

Abstract
It has been known for some considerable time that sustained hypertension changes the circulatory architecture both in the heart and blood vessels. The histopathological alterations are of considerable interest because once they have developed they appear to carry an adverse prognostic risk. In the heart it is apparent that there is hypertrophy. This extends also to the large- and medium-sized blood vessels but at the level of the smaller arteries that contribute to vascular resistance, this is not the case: it is clear that the physiological response to higher pressures is a change in the positional conformation of the pre-existing tissue constituents and as a result of this the lumen is narrowed. This brief review looks at our knowledge in this area and attempts to clarify our understanding of how hypertension brings these about and what happens when these homeostatic mechanisms break down. From a therapeutic perspective it appears imperative to control blood pressure in an attempt to reverse or prevent such alterations to cardiovascular structure. Our knowledge is fast expanding in this field and it is only to be anticipated that as detection methodology improves everyday practice will alter as we profile our patients in terms of structural alterations in the ventricle and blood vessels.
Keywords: resistant artery, eutrophic remodelling, myogenic tone
Introduction
An early cross-sectional study of mild hypertension reported that the predominant hemodynamic feature was a high cardiac output [1] and this observation has been confirmed consistently since. Although not invariably so, the high cardiac output is accompanied by an increased heart rate and several studies have demonstrated that when oxygen consumption is measured, both cardiac output and this parameter are raised [2]. In the mild early stages of essential hypertension the peripheral resistance is low although the crucial point is that it remains appropriately high for the corresponding cardiac output. A 20-year longitudinal study of the haemodynamics of essential hypertension, confirmed the finding of initially increased cardiac index heart rate oxygen consumption and blood pressure with normal peripheral resistance [3]. However, over this period the high cardiac index and normal total peripheral resistance pattern changes to a low cardiac index high resistance pattern. Again, the inappropriately high level of vascular resistance for the increase in cardiac output is a hallmark of early hypertension. Such a haemodynamic profile could be ascribed to a high sympathetic nervous tone with the resulting increased drive to the heart, peripheral circulation and metabolic receptors, which would then promote enhanced oxygen consumption. Other studies demonstrated that autonomic blockade of the heart in mild hypertensive patients restored cardiac output to normal and there was a combination of increased sympathetic tone and decreased parasympathetic activity in such individuals [4, 5]. This combination of increased sympathetic discharge coupled to a reduced parasympathetic activity suggests that the abnormality in essential hypertension is one of integrated function in the medulla oblongata.
Recent data have proposed that this might be a consequence of neurovascular compression on the left ventrolateral medulla. Therefore it should follow that the resulting increased sympathetic activity would be distributed to all innervated organs and vascular beds producing uniform vasoconstriction and the predicted cardiac indices. There is evidence for this in the heart, kidney and skeletal muscle but it has not been confirmed in studies of the hepatomesenteric circulation [6]. In other words, it is difficult to provide evidence for a ubiquitous abnormality of sympathetic function in all vascular beds although the overall integrated haemodynamic profile can be ascribed to increased sympathetic discharge.
Structural changes in the circulation
Since the work of Bright and Johnson it has been recognized that the walls of medium-sized arteries are thickened in hypertension. However, it has to be conceded that their contribution to vascular resistance is small. The histopathological change appears to be hypertrophy, and indeed the heart and medium-sized blood vessels demonstrate this hypertrophic response to blood pressure. In addition, until the heart dilates it is of interest to note that the hypertrophic response takes place at the expense of the ventricular cavity: in other words there is inward encroachment on the chamber space. Therefore in any form of sustained hypertension there is an alteration in the architecture of the circulation that inevitably occurs in consequence. At the level of the resistance artery where the internal diameter of the blood vessels is around 250 μm or less, there is evidence of a reduced lumen diameter and increased media thickness: lumen diameter ratio. This was originally reported in necropsy specimens [7] and subsequently confirmed in segments of artery mounted as isometric ring preparations on wires and in vessels perfused in vitro[8]. Detailed histological analyses more recently carried out have suggested that eutrophic inward remodelling occurs at this point in the circulation (Fig. 1). By this it is meant that there is a narrowing of the vascular lumen without having to invoke a growth response of the arterial wal1 [9]. A small amount of hypertrophy may be observed in some pathological states where hypertrophy may supervene and is an adverse prognostic sign [10].
