

Abstract
Recent reports have suggested that statins induce cell death in certain epithelial cancers and that patients taking statins to reduce cholesterol levels possess lower cancer incidence. However, little is known about the mechanisms of action of different statins or the effects of these statins in gynaecological malignancies. The apoptotic potential of two lipophilic statins (lovastatin and simvastatin) and one hydrophilic statin (pravastatin) was assessed in cancer cell lines (ovarian, endometrial and cervical) and primary cultured cancerous and normal tissues. Cell viability was studied by MTS assays and apoptosis was confirmed by Western blotting of PARP and flow cytometry. The expressions of key apoptotic cascade proteins were analysed. Our results demonstrate that both lovastatin and simvastatin, but not pravastatin, selectively induced cell death in dose- and time-dependent manner in ovarian, endometrial and cervical cancers. Little or no toxicity was observed with any statin on normal cells. Lipophilic statins induced activation of caspase-8 and -9; BID cleavage, cytochrome C release and PARP cleavage. Statin-sensitive cancers expressed high levels of HMG-CoA reductase compared with resistant cultures. The effect of lipophilic statins was dependent on inhibition of enzymatic activity of HMG-CoA reductase since mevalonate pre-incubation almost completely abrogated the apoptotic effect. Moreover, the apoptotic effect involved the inhibition of synthesis of geranylgeranyl pyrophosphate rather than farnesyl pyrophosphate. In conclusion, lipophilic but not hydrophilic statins induce cell death through activation of extrinsic and intrinsic apoptotic cascades in cancerous cells from the human female genital tract, which express high levels of HMG-CoA reductase. These results promote further investigation in the use of lipophilic statins as anticancer agents in gynaecological malignancies.
Keywords: statins, gynaecological cancers, apoptosis, therapy
Introduction
Gynaecological cancers constitute one of the main causes of death from cancer worldwide. Ovarian cancer is the gynaecological malignancy with the highest mortality, mainly because over 75% of the cases are diagnosed at late stage disease [1]. Endometrial and cervical cancers are more frequent than ovarian cancer and the 5-year survival rates for advanced stages are similarly poor [2, 3]. The lower survival of patients at advanced stages is explained by poor or transient response to therapy, making the identification of new-targeted therapies for these diseases a major goal.
Statins, currently used to lower plasma cholesterol levels, are inhibitors of the 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMG-CoA reductase). HMG-CoA reductase catalyses the conversion of HMG-CoA to mevalonic acid, the rate-limiting step in cholesterol biosynthesis [4]. Away from their use in lowering cholesterol, there are other beneficial effects attributed to statins like improvement of endothelial function [5], modulation of inflammatory response [6–8] and reduction of thrombus generation [9]. Further evidence has suggested the pleiotropic effects of statins could extend to prevention and treatment of different cancers [10, 11].
Regarding prevention, several studies have reported a correlation between statins use, for cardiovascular motives, and the reduction of cancer incidence [12, 13]. A retrospective nested case-control study, in close to half a million patients, demonstrated a 48% reduction in renal cell carcinoma, irrespective of age, sex, obesity or tobacco use [14]. However, a recent systematic review of 42 randomized trials failed to demonstrate cancer risk reduction after statin use [15]. In fact, contradictory results were observed, with increased incidence for certain cancers (i.e. melanoma) and reduced incidence for others (i.e. stomach, liver and lymphoma) [15]. The reasons of the discrepancy may include different designs (retrospective versus prospective), insufficient follow-up and more importantly, different type of statins used [16].
Statins belong from two classes: hydrophilic (pravastatin and rosuvastatin) and lipophilic (cerivastatin, simvastatin, lovastatin, fluvastatin and atorvastatin). Lipophilicity of statins improves drug access to different tissues [17]. The more lipophilic statins achieve higher levels of exposure in non-hepatic tissues, while the hydrophilic statins are more hepatoselective [18, 19]. Thus, a differential effect of statins can be predicted among hepatic and non-hepatic tissues. Evidence from randomized controlled clinical trials and in vitro studies support this differential effect of statins depending on its class (lipophilic versus hydrophilic) and the specific tissue assayed [20–22].
Little is known about the effect of statins on gynaecological cancer incidence. A Canadian study, analysing the effect of statins on the incidence of cancer, showed reduction in the majority of cancers, including uterine cancer [20]. In contrast, a recent retrospective cohort study [23] did not show a correlation between statin use and reduced incidence of endometrial or ovarian cancer.
Less information exists regarding the use of statins to treat gynaecological cancers. A retrospective study suggests that statins improved overall survival in patients with epithelial ovarian cancer if they were current statin users when receiving standard therapy [24]. Preliminary in vitro studies suggest that lovastatin, and possibly atorvastatin, could induce cell death in ovarian cancer cell lines [25, 26]. These two statins are members of the lipophilic class of statins. No information has been published about the effect of hydrophilic statins in this type of cancer.
Here, we study whether statins could induce cell death in gynaecological malignancies and explore the molecular mechanisms of this effect. We demonstrate that lipophilic, but not hydrophilic, statins induce cell death in cancer cell lines and primary cultures established from gynaecological cancers, without affecting their normal counterparts. Cell death induced by statins correlates with HMG-CoA expression and can be rescued by the addition of key precursors (mevalonate and geranylgeranyl pyrophosphate) in the synthesis of cholesterol.
Materials and methods
Reagents
Lovastatin and simvastatin were purchased from Calbiochem (Darmstadt, Germany) and pravastatin from Sigma-Aldrich (St. Louis, MO, USA). Lovastatin and simvastatin were prepared in DMSO (Sigma-Aldrich) and pravastatin in distillated water and all stored at –20°C until used. The intermediate metabolites of cholesterol synthesis mevalonate, geranylgeranyl pyrophosphate and farnesyl pyrophosphate were purchased from Sigma-Aldrich and stored at –20°C until co-incubation with statins. The non-selective tetra peptide caspase inhibitor ZVAD-fmk (Enzyme Systems Products, Livermore, CA, USA) was resuspended in DMSO (Sigma-Aldrich) and added to the cells at a final concentration of 50 uM, 30 min. before the addition of statins. Chemotherapeutic drugs, doxorubicin, paclitaxel and cisplatin were kindly supplied by the Cancer Centre of the Pontificia Universidad Católica de Chile.
Tissues collection
Primary tissue from human female reproductive tract was obtained after informed consent (approved by institutional ethical board) from patients undergoing surgery for a benign or malignant condition in the Department of Obstetrics and Gynecology at the Hospital of the Pontificia Universidad Católica de Chile in Santiago, Chile.
