

Abstract
Background
Antibody-mediated rejection (AMR) after lung transplantation remains enigmatic, and there is no consensus on the characteristic clinical, immunological, and histological features.
Methods
We performed a retrospective, single-center cohort study and identified cases of acute AMR based on the presence of circulating donor-specific human leukocyte antigen (HLA) antibodies (DSA), histologic evidence of acute lung injury, C4d deposition, and clinical allograft dysfunction.
Results
We identified 21 recipients with acute AMR based on the above criteria. AMR occurred a median 258 days after transplantation; 7 recipients developed AMR within 45 days of transplantation. All patients had clinical allograft dysfunction, DSA, histology of acute lung injury, and capillary endothelial C4d deposition. Fifteen recipients improved clinically and survived to hospital discharge, but 6 died of refractory AMR. One survivor had BOS at the time of diagnosis of AMR; 13 of the 14 remaining survivors developed chronic lung allograft dysfunction (CLAD) during follow-up. Overall, 15 recipients died during the study period, and the median survival after the diagnosis of AMR was 593 days.
Conclusions
Acute AMR can be a fulminant form of lung rejection, and survivors are at increased risk of developing CLAD. The constellation of acute lung injury, DSA, and capillary endothelial C4d deposition is compelling for acute AMR in recipients with allograft dysfunction. This clinicopathological definition requires validation in a multicenter cohort but may serve as a foundation for future studies to further characterize AMR.
Introduction
Lung transplantation is the ultimate treatment for patients with end-stage lung disease. However, long-term outcomes remain disappointing; the median survival after transplantation is approximately 5.5 years (1, 2). Graft failure as a result of chronic lung allograft dysfunction (CLAD) and bronchiolitis obliterans syndrome (BOS) accounts for over 40% of deaths beyond the first year after transplantation (2). Traditionally, organ rejection has been regarded as a T-cell mediated process. Indeed, standard immunosuppressive therapy, targeting T-cell proliferation and function, has made organ transplantation a clinical reality (3, 4). However, a potential role for antibodies in graft rejection has long been suspected because antibodies to human leukocyte antigens (HLA) are often detected in patients with rejection (5-9). Furthermore, HLA antibodies are known to cause hyperacute, acute, and chronic antibody-mediated rejection (AMR) after kidney transplantation (10).
AMR after lung transplantation remains enigmatic (11). The diagnostic criteria for lung allograft rejection were revised in 2007 classifying four forms of rejection: acute rejection, lymphocytic bronchiolitis, obliterative bronchiolitis, and chronic vascular rejection (12). However, there was no consensus on the histologic and immunologic features of AMR. Nevertheless, although hyperacute rejection, caused by preformed donor-specific antibodies (DSA), is rare, it is a widely accepted form of lung rejection (13-15). This demonstrates that antibodies can cause fulminant lung allograft failure. In addition, multiple reports from different centers have described clinicopathological findings consistent with acute AMR (16-20).
The National Conference to Assess Antibody-Mediated Rejection in Solid Organ Transplantation proposed a general paradigm of humoral responses applicable to all organs to facilitate further study (21). According to these guidelines, AMR is defined by circulating DSA, C4d deposition, tissue pathology, and clinical allograft dysfunction. The International Society for Heart and Lung Transplantation (ISHLT) Pathology Council recently published a summary statement on the pathology of AMR concluding that pulmonary AMR requires: clinical allograft dysfunction, circulating DSA, and pathologic findings (22). In this study, we review a series of cases that fulfill the criteria put forth by both the National Conference and the ISHLT Pathology Council and propose a clinicopathological definition for acute AMR after lung transplantation.
Methods
Study design and case identification
We conducted a retrospective cohort study to characterize the clinicopathological features of acute AMR after lung transplantation. Between 1/1/2004 and 6/30/2012, 484 adults underwent 501 lung transplant procedures at Barnes-Jewish Hospital; 17 patients underwent re-transplantation. We identified 86 recipients who developed acute allograft dysfunction of unclear etiology during this time period (Figure 1). These cases were characterized by dyspnea, hypoxemia, and pulmonary infiltrates without clinical evidence of infection. Although acute cellular rejection was present in 12 of the 86 cases, the severity of rejection (ISHLT grade A1 or A2) did not explain the clinical findings. Among the 86 cases, we identified 21 that fulfilled all criteria for AMR proposed by the National Conference (20) and the Pathology Council (22) including allograft dysfunction, DSA, tissue pathology, and C4d deposition. Of the 65 cases that were excluded, 26 had concomitant DSA, 24 had no DSA, and 15 were not tested. Of the 26 recipients who had DSA, 17 did not have C4d deposition, 3 did not have a lung biopsy performed, and 6 did not have C4d staining performed (Figure 1). Our institutional Human Research Protection Office approved this study as part of our Lung Transplant Registry protocol.