Fig 1.
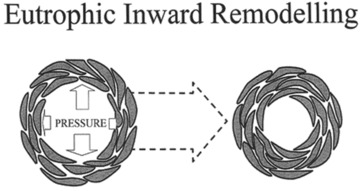
A cartoon representation of small arteries in transfer section. The increase in intraluminal pressure leads to eutrophic inward remodelling. The results of this are clearly demonstrated. The number of smooth muscle cells in both arteries is the same as is their size indicating that there is no hypertrophy but the cells have re-orientated themselves and as a result produced an increased number of smooth cell layers and decreased the external and internal diameters of the vessel. This process appears to be ubiquitously distributed across vascular beds within hypertensive individuals.
The most compelling issue is whether structural changes in these small blood vessels occur before blood pressure rises or in consequence of the hypertension? The corollary of what has been described above, namely the existence of excessive sympathetic nervous activity in the early stages of the development of hypertension being the trigger for the majority of this disorder then it is tempting to regard the small artery as changing shape in consequence. In a study of small artery structural characteristics in first degree offspring of hypertensive patients, in whom one would predict an increasing propensity to developing hypertension, there was no evidence of any alteration despite mean blood pressures being higher [11]. Also, in another study in young genetically hypertension-prone spontaneously hypertensive rats (SHR), where non-constricting ligatures were placed on the iliac artery and the animals allowed to mature until a point when the ligature compressed gradually on the vessel, the pressures in the hind limb were kept low and structural changes were not observed [12]. Therefore the impression gained is that the vascular changes follow rises in pressure. However, most recently it has been reported that mice mutant for Emilin 1, a cysteine-rich secreted glycoprotein expressed in the vascular tree, display hypertension, increased peripheral vascular resistance and reduced blood vessel size. Emilin 1 inhibits TFG-β signalling by binding specifically to proTGF-β precursor and prevents its maturation by furin convertases in the extracellular space [13]. Therefore in this model at least structural changes may precede hypertension. The area remains contentious. The other important issue to consider at this time is whether structural alterations amplify vasoconstrictor responses? Korner and Angus developed a theory with respect to the role of an increased wall: lumen ratio in the resistance vasculature, which maintains the elevated peripheral resistance in hypertension. This is based on calculating resistance from pressure and flow recordings and consequently calculating an arbitrary radius from the fourth route of 1/R (Poiseuille’s law). The calculations indicate a constant narrowing in vessel radius from maximal dilatation to maximal constriction in hypertension, which acts to amplify changes in resistance. This has been very controversial. A number of studies has consistently reported structural changes predicted and described above in small arteries with an internal diameter between 200 and 300 μm from patients with essential hypertension and in various animal models with the disease. Such reports should strengthen this amplifier theory. However, direct supporting evidence needs the measurement of the lumen diameter of blood vessels demonstrated to contribute to the control of resistance in a vascular bed but under extremely rigorous conditions [14]. In this context particular emphasis should be placed on measurement of diameter made at physiological levels of tone. This has been carried out using studies of functional and structural characteristics of small mesenteric arteries cannulated and pressurized in vitro. Such arteries have spontaneous myogenic tone at physiological pressures, which is a major determinant of diameter in the resistance vasculature unlike the upstream mesenteric arteries frequently studied in hypertension research. Distal mesenteric arteries were pressurized to 63% of the mean aortic pressure of each rat. It is the pressure recorded at this level of the mesenteric/intestinal vasculature in both rat strains indicating a location within the resistance vasculature. In the absence of tone, SHR vessels had a reduced lumen diameter although this was of borderline significance. Nevertheless, a clear increase in wall lumen ratio was observed. However, with spontaneous myogenic tone, lumen diameter became identical in the two strains, and importantly remains identical as tone was increased throughout the complete noradrenaline concentration response curve [15]. Such data do not support the hypothesis of an increased wall lumen ratio acting as an amplifier in hypertension.