Primary cultures
Primary cultures of human tissues were established following protocols previously published by our group [27]. Normal ovarian culture, the ovarian tissue was washed with HBSS medium (Hank’s Balanced Salt Solution) without calcium, magnesium or phenol red (Invitrogen, NY, USA), supplement with antibiotics (penicillin 100 U/ml, streptomycin 100 ug/ml) (Invitrogen). Two-mm thick slices containing surface epithelium and underlying stroma were incubated in dispase (Sigma-Aldrich) for 30 min. at 37°C with agitation. Then, ovarian epithelial cells were scraped off from the epithelial side of the slice using a rubber policeman. Cells were suspended in HBSS, pelleted and resuspended in MCDB/M199 medium supplemented with 10% foetal bovine serum (FBS; Invitrogen) plus antibiotics and plated in 10 cm2 tissue culture dishes (Falcon, BD Labware, NJ, USA). Advanced epithelial ovarian cancer cultures were established mainly from ascites. Approximately 400 ml of ascites (per patient) was centrifuged at 2000 g for 5 min. Sixty per cent of the supernatant was discarded and replaced with DMEM/F12 medium (Invitrogen) supplemented with 10% FBS plus antibiotics and transferred to tissue culture dishes. For tissue cultures established from primary tumour of ovarian cancer, the tumour was washed twice in cold phosphate buffer solution (PBS) and then dissected into 1 mm3 cubes and incubated for 1 hr at 37°C (with rocking) in digestion medium containing collagenase-1 (2 mg/ml, Worthington, Lorne Laboratories, Twyford, UK), hyaluronidase I (1 mg/ml, Sigma-Aldrich), DNAase I (0.05 mg/ml, Boehringer Mannheim, Germany), antibiotics/antimycotics (Invitrogen) and prepared in calcium and magnesium free HBSS solution (Invitrogen). The digested medium was then filtered through a 50-μm nylon mesh (PGC Scientific, MD, USA) to separate stromal from epithelial cells. Epithelial cells retained by the filter were washed and resuspended in DMEM/F12 with 10% FBS plus antibiotic/antimycotics and transferred to tissue culture dishes. Primary normal and cancerous endometrial tissue cultures (stroma and epithelium) were derived from tissue obtained from women undergoing hysterectomy with or without bilateral salpingo-oophorectomy (plus surgical staging when indicated in malignant disease). Immediately after hysterectomy, approximately 1–2 cm3 of relevant tissue was removed from the endometrium at the endometrial cavity under sterile conditions and collected into warm saline serum. The stage of the menstrual cycle was determined from patient menstrual history and endometrial histology. The tissue was washed twice in cold PBS and the endometrium separated from the muscular layer. The tissue was dissected into 1 mm3 cubes and incubated in digestion medium containing collagenase type I (2 mg/ml, Worthington, Lorne Laboratories), hyaluronidase type IV (1 mg/ml, Sigma-Aldrich), DNAase I (0.05 mg/ml, Boehringer Mannheim), antibiotics/antimycotics and prepared in calcium and magnesium free HBSS solution. After digestion the majority of the endometrial stromal and immune cells were visualized as individual cells, while cells of endometrial epithelial origin were present as aggregates. This digestion medium was filtered through a 70 μm nylon mesh to separate epithelial from stromal cells. The epithelial cells remaining in clumps in the filter while stromal cells are rescued from filtrate. Each aliquot was centrifuged and resuspended in DMEM/F12 with 10% FBS plus antibiotic/antimycotics and transferred to tissue culture dishes. Primary tissue culture from a recurrent cervical cancer and a uterine sarcoma were established following ascites and endometrial cancer protocols, respectively. Human fallopian tube was obtained from patients undergoing hysterectomy for a benign condition. Immediately after removal the tubes were washed in calcium and magnesium free Hank’s solution (Invitrogen). The fimbria and distal portion of the ampulla were isolated from the tube and placed in sterile DMEM/F12 medium supplemented with 25 mM Hepes at pH 7.4. Each tissue sample was cut into 4- to 8-mm pieces and transferred to a 5 mM EDTA in DMEM/F12 and incubated at 37°C for 45 min. The ciliated epithelium was then mechanically separated form the rest of the tissue using fine forceps. The cell suspension was passed through a tuberculin syringe into a centrifuge tube and spun at 300 g for 5 min. The pellet was resuspended in DMEM supplemented with 44 mM NaHCO3, 10% FBS, penicillin and streptomycin at pH 7.4. The cell suspension was plated in 10 cm2 tissue culture dishes and incubated in a 5% CO2 atmosphere at 37°C until the culture formed a monolayer of cells (approximately 4 days). Myometrial smooth muscle cells were isolated from myometrial tissue obtained from healthy pregnant women undergoing elective caesarean section at term, not in labour. Briefly, the myometrial tissue was minced and incubated in medium containing collagenase (1.5 mg/ml), deoxyribonuclease (0.1 ng/ml) and antibiotics/antimycotics for 4 hrs at 37°C with agitation to disperse the smooth muscle cells. The dispersed cells were separated from non digested tissue by filtration through gauze, centrifuged at 400 g for 10 min. and resuspended in Ham’s DMEM/F12 medium supplemented with 10% FBS plus antibiotics (penicillin and streptomycin), and antimycotics (amphotericin B, 0.25 μg/ml) and plated in tissue culture dishes. Cells were maintained in a 5% CO2 atmosphere at 37°C until confluence (about 7–10 days).
Cell line culture
The ovarian cancer lines A2780 and UCI 101, the ovarian surface epithelial normal cancer cell line HOSE, the endometrial cancer cell line Ishikawa and cervical cancer cell line Hela were maintained in DMEM/F12 media supplemented with 10% FBS plus antibiotic/antimycotics. For protein and RNA experiments, cells were plated at 50% confluence in 10 cm2 and 6 cm2 tissue culture dishes, respectively. Twenty-four hours before statins addition the medium was changed to a charcoal-treated medium supplemented with 5% FBS.
MTS assay
To assess statin-mediated cytotoxicity, cells were plated at 3000–5000 cells per well in 96-well microtitre plates overnight and then incubated for 48 hrs with different concentrations of Statins. Cell viability was assessed by the MTS (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) dye reduction assay as described previously [27]. All experiments were performed in quintuplicate and repeated at least three times.
Western blotting
Cells were harvested in cold PBS, the pellet suspended in lysis buffer (10 mM TRIS-HCl pH 7.4, 150 mM NaCl, 0.5% Triton X-100) for 20 min. at 4°C, sonicated and centrifuged at 14,000 g for 20 min. at 4°C and the pellet discarded. Protein concentration was determined by Bradford assay. One hundred μg of crude membrane extract was loaded in each lane, separated by 10% polyacrylamide gel electrophoresis in the presence of sodium dodecylsulphate, transferred to nitrocellulose membranes and incubated overnight with primary antibody (1:1000; BID, BAD and TRAIL-R2 (Santa Cruz, CA, USA), FLIP (Calbiochem, Darmstadt, Germany), caspase-8 and caspase-9 (Upstate, Lake Placid, NY, USA), cytochrome c (BD Pharmingen, San Jose, CA, USA), MAPK and MAPK-phosphorylated, AKT and AKT-phosphorylated (Cell Signaling, Danvers, MA, USA) and β actin (Sigma-Aldrich)). Secondary antibody, goat anti-mouse or anti-rabbit IgG secondary antibody coupled to hydrogen peroxidase (1:3000, Bio-Rad Labs, Hercules, CA, USA), was applied for 1 hr at room temperature. The reaction was developed with chemiluminescence using ECL Western blot analysis system (NEN, Western lightning, Perkin-Elmer, Waltham, MA, USA).
Flow cytometry analysis
Cell cycle distribution and the detection of a sub-G1 apoptotic peak were analysed by flow cytometry using propidium iodide DNA staining following protocol previously described [28]. Cells treated under experimental conditions were harvested, centrifuged, washed and re-suspended in a cold solution of 1 ml 1× PBS and 4 ml 70% ethanol. The cells were incubated overnight at 4°C, washed in 1× PBS and resuspended in a solution of 250 μl of propidium iodide (50 μg/ml) in 1× PBS and 1 ul RNase (20 μg/ml). Cells were incubated (protected from light) for 15 min. at room temperature before analysis on a FACScan cytometer using the Cell Quest software (Becton Dickinson, Mountain View, CA, USA).
Measurement of caspase activity
Cultured cells were harvested and washed once in cold PBS. After brief centrifugation (3000 rpm, 5 min.), cells were incubated in lysis buffer containing 20 mM Hepes (pH 7.4), 100 mM NaCl, 0.5% (v/v) NP-40 and 10 mM DTT on ice for 30 min. Following centrifugation at 13,000 rpm for 10 min. at 4°C, supernatants were collected, transferred to a 96-well microtitre plate and the corresponding substrate for caspase-8 or -9 added (Calbiochem-Novabiochem Corp., CA, USA). The samples were incubated for 24 hrs at room temperature. Optical density at 405 nm was measured using an ELISA plate reader (EL310 Boots-Celtech). Caspase activity was expressed as percentage from control, which was set at 100%. Statistical analysis was performed using Mann–Whitney analysis and setting statistical significance at P < 0.05.
