

Abstract
Claudins (Cls) are a multigene family of transmembrane proteins with different tissue distribution, which have an essential role in the formation and sealing capacity of tight junctions (TJs). At the level of the blood–brain barrier (BBB), TJs are the main molecular structures which separate the neuronal milieu from the circulatory space, by a restriction of the paracellular flow of water, ions and larger molecules into the brain. Different studies suggested recently significant BBB alterations in both vascular and degenerative dementia types. In a previous study we found in Alzheimer’s disease (AD) and vascular dementia (VaD) brains an altered expression of occludin, a molecular partner of Cls in the TJs structure. Therefore in this study, using an immunohistochemical approach, we investigated the expression of Cl family proteins (Cl-2, Cl-5 and Cl-11) in frontal cortex of aged control, AD and VaD brains. To estimate the number of Cl-expressing cells, we applied a random systematic sampling and the unbiased optical fractionator method. We found selected neurons, astrocytes, oligodendrocytes and endothelial cells expressing Cl-2, Cl-5 and Cl-11 at detectable levels in all cases studied. We report a significant increase in ratio of neurons expressing Cl-2, Cl-5 and Cl-11 in both AD and VaD as compared to aged controls. The ratio of astrocytes expressing Cl-2 and Cl-11 was significantly higher in AD and VaD as compared to aged controls. The ratio of oligodendrocytes expressing Cl-11 was significantly higher in AD and the ratio of oligodendrocytes expressing Cl-2 was significantly higher in VaD as compared to aged controls. Within the cerebral cortex, Cls were selectively expressed by pyramidal neurons, which are the ones responsible for cognitive processes and affected by AD pathology. Our findings suggest a new function of Cl family proteins which might be linked to response to cellular stress.
Keywords: tight junctions, claudins, Alzheimer’ disease, vascular dementia
Introduction
The blood–brain barrier (BBB) is a tightly controlled semi-permeable barrier which prevents the paracellular passage for macromolecules and cells from the blood stream into the brain [1]. It consists primarily of capillary endothelial cells packed together by tight junctions (TJs), together with the adjoining astrocyte end-feet and pericytes. TJs are specialized membrane domains at the most apical region of epithelial and endothelial cells, involved in both prevention of solute and water leakage (barrier function) and maintenance of cellular polarity (fence function) [2]. In the brain of vertebrates there are two types of cells which are known to bear TJs or TJs-like structures: vascular endothelial cells and oligodendrocytes [3]. TJs networks are formed by integral membrane proteins, such as junctional associated membrane protein 1 [4], occludin [5] and the claudin (Cl) family [6]. Occludin and Cls bind to cytoplasmic peripheral membrane proteins, such as ZO-1, ZO-2 or ZO-3, which in turn are connected to the perijunctional actin filament network of the cytoskeleton [1].
Cls are a 20–28 kD multigene family of transmembrane proteins with different tissue distributions, which have an essential role in the formation of TJs strands, and appear to function as the primary seal of the TJs [7]. Among the Cls expressed by brain endothelial cells, Cl-5 is a critical determinant of BBB permeability [8]. Cl-5 is expressed in all tissues, being specifically localized to the endothelia of blood vessels and therefore TJs containing Cl-5 could regulate the permeability of distal vascular territories [9]. However, Cl-1 [10] and Cl-3 [11] were also shown to localize at the BBB level and their expression seems to be altered by various pathological processes. In addition, Cl-1, Cl-2 and Cl-5 are present in the choroid plexus where their expression is regulated by protein kinase C (PKC) [12]. It was suggested that Cls could function by homo- and hetero- oligomerization and a study which employed RNA interference showed that Cl-2, Cl-4 and Cl-7 are permeation operators for the Na+ and Cl− ions. In contrast to other Cls, Cl-2 decreases the tightness of the TJ strands [13] by formation of cation-selective channels [14]. Cl-11 is expressed in TJs of myelin sheaths in brain and of Sertoli cells in testis [15] and it is known as well as oligodendrocyte-specific protein, because it represents a major component of central nervous system (CNS) myelin [16]. Along with other Cl family members, Cl-11 was also found in the epithelial cells of choroid plexus [17]. It was proposed that Cl-11 might be important for the formation and maintenance of myelin and for regulation of oligodendrocytes and Schwann cells proliferation [16].
Considering that the ‘sealing’ role of TJs is determined by the presence of Cls, the latter is presumably important in pathological situations that involve increased vessel permeability [18]. The integrity of the BBB is compromised in different disorders of the human CNS, most likely by injury or dysregulation of TJs [19]. One example where disruption of BBB is thought to play an important role in the pathophysiology is cerebral microangiopathy [20]. Patients with microangiopathy can present with either isolated or diffuse white matter changes that can ultimately lead to impairment of neuronal flow and cognitive decline, characteristic aspects for most of the dementia disorders [21]. There is mounting evidence that Alzheimer’s disease (AD), the most common cause of dementia in the elderly, share vascular risk factors with vascular dementia (VaD), and at least a third of AD cases associates cerebrovascular small vessel disease [22]. In addition to cerebral amyloid angiopathy, AD subjects show profound changes in cerebral microvessels, often independent of amyloid deposition [23]. Moreover, β-amyloid peptide (1–42) might alter BBB integrity by effects on TJs protein complexes [24]. However, based on the current data, the role of TJs proteins is poorly understood in dementia disorders.
Beyond serving the BBB as gatekeepers, TJs proteins could have other physiological functions. Cls have a role in regulation of cell phenotype and growth control and Cl mutations are involved in several human diseases [25]. Furthermore, occludin is expressed at detectable levels in mouse cultures of neurons and astrocytes [26], which do not form TJs. Moreover, we found in a recent study that occludin is overexpressed in neurons and astrocytes of AD and VaD brains as compared to aged controls [27]. Based on the fact that occludin is a molecular partner of Cls in the TJs structure, in this study we aimed to comparatively investigate the expression of Cl-2, Cl-5 and Cl-11 in human brains of aged control cases, AD and VaD.
Material and methods
Brain material
The brain material was obtained from the Huddinge Brain Bank, Stockholm, Sweden in accordance with Swedish law and the permission of the Karolinska University Hospital Ethical Committee. We chose to analyse the cerebral cortex (frontal, Brodmann area 46/9), where specific pathological changes are noted in both AD and VaD [28, 29].
The study was based on evaluation of five aging control, four AD and six VaD post-mortem human brains. The control group included the brains of traffic-accident patients or of patients who had no history of long-term illness or neuropsychiatric disorders. The AD group included brains from patients with clinically and pathologically confirmed AD. Clinical diagnosis was based on combined Diagnostic and Statistical Manual of Mental Disorders (DSM)-III-R [30] and National Institute of Neurological and Communicative Diseases and Stroke/Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) criteria [31]. The definite neuropathological diagnosis of AD was determined by using Consortium to Establish a Registry for Alzheimer’s disease (CERAD) and National Institute on Aging (NIA) – Reagan Institute Criteria [32, 33]. The VaD group included brains from patients with clinical and pathological diagnosis of VaD. The VaD group consisted of two cases of single strategic stroke, three cases with dementia multiinfarct and one case with diffuse white matter changes. Age and sex of the cases are presented in Table 1.
Table 1.