Studies in isolated segments of small artery or in isolated intact vascular beds can provide clear insights into basic mechanisms, which can only be considered as hypothesis generating regarding the relevance of a particular mechanism. Surprisingly few studies have actually been carried out to date in intact conscious animals and overall the results are not consistent with an amplifier hypothesis. In a recent study, blood pressure and total peripheral resistance responses to infusion of the α agonist phenylephrine at two rates at the development phase of hypertension in SHR aged 8–26 weeks was compared with aged matched Wistar Kyoto rats before and after ganglionic blockade. At 16 weeks of age more complete dose–response curves to the α agonist methoxamine were constructed. Total peripheral resistance responses to phenylephrine were significantly less in SHR versus WKY rats at all ages in the study. The higher infusion rate increased mean arterial pressure by approximately 80 mmHg and nearly doubled the TPR. In contrast, blood pressure and TPR responses to methoxamine were enhanced in SHR in low rates of infusion but did not differ at the higher rates. Assuming that methoxamine is the most specific α agonist, these results are suggestive for functional rather than structural changes being contributory to the hyper-responsiveness to modest α receptor stimulation [16]. From a pathophysiological perspective, changes in TPR in response to low/modest stimulation appear to be a relevant parameter of TPR reactivity. More marked stimulation leading to 5- or 6-fold increases in TPR may lead to increased responsiveness but these are unlikely to be relevant for the maintenance or development of most hypertension. In other words, in vivo studies do not actually support the hypothesis that structural changes amplify vasoconstrictor responses throughout the circulation.
Remodelling and hypertrophy in the circulation
To understand how hypertension produces non-hypertrophic changes in small arteries, one must look at the physiological role of the resistance vasculature. At normal pressures, these vessels exhibit a level of contraction (myogenic tone), which is independent of neurohormonal influences and in functioning in this way the response enables arteries to constrict or dilate in response to changes in upstream pressure. This process known as the myogenic response is only observed in smaller resistance arteries, which mediate autoregulation of blood flow and stabilize capillary pressure. This ensures that target organs downstream are supplied with oxygenated blood at a constant flow and pressure. Hypertrophy is observed in vessels which do not possess myogenic tone whereas in smaller resistance arteries, initial increase in pressure will bring about an increased myogenic constriction. If an individual has untreated hypertension then there will be prolonged myogenic constriction as the resistance vasculature endeavours to protect the target organs downstream from pressure-induced damage brought about by an increase in blood flow. Prolonged vasoconstriction will lead to inward eutrophic remodelling and/or a reduced arterial distensibility. The structural difference between large conduit and medium-sized arteries and downstream resistance vessels is apparent in many models of hypertension: for example, in a hypertensive model brought on by chronic nitric oxide synthase inhibition. In addition, the magnitude and duration of an increase in intraluminal pressure plays a role in determining the remodelling response.