RT-PCR
Total RNA was isolated using the Chomczynski method [29]. cDNA was generated using reverse transcriptase (Superscript II, Invitrogen). Semi-quantitative PCR reactions were performed with cDNA generated from cell lines and primary cultured cells, using Taq polymerase (Invitrogen) and the HMGCoA primers sense: 5′-CCGCGGCCACACCCAGAAAGT-3′ and antisense: 5′-GTACATGGGAGGCAAGCAAACAAA-3′ (these primers generate a fragment of 260 bp) (BiosChile, Santiago, Chile). As an internal control and testing the integrity of the starting cDNA, primers amplifying a region of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were used as previously published [30].
Statistical analysis
Statistical analysis was performed using Mann–Whitney analysis and setting the statistical significance at P < 0.05. Interactions between statins and chemotherapeutic drugs were classified using the fractional inhibition method as follows: when expressed as the fractional inhibition of cell viability, additive inhibition produced by both inhibitors (i) occurs when i1,2=i1+i2; synergism when i1,2 > i1+i2; and antagonism when i1,2 < i1+i2[31]. The synergism was confirmed by dose-effect analysis using Calcusyn software (Biososft, Cambridge, UK) [32].
Results
Lipophilic (lovastatin and simvastatin) but not hydrophilic (pravastatin) statins mediate apoptosis in ovarian cancer cell lines
To evaluate if different classes of statins could have a differential effects in ovarian cancer cells we incubated the ovarian cancer cell lines, A2780 and UCI 101, with differing concentrations (0.1–1-10 uM) of two lipophilic statins (lovastatin and simvastatin), one hydrophilic statin (pravastatin) or vehicle (DMSO) for 48 hrs. As shown in Figure 1A, the A2780 cells are sensitive to lovastatin and simvastatin treatment at 1 and 10 uM concentrations. No reduction in cell viability was observed with pravastatin. Similar effect was observed with the UCI 101 cell line (data not shown). To better analyse the response to the different types of statins, and to confirm that the lack of sensitivity with the hydrophilic statin was not due to insufficient time exposure, we performed a time course out to 72 hrs. As depicted in Figure 1B, lovastatin and simvastatin require 48 hrs to significantly lower cell viability in A2780 cells. In contrast, these cells remain insensitive to pravastatin even after 3 days of exposure. To confirm that reduction in cell viability was due to apoptosis, protein extracts from concentration-response curves (0–10 uM) of each statin were analysed for the presence of PARP cleavage by immunoblotting (Fig. 2A). Confirming the results from cell viability assays, the appearance of the cleaved form of PARP protein was observed only with lovastatin and simvastatin (at 1 and 10 uM). To further confirm the induction of apoptosis, we examined the effect of statin exposure on DNA content and cell cycle progression by flow cytometry. In Figure 2B, a significant increase in the per cent of cells in the sub G0/G1 region (representative of dead cells) was observed with increasing concentrations of simvastatin and lovastatin but not pravastatin after 48 hrs of exposure.
Fig 1.
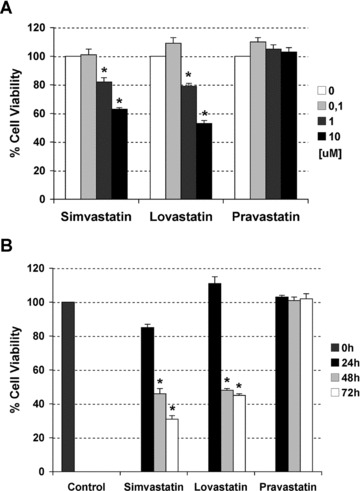
Dose response curve (A) and time course (B) to two lipophilic (lovastatin and simvastatin) and one hydrophilic (pravastatin) statins in A2780 ovarian cancer cells. Cell viability was measured by MTS assays. Data are shown as mean –/+ SD (n= 3). The * indicates statistical significance compared to control (vehicle), (Mann–Whitney test, P-value < 0.05).
Fig 2.
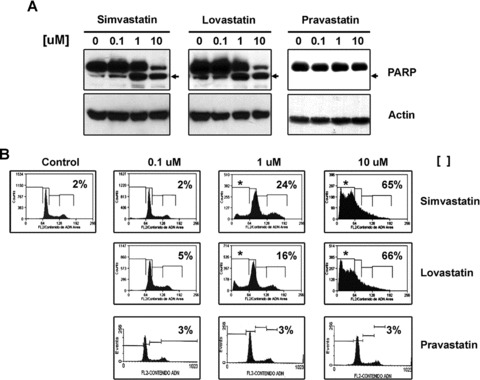
(A) Detection by immunoblotting of the cleaved form of PARP, a demonstration that lipophilic (lovastatin and simvastatin) but not hydrophilic (pravastatin) statins induce apoptosis in concentration-dependent manner in A2780 cells. Actin is used as loading control. (B) Determination of sub G0/G1 region en DNA histogram by FACS in A2780 cells treated under same conditions.
Statins selectively induce cell death in cancerous ovarian epithelial primary tissue cultures
To confirm the effect of statins is not merely an artefact of cell lines, we repeated our experiments in primary cultures of advanced epithelial ovarian cancer (histologically confirmed stage IIIc or IV ovarian cancer). As shown in Figure 3, primary cultures of ovarian cancer cells obtained from four ascites (ovarian ca 1 to 4) and one solid tumour (ovarian ca 5) demonstrated a concentration-dependent reduction (only 10 uM is shown in the figure) in viability in response to lovastatin and simvastatin exposure but not to pravastatin. Apoptosis was confirmed by flow cytometry analysis on three of the cancers (results not shown). Concurrently, we test an ovarian cell line (HOSE) and two primary tissue cultures established from normal ovarian epithelium (one of them mentioned in the figure as normal ovary). Interestingly, the same concentrations of lovastatin and simvastatin did not reduce the viability of epithelial cells isolated from normal ovary.
Fig 3.
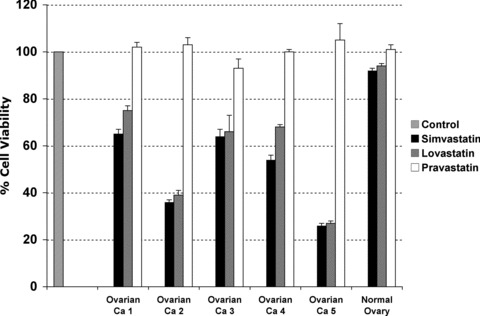
Comparative effects in cell survival of lipophilic (lovastatin and simvastatin) and hydrophilic (pravastatin) statins in primary tissue cultures established from ascites of patients with advanced epithelial ovarian carcinoma (indicated as Ovarian Ca 1 to 5) and from normal ovarian epithelium. Cell viability was measured by MTS assays upon 48 hrs of treatment with each statin at 10 uM. Ovarian Ca = Ovarian cancer sample.
Lovastatin and simvastatin selectively induce apoptosis in cancerous cells from uterine and cervical origin
Two established cancer cell lines, one from endometrial origin (Ishikawa cells) and one from cervical origin (Hela cells), two primary tissue cultures, one from an stage I undifferentiated endometrial sarcoma and one from a recurrent advanced uterine cancer, and primary tissue cultures from normal female human reproductive tissues (including fallopian tube, myometrium, endometrial stroma and epithelium) were incubated with differing concentrations lipophilic (lovastatin and simvastatin) and hydrophilic (pravastatin) statins for 48 hrs and compared to control (DMSO). MTS assays demonstrated a significant reduction in cell viability with both Ishikawa cells and Hela cells (data not shown). As shown by immunoblotting in Figure 4A, a concentration response to each statin in endometrial and cervical cancer cells mirrored the sensitivity previously observed in ovarian cancer cells with lovastatin and simvastatin. As with ovarian cells, no effect was observed with pravastatin in either cell lines. A similar result was observed with one tissue culture established from a recurrent advanced uterine cancer. Conversely, and in concordance with results obtained with normal ovarian epithelium (Fig. 3), all the primary tissue cultures isolated from other normal female human reproductive tissues did not show sensitivity to any statin (Fig. 4B).
