles MDA/mg protein).
The Lp charge was determined by agarose gel electrophoresis. Measurement of the Rf showed a 29% increase for AGE-LDL (0.71 ± 0.06, P < 0.05) and a 10% increase for oxLDL (0.61 ± 0.06, P < 0.05) compared with nLDL (0.55 ± 0.08) (Fig. 1D). The increase in the overall negative charge of AGE-LDL was indicative of the loss of positive charges due the glycation of Lys and Arg residues. All LDLs appeared as a single band indicating the lack of contamination with other proteins.
Regulation of NADPH oxidase activity
To assess further the pro-oxidant effect of AGE-LDL (compared to nLDL) on hSMC, NADPHox activity was determined employing the lucigenin-enhanced chemiluminescence assay. The results revealed that the NADPHox activity was significantly enhanced in hSMC by AGE-LDL (35,530 ± 381 RLU/μg protein, P < 0.05) and oxLDL (34,169 ± 467 RLU/μg protein, P < 0.05); in contrast, the enzyme activity was comparable in control cells (26,970 ± 280 RLU/μg protein) and in cells incubated with nLDL (29,431 ± 400 RLU/μg protein) (Fig. 2A).
Fig 2.
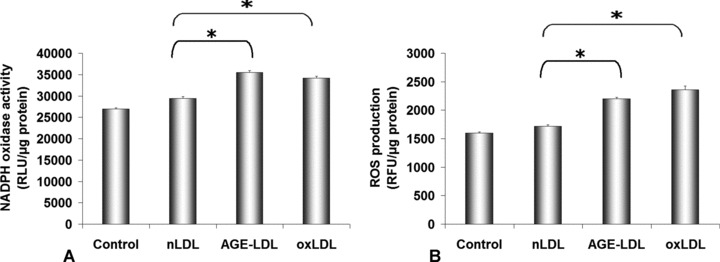
Oxidative stress induced by AGE-LDL incubation with hSMC. NADPH oxidase activity (A) was measured by lucigenin-enhanced chemiluminiscence (A), and reactive oxygen species (ROS) production by dichlorofluoresceine assays (B). 100 μg protein/ml AGE-LDL, oxidized LDL (oxLDL) or native LDL (nLDL) were incubated 24 hrs with hSMC. Control, cells not exposed to LDLs, *P < 0.05, n= 3.
Modulation of intracellular ROS production
The oxidative effect of native and AGE-LDL was analysed in quiescent hSMC incubated for 24 hrs with 100 μg LDLs protein/ml employing DCFA cellular loading. The results showed that the intracellular ROS production increased significantly in cells exposed to AGE-LDL (2198 ± 30 RFU/μg protein, P < 0.05) as compared to control cells (1600 ± 22 RFU/μg protein) or cells incubated with nLDL (1715 ± 28 RFU/μg protein) (Fig. 2B).
Regulation of gene and protein expression for NOX1, NOX4, p22phox and p67phox
To identify which of the NADPHox subunits is implicated in the enhanced enzyme activity, Real-Time PCR was employed to assess the gene expression of NADPHox subunits upon exposure of hSMC to nLDL, AGE-LDL or oxLDL. In comparison with nLDL, AGE-LDL induced a significantly increased level of mRNA for NOX1 (∼80%, P < 0.05), NOX4 (∼60%, P < 0.05) and p67phox (∼60%, P < 0.05) (Fig. 3A–D). Stimulation of hSMC with oxLDL resulted in a higher increase of NOX1, NOX4, p22phox and p67phox mRNA over the AGE-LDL level.
Fig 3.
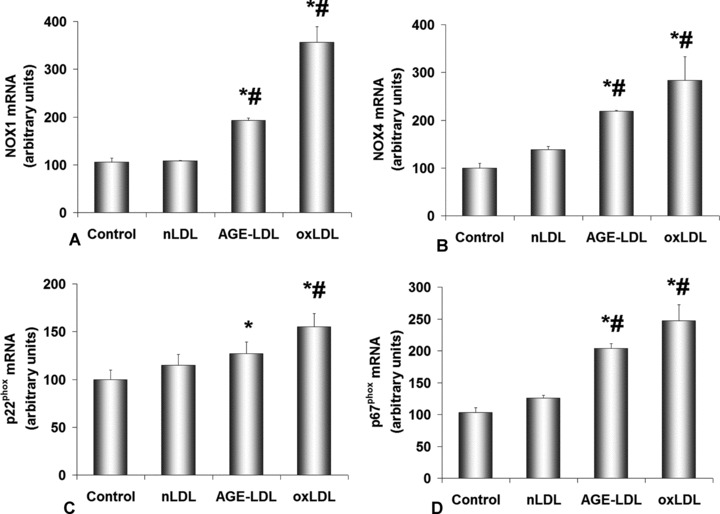
AGE-LDL up-regulates NADPH oxidase subunits expression in hSMC. Expression of mRNA obtained by Real-time PCR for NOX1 (A), NOX4 (B), p22phox (C) and p67phox (D) relative to control cells, not exposed to LDL. GAPDH was the reference gene, AGE-LDL, oxidized LDL (oxLDL) and native LDL (nLDL) concentration was 100 μg/ml and incubation time 24 hrs. Control, cells not exposed to LDL, *P < 0.05 versus control cells, #P < 0.05 versus nLDL, n= 3.