Molecular mechanisms responsible for eutrophic remodelling
Eutrophic inward remodelling is a process of structural adaptation observed in most forms of hypertension including the onset of hypertension and milder hypertensive states. However, a few animal models of hypertension, such as a model developing hypertension independent of the renin angiotensin system (BPH-2 mice), demonstrate hypertrophy as the predominating structural change. Inward eutrophic remodelling is a relatively fast functional adaptation observed after prolonged vasoconstriction and is thought to be an energetically favoured mechanism to preserve a lumen diameter for long periods. The process is also the preferred physiological mechanism by which wall stress can be normalized while maintaining vasomotor tone. Studies of the well-characterized TGR (MREN2) 27 rat which develops fulminant hypertension from 4 weeks of age have demonstrated that eutrophic inward remodelling occurs from 4 weeks and is dependent on the integrin aVβ3, a multifunctional extracellular matrix receptor [17]. However, hypertrophy does begin to appear between 6 and 8 weeks of age. This is important and will be discussed below. The extracellular matrix of resistance arteries is subject to tensile force exerted by blood pressure, which is transferred through integrins across the cell membrane and linked by molecular complexes to the cytoskeleton. Specific integrin subtypes are utilized initially for the mechanotransduction of pressure. Using peptides and specific antibodies, it has been shown that integrins αVβ3 and α5β1 indirectly regulate the myogenic response by influencing the control of calcium flow through ion channels; α5β1 is responsible for the initial calcium influx required to establish vascular tone and αVβ3 mediates force maintenance by calcium sensitization of contractile components. These integrins can form complexes, which regulate cytoskeletal dynamics and maintain a vascular myogenic force at a given pressure (Fig. 2) [18]. This is ameliorated if there is cytoskeletal disruption. Cytoskeletal proteins such as heat-shock protein 27 activated by RhoA kinases have been shown to regulate myogenic tone. It is now clear that RhoA signalling plays a central role in both calcium sensitization and regulation of actin dynamics in small artery remodelling.
Fig 2.
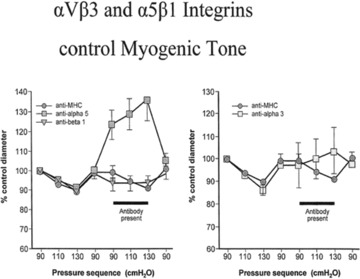
These figures show a percentage control diameter of isolated arterioles exposed to step increments and intraluminal pressure before and after abluminal treatment with a control antibody anti-rat major histocompatibility complex. Anti-α5-integrin function blocking antibody or an anti-β1-integrin function-blocking antibody, or anti-β3-integrin function-blocking antibody. In both cases one can see the amelioration of the myogenic response in consequence implicating these two integrins in the control of myogenic tone (data taken from [17]).
In contrast to molecular signalling mechanisms behind the vascular myogenic response relatively few data are available on the role of integrins and the underlying biochemical pathways of the next stage of vascular adaptation for hypertension. That is, the migration of vascular smooth muscle cells towards a narrow lumen. Remodelling involves a migratory process following prolonged vasoconstriction whereby existing vascular smooth cells reposition themselves in the vascular wall and thereby produce a narrow lumen. A characteristic of migrating cells in vitro is the presence of lamealae podial and filopodial protrusions containing focal adhesion kinase which provide a substrate for other cytosolic proteins such as SrC and interact with actin-associated cytoplasmic components. Recently it has been shown that the migration of vascular smooth muscle cells of arteries in vivo is more subtle and limited to elongation of tape in smooth muscle cells and an increase in cellular overlap [19]. It is thought that cytoskeletal re-arrangements in subsequent force generation play a central role in these changes. The exact cellular signalling system is still uncertain. Integrin αVβ3 is necessary for the pressure-induced inward remodelling process but the rest of the biochemical sensing mechanism is still uncertain. If the physiological response to a raised blood pressure in small arteries is eutrophic inward remodelling then the integrity of the circulation appears to be preserved until this breaks down. As indicated above in the TGR (MREN27) rat there is evidence of the development of vascular wall hypertrophy in small arteries from week 6 onwards. This rat model of hypertension develops a severe