Age and sex of cases analysed in each study group
| Group | Age | Sex |
|---|---|---|
| Control | 90 | F |
| 88 | F | |
| 86 | F | |
| 56 | M | |
| 70 | M | |
| AD | 87 | F |
| 86 | M | |
| 91 | F | |
| 72 | F | |
| VaD | 87 | F |
| 86 | F | |
| 65 | M | |
| 72 | M | |
| 89 | F | |
| 77 | F |
Sectioning and histology
Immunostaining with antibodies against Cl-2, Cl-5 and Cl-11 was performed on buffered formaldehyde-fixed embedded sections (long-fixation time sections were used). A series of thin sections (7 μm) were cut exhaustively in a randomly chosen sagital plane on the microtome. After the deparaffinization and rehydration procedures the sections were rinsed in distilled water for 5 min. and then heated at 80°C for 20 min in 0.1 M citrate buffer of pH 9. After cooling, the sections were incubated with the primary antibody solution overnight (approximately 16 hrs) at 4°C. The primary antibodies were rabbit polyclonals against Cl-2 (dilution 1:50), Cl-5 (dilution 1:100) or Cl-11 (dilution 1:200) and were purchased from Zymed Laboratories, Inc. (South San Francisco, CA, USA). These primary antibodies were tested in Western blots analysis of rat brain homogenates and no unspecific binding was noted. Thereafter, the sections were treated with a biotinylated secondary antibody (1:300, Vector Laboratories, Burlingame, CA, USA) and with the avidin–biotin-peroxidase complex kit (Vector) with 3,3′-diaminobenzidine-4HCl/H2O2 (DAB, Sigma, St. Louis, MO, USA) as a substrate. Next, the sections were counterstained with haematoxylin–eosin stain for background. Two types of negative controls slides were run for all the sections. The negative controls were run in an identical manner except the incubation with the primary antibody. For the first set of negative controls we omitted the primary antibody, whereas for the second set we added instead of the primary antibody an unspecific immunoglobulin serum at the same concentration. The sections were then mounted and examined under a Nikon microscope.
Quantification of Cl-expressing brain cells
To estimate the number of Cl-expressing neurons, astrocytes and oligodendrocytes we applied two-dimension quantification with random systematic sampling and the unbiased optical fractionator method [34], similar to a previous study [35]. In brief, the area of interest was delineated in low magnification (×2.5) using the cursor. Due to the clear anatomical borders, we were able to distinguish the grey matter from the white matter. A meander sampling function of the GRID v2.0 program (Olympus, A/S, Bellarup, Denmark) was used for stepping through the delineated region with a chosen counting frame. Then, a 100× oil-immersion objective with a numerical aperture of 1.40 was moved into place and the appropriate counting frame superimposed on the screen. The desired horizontal and vertical step lengths, assisted by a highly precise servo-controlled motorized microscopy stage, were dimensioned for the appropriate distance (231.22 μm [x-step]× 231.22 μm [y-step]) in between the counting frames (1603.9 μm2). The cells in the space were counted by an optical dissector probe (z-axis). The optical dissector uses the simple rule that a cell is counted if its body cell is in the counter-frame. This procedure ensured the selection of a systematic random sample of sections. The number of cells was expressed as a number per squared millimetre. The neurons could be easily distinguished from the other types of cells due to the presence of the round nucleus with visible cytoplasm and a single large nucleolus, no heterochromatin. Oligodendrocytes were identified by smaller round or oval dark nuclei with dense chromatin, and they are usually close to the neurons. The astrocytes were recognized as multipolar cells with bigger, paler and less densely packed heterochromatin, without a clear nucleolus and showing a patchy pattern of granules in a rim bellow the nuclear membrane.
Statistical analysis
All statistical analyses of the results were performed using Statistica 8.0 software (Statsoft, Tulsa, OK, USA). The neuronal, astrocyte or oligodendrocyte ratio was defined as (number of Cl-expressing cells / total number of cells of respective type) × 100. Differences within individual cell type were analysed by two-way factorial ANOVA, followed by Fisher’s LSD post hoc test. For all tests, the significance threshold was set to 0.05. All results are given as mean ± standard deviation (S.D.).
Results
Expression of claudins in neurons
Our first observation was that selected neurons, in the frontal cortex from aged controls expressed Cl-2, Cl-5 and Cl-11 in detectable amounts (Fig. 1). Expression of Cl-2, Cl-5 and Cl-11 was increased in neurons from frontal cortex of AD and VaD brains as compared to control brains (Table 2 and Fig. 1). The neurons which expressed Cls were in all cases predominantly pyramidal. The neuronal fibres were also stained more intensely in AD and VaD as compared to control (Fig. 1).
Fig 1.
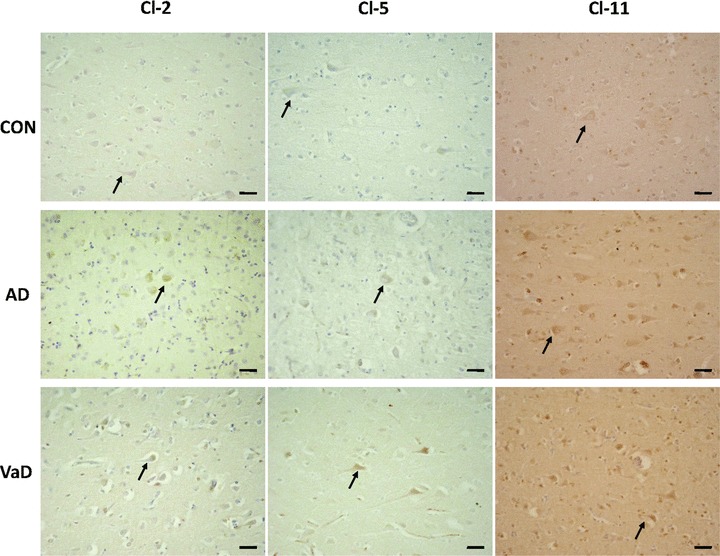
Expression of Cl-2, Cl-5 and Cl-11 in control (CON), AD and VaD brains, in the frontal cortex, Brodmann area 46/9. Cl-expressing cells are brownish, as a result of the immunohistochemical staining method with anti-Cl sera detected with avidin–biotin-peroxidase complex kit and DAB substrate. Expression of Cl-2, Cl-5 and Cl-11 was increased in neurons and neuronal fibres in AD and VaD brains, as compared to control brains. Expression of Cls was noticed mainly in the pyramidal neurons in CON, AD and VaD. Arrows are indicating Cl-expressing neurons. Bars: 30 μm.
Table 2.
Quantification of total number of neurons and Cl-expressing neurons (positive neurons) in the frontal cortex region in control (CON, five cases), AD (four cases) and VaD (six cases) groups. All values were truncated to two decimals. S.D. = standard deviation
| Group | Total number of neurons | Number of positive neurons | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cl-2 | Cl-5 | Cl-11 | Cl-2 | Cl-5 | Cl-11 | |||||||
| Mean | S.D. | Mean | S.D. | Mean | S.D. | Mean | S.D. | Mean | S.D. | Mean | S.D. | |
| CON | 33.47 | 4.79 | 31.33 | 4.82 | 32.53 | 5.78 | 2.47 | 0.73 | 3.20 | 4.56 | 7.80 | 2.64 |
| AD | 32.17 | 3.87 | 33.58 | 2.20 | 32.58 | 0.32 | 17.42 | 2.63 | 13.33 | 0.98 | 23.33 | 3.02 |
| VaD | 27.00 | 4.49 | 27.83 | 3.13 | 28.44 | 2.93 | 10.44 | 3.12 | 7.39 | 1.48 | 19.44 | 2.93 |
Subsequently, we used a quantitative approach in order to estimate the number of Cl-expressing neurons in control, AD and VaD groups. Table 2 presents comparatively the quantitative data of neurons/mm2 and Cl-2, Cl-5 and Cl-11+ neurons/mm2 in control, AD and VaD groups, respectively. The number of neurons did not significantly differ between the groups analysed. The area chosen for counting was similar in all cases (with around 100 counting frames/area) and there was no significant difference in the density (number of neurons/area) between the cases analysed. Qualitatively, we found no areas with marked heterogeneity in distribution of Cls. The ratios of Cl-2, Cl-5 and Cl-11-expressing neurons were significantly higher in the AD group (54.02 ± 1.77, P < 0.05, 39.82 ± 3.75, P < 0.05 and 71.62 ± 9.19, P < 0.05, respectively) and in the VaD group (38.07 ± 6.97, P < 0.05, 26.56 ± 4.44, P < 0.05 and 68.34 ± 7.19, P < 0.05, respectively) as compared to the control group (7.30 ± 1.36, 9.62 ± 13.08 and 23.77 ± 5.00, respectively), as shown in Fig. 2. The ratio of Cl-2-expressing neurons was significantly higher in the AD group as compared to the VaD group (P < 0.05). The ratios of Cl-5 and Cl-11-expressing neurons did not differ significantly between AD and VaD groups.