Western blotting employing anti-NOX1 and anti-NOX4 antibodies revealed a significant increase in the protein expression of NOX1 for AGE-LDL (0.54 ± 0.03, P < 0.05) exposed hSMC relative to control cells (0.33 ± 0.04). The data are in good agreement with the increase of NOX1 gene expression. The protein expression of NOX4 was significantly augmented in hSMC incubated with oxLDL (0.69 ± 0.12, P < 0.05), while AGE-LDL induced a smaller increase (0.48 ± 0.13) compared to control cells (0.34 ± 0.03).
AGE-LDL induces lipid loading of hSMC
To determine the lipid accumulation in hSMC following exposure to AGE-LDL, nLDL or oxLDL, the cells were stained with lipid specific fluorochrome, Nile Red. Fluorescence microscopy revealed a considerably increased number of fluorescent lipid droplets in cells incubated with AGE-LDL compared to control cells or cells incubated with nLDL (Fig. 4 A–C) or oxLDL (Fig. 4D). Enzymatic measurement of the intracellular cholesteryl esters (CE) accumulation in cells incubated for 24 hrs with 25, 50, 100 and 150 μg/ml AGE-LDL indicated that the maximum level of cholesterol was at 100 μg/ml glycated LDL in the medium. Time-course determination of the intracellular CE accumulation at 12, 24, 36 and 48 hrs indicated that the maximum level of cholesterol was at 24 hrs incubation, and the CE/TC ratio increased in cells exposed to 100 μg/ml AGE-LDL (30.08 ± 5.54, P < 0.05) compared to nLDL (16.68 ± 3.67) or oxLDL (20.06 ± 2.54). These data corroborate well with those obtained by lipid staining.
Fig 4.
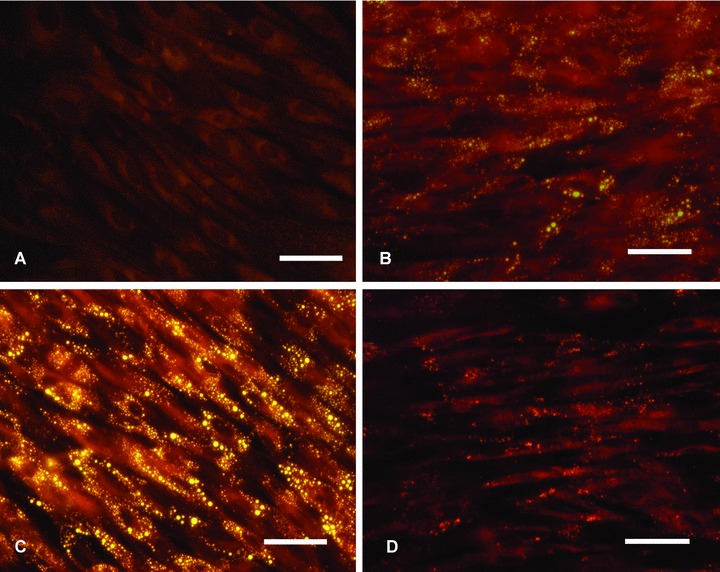
AGE-LDL induces lipid accumulation in hSMC. The cells were incubated with 100 μg/ml AGEL-DL, oxidized LDL (oxLDL) or native LDL (nLDL) for 24 hrs, fixed in 2% formaldehyde and stained with Nile Red (yellow fluorescence). (A) Control, cells not exposed to LDL, (B) cells exposed to nLDL, (C) AGE-LDL and (D) oxLDL; bar 5 25 mm, n= 3.
LDL receptors, CD36 and RAGE gene expression
The gene expression of LDL-R, LRP1, CD36 and RAGE in hSMC incubated with AGE-LDL or nLDL for 24 hrs was quantified by quantitative PCR.
The gene expression of LDL-R was significantly inhibited by exposure of cells both to nLDL (18 ± 2, P < 0.001) and to AGE-LDL (28 ± 7, P < 0.01), compared to control hSMC (considered 100) (Fig. 5A).
Fig 5.