Figure 1. Study design and case selection. AMR, antibody-mediated rejection; DSA, donor-specific anti-human leukocyte antigen antibody.

Clinical management
We have previously detailed our general management protocol (23). Briefly, all patients are screened for preformed HLA antibodies using the LABScreen® Single Antigen assay before transplantation, and donor lungs are accepted only if a virtual crossmatch with all previously identified antibodies is compatible; a direct crossmatch is performed at the time of transplantation. After transplantation, recipients are screened for DSA using the LABScreen® Single Antigen assay at the time of surveillance bronchoscopy and if they develop signs of allograft dysfunction, including dyspnea, pulmonary infiltrates, hypoxemia, or a decrement in lung function. Our center's HLA lab defines DSA positivity as donor-specific reactivity with a mean fluorescence intensity (MFI) ≥ 2000. In general, we evaluate allograft dysfunction with chest imaging, blood assay for cytomegalovirus (CMV) polymerase chain reaction (PCR), nasopharyngeal swab for direct fluorescent antibody (DFA) staining for respiratory viruses, and bronchoscopy with transbronchial lung biopsies and bronchoalveolar lavage (BAL) for microbiological studies.
Bronchoscopy and pathology
In general, we obtain at least 10 transbronchial biopsies during each bronchoscopy to submit at least 5 specimens of alveolated parenchyma for histologic interpretation. Three levels are cut from the paraffin block, and sections are stained with hematoxylin and eosin (H&E). A pathologist interpreted the histology according to ISHLT guidelines (12). When there was clinical concern for AMR, C4d evaluation was performed using immunohistochemistry. This technique tends to show less background staining compared with immunofluorescence. A rigorous comparison of these two techniques has not been performed. However, the diminished background staining observed using immunohistochemistry creates a more readily interpretable stain. C4d was considered positive when staining in a circumferential capillary sub-endothelial pattern was present.
Statistical analysis
We characterized patient demographics using descriptive statistics. Continuous variables are represented as mean±standard deviation. We evaluated freedom from CLAD and survival after the diagnosis of AMR using the Kaplan-Meier method and groups were compared using the log-rank test. We conducted the statistical analysis using GraphPad Prism 5.0f for Mac OSX (GraphPad Software, San Diego,CA).
Results
During the study period, we identified 21 cases that fulfilled all criteria for AMR proposed by the National Conference and the ISHLT Pathology Council. Follow-up was complete through 12/31/2012. Patients' demographics were typical of a cohort of adult lung transplant recipients at our center (Table 1). The control and AMR groups were generally similar, but the AMR group was younger (Table 1). All 21 recipients had negative virtual and direct crossmatches at the time of transplantation. All recipients were treated with induction and triple-drug maintenance immunosuppression (Table 2).
Table 1. Patient demographics.
| Demographics | Controls, n = 480 N (%) | AMR, n = 21 N (%) | p | |
|---|---|---|---|---|
| Gender | Female | 208 (43%) | 12 (57%) | 0.26 |
| Male | 272 (57%) | 9 (43%) | ||
| Age at time of transplant | 51.4 ± 14.1 years | 42.0 ± 16.2 years | <0.01 | |
| Type of Transplant | Bilateral | 458 (95%) | 20 (95%) | 0.94 |
| Single | 19 (4%) | 1 (5%) | ||
| Heart-Lung | 3 (1%) | 0 (0%) | ||
| Diagnosis | COPD | 135 (28%) | 6 (29%) | 0.21 |
| A1AT Deficiency | 28 (6%) | 0 (0%) | ||
| Pulmonary fibrosis | 161 (34%) | 7 (33%) | ||
| CF | 89 (19%) | 5 (24%) | ||
| BOS / Retransplant | 15 (3%) | 2 (10%) | ||
| PAH | 9 (2%) | 0 (0%) | ||
| Other | 43 (9%) | 1 (5%) | ||
| CMV Status* | D+ / R- | 128 (27%) | 9 (43%) | 0.26 |
| D+ or - / R+ | 289 (60%) | 10 (48%) | ||
| D- / R- | 63 (13%) | 2 (10%) | ||
| Allosensitized prior to transplant | 136 (28%) | 7 (33%) | 0.63 |
A1AT: alpha-1-antitrypsin, BOS: bronchiolitis obliterans syndrome, CF: cystic fibrosis, COPD: chronic obstructive pulmonary disease, PAH: pulmonary arterial hypertension
D+ / R-: donor positive/recipient negative, D+ or- / R+: donor positive or negative, recipient negative, D- / R-: donor negative/recipient negative
Table 2.