Fig 2.
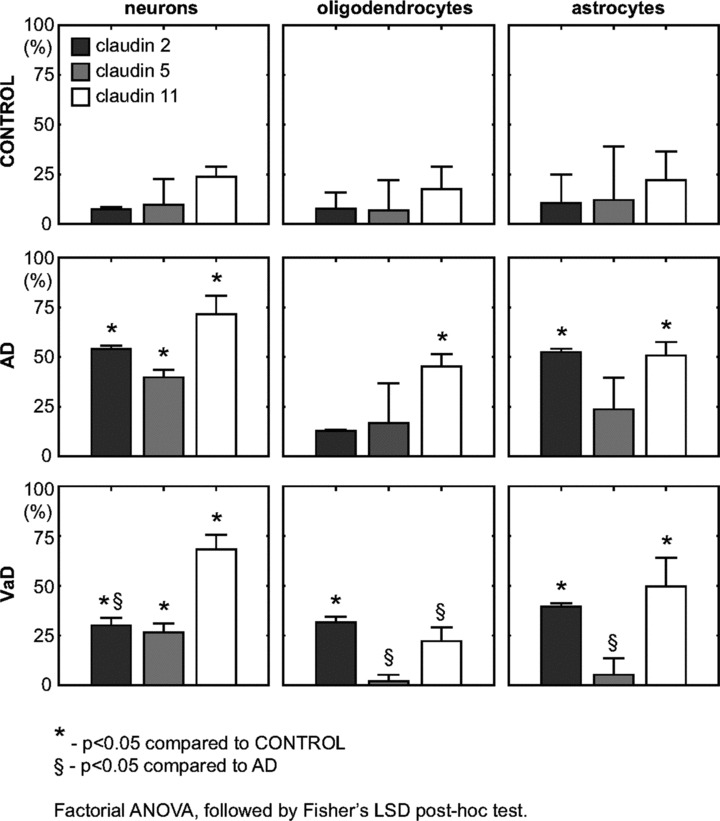
Ratios of Cl-2, Cl-5 and Cl-11-expressing cells in control, AD and VaD brains, in the frontal cortex, Brodmann area 46/9. The neuronal, astrocyte or oligodendrocyte ratio was defined as (number of Cl-expressing cells / total number of cells) × 100. To compare the cell ratio between controls, AD and VaD (two by two) factorial ANOVA has been used, followed by the Fisher’s LSD post hoc test. Error bars mark the S.D.
Expression of claudins in oligodendrocytes
Further on, we evaluated the expression of Cl-2, Cl-5 and Cl-11 in oligodendrocytes in the frontal region of aged control, AD and VaD brains. In the grey matter of the frontal cortex, selected oligodendroglial cells expressed Cl-2, Cl-5 and Cl-11 in all AD, VaD and control brains (Fig. 3). In order to estimate the number of Cl-expressing oligodendrocytes in control, AD and VaD groups we used the same quantitative approach as described for the neurons. Table 3 presents comparatively the quantitative data of oligodendrocytes/mm2 and Cl-2, Cl-5 and Cl-11+ oligodendrocytes/mm2 in control, AD and VaD groups, respectively. As shown in Fig. 2, the ratio of Cl-2-expressing oligodendrocytes was significantly higher in the VaD group (17.61 ± 11.01, P < 0.05), but not in the AD group (12.77 ± 0.71), as compared to the control group (7.74 ± 8.16). The ratio of Cl-5-expressing oligodendrocytes did not significantly differ in the AD (16.75 ± 20.08) and VaD (2.03 ± 3.17) groups as compared to the control group (6.80 ± 15.21). However, the ratio of Cl-5-expressing oligodendrocytes was significantly lower in VaD group as compared to the AD group (P < 0.05). The ratio of Cl-11-expressing oligodendrocytes was significantly higher in the AD group (45.32 ± 6.21, P < 0.05), but not in the VaD group (22.18 ± 6.91), as compared to the control group (17.62 ± 11.18). Similar to Cl-5 ratios, the ratio of Cl-11-expressing oligodendrocytes was significantly lower in VaD group as compared to the AD group (P < 0.05).
Fig 3.
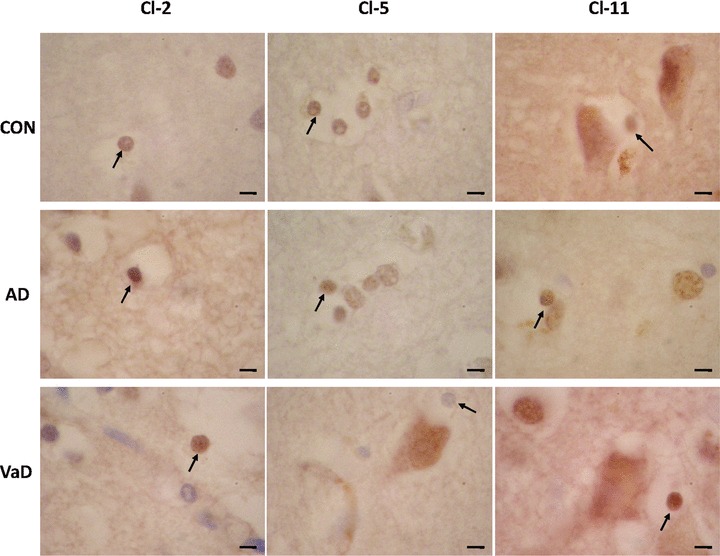
Expression of Cl-2, Cl-5 and Cl-11 in oligodendrocytes in control (CON), AD and VaD brains, in the frontal cortex, Brodmann area 46/9. Cl-expressing cells are brownish, as a result of the immunohistochemical staining method with anti-Cl sera detected with avidin–biotin-peroxidase complex kit and DAB substrate. Oligodendrocytes are indicated by arrows. Bars: 5 μm.
Table 3.
Quantification of total number of oligodendrocytes and Cl-expressing oligodendrocytes (positive oligodendrocytes) in the frontal cortex region in control (CON, five cases), AD (four cases) and VaD (six cases) groups. All values were truncated to two decimals. S.D. = standard deviation
| Group | Total number of oligodendrocytes | Number of positive oligodendrocytes | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cl-2 | Cl-5 | Cl-11 | Cl-2 | Cl-5 | Cl-11 | |||||||
| Mean | S.D. | Mean | S.D. | Mean | S.D. | Mean | S.D. | Mean | S.D. | Mean | S.D. | |
| CON | 53.47 | 4.27 | 54.47 | 5.29 | 57.60 | 5.58 | 4.27 | 4.65 | 3.40 | 7.60 | 10.13 | 6.38 |
| AD | 57.25 | 4.72 | 46.50 | 1.91 | 53.50 | 7.10 | 7.33 | 0.98 | 7.75 | 9.29 | 23.92 | 1.97 |
| VaD | 43.78 | 5.29 | 39.44 | 1.99 | 47.44 | 5.62 | 7.78 | 4.96 | 0.78 | 1.22 | 10.67 | 3.92 |
Expression of claudins in astrocytes
For evaluation of Cl expression in astrocytes we followed the same procedure as for neurons and oligodendrocytes. In the frontal cortex examined, we found astrocytes expressing Cl-2, Cl-5 and Cl-11 in all AD, VaD and control brains (Fig. 4). Table 4 presents comparatively the quantitative data of astrocytes/mm2 and Cl-2, Cl-5 and Cl-11+ astrocytes/mm2 in control, AD and VaD groups, respectively. As Fig. 2 shows, the ratios of Cl-2 and Cl-11-expressing astrocytes were significantly higher in the AD group (52.47 ± 1.67, P < 0.05 and 50.79 ± 6.76, P < 0.05) and in the VaD group (37.75 ± 6.86, P < 0.05 and 49.73 ± 14.36, P < 0.05) as compared to the control group (10.51 ± 14.45 and 22.00 ± 14.43). Similar to the result obtained for oligodendrocytes, the ratio of Cl-5-expressing astrocytes did not significantly differ in the AD (23.74 ± 16.01) and VaD (5.28 ± 8.22) groups as compared to the control group (12.08 ± 27.01). Again as for the oligodendrocytes, the ratio of Cl-5-expressing astrocytes was significantly lower in VaD group as compared to the AD group (P < 0.05).