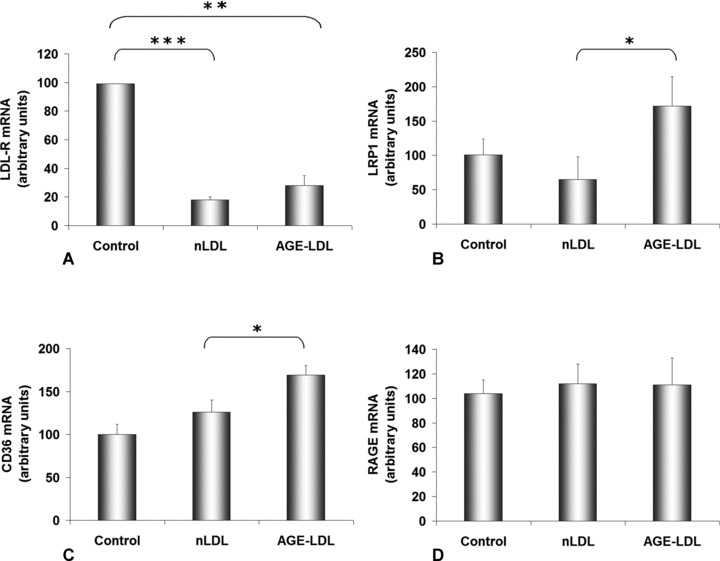
AGE-LDL modulates the gene expression of LDL receptors in hSMC. Cells were incubated for 24 hrs with 100 μg/ml native LDL (nLDL) or AGE-LDL and then subjected to quantitative PCR for LDL receptor (LDL-R, A), LDL receptor-related protein 1 (LRP1, B), scavenger receptor CD36 (C) and AGE-receptor (RAGE, D); 28S rRNA was the reference gene. Control, cells not exposed to LDL, *P < 0.05, **P < 0.01, ***P < 0.001, n= 3.
The LRP1 gene expression increased 2.6-fold (172 ± 43, P < 0.05) in AGE-LDL-exposed cells compared to cells incubated with nLDL (65 ± 33) (Fig. 5B).
AGE-LDL determined an increase of the gene expression for scavenger receptor CD36 with 35% (169 ± 11, P < 0.05) above the values obtained for nLDL-exposed cells (126 ± 14) (Fig. 5C).
Results of the quantification of RAGE mRNA indicated a 10% increase of the gene expression in both LDLs exposed cells compared to control ones, but this was not statistically significant (Fig. 5D).
LRP1, CD36 and RAGE protein expression
Incubation of hSMC with AGE-LDL (24 hrs) induced a 32% increase of LRP1 protein expression (0.90 ± 0.13, P < 0.05) relative to nLDL (0.68 ± 0.08), whereas oxLDL caused a 57% (1.07 ± 0.04, P < 0.05) increase (Fig. 6A); the data are in good accordance with the raise of the gene expression of LRP1.
Fig 6.
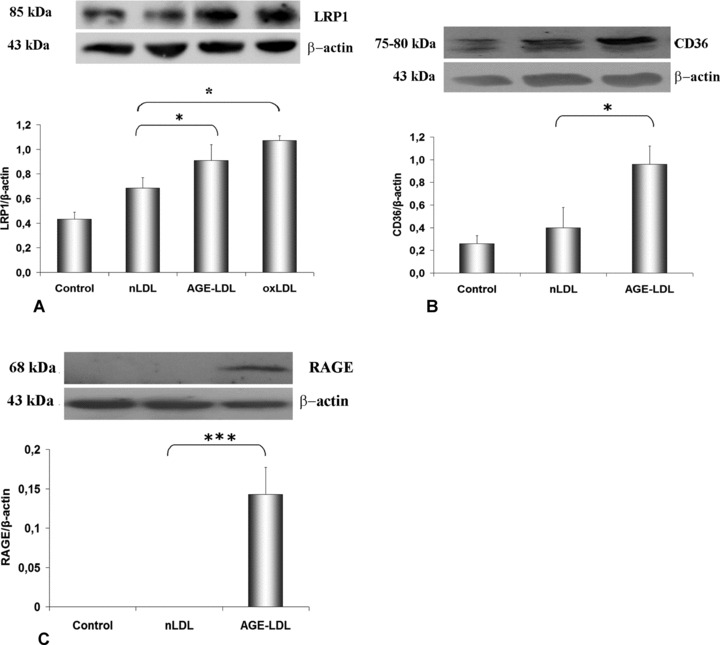
AGE-LDL promotes changes in LDL receptors protein expression in hSMC. Western blots and quantification of protein expression in cells incubated for 24 hrs with 100 μg/ml native LDL (nLDL), AGE-LDL or oxidized LDL (oxLDL): (A) LRP1; (B) CD36; (C) RAGE; b-actin was the reference protein. Control, cells not exposed to LDL, *P < 0.05, ***P < 0.001, n= 3.
In similar experimental conditions, the results showed that AGE-LDL induced a significantly increased protein expression of CD36 (0.96 ± 0.02, P < 0.05) compared to nLDL (0.40 ± 0.18), as illustrated in the blots and corresponding histograms (Fig. 6B). The augmentation of the CD36 protein expression was in good agreement with the measured increase of the CD36 mRNA. Under the same conditions, AGE-LDL induced an increased expression of RAGE protein (0.14 ± 0.03, P < 0.001) compared to nLDL (Fig. 6C).
MCP-1 gene and protein expression
The results obtained by quantitative PCR showed that upon incubation of hSMC with AGE-LDL, the gene expression of MCP-1 increased 1.6-fold (256 ± 35, P < 0.05) above control values, whereas the effect of nLDL was significantly lower (158 ± 5) (Fig. 7A).