form of the disorder and indeed dams are unable to breed if they do not receive antihypertensive medication. Therefore against this background it would appear that the breakdown of autoregulation (at the myogenic tone) is associated with the vascular wall developing a growth response (hypertrophy) in an attempt to offset the increased wall stress. Recent work on the small blood vessels of patients with type 2 (maturity onset) diabetes mellitus has demonstrated that there is vascular wall hypertrophy. These patients were selected as already having evidence of downstream target organ damage because they demonstrated microproteinuria and their myogenic tone was disordered [20]. In other words, the onset of hypertrophy is a consequence of disruption of normal myogenic tone and the delivery of blood at a higher perfusion pressure causing cellular damage. In the kidney, this would inevitably lead to a loss of filtration capability and protein leak. In terms of cardiovascular risk, recent data from Italy have demonstrated that there is an increased risk of development of cardiovascular events in patients whose small arteries demonstrate hypertrophy rather than eutrophic inward remodelling (Figs 3 and 4) [21]. Recent in vitro studies support these considerations. It has been demonstrated that the myogenic response of the middle cerebral artery from pre-stroke spontaneously hypertensive stroke-prone rat (SHRSP) are impaired compared with the SHR [22]. This observation would explain the deranged cerebral autoregulation that has been observed in the SHRSP before stroke occurs. Also this is associated with a re-distribution of collagen throughout the blood vessel wall. Other studies have demonstrated that the myogenic component of renal autoregulation is impaired in the fawn hooded rat (the tubuloglomerular feedback component of renin autoregulation is unchanged) compared with controls thereby causing glomerular hypertension and hyperfiltration, which explains why the kidneys are susceptible to the deleterious effects of moderate hypertension [23]. Furthermore, the Brown Norway rat is normotensive but has a greater than normal life expectancy. However, when hypertension is induced, these animals have a high incidence of cerebral haemorrhage and mortality compared with the Long Evans rat [24]. Also the Brown Norway rat is very sensitive to hypertension-induced renal injury and recently the myogenic component of renal autoregulation has been found to be abnormal in normotensive Brown Norway rats [24]. Therefore it seems reasonable to speculate that the cerebral vessels from the Brown Norway animals exhibit weaker myogenic responses compared with cerebral vessels from Wistar rats, which would explain the susceptibility of the Brown Norway rat to hypertension-induced cerebral haemorrhage. Inhibition of the renin angiotensin system markedly delays the development of cerebral haemorrhage and mortality in salt-loaded SHRSP [25, 26]. In the fawn hooded rat early angiotensin-converting enzyme (ACE) inhibition prevents renal damage and this protection is associated with a normalization of glomerular pressure [27]. The protective effect of ACE inhibition in the kidney has been presumed to be a consequence of an inhibition of angiotensin II induced efferent arteriolar myogenic tone. Of course it could be argued that the effects of blood pressure lowering would be important on stroke development and, in consequence, ACE inhibition is working by its antihypertensive effects. However, dexamethasone or thyroxine increased blood pressure in the SHRSP to a greater extent than salt loading but stroke does not occur [28]. Also, the anti-stroke effect of captopril on salt-loaded SHRSP which occurs without an antihypertensive effect is unchanged when blood pressure is increased with dexamethasone [28]. Therefore it seems that the renin angiotensin system inhibition improves myogenic responses and survival in salt-loaded SHRSP largely independent of changes in blood pressure although this remains to be confirmed.
Fig 3.
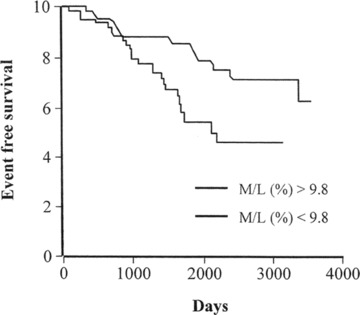
Event-free survival of patients who have undergone arteriola biopsy. Individuals with a medial lumen ration greater than the medium of 9.8% show a larger number of cardiovascular events compared with those with smaller wall thickening v (data from [10]).
Fig 4.
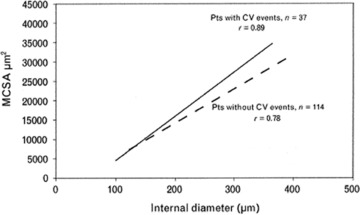
A further analysis of the data from reference 17 indicating that those individuals with marked vascular hypertrophy rather than eutrophic inward remodelling have an increased number of cardiovascular events (data taken from [19]).