Fig 4.
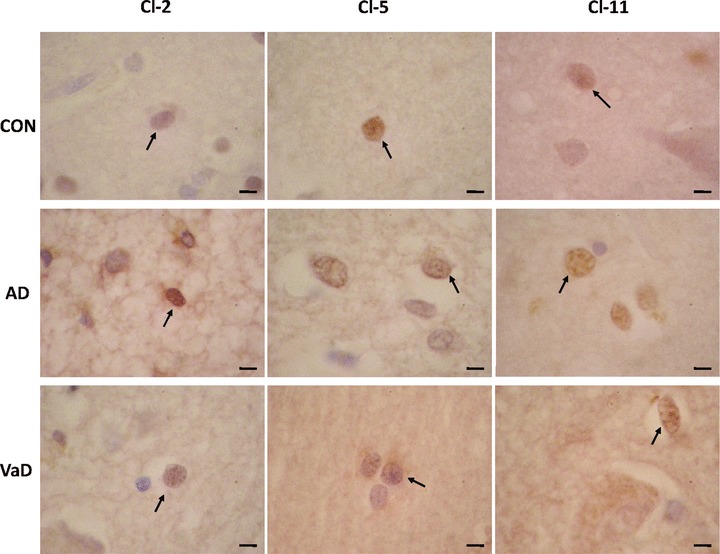
Expression of Cl-2, Cl-5 and Cl-11 in astrocytes in control (CON), AD and VaD brains, in the frontal cortex, Brodmann area 46/9. Cl-expressing cells are brownish, as a result of the immunohistochemical staining method with anti-Cl sera detected with avidin–biotin-peroxidase complex kit and DAB substrate. Astrocytes are indicated by arrows. Bars: 5 μm.
Table 4.
Quantification of total number of astrocytes and Cl-expressing astrocytes (positive astrocytes) in the frontal cortex region in control (CON, five cases), AD (four cases) and VaD (six cases) groups. All values were truncated to two decimals. S.D. = standard deviation
| Group | Total number of astrocytes | Number of positive astrocytes | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cl-2 | Cl-5 | Cl-11 | Cl-2 | Cl-5 | Cl-11 | |||||||
| Mean | S.D. | Mean | S.D. | Mean | S.D. | Mean | S.D. | Mean | S.D. | Mean | S.D. | |
| CON | 35.40 | 2.77 | 37.53 | 8.17 | 38.47 | 2.29 | 3.67 | 5.16 | 4.07 | 9.09 | 8.40 | 5.47 |
| AD | 33.92 | 3.06 | 40.17 | 4.85 | 43.33 | 6.02 | 17.83 | 2.13 | 9.75 | 7.28 | 21.83 | 2.74 |
| VaD | 31.00 | 4.27 | 34.83 | 1.44 | 35.61 | 4.06 | 11.50 | 1.07 | 1.89 | 2.93 | 17.78 | 5.63 |
Expression of claudins in cerebral vessels
At the level of brain microvasculature in AD, VaD and control brains we observed that expression of Cls localized selectively in some of the endothelial cells or in the media layer (Fig. 5). The level of expression of Cl-2, Cl-5 and Cl-11 was variable from one vessel to another in all cases studied and no evident qualitative difference between AD, VaD and aged control groups was observed.
Fig 5.
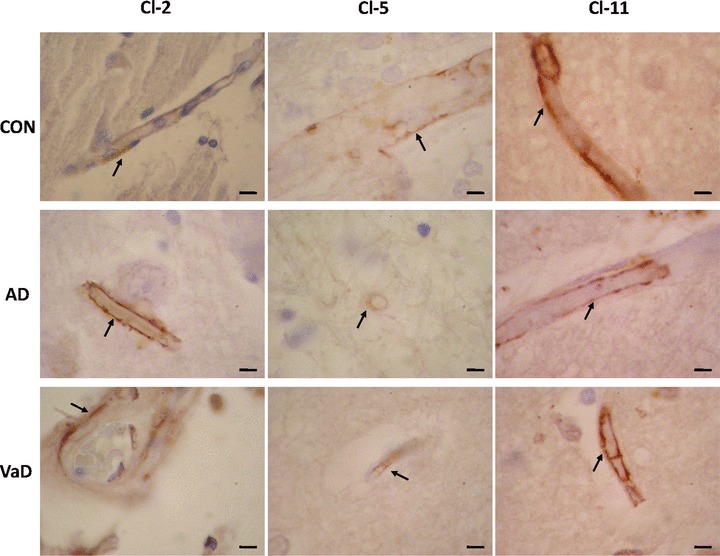
Expression of Cl-2, Cl-5 and Cl-11 in brain microvessels in control (CON), AD and VaD brains, in the frontal cortex, Brodmann area 46/9. Cl-expressing cells are brownish, as a result of the immunohistochemical staining method with anti-Cl sera detected with avidin–biotin-peroxidase complex kit and DAB substrate. Endothelial cells expressing Cl-2, Cl-5 or Cl-11 are indicated by arrows. Bars: 5 μm.
Discussion
To our knowledge, this is the first report on different Cl isoforms being expressed in the human brain, not only by endothelial cells and oligodendrocytes, but also by astrocytes and neurons. From the Cl family of proteins, Cl-1 [10], Cl-3 [11] and Cl-5 [8] are known to localize to the brain endothelial cells. In addition to Cl-5, we found Cl-2 and Cl-11 to be expressed by endothelial cells in aged controls, as well as in AD and VaD brains. We were able to identify microvessels in the frontal cortex expressing Cl-2, Cl-5 or Cl-11 in various amounts in all brains investigated. We noted no qualitative difference in the distribution or amount of expression of these proteins in vessels between AD, VaD and aged control brains. However, we found significant changes in the expression pattern of Cls which differentiate the AD and VaD from control brains. Thus, Cl-2 or Cl-5 is expressed by approximately one tenth and Cl-11 by one fourth of cortical frontal neurons in aged control brains, cells which have not been previously known to form TJs. In AD and VaD brains two to eight times more neurons in the frontal cortex expressed these Cl isoforms in detectable amounts. These Cl-expressing neurons were mainly of pyramidal type, which are involved in cognition and are typically affected by AD pathology. Importantly, while Cl-5 and Cl-11 are known to strengthen the TJs and to increase the transendothelial resistance [41], Cl-2 induces a leaky strand type [13]. Therefore, the permeability of the BBB is presumably decided by a complex interplay of differential Cl isoform regulation, suggesting that increased Cl expression might be a part of the cellular response to stress.
Cls are expressed in a tissue-dependent manner and more than two different Cl isoforms are typically found in the most cell types, conceivably influencing the TJs barrier function properties [36]. Expression of Cls in endothelial cells seems to be regulated by both blood stream generated factors, and signals derived from astrocytes and pericytes [37–39]. However, regulation of Cls expression might be operated by different signalling systems in different cell types. Ageing itself decreases expression of Cls and occludin at the level of brain microvessels, altering the BBB permeability [40]. Disruption of BBB is now increasingly documented in ageing, vascular and degenerative diseases and this disruption might contribute to the progression of the pathology [1].