Fig 4.
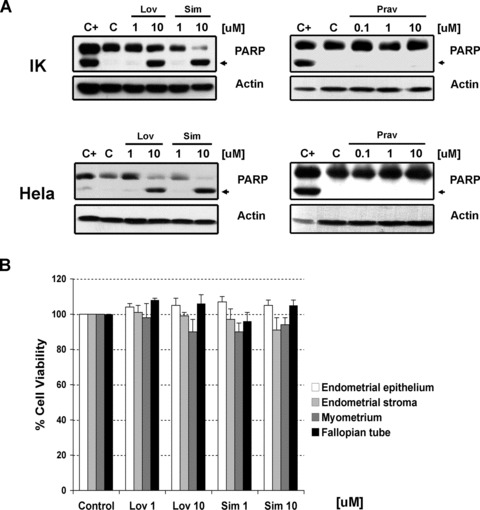
(A) Detection by immunoblotting of the cleaved form of PARP after treatment with increasing concentrations of lipophilic (lovastatin and simvastatin) or hydrophilic (pravastatin) statins in Ishikawa (indicated as IK) and Hela cells. Actin is used as loading control. C+ stands for positive control. (B) Absence of effects in cell viability of different concentrations (1–10 uM) of lovastatin (Lov) and simvastatin (Sim) in tissue cultures established from normal gynaecological origin tissues. Cell viability was measured by MTS assays upon 48 hrs of treatment.
Statins induce caspase-mediated apoptosis through activation of intrinsic and extrinsic cascades
To characterize the mechanisms involved in statin-mediated apoptosis, we further analysed statin effect on the A2780 ovarian cancer cell line. First at all, we investigated if the cell death could involve the activation of caspase cascade. Figure 5A shows an increase in both caspase-8 (the major caspase initiator of the extrinsic pathway) and caspase-9 (a caspase initiator of intrinsic pathway) activity after 24 hrs of incubation with lovastatin or simvastatin (1 and 10 μM). The non-selective caspase inhibitor ZVAD-fmk reverted the loss of cell viability induced by statins, proving lovastatin- and simvastatin-mediated apoptosis occurs through caspase-dependent mechanisms (Fig. 5B). To further investigate the use of both the intrinsic and extrinsic apoptotic pathways, the expression of key proteins from each pathway were analysed. We found that increasing concentrations of statins decrease levels of FLIP protein (an inhibitor of the DISC complex) and pro-caspase 8 but increase the cleaved form (active caspase 8), thus indicating the activation of extrinsic pathway. On the other hand, a decrease in cytosolic BAD, release of cytochrome c from mitochondria and the cleavage of caspase-9 also occur upon treatment with lovastatin and simvastatin, indicating activation of the intrinsic pathway (Fig. 6). The decrease in BID protein suggests a communication between both pathways, given that BID is cleaved after activation of extrinsic pathway and facilitates the release of proteins involved in the intrinsic pathway.
Fig 5.
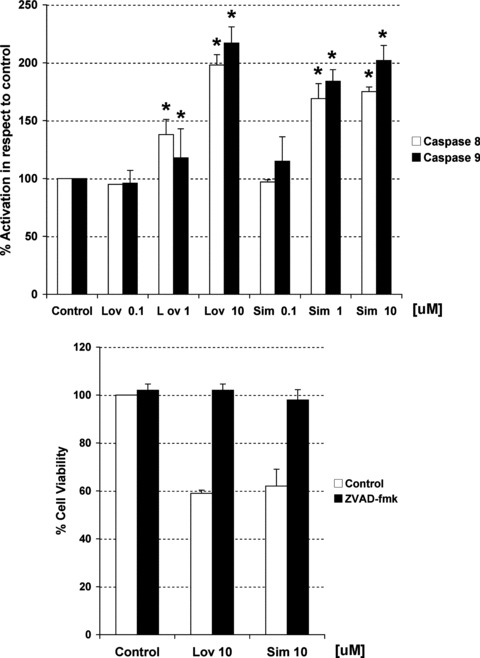
(A) Measurement of caspase-8 and -9 activities through an in vitro caspase assay in A2780 cells after 48 hrs of treatment with increasing concentrations of lovastatin (Lov) and simvastatin (Sim). Activity is expressed as per cent from control. (B) Effects in cell viability of both statins in the presence or absence of a non-selective caspase inhibitor (ZVAD-fmk). Data are shown as mean –/+ SD (n= 3). The * indicates statistical significance compared to control (vehicle), (Mann–Whitney test, P-value < 0.05).
Fig 6.
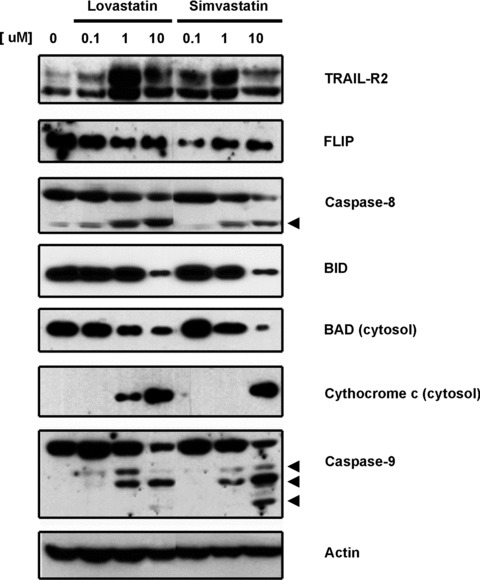
Determination by immunoblotting of expression levels of different proteins involved in the extrinsic (TRAIL-R2, FLIP, cleaved caspase-8 and BID) and intrinsic apoptotic cascade (BAD, cytochrome c release and cleaved caspase-9) after incubation with increasing concentrations of lipophilic statins for 24 hrs. Actin was used as loading control.
Differential expression of HMG-CoA reductase between statin-sensitive and -resistant cells
It has been suggested in previous publications that some cancer cells depend on the high levels of mevalonate and other intermediate metabolites in the biosynthesis of cholesterol to mediate their cell proliferation and evasion of apoptotic signals [33]. Thus, the depletion of these metabolites would lead to preferential cell death in cancerous cells as opposed to normal cells. We analysed the levels of HMG-CoA reductase (the key enzyme, inhibited by statins, directly responsible for mevalonate levels) in all the cell lines and primary tissue cultures established from female human reproductive tissues. Interestingly, all the cells sensitive to statin-mediated apoptosis expressed higher levels of HMG-CoA reductase compared with those non-sensitive to statin effect (including all the normal cells and the primary tissue culture from the undifferentiated endometrial sarcoma) (Fig. 7).
Fig 7.
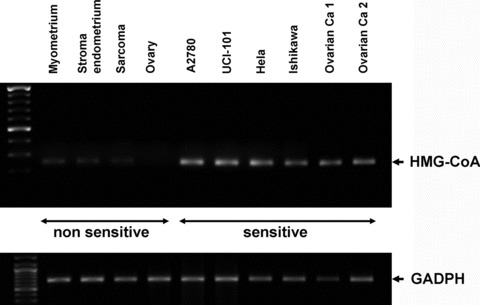
Determination of basal mRNA levels of HMG-CoA reductase in cell lines and primary tissue cultures from gynaecological origin considered sensitive (ovarian: A2780, UCI-101, ca ovary 1–2; cervical: Hela; endometrial: Ishikawa) and non-sensitive (normal myometrium, endometrial stroma, ovarian tissues and uterine sarcoma) to statin-mediated cell death through RT-PCR. GADPH is shown as loading control.