Small artery function
There is little evidence to suggest that hypertension is associated with abnormalities of contractile function. Both in vitro and in vivo studies have suggested that contraction is normal although there is controversy about whether the structural alterations in the vascular wall lead to exaggerated constriction and vascular amplification (see above). This has been the subject of intense debate over a number of years although some work in intact animals really seems to suggest that vascular amplification seen in isolated vascular beds is not something which is observed when the whole of the circulation is integrated and examined. Again it is controversial as to whether vascular relaxation is abnormal in hypertension. The problem with interpreting studies which have been published is that many other risk factors are often abnormal and accompany hypertension. For example, there is often associated dyslipidaemia and there is clear evidence that oxidized low-density lipoprotein (LDL) can reduce the bioavailability of nitric oxide and as a result of this there is evidence of abnormal endothelium dependent dilator function which has been reported in patients with high blood pressure and dyslipidaemia, patients with dyslipidaemia and coronary artery disease or the subcutaneous vasculature of patients with hypercholesterolaemia [29]. In addition endothelial function is recognized to decline as individuals age and therefore it is obviously complex to dissect out whether endothelial function is abnormal as a result of hypertension per se or as a result of other demographic abnormalities and the cohorts being examined. The overall impression that one is left with is that it is the level of oxidized LDL that is important in the bloodstream of individuals with hypertension and that blood pressure per se is not responsible for endothelial dysfunction. With regard to improvement in endothelial function the use of statins has been demonstrated to restore endothelial integrity to near normal as soon as the cholesterol levels are improved [29]. There is also evidence that the use of ACE inhibitors or angiotensin receptor blockers can also ameliorate abnormal endothelial function. This is because hypercholesterolaemia is associated with an increased expression of a type 1 angiotensin receptor (ATl) and that the binding of angiotensin II to the ATl receptor is associated with an increase in oxidative stress and a reduced bioavailability of nitric oxide. Animal experiments have demonstrated that the use of angiotensin receptor blockers independent of their antihypertensive effect can be associated with an improvement in endothelial function and a reduction in plaque load throughout the vasculature. This has also been demonstrated in a non-human primate. Of course, longitudinal experiments in human beings are awaited but it is clear that endothelial function can be improved with the use of angiotensin receptor antagonists and ACE inhibitors and recent studies have demonstrated in human beings that the combination of an angiotensin receptor blocker and a statin is an extremely powerful one for improving endothelial function.
Conclusions
The molecular basis of the physiological remodelling response to hypertension is slowly being understood. The replacement of eutrophic inward remodelling by a pathological change such as hypertrophy appears to herald the development of an increased risk of circulatory disease. Future research should look at why this occurs and whether effective antihypertensive medication can reverse this and improve outcome for individual patients. The obvious attraction is that the identification of the, at risk patient will mean that scarce healthcare resource can be targeted at such individuals with maximum benefit in terms of circulatory protection. The challenge ahead is to identify those that are going to develop hypertrophic remodelling against a background of sustained high blood pressure and more indirect measurements of structural changes in blood vessels are urgently required.
Acknowledgments
We thank The Wellcome Trust for funding this work, as well as Ingenious Hypercare and the Manchester Wellcome Trust Clinical Research Facility where many of the human studies were carried out. We also thank Maureen Speed for her preparation of this manuscript.
Conflict of interest
The authors state that there is no conflict of interest with the view state in this paper.