Alteration of the brain microvessels is common to both AD and VaD [42] and chronic brain hypoperfusion as well [43]. A recent report demonstrated that in rats exposed to cerebral hypoperfusion, an up-regulation of Cl-3, which accompanies the BBB breakdown, occurs in endothelial cells [44]. The profile of Cl isoforms expression can be changed by a reduced blood flow not only in the brain, but in other organs as well, such as kidney, where levels of Cl-7 have been found increased in an aging kidney rat model [45]. Ischemia and hypoxia are able to trigger a multitude of cellular responses, including changes in gene expression [46]. Our findings of increased level of Cls in neurons and, to some extent, in glial cells as well, might be explained by hypoxia-induced gene regulation. Moreover, as recently suggested for occludin, the increased levels of Cls found in dementia brains might reflect an autophagy of the TJ proteins by neighbouring cells after the BBB breakdown triggered by chronic hypoxia [47].
Several studies indicated a disturbance of function of the hypothalamic–pituitary–adrenocortical (HPA) axis in both AD and VaD, with increased plasma and urinary cortisol levels, as compared to aged controls [48, 49]. Besides increasing the amyloid-β and τ pathology, at least in an AD mouse model [50] and triggering neuronal cell death [51], glucocorticoids (GC) have been shown to up-regulate expression of Cl-5 in murine brain [52] and of other Cl isoforms in cultures from Atlantic Salmon gill [53]. Therefore, our findings of increased expression of different Cl isoforms in AD and VaD brains might be explained by the effect of an improper HPA axis functioning. Moreover, the different profile of Cl isoforms expression in different cell types could be explained by a recent hypothesis on the combined effect of GC and brain ageing, which states that ageing selectively increases GC efficacy in neurons while decreasing GC efficacy in glial cells, by an age-related phenotype change [54]. A GC-related susceptibility for AD, based on a genetic variation in the GC system, has been reported as well [55].
We found that expression of Cl-5 is significantly increased in neurons, but not in oligodendrocytes and astrocytes, in AD and VaD brains as compared to aged control brains. Based on experiments in endothelial cell cultures it has been suggested that β-amyloid does not influence directly the expression of Cl-5 [56]. One recent study [39] reported that Cl-5 and occludin are down-regulated by vascular endothelial growth factor (VEGF)-A, released by astrocytes. VEGF-A signalling is known to be involved as well in the ischemic preconditioning of neurons, by activation of cAMP response element-binding protein pathway [57] and in neuroprotection against excitotoxicity [58]. Therefore, a decrease in VEGF-A availability could explain both the increased Cl-5 expression in neurons in AD and VaD found in this study and the increase in occludin expression, reported before [27]. Furthermore, diminished VEGF activity in brain microvessels was found in areas of AD brains with pathological hallmarks of the disease [59] and VEGF is significantly decreased in serum of AD patients [60].
Transforming growth factor (TGF)-β is a member of a family of dimeric polypeptide growth factors involved in cellular differentiation, proliferation, anti-inflammatory signalling, reparative and neuroprotective functions. Increased levels of TGF-β seem to enhance the AD pathology and an experimental blockade of TGF-β-Smad 2/3 signalling in an AD transgenic mouse model resulted in a 90% clearance of β-amyloid brain deposits [61]. TGF-β is elevated in plasma [62] and in CSF [63] of AD and VaD patients and its expression is induced following cerebral ischemia with neuroprotective consequences [64]. In addition, TGF-β had been found to induce expression of Cl-2 and occludin through activation of ERK/MAPK signalling [65]. Therefore, our finding of increased Cl-2 expression in AD and VaD brains, in both neurons and astrocytes, is in the line with the above mentioned studies and might be an effect of TGF-β increased signalling. Interestingly enough, the decrease of neuronal TGF-β signalling in a mouse AD model resulted in neurodegeneration, β-amyloid accumulation and dendritic loss, which suggests that the increased TGF-β level found in dementia patients is part of an endogenous protective response. Consequently, it is possible that overexpression of Cls in brain cells in AD and VaD to be a downstream result, at the phenotype level, of this protective response. However, a limitation of our study remains the low number of brains analysed and therefore confirmation of these mechanisms in cell culture systems or animal models would be important.
Similar to TGF-β, the level of basic fibroblast growth factor-2 (FGF-2) is markedly elevated in AD brains [66] and, on the other hand, in cortical organotypic slice cultures of mice, FGF-2 had been shown to stimulate expression of Cl-5 and Cl-3 and to con serve the integrity of TJs [67]. Therefore, the high FGF levels might trigger as well an increase in Cls expression, with a possible protective role.
PKC is a potent modulator of the non-amyloidogenic pathway of the amyloid precursor protein (APP) processing, being involved as well in synaptic remodelling and induction of protein synthesis. PKC levels are significantly decreased in AD and VaD brains [68]. PKC activation had been reported to down-regulate Cl-2 [12, 69] and to phosphorylate and down-regulate Cl-5, with subsequent alteration of TJs properties [12, 70]. Therefore, the general reduction in PKC activity found in dementia brains could account for the increased levels of Cls found in our study. Moreover, phospholipase D (PLD) expression and activity are up-regulated in AD brains [71]. Phosphatidic acid (PA), a product of PLD, and lysophosphatidic acid (LPA) have been found significantly increased in AD brains as well [72, 73]. PLD and its end-products seem to have multiple links to AD pathogenesis. Although PLD1 colocalizes and interacts with APP [71] and promotes the trafficking and cell surface accumulation of presenilin 1/γ secretase complex [74], PLD 2 has been reported to regulate endothelial barrier permeability through cytoskeleton reorganization and occludin down-regulation, in cell culture experiments [75]. LPA, which in contrast to PA [76] has a pro-apoptotic effect on neurons [77], is able to increase the permeability of TJs without triggering a relocalization of TJs-associated proteins. All these PLD pathway effects might participate to alteration of BBB properties in AD patients. However, no studies explored the effect of PLD on expression of Cls in neurons or glial cells.
Cl-11 was classically known to be localized to TJs of myelin sheaths in brain and of Sertoli cells in testis [9]. The expression of Cl-11 in endothelial cells was recently reported in human corpum cavernosum [78]. In line with this study, we report here, presumably for the first time, expression of Cl-11 in human brain endothelial cells. Moreover, we found that Cl-11 is expressed by a large population of neurons, oligodendrocytes and astrocytes in ageing controls and significantly by more of these cells in AD and VaD brains. Both estrogens and testosterone have neuroprotective roles against AD-related insults, being also able to reduce β-amyloid accumulation [79]. Meanwhile, testosterone has been also reported to be an important operator of Cl-11 gene expression, physiological concentrations increasing the amount of Cl-11 mRNA two- to threefold within 3 days in rat Sertoli cells in vitro[80]. However the same study demonstrated that estradiol had no effect on Cl-11 expression. Serum androgen levels are not different in male aged controls and AD patients [81] and we found no difference in Cl-11 expression between male and female brains. In addition, Kaitu’u-Lino et al. demonstrated that gonadotropins stimulate Cl-11 expression as well and follicle stimulating hormone (FSH) and luteinizing hormone (LH) serum levels have been found significantly increased in patients with dementia [82]. LH is also involved in regulation of presenilin 1 and 2 genes [83], which are the responsible for amyloid processing [84]. LH and FSH could be important players in the pathogenesis of dementia, since their receptors follow a regional pattern of the neuronal populations affected in AD [85] and could operate as well the increased expression of Cl-11 in dementia brains reported in our study.