Fig 7.
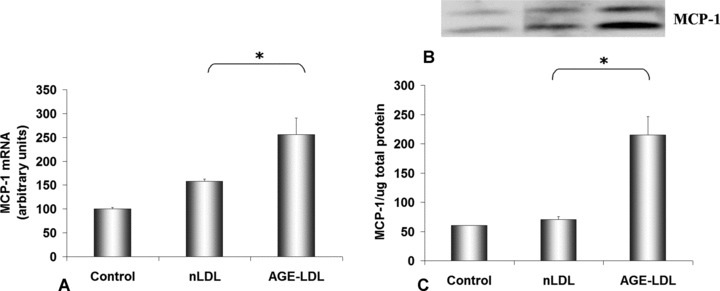
AGE-LDL stimulates MCP-1 gene and protein expression in hSMC. Cells were incubated for 24 hrs with 100 μg/ml native LDL (nLDL) or AGE-LDL and then subjected to quantification of MCP-1 gene expression relative to 28S rRNA (A), and Western blot of soluble MCP-1 protein relative to the total protein in the medium (B). Control, cells not exposed to LDL, *P < 0.05, n= 3.
A significant increase of MCP-1 protein released in the culture medium was detected after hSMC were incubated with AGE-LDL (215 ± 31, P < 0.05) compared to nLDL (70 ± 5) (Fig. 7B). In contrast, analysis of the protein expression of MCP-1 in cell lysates revealed no specific band on the Western blot.
The effect of AGE-LDL on hSMC viability, metabolic competence and proliferation
Viability of hSMC after exposure (24 hrs) to AGE-LDL, nLDL or oxLDL evaluated by the MTT test showed that the cells were not affected by the exposure to either of the LDLs, as compared to the viability of control cells (data not shown).
The energy metabolism test showed no significant change in ATP/DNA content when hSMC were incubated with AGE-LDL (1729 ± 151 RFU/μg DNA) relative to nLDL (1863 ± 84 RFU/μg DNA). The ATP content of cells incubated with oxLDL significantly decreased (1102 ± 257 RFU/μg DNA) as compared to nLDL. Results on the metabolic competence of treated cells indicated no changes in glucose uptake when cells were incubated with AGE-LDL (29.26 ± 1.47 mg glucose consumed /μg DNA) or oxLDL (28.85 ± 0.63 mg glucose consumed /μg DNA) compared to control cells (29.36 ± 0.84 mg glucose consumed / mg DNA) or cells incubated with nLDL (28.94 ± 0.84 mg glucose consumed / mg DNA).
Determination of the cellular DNA content of hSMC incubated for 24 hrs with 25, 50, 100 and 150 μg/ml AGE-LDL indicated that the maximum proliferation rate was at 100 μg/ml glycated LDL in the medium. Time-course determination of the cellular DNA content of hSMC incubated with 100 mg/ml AGE-LDL for 12, 24, 36 and 48 hrs indicated a maximum rate of proliferation at 24 hrs. Results showed that AGE-LDL induced a 30% increase (49.57 ± 4.42 μg DNA, P < 0.01) of the cells proliferation rate at 24 hrs, while oxLDL induced a 48% increase (56.87 ± 3.84 μg DNA, P < 0.001) relative to the cells exposed to nLDL (38.45 ± 1.44 μg DNA) (Fig. 8).
Fig 8.
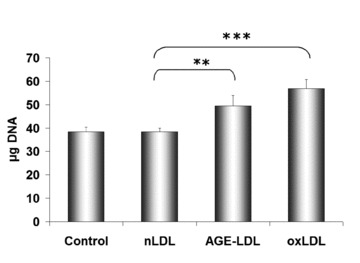
Increased proliferation rate of hSMC induced by AGE-LDL. Cells were incubated for 24 hrs with 100 μg protein/ml AGE-LDL, oxidized LDL (oxLDL) or native LDL (nLDL). Cell proliferation was assessed by Hoechst 33258 fluorimetric assay. Control, cells not exposed to LDL, **P < 0.01, ***P < 0.001, n= 3.
Discussion
In diabetes, dysfunction of the cells of the vessel wall may be in part the result of their interaction with glycated proteins, such as AGE-LDL, present concurrently in the plasma and in the intima. AGE-LDL is detected both in hyperlipidaemic and hyperglycaemic patients’ plasma [28, 29]. The effect of AGE-LDL on vascular SMC is not known. To detect the alterations generated by AGE-LDL on hSMC, we looked for the NADPHox activity and the intracellular ROS production, the accumulation of lipid droplets and the implication of specific receptors (LRP1, CD36, RAGE), the induction of MCP-1 and the modulation of the proliferation rate.