References
- 1.Widimski J, Fejfarova MH, Fejfar Z. Changes of cardiac output in hypertensive disease. Cardilogica. 1957;31:381–9. doi: 10.1159/000165747. [DOI] [PubMed] [Google Scholar]
- 2.Julius S, Petrin J. Autonomic nervous and behavioural factors in hypertension. In: Laragh JH, Brenner BM, editors. Hypertension: pathophys diagn manage. New York: Raven Press; 1990. pp. 2083–90. [Google Scholar]
- 3.Lund-Johansen P. Twenty year follow-up of haemodynamics in essential hypertension during rest and exercise. Hypertension. 1991;18:54–61. doi: 10.1161/01.hyp.18.5_suppl.iii54. [DOI] [PubMed] [Google Scholar]
- 4.Julius S, Pascul A, Sannerstedte R, et al. Relationship between cardiac output and peripheral resistance in borderline hypertension. Circ. 1971;43:382–90. doi: 10.1161/01.cir.43.3.382. [DOI] [PubMed] [Google Scholar]
- 5.Esler M, Julius S, Zweifler A, et al. Mild high renin essential hypertension: neurogenic hypertension. N Engl J Med. 1977;296:405–11. doi: 10.1056/NEJM197702242960801. [DOI] [PubMed] [Google Scholar]
- 6.Esler M, Jennins G, Korner P, et al. Assessment of human sympathetic nervous system activity from measurements of norepinephrine turnover. Hypertension. 1988;11:3–20. doi: 10.1161/01.hyp.11.1.3. [DOI] [PubMed] [Google Scholar]
- 7.Bright R. Tabular view of the morbid appearances in 100 cases connected with albuminous urine with observations. Guy’s Hosp Reports. 1836;1:380–400. [Google Scholar]
- 8.Heagerty AM, Aalkjaer C, Bund SJ, et al. Small artery structure in hypertension: dual processes of remodelling and growth. Hypertension. 1993;21:391–7. doi: 10.1161/01.hyp.21.4.391. [DOI] [PubMed] [Google Scholar]
- 9.Korsgaard N, Aalkjaer C, Heagerty AM, et al. Histology of subcutaneous small arteries from patients with essential hypertension. Hypertension. 1993;22:523–6. doi: 10.1161/01.hyp.22.4.523. [DOI] [PubMed] [Google Scholar]
- 10.Rizzoni D, Porteri E, Boarig E, et al. Prognostic significance of small artery structure in hypertension. Circ. 2003;108:2230–5. doi: 10.1161/01.CIR.0000095031.51492.C5. [DOI] [PubMed] [Google Scholar]
- 11.Aalkjaer C, Heagerty AM, Bailey I, et al. Studies of isolated resistance vessels from offspring of essential hypertensive patients. Hypertension. 1987;9:155–8. doi: 10.1161/01.hyp.9.6_pt_2.iii155. [DOI] [PubMed] [Google Scholar]
- 12.Bund SJ, West KP, Heagerty AM. Effects of protection from pressure on resistance artery morphology and reactivity in spontaneously hypertensive Wistar Kyoto rats. Circ Res. 1991;68:1230–40. doi: 10.1161/01.res.68.5.1230. [DOI] [PubMed] [Google Scholar]
- 13.Zacchigna L, Vecchione C, Notte A, et al. Emilin 1 links TGG-β maturation to blood pressure homeostasis. Cell. 2006;124:929–42. doi: 10.1016/j.cell.2005.12.035. [DOI] [PubMed] [Google Scholar]
- 14.Izzard AS, Heagerty AM, Leenen FHH. The amplifier hypothesis: permission to dissent. J Hypertens. 1999;17:1667–9. doi: 10.1097/00004872-199917120-00002. [DOI] [PubMed] [Google Scholar]
- 15.Izzard AS, Bund SJ, Heagerty AM. Increased wall-lumen ratio of mesenteric vessels from the spontaneously hypertensive rat is not associated with increased contractility under isobaric conditions. Hypertension. 1996;28:604–8. doi: 10.1161/01.hyp.28.4.604. [DOI] [PubMed] [Google Scholar]