In conclusion we describe, to our knowledge for the first time, the pattern of expression of Cl-2, Cl-5 and Cl-11 in neurons, astrocytes, oligodendrocytes and microvessels in human brains. In both AD and VaD, significantly more neurons in the frontal cortex express all Cl isoforms examined as compared to controls. More astrocytes in both AD and VaD brains express Cl-2 and Cl-11 as compared to aged control brains. More oligodendrocytes express Cl-11 in AD and Cl-2 in VaD as compared to controls. This complex up-regulation of expression of different Cl isoforms in demented brains might be a result of an endogenous protective response. Our findings could be important to elucidate new pathogenic pathways in dementia disorders and to add new functions to the heterogeneous Cl family of proteins.
Acknowledgments
This study has been supported by Marie Curie–Eurogendis fellowship program, Swedish Brain Power Program and Alzheimer Foundation, Sweden and by the Romanian Ministry of Education and Research, the National Innovation, Research and Development Plan – grants no. 41–013/2007, 61–019-P2/2007 and PN 06.26–01.01/2007. We thank Inga Volkmann for excellent technical support, Anca Catrina for providing us the unspecific immunoglobulin serum for the negative controls and Cătălin Manole for critically reading the manuscript. Our gratefulness is towards the families that donated the brain material for scientific research.
Disclosure
The scientific work of Dr. Nenad Bogdanovic and results in this paper are not subject of commercial interest but pure academic as a part of his academic position at Karolinska Institutet in Sweden.
References
- 1.Popescu BO, Toescu EC, Popescu LM, et al. Blood-brain barrier alterations in ageing and dementia. J Neurol Sci. 2009;283:99–106. doi: 10.1016/j.jns.2009.02.321. [DOI] [PubMed] [Google Scholar]
- 2.Schneeberger EE, Lynch RD. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol. 2004;286:C1213–28. doi: 10.1152/ajpcell.00558.2003. [DOI] [PubMed] [Google Scholar]
- 3.Rubin LL. The blood-brain barrier in and out of cell culture. Curr Opin Neurobiol. 1991;1:360–3. doi: 10.1016/0959-4388(91)90053-a. [DOI] [PubMed] [Google Scholar]
- 4.Martin-Padura I, Lostaglio S, Scheneemann M, et al. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and regulates monocytes transmigration. J Cell Biol. 1998;142:117–27. doi: 10.1083/jcb.142.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Furuse M, Hirase T, Itoh M, et al. Occludin: a novel integral membrane protein localizing at the tight junctions. J Cell Biol. 1993;123:1777–88. doi: 10.1083/jcb.123.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sonoda N, Furuse M, Sasaki H, et al. Clostridium perfringens enterotoxin fragment removes specific claudins from tight junction strands: evidence for direct involvement of claudins in tight junction barrier. J Cell Biol. 1999;147:195–204. doi: 10.1083/jcb.147.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsukita S, Furuse M, Itoh M. Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol. 2001;2:285–93. doi: 10.1038/35067088. [DOI] [PubMed] [Google Scholar]
- 8.Nitta T, Hata M, Gotoh S, et al. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol. 2003;161:653–60. doi: 10.1083/jcb.200302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morita K, Furuse M, Fujimoto K, et al. Claudin multigene family encoding four-transmembrane domain protein components of tight junction strands. Proc Natl Acad Sci USA. 1999;96:511–6. doi: 10.1073/pnas.96.2.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huber JD, Witt KA, Hom S, et al. Inflammatory pain alters blood-brain barrier permeability and tight junctional protein expression. Am J Physiol Heart Circ Physiol. 2001;280:H1241–8. doi: 10.1152/ajpheart.2001.280.3.H1241. [DOI] [PubMed] [Google Scholar]
- 11.Wolburg H, Wolburg-Buchholz K, Kraus J, et al. Localization of claudin-3 in tight junctions of the blood-brain barrier is selectively lost during experimental autoimmune encephalomyelitis and human glioblastoma multiforme. Acta Neuropathol. 2003;105:586–92. doi: 10.1007/s00401-003-0688-z. [DOI] [PubMed] [Google Scholar]
- 12.Lippoldt A, Liebner S, Andbjer B, et al. Organization of choroid plexus epithelial and endothelial cell tight junctions and regulation of claudin-1, -2 and -5 expression by protein kinase C. Neuro Report. 2000;11:1427–31. doi: 10.1097/00001756-200005150-00015. [DOI] [PubMed] [Google Scholar]
- 13.Furuse M, Furuse K, Sasaki H, et al. Converison of Zonulae occludentes from tight to leaky strand type by introducing claudin-2 into Madin-Darby canine kidney cells. J Cell Biol. 2001;153:263–72. doi: 10.1083/jcb.153.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amasheh S, Meiri N, Gitter AH, et al. Claudin-2 expression induces cation-selective channels in tight junctions of epithelial cells. J Cell Sci. 2002;115:4969–76. doi: 10.1242/jcs.00165. [DOI] [PubMed] [Google Scholar]
- 15.Morita K, Sasaki H, Fujimoto K, et al. Claudin-11/OSP- based tight junctions of myelin sheaths in brain and Sertoli cells in testis. J Cell Biol. 1999;145:579–88. doi: 10.1083/jcb.145.3.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bronstein JM, Tiwari-Woodruff S, Buznikov AG, et al. Involvement of OSP/claudin-11 in oligodendrocyte membrane interactions: role in biology and disease. J Neurosci Res. 2000;59:706–11. doi: 10.1002/(SICI)1097-4547(20000315)59:6<706::AID-JNR2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 17.Wolburg H, Wolburg-Buchholz K, Liebner S, et al. Claudin-1, claudin-2 and claudin-11 are present in tight junctions of choroid plexus epithelium of the mouse. Neurosci Lett. 2001;307:77–80. doi: 10.1016/s0304-3940(01)01927-9. [DOI] [PubMed] [Google Scholar]
- 18.Miyamori H, Takino T, Kobayashi Y, et al. Claudin promotes activation of Pro-MMP-2 mediated by membrane-type matrix metalloproteinases. J Biol Chem. 2001;276:28204–11. doi: 10.1074/jbc.M103083200. [DOI] [PubMed] [Google Scholar]
- 19.Rubin LL, Staddon JM. The cell biology of the blood-brain barrier. Annu Rev Neurosci. 1999;22:11–28. doi: 10.1146/annurev.neuro.22.1.11. [DOI] [PubMed] [Google Scholar]
- 20.Nag S. Patophysiology of blood-brain barier breakdown. Methods Mol Med. 2003;89:97–119. doi: 10.1385/1-59259-419-0:97. [DOI] [PubMed] [Google Scholar]
- 21.Kreuter J. Neuroparticulate systems for brain delivery of drugs. Adv Drug Deliv Rev. 2001;47:65–81. doi: 10.1016/s0169-409x(00)00122-8. [DOI] [PubMed] [Google Scholar]
- 22.Kalaria RN. Vascular factors in Alzheimer’s disease. Int Psychogeriatr. 2003;15:47–52. doi: 10.1017/S1041610203008950. [DOI] [PubMed] [Google Scholar]
- 23.Thal DR, Griffin WS, Braak H. Parenchymal and vascular Abeta-deposition and its effects on the degeneration of neurons and cognition in Alzheimer’s disease. J Cell Mol Med. 2008;12:1848–62. doi: 10.1111/j.1582-4934.2008.00411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marco S, Skaper S. Amyloid beta-peptide (1–42) alters tight junction protein distribution and expression in brain microvessel endothelial cells. Neurosci Lett. 2006;401:219–24. doi: 10.1016/j.neulet.2006.03.047. [DOI] [PubMed] [Google Scholar]