We chose to incubate the cells with 10 mg AGE-LDL protein/dl culture medium that is in the physiological range (9.3 mg/dl glycated LDL apoB in the serum of patients with diabetes versus 4.8 mg/dl in controls) [28]. Interestingly, Akanji et al.[29] reported that serum-glycated LDL level is increased in patients with hyperlipidaemia without diabetes (6.12 mg/dl), and further increased in patients with both hyperlipidaemia and diabetes (7.82 mg/dl) compared with 2.74 mg/dl in healthy subjects. It has been suggested that glycated LDL apoB could be utilized as an index of medium-term glycaemic control as it correlates with glucose, fructosamine and glycated haemoglobin (HbA1c) in patients with diabetes [30].
It was reported that in the vasculature, the up-regulation of NADPHox activity is often associated with the progression of vascular disorders related to hypertension, diabetes or obesity, and is correlated with increase in NOX1 mRNA level. Unlike NOX1, which requires regulatory subunits for its activity, NOX4 produces ROS constitutively and changes in its gene expression affect directly the NOX4 activity [31].
In our experiments, the pro-oxidant potential of AGE-LDL on hSMC was substantiated by the pronounced increase of the gene expression of NADPHox subunits (NOX1, NOX4, p22phox, p67phox) compared to cells exposed to nLDL, and this correlated well with an enhanced superoxide production driven by the activated NADPHox. The demonstrated AGE-LDL stimulated protein expression of RAGE may be part of the molecular mechanism accountable for the increased ROS production in hSMC by activation of protein kinase C and the increased activity of NADPHox, considered to be key events by which AGEs trigger ROS. Our data indicate that AGE-LDL induces up-regulation of NOX1 and NOX4; these may have an important role in promoting vascular SMC growth, proliferation and differentiation [32]. The p22phox subunit is essential for the NADPHox activity and it was demonstrated [33] that p22phox is more abundant in advanced atherosclerosis plaques than in non-atherosclerotic arteries, suggesting a correlation between p22phox expression, superoxide production and the severity of atherosclerotic lesions. In previous studies, we demonstrated that NF-κB and AP-1 regulate p22phox gene expression in hSMC [34, 35]. The p67phox is regulated also by the same transcription factors [36, 37]. We can suggest that the regulation of p22phox and p67phox expression by AGE-LDL in hSMC may be mediated through an NF-κB or AP-1-dependent mechanism.
AGE-LDL induced lipid loading and cholesteryl esters accumulation in hSMC, having an optimum dose at 100 μg/ml AGE-LDL and 24 hrs incubation, in good agreement with Tirziu et al. and Llorente-Cortes et al.[21, 38]. To uncover the possible mechanisms involved in this process, we searched for the expression of native and modified LDL receptors. We report here that AGE-LDL induces concurrently a decrease in the gene expression for the LDL receptor, and a significant increase in LRP1 expression. To the best of our knowledge, this is the first report indicating the significant augmentation of LRP1 gene and protein expression induced by AGE-LDL in vascular hSMC. The decreased recognition of glycated LDL by the LDL-R correlates directly with the degree of apoB glycation, and may explain the increased intimal retention of glycated Lp [39], formation of AGE products and the activation of alternative uptake mechanisms (non-LDL-R pathways) in vascular cells of diabetic patients [40]. It was reported that in vitro aggregated LDL stimulates LRP1 expression and is internalized by the LRP1 pathway [38].
In diabetic patients, chronic hyperglycaemia enhances glucose-induced LDL glycoxidation, both in circulation and in the vessel wall. We have demonstrated the increased expression of CD36 in hSMC exposed to AGE-LDL. This result adds to the previous reports that AGE-proteins are recognized by the CD36 receptor [41] and its protein expression is higher in type 2 diabetic rat aortas than in controls [42].
AGE-LDL induced increased protein expression of RAGE in hSMC, without affecting the RAGE gene expression. It was reported that RAGE mRNA levels do not appear to correlate closely with levels of RAGE antigen, suggesting that there are likely to be multiple factors regulating translation, post-translational processing, and degradation of RAGE [43].
Taken together our data, we can predict that the AGE-LDL-induced augmented expression of LRP1, CD36, and RAGE in hSMC contributes to the increased uptake of modified lipoproteins in the neointima and the progression of diabetic atherosclerosis.
Data from this study show that AGE-LDL generates an increase of MCP-1 gene expression in hSMC and of MCP-1 protein released from the cells in the culture medium. It was reported that AGE-LDL induces expression of MCP-1 receptor (CCR2) in monocytes and thus can stimulate the chemotactic response elicited by MCP-1 [44]. Thus, we can suggest that AGE-LDL-induced expression of MCP1 in hSMC and that of the matching receptor on monocytes/macrophages contributes significantly to the infiltration of plasma monocytes into the neointima and to the interaction between SMC and macrophages within the atherosclerotic plaque.
In our experiments, the energy metabolism of hSMC, expressed as intracellular ATP/μg DNA content, was not affected by exposure to AGE-LDL relative to nLDL, while in cells incubated with oxLDL the level was decreased, in good agreement with previous data [45]. The metabolic competence of hSMC showed no change in cellular glucose/mg DNA consumption when cells were incubated with AGE-LDL or oxLDL. A similar report on the effect of AGE-proteins on hSMC glucose metabolism was presented by Ballinger [46] on AGE-BSA-treated macrophages.