Induction and maintenance immunosuppression at the time of the diagnosis of acute antibody-mediated rejection.
| N (% of cohort) | |
|---|---|
| Induction immunosuppression | |
| Basiliximab | 14 (67%) |
| Equine anti-thymocyte globulin | 7 (33%) |
| Maintenance Immunosuppression | |
| Tacrolimus, mycophenolate mofetil, prednisone | 12 (57%) |
| Tacrolimus, azathioprine, prednisone | 4 (19%) |
| Cyclosporine, mycophenolate mofetil, prednisone | 1 (5%) |
| Cyclosporine, azathioprine, prednisone | 2 (10%) |
| Tacrolimus, sirolimus, prednisone | 2 (10%) |
Recipients developed AMR a median 258 days (mean 364±402) after transplantation. Fourteen recipients presented in the first year after transplantation and 7 presented beyond the first year. All recipients were hospitalized for breathlessness and hypoxemia at presentation. Chest radiographs demonstrated diffuse pulmonary infiltrates in all cases, and 14 of the 21 recipients required invasive mechanical ventilation.
In all cases, we established the diagnosis of acute AMR based on the combination of histologic findings, presence of DSA, and exclusion of other causes of graft dysfunction. Acute lung injury (ALI) was the predominant pathologic finding on H&E. This varied in severity from a neutrophilic pneumonitis to diffuse alveolar damage (DAD) with widespread hyaline membrane formation (Figure 2A). Frank capillary injury with fibrinoid necrosis and peri-capillary neutrophilic infiltration was seen in 11 cases (Figure 2B). This finding was conspicuous in some cases, but the presence of a focal infiltrate of neutrophils within alveolar septa was characteristic (Figure 2C). Notably, capillary injury was not detected in 10 cases; acute pneumonitis with alveolar damage and fibrin exudates were the characteristic findings in those cases. While these findings are non-specific, the diagnosis of AMR in cases where capillary injury was not seen was supported by capillary endothelial C4d deposition and circulating DSA. In all 21 cases, C4d deposition was detected in a circumferential pattern in the capillary endothelium (Figure 2D). Four recipients had concomitant acute cellular rejection (2 had ISHLT grade A1, 1 had A2, and 1 had A3), 1 recipient had concomitant obliterative bronchiolitis; there was no evidence of cellular rejection in the remaining 16 cases.
Figure 2.
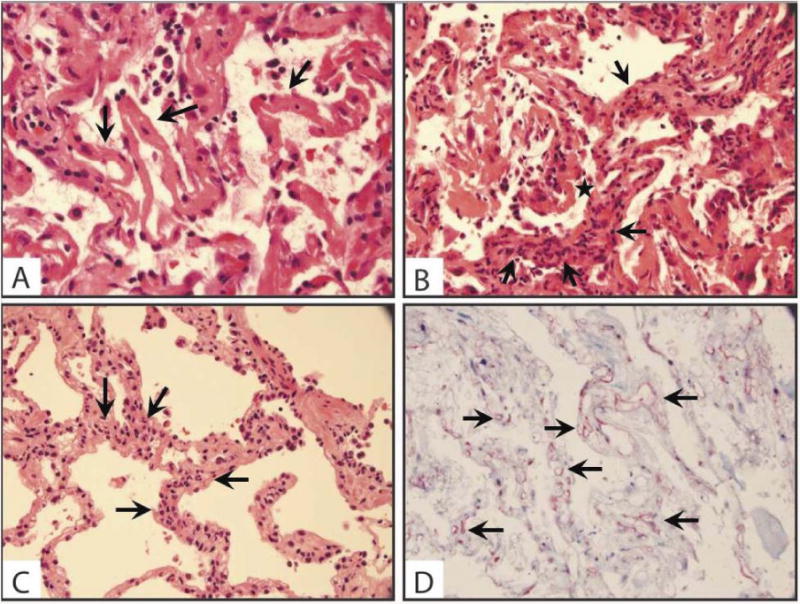
Histopathological findings in AMR. (A) Diffuse alveolar damage with hyaline membrane (arrows) formation (original magnification 400×) (B) Capillary injury with capillaritis (arrows) and hyaline membrane (star) formation (original magnification 400×) (C) Neutrophilic infiltrate (arrows) of the alveolar septa (original magnification 400×) (D) Circumferential C4d staining (arrows) of the capillary endothelium (original magnification 400×)