Mevalonate and geranylgeranyl pyrophosphate but not farnesyl pyrophosphate rescue cancer cells from statin-mediated apoptosis
To confirm that cell death induced by lipophilic statins in gynaecological cancers is due to the decrease in the synthesis of mevalonate (through inhibition of HMGCoA reductase by statins), we analysed if statin-mediated cell death could be rescued by the supplementation of mevalonate in A2780 and Hela cells. In accordance with our theory, we demonstrated that mevalonate supplementation prevented the loss of cell viability induced by statins in A2780 cells (Fig. 8A). Figure 8B shows by immunoblotting the absence of PARP cleavage upon supplementation with mevalonate in both A2780 as well as Hela cells confirming the importance of mevalonate for the sensitivity of these cells to statins. Previous reports in endothelial and lymphoblast cells have suggested that the two isoprenes, farnesyl and geranylgeranyl pyrophosphates, can be involved in the ability of evading cell death of cancerous cells [21, 34]. Both isoprenes are located downstream to mevalonate in the cholesterol pathway. They act as lipophilic anchors on the cell membrane for both attachment and biological activity of small-GTP binding proteins (i.e. Rho family - Rac 1 and Cdc42-, and Ras) and activate other functional proteins involved in both the cell cycle and cell proliferation [35]. In vitro studies in ovarian cancer cells have shown that farnesyl and geranylgeranyl pyrophosphate could revert the effect in cell viability induced by lovastatin [25, 26]. Therefore, we decided to investigate if any of these isoprenes could affect cell death induced by simvastatin in cell lines and primary tissue cultures from gynaecological cancers. As shown in Figure 9, the addition of geranylgeranyl but not farnesyl pyrophosphate almost completely abrogates the effect in cell viability induced by both statins in the A2780 cells and Hela cells. Similar effect was observed with two primary cultures established from ovarian cancer (indicated as ovarian ca 6 in the Fig. 9) and uterine cancer (data not shown).
Fig 8.
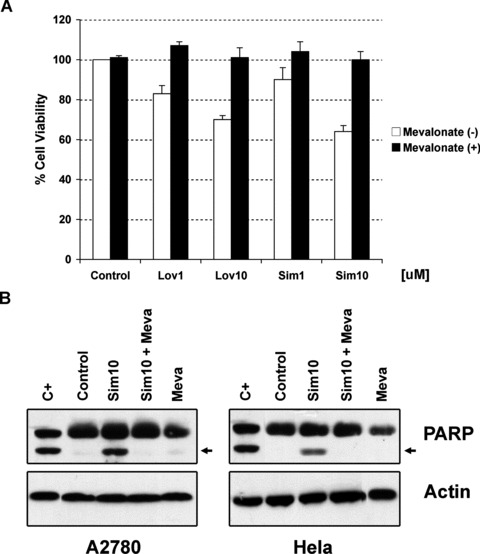
(A) Effects of increasing concentrations of lipophilic statins in cell viability of A2780 cells in presence or absence of mevalonate (100 uM). (B) Detection by immunoblotting of the cleaved form of PARP in A2780 cells under basal conditions (control) and after treatment with simvastatin (sim10), mevalonate (meva) or their combination for 48 hrs. Actin is shown as loading control. C+ stands for positive control.
Fig 9.
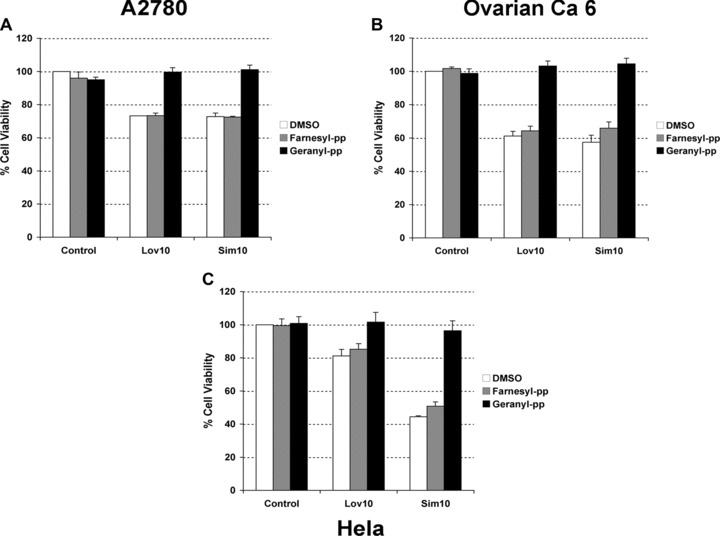
(A) Effect of lipophilic (lovastatin: Lov10; simvastatin: Sim10) statins used at 10 uM for 48 hrs in the presence or absence of vehicle (DMSO), geranylgeranyl (geranyl-pp, 10 uM) or farnesyl (farnesyl-pp, 10 uM) pyrophosphates in two cell lines [A2780 (A) and Hela (C)] and one cancerous primary tissue culture [ca ovary 6 (B)]. Cell viability was measured by MTS assay.
Synergistic effect on cell viability by the combination of statins and chemotherapies in primary tissue cultures of ovarian and uterine cancers
A recent retrospective study in patients with ovarian cancer, which were treated with surgery plus chemotherapy, demonstrated that patients using statins during their treatment experienced better survival than those non-users [24]. This prompted us to investigate the effects of statins used in combination with chemotherapy in gynaecological cancers. We also wanted to compare the effect of statins used alone with current chemotherapies, both in the capacity to kill tumour cells as in the collateral effects on normal tissues. We analysed the effect of lovastatin and simvastatin in the absence or presence of the chemotherapeutic reagents doxorubicin, cisplatin and paclitaxel, in the immortalized non-cancerous cell line HOSE, in A2780 ovarian cancer cells and in three primary tissue cultures, two established from advanced ovarian cancer (from primary tumour and ascites) and the other from a recurrent uterine cancer. As shown in Figure 10A, chemotherapy drastically reduces the cell viability of cells of both normal and cancerous ovarian origin. In contrast, lovastatin and simvastatin demonstrate minimal effects on normal tissue but reduced by 50% cell viability of primary tissue cultures from ovarian cancer. Statins in combination with the different chemotherapeutic reagents, at similar concentrations, resulted in an additive effect on cell viability in both A2780 cells (Fig. 10B) and cancerous primary tissue cultures (data not shown). However, a synergistic effect is observed if same concentrations of statins are combined with lower concentrations of chemotherapies (Fig. 10C). This synergism was confirmed by dose-effect analysis (data not shown). More importantly, when ovarian cancer cells were treated with statins in combination with chemotherapies to which they were resistant an augmentation in cell death effect was observed over a wide range of concentrations (using statins from 0.25 uM to 2.5 uM in a ratio 1:5 with the chemotherapeutic drug).
Fig 10.