Incubation of AGE-LDL with hSMC induced a significant increase in the proliferation rate (30%) compared to cells exposed to nLDL. This effect can be correlated with the AGE-LDL-induced NOX4 up-regulation, which is known to have an important role in promoting vascular SMC proliferation. This result corroborates and extends previous data showing that glycated LDL directly stimulates SMC proliferation [47] and AGEs induce proliferation of cultured rat vascular SMC [48]. We suggest that AGE-LDL, by promoting the proliferation of SMC in the neointima, contributes to the accelerated atherosclerotic plaque formation in diabetic patients.
In summary, the results of this study indicate that AGE-LDL activates hSMC by up-regulation of receptors such as LRP1, CD36, RAGE and the subsequent lipid accumulation, determines a pro-oxidant state by activation of NADPHox, and a pro-inflammatory state expressed by the induction of MCP-1. Understanding the outcome of the association of irreversible glycation and oxidation of LDL, two critical processes occurring in diabetes, will guide the design of novel or complementary therapeutic strategies to prevent or treat this disease.
Acknowledgments
This project was supported by a grant from the Romanian Ministry of Education and Research (PNII #41-067/2007). The authors thank Dr. Loredan Stefan Niculescu, Mrs Ioana Andreescu, Mrs. Ioana Manolescu and Ms. Cristina Dobre for their excellent technical assistance.
References
- 1.Simionescu M, Popov D, Sima A, et al. Pathobiochemistry of combined diabetes and atherosclerosis studied on a novel animal model. The hyperlipemic-hyperglycemic hamster. Am J Pathol. 1996;148:997–1014. [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen MP, Ziyadeh FN, Chen S. Amadori-modified glycated serum proteins and accelerated atherosclerosis in diabetes: pathogenic and therapeutic implications. J Lab Clin Med. 2006;147:211–9. doi: 10.1016/j.lab.2005.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shiu SW, Tan KC, Wong Y, et al. Glycoxidized LDL increases lectin-like oxidized low density lipoprotein receptor-1 in diabetes mellitus. Atherosclerosis. 2009;203:522–7. doi: 10.1016/j.atherosclerosis.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 4.Younis N, Sharma R, Soran H, et al. Glycation as an atherogenic modification of LDL. Curr Opin Lipidol. 2008;19:378–84. doi: 10.1097/MOL.0b013e328306a057. [DOI] [PubMed] [Google Scholar]
- 5.Stitt AW, He C, Friedman S, et al. Elevated AGE-modified ApoB in sera of euglycemic, normolipidemic patients with atherosclerosis: relationship to tissue AGEs. Mol Med. 1997;3:617–27. [PMC free article] [PubMed] [Google Scholar]
- 6.Doran AC, Meller N, McNamara CA. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:812–9. doi: 10.1161/ATVBAHA.107.159327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bucala R, Makita Z, Vega G, et al. Modification of low density lipoprotein by advanced glycation end products contributes to the dyslipidemia of diabetes and renal insufficiency. Proc Natl Acad Sci USA. 1994;91:9441–5. doi: 10.1073/pnas.91.20.9441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sima A, Stancu C. Modified lipoproteins accumulate in human coronary atheroma. J Cell Mol Med. 2002;6:110–1. doi: 10.1111/j.1582-4934.2002.tb00316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sima A, Popov D, Starodub O, et al. Pathobiology of the heart in experimental diabetes: immunolocalization of lipoproteins, immunoglobulin G, and advanced glycation endproducts proteins in diabetic and/or hyperlipidemic hamster. Lab Invest. 1997;77:3–18. [PubMed] [Google Scholar]
- 10.Petry A, Djordjevic T, Weitnauer M, et al. NOX2 and NOX4 mediate proliferative response in endothelial cells. Antioxid Redox Signal. 2006;8:1473–84. doi: 10.1089/ars.2006.8.1473. [DOI] [PubMed] [Google Scholar]
- 11.Soro-Paavonen A, Watson AM, Li J, et al. Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes. 2008;57:2461–9. doi: 10.2337/db07-1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yin CC, Huang KT. H2O2 but not O2- elevated by oxidized LDL enhances human aortic smooth muscle cell proliferation. J Biomed Sci. 2007;14:245–54. doi: 10.1007/s11373-006-9132-4. [DOI] [PubMed] [Google Scholar]