All patients had DSA at the time of AMR diagnosis; 2 to class I HLA only, 15 to class II HLA only, and 4 to both class I and class II HLA. Of note, 17 (81%) recipients had DSA to the DQ locus and 8 (38%) to the DR locus. In contrast, 3 (14%) had DSA to the A locus, 2 (10%) to the B locus, and 2 (10%) to the C locus. At the time of AMR diagnosis, the mean MFI of the immunodominant DSA was 5757±3062 (median: 4729, range: 2188-14232). There was no significant difference between the mean MFI of class I DSA (5583±1646) and to class II DSA (5100±2677) (p=0.53). We evaluated 18 patients' serum samples obtained at the time of AMR diagnosis with the C1q assay retrospectively; samples were not available for the remaining 3 patients. The C1q assay was positive in 12 patients (mean MFI=24560±5972), negative in 3 patients, and had high background reactivity such that the results were uninterpretable in 3 patients.
Two recipients had positive BAL fluid cultures for bacterial organisms (Pseudomonas aeruginosa in both); the remaining recipients had negative BAL fluid bacterial cultures. Three recipients had positive BAL fluid cultures for Candida species; the remaining patients had negative fungal cultures. All recipients had negative BAL fluid DFA stains for community-acquired respiratory viruses and negative BAL fluid cultures for CMV. Lastly, all recipients had negative CMV blood PCR assays when AMR was diagnosed.
Our approach to treatment evolved over time and the regimen was individualized based on severity of illness, clinical course, and response to therapy (Table 3). With treatment, DSA became undetectable or cleared in 9 (43%) patients within a mean 83±77 days, but 12 (57%) had persistent DSA. Five of the 15 (33%) recipients who had DSA to class II antigens cleared the DSA compared to 2 of the 2 (100%) who had DSA to class I antigens and 2 of the 4 (50%) who had DSA to class I and class II antigens (p=0.19). Of note, 5 of the 17 (29%) recipients who had DSA to the HLA DQ locus cleared the DSA compared to 4 of 4 (100%) recipients who had DSA to a non-DQ locus (p=0.01). Fifteen (71%) recipients improved clinically with resolution of breathlessness, hypoxemia, and pulmonary infiltrates, and were discharged from the hospital. Eight of the 14 (57%) recipients who required invasive mechanical ventilation were successfully liberated from ventilatory support and were discharged home. Six (29%) recipients died of refractory AMR during the index hospitalization, and all had C1q-positive DSA. The remaining 6 patients with C1q-positive DSA improved and were discharged from the hospital. DSA clearance was associated with clinical improvement; all 9 recipients who cleared the DSA improved clinically, while 6 of the 12 (50%) who had persistent DSA improved and 6 (50%) died of refractory AMR (p=0.012). There was no significant association between the time of onset of AMR after transplantation and clinical improvement; 11 of the 14 (79%) recipients who developed AMR in the first year after transplantation improved clinically compared to 4 of the 7 (57%) recipients who developed AMR beyond the first year (p=0.31). Eleven of the 17 (65%) recipients who had DSA to the DQ locus improved compared to 4 of the 4 (100%) recipients who had DSA to a non-DQ locus (p=0.16).
Table 3.