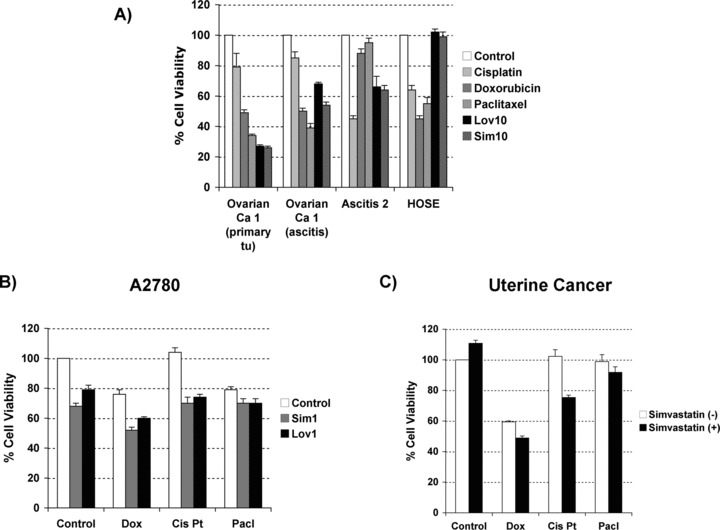
(A) Comparative effects in cell survival between treatment with lipophilic statins (lovastatin: lov; simvastatin: sim; used at 10 uM) and chemotherapies (doxorubicin: dox; cisplatin: cis Pt; and paclitaxel: pacl; all used at 5 uM) in three cancerous primary tissue cultures and an immortalized ovarian cell line (HOSE). (B) Effects in cell viability by the combination of statins (lovastatin: lov, and simvastatin: sim, at 1 uM) and different chemotherapies (at 5 uM) in A2780 cells. (C) Similar experiment in a primary tissue culture from recurrent uterine cancer but using lower concentrations of chemotherapies (at 1 uM). Cell viability was measured by MTS assays upon 48 hrs of treatment.
Discussion
In recent years, the potential benefit of statins to reduce the incidence of a wide range of cancers has been discussed [11, 13, 15, 16, 23]. In addition to its chemopreventive effects, a potential role as therapeutic tool has been postulated for statins, based principally on in vitro studies [21, 26, 36]. Regarding gynaecological cancers, the evidence for either chemoprevention or therapeutic usefulness of statins is scarce. To date, three studies have addressed statin use in ovarian cancer and no information exists in relation to other gynaecological malignancies [24–26]. In the present study, we examined the effects of statins in different cancers originating from the female genital tract and explored the mechanisms explaining the effects of statins. To have a closer approximation to in vivo tumour behaviour, we assessed commercially available cell lines and primary cultures established from fresh cancer samples.
We demonstrated that ovarian, endometrial and cervical cancer cells undergo apoptosis in the presence of lipophilic (i.e. lovastatin and simvastatin) but not hydrophilic statins (i.e. pravastatin). This difference has been previously demonstrated in other systems where lipophilic but not hydrophilic statins induce apoptosis in osteocarcinoma cells [37]. This differential response may explain the contradictory results among different papers, including those in ovarian cancer [23], in which all classes of statins have been included together. The difference between lipophilic and hydrophilic statins may lay on their differing chemical structures, pharmacokinetic profiles and lipid-modifying efficacy. The chemical structures of statins govern their water solubility, which in turn influences their absorption, distribution, metabolism and excretion by different tissues [38]. Lovastatin is derived from fungal metabolites while simvastatin and pravastatin correspond to chemical modifications of lovastatin. Lovastatin and simvastatin are synthesized and administered as inactive lactone pro-drugs, a condition that increases their lipophilicity about 100 times compared with pravastatin. Based in their higher lipophilicity, lovastatin and simvastatin easily pass the cellular membrane through passive diffusion and are metabolized to their active open hydroxy-acid forms by cytochrome P450 3A4 (CYP3A4) [39]. In contrast, pravastatin, which is administered as the active open hydroxy-acid form, does not pass the cellular membrane requiring a transporter [17]. Those transporters not only mediate the entrance but also the exit of pravastatin from the cells. These transporters include among others the organic anion-transporting polypeptide OATP1B1 and the multi-drug resistance-associated protein MRP-2 [40, 41]. In vitro pharmacological studies have shown that the hydrophilic pravastatin has lower inhibitory effect in HMG-CoA reductase activity in most tissues (excluding liver, intestine and kidney) [17]. In our study, we demonstrated the absence of effect of pravastatin on the ovarian cancer cell line A2780 that are known to express MRP-2. This transporter may extrude pravastatin from the cell [42] generating low concentration of the drug inside the cell and thus explaining the absence of cytotoxicity.
The sensitivity to lovastatin and simvastatin differs depending on the tumour type. Here we found that sarcoma cells did not experience cell death after incubation with this statins. Difference in tissue response has been previously reported by others showing that lipophilic statins promote proliferation and invasion of vascular smooth muscle cells [43, 44] while inducing apoptosis in human hepatocytes [45] and osteocarcinoma cells [37]. Besides tumour type, of great interest is to know if the response to lovastatin and simvastatin correlates to tumour stage/grade. Our reduced sample size prevents a correct opinion.
We demonstrated that cell death induced by statins is mediated by activation of caspase cascade (Fig. 5). The complete abrogation of cell death using non-selective caspase inhibitors and the absence of AIF release (data not shown) support that the mechanism is mainly caspase-dependent. The mechanism of statins action is through activation both extrinsic and intrinsic pathways (Fig. 6). The activation of both pathways can be explained by crosstalk between pathways as a decrease in total BID is observed. An interesting result was the increase in protein expression of one of the members of TNF-death receptor family, the death receptor TRAIL-2, upon treatment with 1 uM of both lipophilic statins. This finding agrees with a previous study reporting that both lovastatin and simvastatin increased sensitivity to TRAIL-mediated apoptosis [46]. Our preliminary results in ovarian cancer cells suggest at least an additive effect for the combination of statins and TRAIL (data not published). Current evidence suggests, like our data, that statin would induce apoptosis of ovarian cancer cells through the expression of small GTPase family members, Jun N-terminal kinases (JNK), involved in anchorage-independent growth, and through increased expression of the pro-apoptotic protein Bim [26].
We demonstrated that tissues sensitive to statins correlates to levels of HMG-CoA evaluated by mRNA (Fig. 7). These higher of levels HMG-CoA in sensitive cells could reflect the dependence for survival on enzyme activity and synthesis of metabolites downstream in the cholesterol pathway [33]. The ability of mevalonate supplementation to rescue cancer cells from death further confirms the relevance of HMG-CoA reductase activity for cancer cell survival. Mevalonate is a precursor of isoprenes geranylgeranyl (GPP) and farnesyl (FPP) pyrophosphates, both involved in cell growth and proliferation, through frenylation of small-GTP binding proteins (such as Rho-A and Ras) [4]. We demonstrated that co-incubation with GPP but not FPP completely reverses the loss in viability in ovarian, endometrial and cervical cancer cells, suggesting that geranylgeranylated proteins (GPP proteins) are also important to the survival of these cancer cells (Fig. 9). In addition, the disruption of localization and function of GPP proteins affects crosstalk and activation of other signal transduction pathways involved in growth and survival of cancer cells and exposes the cells to a pro-apoptotic environment [21, 47, 48]. In this sense, changes in cholesterol concentrations within membrane domains lead to recruitment, detachment or retention at these sites of protein kinases required in cell growth, proliferation and invasiveness [48, 49]. For example, in osteoblasts, the mechanism by which lovastatin promotes differentiation involves rapid activation of Ras, which associates with and activates PI3K in the plasma membrane, which in turn regulates Akt and MAPK activities [47]. Recently, Liu et al. have shown that statin-mediated apoptosis in ovarian cancer cells involved an induction in expression of Rac 1 and Cdc42 and subsequently JNK activity [26]. Upon treatment with both lipophilic statins, we observed a decrease in phosphorylated forms of AKT and MAPK supporting a role of these signalling pathways in cell survival and statin-mediated apoptosis (data not shown). Therefore, statins not only affect the prenylation of GTPases but also the activity of protein kinases interacting with them, critical for cell survival. The RhoA activity, whose expression is significantly increased in advanced ovarian carcinomas and has been involved in the pathophysiology of intraperitoneal dissemination, is also reduced upon statin exposure [50].