- 13.Li AC, Glass CK. The macrophage foam cell as a target for therapeutic intervention. Nat Med. 2002;8:1235–42. doi: 10.1038/nm1102-1235. [DOI] [PubMed] [Google Scholar]
- 14.Dragomir E, Manduteanu I, Calin M, et al. High glucose conditions induce upregulation of fractalkine and monocyte chemotactic protein-1 in human smooth muscle cells. Thromb Haemost. 2008;100:1155–65. [PubMed] [Google Scholar]
- 15.Nistor A, Simionescu M. Uptake of low density lipoproteins by the hamster lung. Interactions with capillary endothelium. Am Rev Respir Dis. 1986;134:1266–72. doi: 10.1164/arrd.1986.134.6.1266. [DOI] [PubMed] [Google Scholar]
- 16.Nivoit P, Wiernsperger N, Moulin P, et al. Effect of glycated LDL on microvascular tone in mice: a comparative study with LDL modified in vitro or isolated from diabetic patients. Diabetologia. 2003;46:1550–8. doi: 10.1007/s00125-003-1225-2. [DOI] [PubMed] [Google Scholar]
- 17.Murtiashaw MH, Young JE, Strickland AL, et al. Measurement of nonenzymatically glucosylated serum protein by an improved thiobarbituric acid assay. Clin Chim Acta. 1983;130:177–87. doi: 10.1016/0009-8981(83)90115-8. [DOI] [PubMed] [Google Scholar]
- 18.Markwell MA, Haas SM, Bieber LL, et al. A modification of the Lowry procedure to simplify protein determination in membrane and lipoprotein samples. Anal Biochem. 1978;87:206–10. doi: 10.1016/0003-2697(78)90586-9. [DOI] [PubMed] [Google Scholar]
- 19.Krishnamurti U, Rondeau E, Sraer JD, et al. Alterations in human glomerular epithelial cells interacting with nonenzymatically glycosylated matrix. J Biol Chem. 1997;272:27966–70. doi: 10.1074/jbc.272.44.27966. [DOI] [PubMed] [Google Scholar]
- 20.Fogelman AM, Shechter I, Seager J, et al. Malondialdehyde alteration of low density lipoproteins leads to cholesteryl ester accumulation in human monocyte-macrophages. Proc Natl Acad Sci USA. 1980;77:2214–8. doi: 10.1073/pnas.77.4.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tirziu D, Jinga VV, Serban G, et al. The effects of low density lipoproteins modified by incubation with chondroitin 6-sulfate on human aortic smooth muscle cells. Atherosclerosis. 1999;147:155–66. doi: 10.1016/s0021-9150(99)00187-2. [DOI] [PubMed] [Google Scholar]
- 22.Gamble W, Vaughan M, Kruth HS, et al. Procedure for determination of free and total cholesterol in micro- or nanogram amounts suitable for studies with cultured cells. J Lipid Res. 1978;19:1068–70. [PubMed] [Google Scholar]
- 23.Rago R, Mitchen J, Wilding G. DNA fluorometric assay in 96-well tissue culture plates using Hoechst 33258 after cell lysis by freezing in distilled water. Anal Biochem. 1990;191:31–4. doi: 10.1016/0003-2697(90)90382-j. [DOI] [PubMed] [Google Scholar]
- 24.Ungvari Z, Csiszar A, Edwards JG, et al. Increased superoxide production in coronary arteries in hyperhomocysteinemia: role of tumor necrosis factor-alpha, NAD(P)H oxidase, and inducible nitric oxide synthase. Arterioscler Thromb Vasc Biol. 2003;23:418–24. doi: 10.1161/01.ATV.0000061735.85377.40. [DOI] [PubMed] [Google Scholar]
- 25.Skatchkov MP, Sperling D, Hink U, et al. Validation of lucigenin as a chemiluminescent probe to monitor vascular superoxide as well as basal vascular nitric oxide production. Biochem Biophys Res Commun. 1999;254:319–24. doi: 10.1006/bbrc.1998.9942. [DOI] [PubMed] [Google Scholar]
- 26.Li JM, Mullen AM, Yun S, et al. Essential role of the NADPH oxidase subunit p47(phox) in endothelial cell superoxide production in response to phorbol ester and tumor necrosis factor-alpha. Circ Res. 2002;90:143–50. doi: 10.1161/hh0202.103615. [DOI] [PubMed] [Google Scholar]
- 27.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 28.Tames FJ, Mackness MI, Arrol S, et al. Non-enzymatic glycation of apolipoprotein B in the sera of diabetic and non-diabetic subjects. Atherosclerosis. 1992;93:237–44. doi: 10.1016/0021-9150(92)90260-n. [DOI] [PubMed] [Google Scholar]
- 29.Akanji AO, Abdella N, Mojiminiyi OA. Determinants of glycated LDL levels in nondiabetic and diabetic hyperlipidaemic patients in Kuwait. Clin Chim Acta. 2002;317:171–6. doi: 10.1016/s0009-8981(01)00792-6. [DOI] [PubMed] [Google Scholar]
- 30.Cohen MP, Jin Y, Lautenslager GT. Increased plasma glycated low-density lipoprotein concentrations in diabetes: a marker of atherogenic risk. Diabetes Technol Ther. 2004;6:348–56. doi: 10.1089/152091504774198043. [DOI] [PubMed] [Google Scholar]