- 16.Leenen FHH, Yuan B, Tsoporis J, et al. Arterial hypertrophy and pressure responsiveness during development of hypertension in spontaneously hypertensive rats. J Hypertens. 1994;12:23–32. [PubMed] [Google Scholar]
- 17.Heerkens E, Shaw L, Ryding A, et al. Alpha V integrins are necessary for eutrophic inward remodelling of small arteries in hypertension. Hypertension. 2006;47:281–7. doi: 10.1161/01.HYP.0000198428.45132.02. [DOI] [PubMed] [Google Scholar]
- 18.Martinez-Lemus LA, Crow T, Davis MJ, et al. ανβ3 and α5β1-integrin blockade inhibits myogenic constriction of skeletal muscle resistance arterioles. Am J Physiol-Heart C. 2005;289:H322–9. doi: 10.1152/ajpheart.00923.2003. [DOI] [PubMed] [Google Scholar]
- 19.Heerkens EH, Izzard AS, Heagerty AM. Integrins, vascular remodelling and hypertension. Hypertension. 2007;49:1–4. doi: 10.1161/01.HYP.0000252753.63224.3b. [DOI] [PubMed] [Google Scholar]
- 20.Schofield I, Malik R, Izzard AS, et al. Vascular structural and functional changes in Type 2 diabetes mellitus: evidence for the role of abnormal myogenic responsiveness and dyslipidaemia. Circ. 2002;106:3037–43. doi: 10.1161/01.cir.0000041432.80615.a5. [DOI] [PubMed] [Google Scholar]
- 21.Izzard AS, Rizzoni D, Agabeti-Rosei E, et al. Small artery structure in hypertension: adaptive changes and target organ damage. J Hypertens. 2005;23:247–50. doi: 10.1097/00004872-200502000-00002. [DOI] [PubMed] [Google Scholar]
- 22.Izzard AS, Horton S, Heerkens EH, et al. Middle cerebral artery structure and distensibility during developing and established phases of hypertension in the spontaneously hypertensive rat. J Hypertens. 2006;24:875–80. doi: 10.1097/01.hjh.0000222757.54111.06. [DOI] [PubMed] [Google Scholar]
- 23.Van Dokkum RPE, Sun CW, Provoost AP, et al. Altered renal haemodynamics and impaired myogenic responses in the Fawn Hooded rat. Am J Physiol. 1999;276:R855–63. doi: 10.1152/ajpregu.1999.276.3.R855. [DOI] [PubMed] [Google Scholar]
- 24.Capdeville M, Cutard M, Osborne-Pellegrin MJ. Spontaneous rupture of the internal elastic laminar in the rat: the manifestation of a genetically determined factor which may be linked to vascular fragility. Blood Vessels. 1989;26:197–212. doi: 10.1159/000158768. [DOI] [PubMed] [Google Scholar]
- 25.Wang X, Ajikobi DO, Salevsky FC, et al. Impaired myogenic autoregulation in the kidneys in the Brown Norway rat. Am J Physiol. 2000;278:F962–9. doi: 10.1152/ajprenal.2000.278.6.F962. [DOI] [PubMed] [Google Scholar]
- 26.Wang H, Smeda JS, Lee RKNW. Prevention of stroke and preservation of functions of cerebral arteries by treatment with perindopril in stroke-prone hypertensive rat. Can J Phys Pharm. 1998;76:26–34. [PubMed] [Google Scholar]
- 27.Verseput GH, Provoost AP, Braam BB, et al. Angiotensin converting enzyme inhibition in the prevention and treatment of chronic renal damage in the hypertensive Fawn Hooded rat. JAm Soc Nephrol. 1997;8:249–59. doi: 10.1681/ASN.V82249. [DOI] [PubMed] [Google Scholar]
- 28.Nagaoke A, Shino A, Twatsuka H. Accelerating effects of dexamethasone and thyroxine on hypertension without accompanying stroke in stroke-prone spontaneously hypertensive rats. Life Sc. 1979;24:71–8. doi: 10.1016/0024-3205(79)90282-0. [DOI] [PubMed] [Google Scholar]
- 29.Goode GK, Heagerty AM. In-vitro responses of human peripheral small arteries in hypercholesterolaemia and effects of therapy. Circ. 1995;91:2898–903. doi: 10.1161/01.cir.91.12.2898. [DOI] [PubMed] [Google Scholar]