As described above, our results suggest that statins not only affect GTPases and metastatic potential but also other proteins involved in the apoptotic cascade at different levels such as AKT, ERK, FLIP and death receptors. A major effect is the activation of the intrinsic cascade or mitochondrial pathway, same way used by chemotherapeutic drugs to induce cell death. This effect becomes more relevant taking in account that a major mechanism contributing to resistance is the abnormal content of cholesterol at the mitochondrial membrane [51]. As shown in Figure 10, statins can sensitize ovarian cancer cells to different chemotherapies and thus may allow the use of lower concentrations of chemotherapy with the benefit of less toxicity to normal tissues. These results are consistent with previous work from Holstein et al. who demonstrated by isobologram analysis a synergistic interaction between lovastatin and paclitaxel in leukaemia cells [52].
Our present work demonstrates that in vitro lipophilic statins can be as effective as currently chemotherapeutic drugs, to induce cell death. At lower concentrations, statins can enhance the effect of several of these treatments reducing the risk of experience side effects. These results have prompted other researchers and us to consider the inclusion of lipophilic statins in future clinical trials of gynaecological cancer treatment. Those studies will allow understanding the potential benefits of statins as chemopreventive reagent or as therapeutic tool for gynaecological malignancies.
Acknowledgments
This work was supported by FONDECYT 1080163 (MC) and 1060495 (GO).
References
- 1.Heintz AP, Odicino F, Maisonneuve P, et al. Carcinoma of the ovary. FIGO 6th Annual Report on the Results of Treatment in Gynecological Cancer. Int J Gynaecol Obstet. 2006;95:S161–92. doi: 10.1016/S0020-7292(06)60033-7. [DOI] [PubMed] [Google Scholar]
- 2.Creasman WT, Odicino F, Maisonneuve P, et al. Carcinoma of the corpus uteri. FIGO 6th Annual Report on the Results of Treatment in Gynecological Cancer. Int J Gynaecol Obstet. 2006;95:S105–43. doi: 10.1016/S0020-7292(06)60031-3. [DOI] [PubMed] [Google Scholar]
- 3.Quinn MA, Benedet JL, Odicino F, et al. Carcinoma of the cervix uteri. FIGO 6th Annual Report on the Results of Treatment in Gynecological Cancer. Int J Gynaecol Obstet. 2006;95:S43–103. doi: 10.1016/S0020-7292(06)60030-1. [DOI] [PubMed] [Google Scholar]
- 4.Wang CY, Liu PY, Liao JK. Pleiotropic effects of statin therapy: molecular mechanisms and clinical results. Trends Mol Med. 2008;14:37–44. doi: 10.1016/j.molmed.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beckman JA, Creager MA. The nonlipid effects of statins on endothelial function. Trends Cardiovasc Med. 2006;16:156–62. doi: 10.1016/j.tcm.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 6.Schonbeck U, Libby P. Inflammation, immunity, and HMG-CoA reductase inhibitors: statins as antiinflammatory agents. Circulation. 2004;109:II18–26. doi: 10.1161/01.CIR.0000129505.34151.23. [DOI] [PubMed] [Google Scholar]
- 7.Forrester JS, Libby P. The inflammation hypothesis and its potential relevance to statin therapy. Am J Cardiol. 2007;99:732–8. doi: 10.1016/j.amjcard.2006.09.125. [DOI] [PubMed] [Google Scholar]
- 8.Arnaud C, Mach F. Pleiotropic effects of statins in atherosclerosis: role on endothelial function, inflammation and immunomodulation. Arch Mal Coeur Vaiss. 2005;98:661–6. [PubMed] [Google Scholar]
- 9.Undas A, Celinska-Lowenhoff M, Domagala TB, et al. Early antithrombotic and anti-inflammatory effects of simvastatin versus fenofibrate in patients with hypercholesterolemia. Thromb Haemost. 2005;94:193–9. doi: 10.1160/TH05-01-0067. [DOI] [PubMed] [Google Scholar]
- 10.Graaf MR, Beiderbeck AB, Egberts AC, et al. The risk of cancer in users of statins. J Clin Oncol. 2004;22:2388–94. doi: 10.1200/JCO.2004.02.027. [DOI] [PubMed] [Google Scholar]
- 11.Sassano A, Platanias LC. Statins in tumor suppression. Cancer Lett. 2008;260:11–9. doi: 10.1016/j.canlet.2007.11.036. [DOI] [PubMed] [Google Scholar]
- 12.Friis S, Poulsen AH, Johnsen SP, et al. Cancer risk among statin users: a population-based cohort study. Int J Cancer. 2005;114:643–7. doi: 10.1002/ijc.20758. [DOI] [PubMed] [Google Scholar]
- 13.Blais L, Desgagne A, LeLorier J. 3-Hydroxy-3-methylglutaryl coenzyme A reductase inhibitors and the risk of cancer: a nested case-control study. Arch Intern Med. 2000;160:2363–8. doi: 10.1001/archinte.160.15.2363. [DOI] [PubMed] [Google Scholar]
- 14.Khurana V, Caldito G, Ankem M. Statins might reduce risk of renal cell carcinoma in humans: case-control study of 500,000 veterans. Urology. 2008;71:118–22. doi: 10.1016/j.urology.2007.08.039. [DOI] [PubMed] [Google Scholar]
- 15.Kuoppala J, Lamminpaa A, Pukkala E. Statins and cancer: a systematic review and meta-analysis. Eur J Cancer. 2008;44:2122–32. doi: 10.1016/j.ejca.2008.06.025. [DOI] [PubMed] [Google Scholar]
- 16.Moorman PG, Hamilton RJ. Statins and cancer risk: what do we know and where do we go from here. Epidemiology. 2007;18:194–6. doi: 10.1097/01.ede.0000254699.31405.e2. [DOI] [PubMed] [Google Scholar]
- 17.Hamelin BA, Turgeon J. Hydrophilicity/lipophilicity: relevance for the pharmacology and clinical effects of HMG-CoA reductase inhibitors. Trends Pharmacol Sci. 1998;19:26–37. doi: 10.1016/s0165-6147(97)01147-4. [DOI] [PubMed] [Google Scholar]
- 18.White CM. Pharmacological effects of HMG CoA reductase inhibitors other than lipoprotein modulation. J Clin Pharmacol. 1999;39:111–8. doi: 10.1177/00912709922007642. [DOI] [PubMed] [Google Scholar]
- 19.Pfefferkorn JA, Song Y, Sun KL, et al. Design and synthesis of hepatoselective, pyrrole-based HMG-CoA reductase inhibitors. Bioorg Med Chem Lett. 2007;17:4538–44. doi: 10.1016/j.bmcl.2007.05.096. [DOI] [PubMed] [Google Scholar]
- 20.Karp I, Behlouli H, Lelorier J, et al. Statins and cancer risk. Am J Med. 2008;121:302–9. doi: 10.1016/j.amjmed.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 21.Cafforio P, Dammacco F, Gernone A, et al. Statins activate the mitochondrial pathway of apoptosis in human lymphoblasts and myeloma cells. Carcinogenesis. 2005;26:883–91. doi: 10.1093/carcin/bgi036. [DOI] [PubMed] [Google Scholar]
- 22.Katz MS. Therapy insight: potential of statins for cancer chemoprevention and therapy. Nat Clin Pract Oncol. 2005;2:82–9. doi: 10.1038/ncponc0097. [DOI] [PubMed] [Google Scholar]
- 23.Yu O, Boudreau DM, Buist DS, et al. Statin use and female reproductive organ cancer risk in a large population-based setting. Cancer Causes Control. 2008 doi: 10.1007/s10552-008-9271-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elmore RG, Ioffe Y, Scoles DR, et al. Impact of statin therapy on survival in epithelial ovarian cancer. Gynecol Oncol. 2008;111:102–5. doi: 10.1016/j.ygyno.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 25.Han Z, Wyche JH. Lovastatin induces apoptosis in a metastatic ovarian tumour cell line. Cell Death Differ. 1996;3:223–8. [PubMed] [Google Scholar]