Treatment regimens administered for acute antibody mediated rejection.
| Patient | ATG | IVIG dose (number of doses) | Rituximab | Plasma Exchange number of treatments | Bortezomib dose (number of doses) | Eculizumab | Survived to discharge |
|---|---|---|---|---|---|---|---|
| 1 | Yes* | None | - | Not done | - | - | Yes |
| 2 | - | 1 g/kg (1) | 375 mg/m2 | 12 | - | - | Yes |
| 3 | - | 1 g/kg (1) | 375 mg/m 2 | Not done | - | - | Yes |
| 4 | - | 1 g/kg (1) | 375 mg/m2 | Not done | - | - | Yes |
| 5 | - | 0.5 g/kg (2) | 375 mg/m2 | 10 | - | - | No |
| 6 | - | 0.5 g/kg (1) | 375 mg/m2 | Not done | - | - | No |
| 7 | - | 0.5 g/kg (1) | 375 mg/m2 | 5 | - | - | Yes |
| 8 | - | 0.5 g/kg (2) | 375 mg/m2 | Not done | - | - | Yes |
| 9 | - | 1 g/kg (1) | 375 mg/m2 | Not done | - | - | Yes |
| 10 | - | 0.5 g/kg (2) | 375 mg/m2 | Not done | - | - | Yes |
| 11 | - | 0.5 g/kg (1) | - | 3 | - | - | Yes |
| 12 | - | 0.5 g/kg (1) | 375 mg/m 2 | Not done | - | - | Yes |
| 13 | - | 0.5 g/kg (1) | 375 mg/m2 | Not done | - | - | Yes |
| 14 | - | 1 g/kg (1) | 375 mg/m2 | Not done | - | - | No |
| 15 | - | 0.5 g/kg (2) | - | Not done | - | - | Yes |
| 16 | - | 0.5 g/kg (3) | 375 mg/m2 | 5 | - | - | No |
| 17 | - | 0.5 g/kg (1) | 375 mg/m2 | Not done | - | - | Yes |
| 18 | - | 1 g/kg (2) | 375 mg/m2 | 5 | 1.3 mg/m2(8) | - | Yes |
| 19 | - | 0.5 g/kg (1) | 375 mg/m2 | 1 | 1.3 mg/m2(1) | - | No |
| 20 | - | 0.5 g/kg(1) 1 g/kg (2) | 375 mg/m2 | Not done | - | - | Yes |
| 21 | - | 1 g/kg (3) | 375 mg/m2 | 5 | 1.3 mg/m2(4) | Yes** | No |
ATG: antithymocyte globulin, IVIG: intravenous immune globulin
(-): Not given
ATG dosing: 1 mg/kg/day for 5 days
Eculizumab dosing: 1200 mg IV Day 1, 900 mg IV Day 8, 900 mg IV Day 15
Among the 15 recipients who improved and were discharged from the hospital, 1 had pre-existing BOS and was excluded from the analysis of freedom from CLAD. Thirteen of the remaining 14 (93%) recipients developed CLAD during the study follow-up, a mean 389±137 days (median 114 days) after the diagnosis of AMR (Figure 3A). In fact, 10 of the 14 (71%) recipients developed CLAD within one year of the diagnosis of AMR. There was no significant difference in the incidence of CLAD after the diagnosis of AMR between those who cleared and who had persistent DSA (Figure 3B, log rank p=0.27). Furthermore, survival after the diagnosis of AMR was poor. Fifteen of the 21 (71%) recipients died during the study period. 10 died within one year of the diagnosis of AMR (Figure 4A); 6 patients died of refractory AMR and 9 of CLAD. However, recipients who cleared the DSA had a significantly better survival after the diagnosis of AMR than those with persistent DSA (Figure 4B, log rank p<0.0001); 3 of the 9 (33%) recipients who cleared the DSA died during the study period compared to all 12 (100%) recipients who had persistent DSA.
Figure 3.