- 31.Lassegue B, Clempus RE. Vascular NAD(P)H oxidases: specific features, expression, and regulation. Am J Physiol Regul Integr Comp Physiol. 2003;285:R277–97. doi: 10.1152/ajpregu.00758.2002. [DOI] [PubMed] [Google Scholar]
- 32.Zhang L, Sheppard OR, Shah AM, et al. Positive regulation of the NADPH oxidase NOX4 promoter in vascular smooth muscle cells by E2F. Free Radic Biol Med. 2008;45:679–85. doi: 10.1016/j.freeradbiomed.2008.05.019. [DOI] [PubMed] [Google Scholar]
- 33.Azumi H, Inoue N, Takeshita S, et al. Expression of NADH/NADPH oxidase p22phox in human coronary arteries. Circulation. 1999;100:1494–8. doi: 10.1161/01.cir.100.14.1494. [DOI] [PubMed] [Google Scholar]
- 34.Manea A, Manea SA, Gafencu AV, et al. Regulation of NADPH oxidase subunit p22(phox) by NF-κB in human aortic smooth muscle cells. Arch Physiol Biochem. 2007;113:163–72. doi: 10.1080/13813450701531235. [DOI] [PubMed] [Google Scholar]
- 35.Manea A, Manea SA, Gafencu AV, et al. AP-1-dependent transcriptional regulation of NADPH oxidase in human aortic smooth muscle cells: role of p22phox subunit. Arterioscler Thromb Vasc Biol. 2008;28:878–85. doi: 10.1161/ATVBAHA.108.163592. [DOI] [PubMed] [Google Scholar]
- 36.Gauss KA, Nelson-Overton LK, Siemsen DW, et al. Role of NF-κappaB in transcriptional regulation of the phagocyte NADPH oxidase by tumor necrosis factor-alpha. J Leukoc Biol. 2007;82:729–41. doi: 10.1189/jlb.1206735. [DOI] [PubMed] [Google Scholar]
- 37.Gauss KA, Bunger PL, Quinn MT. AP-1 is essential for p67(phox) promoter activity. J Leukoc Biol. 2002;71:163–72. [PubMed] [Google Scholar]
- 38.Llorente-Cortes V, Otero-Vinas M, Camino-Lopez S, et al. Cholesteryl esters of aggregated LDL are internalized by selective uptake in human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2006;26:117–23. doi: 10.1161/01.ATV.0000193618.32611.8b. [DOI] [PubMed] [Google Scholar]
- 39.Moro E, Alessandrini P, Zambon C, et al. Is glycation of low density lipoproteins in patients with Type 2 diabetes mellitus a LDL pre-oxidative condition. Diabet Med. 1999;16:663–9. doi: 10.1046/j.1464-5491.1999.00136.x. [DOI] [PubMed] [Google Scholar]
- 40.Lyons TJ. Lipoprotein glycation and its metabolic consequences. Diabetes. 1992;41:67–73. doi: 10.2337/diab.41.2.s67. [DOI] [PubMed] [Google Scholar]
- 41.Ohgami N, Nagai R, Ikemoto M, et al. Cd36, a member of the class b scavenger receptor family, as a receptor for advanced glycation end products. J Biol Chem. 2001;276:3195–202. doi: 10.1074/jbc.M006545200. [DOI] [PubMed] [Google Scholar]
- 42.De Oliveira Silva C, Delbosc S, Arais C, et al. Modulation of CD36 protein expression by AGEs and insulin in aortic VSMCs from diabetic and non-diabetic rats. Nutr Metab Cardiovasc Dis. 2008;18:23–30. doi: 10.1016/j.numecd.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 43.Brett J, Schmidt AM, Yan SD, et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol. 1993;143:1699–712. [PMC free article] [PubMed] [Google Scholar]
- 44.Isoda K, Folco E, Marwali MR, et al. Glycated LDL increases monocyte CC chemokine receptor 2 expression and monocyte chemoattractant protein-1-mediated chemotaxis. Atherosclerosis. 2008;198:307–12. doi: 10.1016/j.atherosclerosis.2007.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sukhanov S, Higashi Y, Shai SY, et al. Novel effect of oxidized low-density lipoprotein: cellular ATP depletion via downregulation of glyceraldehyde-3-phosphate dehydrogenase. Circ Res. 2006;99:191–200. doi: 10.1161/01.RES.0000232319.02303.8c. [DOI] [PubMed] [Google Scholar]
- 46.Ballinger ML, Thomas MC, Nigro J, et al. Glycated and carboxy-methylated proteins do not directly activate human vascular smooth muscle cells. Kidney Int. 2005;68:2756–65. doi: 10.1111/j.1523-1755.2005.00746.x. [DOI] [PubMed] [Google Scholar]
- 47.Cho HM, Choi SH, Hwang KC, et al. The Src/PLC/PKC/MEK/ERK signaling pathway is involved in aortic smooth muscle cell proliferation induced by glycated LDL. Mol Cells. 2005;19:60–6. [PubMed] [Google Scholar]
- 48.Wu S, Song T, Zhou S, et al. Involvement of Na+/H+ exchanger 1 in advanced glycation end products-induced proliferation of vascular smooth muscle cell. Biochem Biophys Res Commun. 2008;375:384–9. doi: 10.1016/j.bbrc.2008.08.008. [DOI] [PubMed] [Google Scholar]