- 26.Liu H, Liang SL, Kumar S, et al. Statins induce apoptosis in ovarian cancer cells through activation of JNK and enhancement of Bim expression. Cancer Chemother Pharmacol. 2009;63:997–1005. doi: 10.1007/s00280-008-0830-7. [DOI] [PubMed] [Google Scholar]
- 27.Sadarangani A, Kato S, Espinoza N, et al. TRAIL mediates apoptosis in cancerous but not normal primary cultured cells of the human reproductive tract. Apoptosis. 2007;12:73–85. doi: 10.1007/s10495-006-0492-z. [DOI] [PubMed] [Google Scholar]
- 28.Kato S, Sadarangani A, Lange S, et al. The oestrogen metabolite 2-methoxyoestradiol alone or in combination with tumour necrosis factor-related apoptosis-inducing ligand mediates apoptosis in cancerous but not healthy cells of the human endometrium. Endocr Relat Cancer. 2007;14:351–68. doi: 10.1677/ERC-07-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–9. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 30.Kato S, Pinto M, Carvajal A, et al. Tissue factor is regulated by epidermal growth factor in normal and malignant human endometrial epithelial cells. Thromb Haemost. 2005;94:444–53. doi: 10.1160/TH05-01-0066. [DOI] [PubMed] [Google Scholar]
- 31.Webb JL. Effects of more than one inhibitor. In: Webb JL, editor. Enzymes and metabolic inhibitors. First ed. New York: Academic Press; 1963. pp. 487–512. [Google Scholar]
- 32.Chou TC, Motzer RJ, Tong Y, et al. Computerized quantitation of synergism and antagonism of taxol, topotecan, and cisplatin against human teratocarcinoma cell growth: a rational approach to clinical protocol design. J Natl Cancer Inst. 1994;86:1517–24. doi: 10.1093/jnci/86.20.1517. [DOI] [PubMed] [Google Scholar]
- 33.Notarnicola M, Messa C, Pricci M, et al. Up-regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in left-sided human colon cancer. Anticancer Res. 2004;24:3837–42. [PubMed] [Google Scholar]
- 34.Li X, Liu L, Tupper JC, et al. Inhibition of protein geranylgeranylation and RhoA/RhoA kinase pathway induces apoptosis in human endothelial cells. J Biol Chem. 2002;277:15309–16. doi: 10.1074/jbc.M201253200. [DOI] [PubMed] [Google Scholar]
- 35.Bustelo XR, Sauzeau V, Berenjeno IM. GTP-binding proteins of the Rho/Rac family: regulation, effectors and functions in vivo. Bioessays. 2007;29:356–70. doi: 10.1002/bies.20558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ukomadu C, Dutta A. p21-dependent inhibition of colon cancer cell growth by mevastatin is independent of inhibition of G1 cyclin-dependent kinases. J Biol Chem. 2003;278:43586–94. doi: 10.1074/jbc.M307194200. [DOI] [PubMed] [Google Scholar]
- 37.Fromigue O, Hay E, Modrowski D, et al. RhoA GTPase inactivation by statins induces osteosarcoma cell apoptosis by inhibiting p42/p44-MAPKs-Bcl-2 signaling independently of BMP-2 and cell differentiation. Cell Death Differ. 2006;13:1845–56. doi: 10.1038/sj.cdd.4401873. [DOI] [PubMed] [Google Scholar]
- 38.Schachter M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: an update. Fundam Clin Pharmacol. 2005;19:117–25. doi: 10.1111/j.1472-8206.2004.00299.x. [DOI] [PubMed] [Google Scholar]
- 39.Shitara Y, Sugiyama Y. Pharmacokinetic and pharmacodynamic alterations of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors: drug-drug interactions and interindividual differences in transporter and metabolic enzyme functions. Pharmacol Ther. 2006;112:71–105. doi: 10.1016/j.pharmthera.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 40.Watanabe T, Kusuhara H, Maeda K, et al. Physiologically based pharmacokinetic modeling to predict transporter-mediated clearance and distribution of pravastatin in humans. J Pharmacol Exp Ther. 2009;328:652–62. doi: 10.1124/jpet.108.146647. [DOI] [PubMed] [Google Scholar]
- 41.Kusuhara H, Sugiyama Y. Role of transporters in the tissue-selective distribution and elimination of drugs: transporters in the liver, small intestine, brain and kidney. J Control Release. 2002;78:43–54. doi: 10.1016/s0168-3659(01)00480-1. [DOI] [PubMed] [Google Scholar]
- 42.Hector S, Nava ME, Clark K, et al. Characterization of a clonal isolate of an oxaliplatin resistant ovarian carcinoma cell line A2780/C10. Cancer Lett. 2007;245:195–204. doi: 10.1016/j.canlet.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 43.Guijarro C, Blanco-Colio LM, Massy ZA, et al. Lipophilic statins induce apoptosis of human vascular smooth muscle cells. Kidney Int Suppl. 1999;71:S88–91. [PubMed] [Google Scholar]
- 44.Turner NA, Midgley L, O’Regan DJ, et al. Comparison of the efficacies of five different statins on inhibition of human saphenous vein smooth muscle cell proliferation and invasion. J Cardiovasc Pharmacol. 2007;50:458–61. doi: 10.1097/FJC.0b013e318123767f. [DOI] [PubMed] [Google Scholar]
- 45.Kubota T, Fujisaki K, Itoh Y, et al. Apoptotic injury in cultured human hepatocytes induced by HMG-CoA reductase inhibitors. Biochem Pharmacol. 2004;67:2175–86. doi: 10.1016/j.bcp.2004.02.037. [DOI] [PubMed] [Google Scholar]
- 46.Jin Z, Dicker DT, El-Deiry WS. Enhanced sensitivity of G1 arrested human cancer cells suggests a novel therapeutic strategy using a combination of simvastatin and TRAIL. Cell Cycle. 2002;1:82–9. [PubMed] [Google Scholar]
- 47.Ghosh-Choudhury N, Mandal CC, Choudhury GG. Statin-induced Ras activation integrates the phosphatidylinositol 3-kinase signal to Akt and MAPK for bone morphogenetic protein-2 expression in osteoblast differentiation. J Biol Chem. 2007;282:4983–93. doi: 10.1074/jbc.M606706200. [DOI] [PubMed] [Google Scholar]
- 48.Calleros L, Lasa M, Rodriguez-Alvarez FJ, et al. RhoA and p38 MAPK mediate apoptosis induced by cellular cholesterol depletion. Apoptosis. 2006;11:1161–73. doi: 10.1007/s10495-006-6980-3. [DOI] [PubMed] [Google Scholar]
- 49.Kofler S, Schlichting C, Jankl S, et al. Dual mode of HMG-CoA reductase inhibition on dendritic cell invasion. Atherosclerosis. 2008;197:105–10. doi: 10.1016/j.atherosclerosis.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 50.Horiuchi A, Kikuchi N, Osada R, et al. Overexpression of RhoA enhances peritoneal dissemination: RhoA suppression with Lovastatin may be useful for ovarian cancer. Cancer Sci. 2008;99:2532–9. doi: 10.1111/j.1349-7006.2008.00977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Montero J, Morales A, Llacuna L, et al. Mitochondrial cholesterol contributes to chemotherapy resistance in hepatocellular carcinoma. Cancer Res. 2008;68:5246–56. doi: 10.1158/0008-5472.CAN-07-6161. [DOI] [PubMed] [Google Scholar]
- 52.Holstein SA, Hohl RJ. Synergistic interaction of lovastatin and paclitaxel in human cancer cells. Mol Cancer Ther. 2001;1:141–9. [PubMed] [Google Scholar]