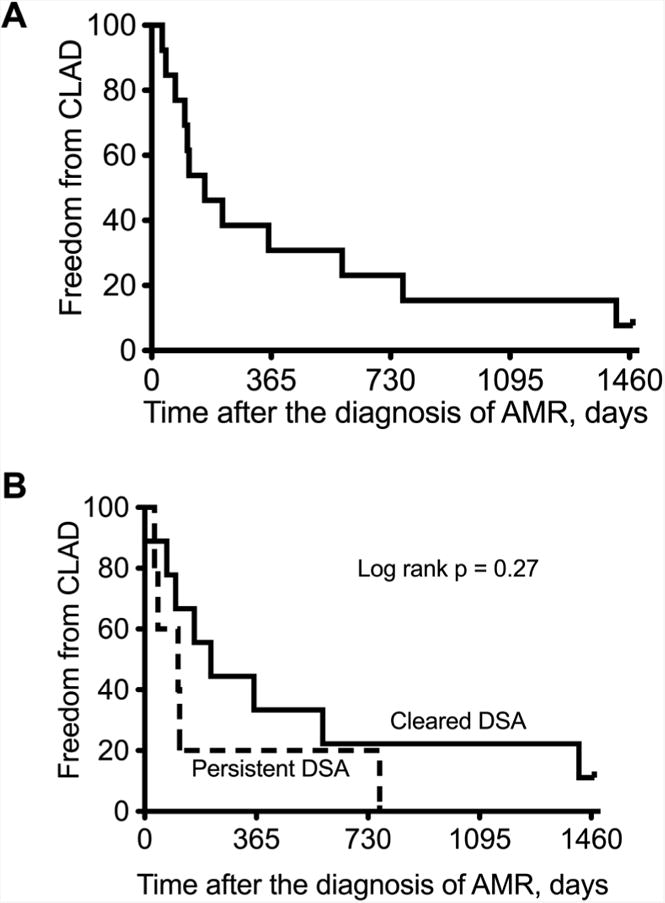
Kaplan-Meier curves of freedom from chronic lung allograft dysfunction (CLAD) after diagnosis of antibody mediated rejection (AMR): (A) All patients (B) Comparing those who cleared donor specific antibodies (DSA) versus those with persistent DSA.
Figure 4.
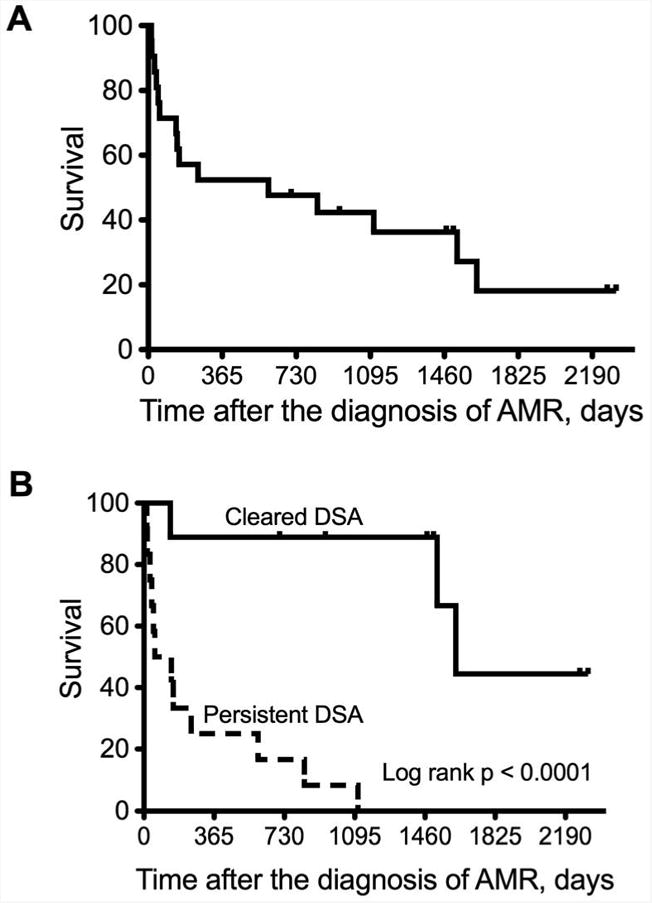
Kaplan-Meier curves of survival after diagnosis of antibody-mediated rejection (AMR): (A) All patients (B) Comparing those who cleared donor specific antibodies (DSA) versus those with persistent DSA.
The 21 recipients who developed AMR had a significantly better survival than the 26 who developed allograft dysfunction and DSA but did not have C4d deposition (Figure 5, log rank p=0.0001). However, we note that it is not clear that the 26 patients with allograft dysfunction and DSA all had the same syndrome and believe that this is a heterogeneous group with different causes of allograft dysfunction. Furthermore, the diagnosis of AMR was not suspected prospectively in many of these patients and antibody-directed therapy was not implemented.
Figure 5.