- 25.Findley MK, Koval M. Regulation and roles for claudin-family tight junction proteins. IUBMB Life. 2009;61:431–7. doi: 10.1002/iub.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bauer H, Stelzhammer W, Fuchs R, et al. Astrocytes and Neurons Express the Tight Junction-Specific Protein Occludin in vitro. Exp Cell Res. 1999;250:434–8. doi: 10.1006/excr.1999.4558. [DOI] [PubMed] [Google Scholar]
- 27.Romani?an MO, Popescu BO, Winblad B, et al. Occludin is overexpressed in Alzheimer’s disease and vascular dementia. J Cell Mol Med. 2007;11:569–79. doi: 10.1111/j.1582-4934.2007.00047.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bailey TL, Rivara CB, Rocher AB, et al. The nature and effects of cortical microvascular pathology in aging and Alzheimer’s disease. Neurol Res. 2004;26:573–8. doi: 10.1179/016164104225016272. [DOI] [PubMed] [Google Scholar]
- 29.Viswanathan A, Gray F, Bousser MG, et al. Cortical neuronal apoptosis in CADASIL. Stroke. 2008;37:2690–5. doi: 10.1161/01.STR.0000245091.28429.6a. [DOI] [PubMed] [Google Scholar]
- 30.American Psychiatric Association. Committee on Nomenclature and Statistics. Diagnostical and statistical manual of mental disorders. 4th ed. Washington, DC: American Psychiatric Association; 1994. pp. 133–45. [Google Scholar]
- 31.McKhann G, Drachmann D, Folstein M, et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work group under the auspices of the Department of Health and Human Services. Neurology. 1984;34:939–44. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 32.Bogdanovic N, Morris JH. Diagnostic criteria for Alzheimer’s disease in multicentre brain banking. In: Cruz-Sanches FF, Ravid R, Cuzner ML, editors. Neuropathological diagnostic criteria for brain banking. Amsterdam: IOS Press; 1995. pp. 20–9. [Google Scholar]
- 33.National Institute on Aging and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer Disease. Consensus recommendations for the post-mortem diagnosis of Alzheimer’s disease. Neurobiol Aging. 1997;18:S1–2. [PubMed] [Google Scholar]
- 34.West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in the subdivisions of rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–97. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- 35.Lasn H, Winblad B, Bogdanovic N. Neuroglia in the inferior olivary nucleus during normal aging and Alzheimer’s disease. J Cell Mol Med. 2006;10:145–56. doi: 10.1111/j.1582-4934.2006.tb00296.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Förster C. Tight junctions and the modulation of barrier function in disease. Histochem. Cell Biol. 2008;130:55–70. doi: 10.1007/s00418-008-0424-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kröll S, El-Gindi J, Thanabalasundaram G, et al. Control of the blood-brain barrier by glucocorticoids and the cells of the neurovascular unit. Ann NY Acad Sci. 2009;1165:228–39. doi: 10.1111/j.1749-6632.2009.04040.x. [DOI] [PubMed] [Google Scholar]
- 38.Ronaldson PT, Demarco KM, Sanchez-Covarrubias L, et al. Transforming growth factor-beta signaling alters substrate permeability and tight junction protein expression at the blood-brain barrier during inflammatory pain. J Cereb Blood Flow Metab. 2009;29:1084–98. doi: 10.1038/jcbfm.2009.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Argaw AT, Gurfein BT, Zhang Y, et al. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc Natl Acad Sci USA. 2009;106:1977–82. doi: 10.1073/pnas.0808698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bake S, Friedman JA, Sohrabji F. Reproductive age-related changes in the blood brain barrier: expression of IgG and tight junction proteins. Microvasc Res. 2009;78:413–24. doi: 10.1016/j.mvr.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Furuse M, Tsukita S. Claudins in occluding junctions of humans and flies. Trends Cell Biol. 2006;16:181–8. doi: 10.1016/j.tcb.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 42.Farkas E, Luiten PG. Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog Neurobiol. 2001;64:575–611. doi: 10.1016/s0301-0082(00)00068-x. [DOI] [PubMed] [Google Scholar]
- 43.De Jong GI, De Vos RAI, Jansen Steur ENH, et al. Cerebrovascular hypoperfusion: a risk factor for Alzheimer’s disease. Ann NY Acad Sci. 1997;826:56–74. doi: 10.1111/j.1749-6632.1997.tb48461.x. [DOI] [PubMed] [Google Scholar]
- 44.Shin JS, Hyun SY, Kim DH, et al. Chronic hypoperfusion increases claudin-3 immunoreactivity in rat brain. Neurosci Lett. 2008;445:144–8. doi: 10.1016/j.neulet.2008.08.082. [DOI] [PubMed] [Google Scholar]
- 45.Chen G, Bridenbaugh EA, Akintola AD, et al. Increased susceptibility of aging kidney to ischemic injury: identification of candidate genes changed during aging, but corrected by caloric restriction. Am J Physiol Renal Physiol. 2007;293:F1272–81. doi: 10.1152/ajprenal.00138.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van Elzen R, Moens L, Dewilde S. Expression profiling of the cerebral ischemic and hypoxic response. Expert Rev Proteomics. 2008;5:263–82. doi: 10.1586/14789450.5.2.263. [DOI] [PubMed] [Google Scholar]
- 47.Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 48.Gottfries CG, Balldin J, Blennow K, et al. Hypothalamic dysfunction in dementia. J Neural Transm Suppl. 1994;43:203–9. [PubMed] [Google Scholar]
- 49.Maeda K, Tanimoto K, Terada T, et al. Elevated urinary free cortisol in patients with dementia. Neurobiol Aging. 1991;12:161–3. doi: 10.1016/0197-4580(91)90055-o. [DOI] [PubMed] [Google Scholar]
- 50.Green KN, Billi$ngs LM, Roozendaal B, et al. Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer’s disease. J Neurosci. 2006;26:9047–56. doi: 10.1523/JNEUROSCI.2797-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Popescu BO, Popescu LM. Neuronal apoptosis triggered by anti-Fas (CD95, APO-1) antibody is enhanced by dexamethasone. J Med Biochem. 2000;4:1–14. [Google Scholar]
- 52.Burek M, Förster CY. Cloning and characterization of the murine claudin-5 promoter. Mol Cell Endocrinol. 2009;298:19–24. doi: 10.1016/j.mce.2008.09.041. [DOI] [PubMed] [Google Scholar]
- 53.Tipsmark CK, J?rgensen C, Brande-Lavridsen N, et al. Effects of cortisol, growth hormone and prolactin on gill claudin expression in Atlantic salmon. Gen Comp Endocrinol. 2009;163:270–7. doi: 10.1016/j.ygcen.2009.04.020. [DOI] [PubMed] [Google Scholar]
- 54.Landfield PW, Blalock EM, Chen KC, et al. A new glucocorticoid hypothesis of brain aging: implications for Alzheimer’s disease. Curr Alzheimer Res. 2007;4:205–12. doi: 10.2174/156720507780362083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.De Quervain DJ, Poirier R, Wollmer MA, et al. Glucocorticoid-related genetic susceptibility for Alzheimer’s disease. Hum Mol Genet. 2004;13:47–52. doi: 10.1093/hmg/ddg361. [DOI] [PubMed] [Google Scholar]
- 56.Tai LM, Holloway KA, Male DK, et al. Amyloid-beta-induced occludin down-regulation and increased permeability in human brain endothelial cells is mediated by MAPK activation. J Cell Mol Med. 2009 doi: 10.1111/j.1582-4934.2009.00717.x. DOI: 10.1111/j.1582-4934.2009.00717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee HT, Chang YC, Tu YF, et al. VEGF-A/VEGFR-2 signaling leading to cAMP response element-binding protein phosphorylation is a shared pathway underlying the protective effect of preconditioning on neurons and endothelial cells. J Neurosci. 2009;29:4356–68. doi: 10.1523/JNEUROSCI.5497-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moser KV, Humpel C. Vascular endothelial growth factor counteracts NMDA-induced cell death of adult cholinergic neurons in rat basal nucleus of Meynert. Brain Res Bull. 2005;65:125–31. doi: 10.1016/j.brainresbull.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 59.Provias J, Jeynes B. Neurofibrillary tangles and senile plaques in Alzheimer’s brains are associated with reduced capillary expression of vascular endothelial growth factor and endothelial nitric oxide synthase. Curr Neurovasc Res. 2008;5:199–205. doi: 10.2174/156720208785425729. [DOI] [PubMed] [Google Scholar]
- 60.Mateo I, Llorca J, Infante J, et al. Low serum VEGF levels are associated with Alzheimer’s disease. Acta Neurol Scand. 2007;116:56–8. doi: 10.1111/j.1600-0404.2006.00775.x. [DOI] [PubMed] [Google Scholar]
- 61.Town T, Laouar Y, Pittenger C, et al. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14:681–687. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Malaguarnera L, Motta M, Di Rosa M, et al. Interleukin-18 and transforming growth factor-beta 1 plasma levels in Alzheimer’s disease and vascular dementia. Neuropathology. 2006;26:307–12. doi: 10.1111/j.1440-1789.2006.00701.x. [DOI] [PubMed] [Google Scholar]
- 63.Tarkowski E, Issa R, Sjögren M, et al. Increased intrathecal levels of the angiogenic factors VEGF and TGF-beta in Alzheimer’s disease and vascular dementia. Neurobiol Aging. 2002;23:237–43. doi: 10.1016/s0197-4580(01)00285-8. [DOI] [PubMed] [Google Scholar]
- 64.Hu Z, Fan J, Zeng L, et al. Transient cerebral ischemia leads to TGF-beta2 expression in Golgi apparatus organelles. Curr Neurovasc Res. 2008;5:178–184. doi: 10.2174/156720208785425693. [DOI] [PubMed] [Google Scholar]
- 65.Howe KL, Reardon C, Wang A, et al. Transforming growth factor-beta regulation of epithelial tight junction proteins enhances barrier function and blocks enterohemorrhagic Escherichia coli O157:H7-induced increased permeability. Am J Pathol. 2005;167:1587–97. doi: 10.1016/s0002-9440(10)61243-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stopa EG, Gonzalez AM, Chorsky R, et al. Basic fibroblast growth factor in Alzheimer’s disease. Biochem Biophys Res Commun. 1990;171:690–6. doi: 10.1016/0006-291x(90)91201-3. [DOI] [PubMed] [Google Scholar]
- 67.Bendfeldt K, Radojevic V, Kapfhammer J, et al. Basic fibroblast growth factor modulates density of blood vessels and preserves tight junctions in organotypic cortical cultures of mice: a new in vitro model of the blood-brain barrier. J Neurosci. 2007;27:3260–7. doi: 10.1523/JNEUROSCI.4033-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cowburn RF, O’Neill C, Bonkale WL, et al. Receptor-G-protein signalling in Alzheimer’s disease. Biochem Soc Symp. 2001;67:163–75. doi: 10.1042/bss0670163. [DOI] [PubMed] [Google Scholar]
- 69.Angelow S, Zeni P, Höhn B, et al. Phorbol ester induced short- and long-term permeabilization of the blood-CSF barrier in vitro. Brain Res. 2005;1063:168–79. doi: 10.1016/j.brainres.2005.09.058. [DOI] [PubMed] [Google Scholar]
- 70.Stamatovic SM, Dimitrijevic OB, Keep RF, et al. Protein kinase Calpha-RhoA cross-talk in CCL2-induced alterations in brain endothelial permeability. J Biol Chem. 2006;281:8379–88. doi: 10.1074/jbc.M513122200. [DOI] [PubMed] [Google Scholar]
- 71.Jin JK, Ahn BH, Na YJ, et al. Phospholipase D1 is associated with amyloid precursor protein in Alzheimer’s disease. Neurobiol Aging. 2007;28:1015–27. doi: 10.1016/j.neurobiolaging.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 72.Jin JK, Kim NH, Lee YJ, et al. Phospholipase D1 is up-regulated in the mitochondrial fraction from the brains of Alzheimer’s disease patients. Neurosci Lett. 2006;407:263–7. doi: 10.1016/j.neulet.2006.08.062. [DOI] [PubMed] [Google Scholar]
- 73.Umemura K, Yamashita N, Yu X, et al. Autotaxin expression is enhanced in frontal cortex of Alzheimer-type dementia patients. Neurosci Lett. 2006;400:97–100. doi: 10.1016/j.neulet.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 74.Liu Y, Zhang YW, Wang X, et al. Intracellular trafficking of presenilin 1 is regulated by beta-amyloid precursor protein and phospholipase D1. J Biol Chem. 2009;284:12145–52. doi: 10.1074/jbc.M808497200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zeiller C, Mebarek S, Jaafar R, et al. Phospholipase D2 regulates endothelial permeability through cytoskeleton reorganization and occludin downregulation. Biochim Biophys Acta. 2009;1793:1236–49. doi: 10.1016/j.bbamcr.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 76.Popescu AT, Vidulescu C, Stanciu CL, et al. Selective protection by phosphatidic acid against staurosporine-induced neuronal apoptosis. J Cell Mol Med. 2002;6:433–438. doi: 10.1111/j.1582-4934.2002.tb00523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Steiner MR, Holtsberg FW, Keller JN, et al. Lysophosphatidic acid induction of neuronal apoptosis and necrosis. Ann NY Acad Sci. 2000;905:132–41. doi: 10.1111/j.1749-6632.2000.tb06545.x. [DOI] [PubMed] [Google Scholar]
- 78.Wessells H, Sullivan CJ, Tsubota Y, et al. Transcriptional profiling of human cavernosal endothelial cells reveals distinctive cell adhesion phenotype and role for claudin 11 in vacular barrier function. Physiol Genomics. 2009;39:100–8. doi: 10.1152/physiolgenomics.90354.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pike CJ, Carroll JC, Rosario ER, et al. Protective actions of sex steroid hormones in Alzheimer’s disease. Front Neuroendocrinol. 2009;30:239–58. doi: 10.1016/j.yfrne.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kaitu’u-Lino TJ, Sluka P, Foo CF, et al. Claudin-11 expression and localisation is regulated by androgens in rat Sertoli cells in vitro. Reproduction. 2007;133:1169–79. doi: 10.1530/REP-06-0385. [DOI] [PubMed] [Google Scholar]
- 81.Ponholzer A, Madersbacher S, Rauchenwald M, et al. Serum androgen levels and their association to depression and Alzheimer dementia in a cohort of 75-year-old men over 5 years: results of the VITA study. Int J Impot Res. 2009;21:187–91. doi: 10.1038/ijir.2009.10. [DOI] [PubMed] [Google Scholar]
- 82.Bowen RL, Isley JP, Atkinson RL. An association of elevated serum gonadotropin concentrations and Alzheimer disease. J Neuroendocrinol. 2000;12:351–4. doi: 10.1046/j.1365-2826.2000.00461.x. [DOI] [PubMed] [Google Scholar]
- 83.Rimon E, Sasson R, Dantes A, et al. Gonadotropin-induced gene regulation in human granulosa cells obtained from IVF patients: modulation of genes coding for growth factors and their receptors and genes involved in cancer and other diseases. Int J Oncol. 2004;24:1325–38. doi: 10.3892/ijo.24.5.1325. [DOI] [PubMed] [Google Scholar]
- 84.Popescu BO, Ankarcrona M. Neurons bearing presenilins: weapons for defense or suicide. J Cell Mol Med. 2000;4:249–261. doi: 10.1111/j.1582-4934.2000.tb00124.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Casadesus G, Garrett MR, Webber KM, et al. The estrogen myth: potential use of gonadotropin-releasing hormone agonists for the treatment of Alzheimer’s disease. Drugs R D. 2006;7:187–93. doi: 10.2165/00126839-200607030-00004. [DOI] [PubMed] [Google Scholar]