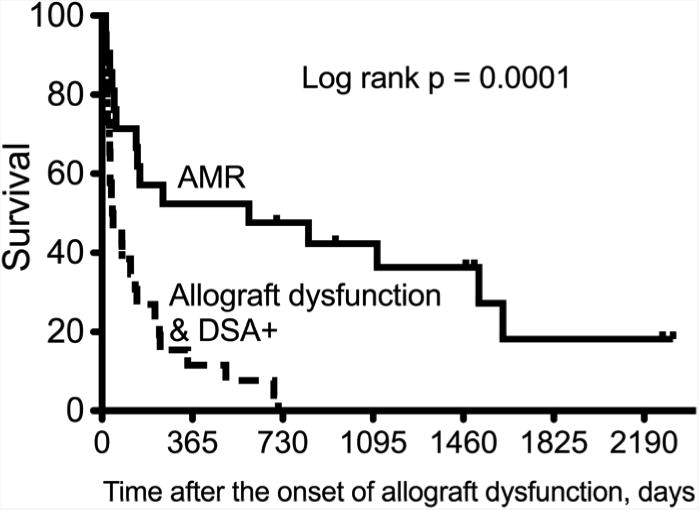
Kaplan-Meier curves of survival comparing those with antibody mediated rejection (AMR) by all four criteria versus those with allograft dysfunction and donor specific antibodies (DSA) without C4d deposition. Note that it is not clear that the 26 patients without C4 deposition all had the same syndrome, but likely represents a heterogeneous group with different causes of allograft dysfunction.
Discussion
In this study, we characterize the clinicopathological findings and outcomes of 21 cases of acute AMR after lung transplantation. These cases met criteria for AMR proposed by the National Conference and the ISHLT Pathology Council. Importantly, these cases illustrate that antibodies can directly injure the lung allograft and acute AMR may be a reversible cause of allograft failure. However, a high index of suspicion is necessary to establish the diagnosis and requires a multidisciplinary approach; testing for HLA antibodies is critical to making the diagnosis. Furthermore, the histologic findings of ALI or DAD are non-specific. Capillary endothelial C4d deposition underscores the role of complement in mediating the ALI in those cases and confirms the diagnosis of AMR. Additionally, C1q positivity in the majority of cases corroborates the paradigm of complement activation by DSA resulting in ALI. However, C4d immunostaining of lung tissue has been fraught with practical problems and inconsistent results (24, 25). A recent study of the pathology associated with DSA found no correlation between C4d or C3d deposition and DSA or morphologic features of AMR (25). Additionally, cases of AMR without C4d deposition are increasingly recognized in kidney transplantation (26). Nonetheless, we included C4d deposition as a criterion for the diagnosis in this cohort to develop a stringent definition of acute AMR in lung transplantation, recognizing that C4d deposition may be inconspicuous or absent in some cases that are otherwise consistent with AMR. A strict definition is likely more specific although some cases may be missed. However, we believe that specificity is critical when describing a new syndrome and propose that the definition can be refined over time by adding modifiers such as “suspicious for AMR” based on specific features. Nevertheless, this definition needs to be validated in a multicenter study.
The majority of patients in this cohort had class II DSA, often directed at the DQ locus. This is similar to our previous report of lung recipients who developed DSA without allograft dysfunction and reports of AMR and DSA associated with poor allograft survival after kidney transplantation (23, 27-30). However, reasons for the predominance of class II DSA in this clinical setting are unknown, and we cannot glean any insights into the underlying immunobiology from our data. However, proinflammatory cytokines can induce pulmonary endothelial cells to upregulate and express class II HLA molecules (31). Indeed, endothelial cell expression of class II HLA molecules was demonstrated in over 60% of lung allograft biopsies in a previous study (32). These findings are consistent with the paradigm that endothelial cells are the initial focal point of antibody-mediated injury.
There are inherent limitations to this study. Although we identified 21 cases among 484 recipients over an 8-year period, the incidence of acute AMR may be higher. It is likely that less severe cases were unrecognized, and that some cases of unexplained allograft dysfunction that were excluded may have represented AMR. However, we sought to characterize a convincing series of cases to develop a preliminary definition that may serve as a foundation for future studies. Nonetheless, it is possible that some cases of AMR may be subclinical as most cases of acute cellular rejection are. Indeed, recipients with DSA have been found to have pathology suspicious for AMR on routine surveillance biopsies (25). This suggests that subclinical AMR may explain the association between DSA and CLAD development. In addition, there may be different syndromes of AMR with unique features, natural histories, and responses to treatment. An additional limitation of this study is that C4d deposition may be an impractical component of the definition as the interpretation of C4d staining in the lung has been difficult. Furthermore, the inter-reader reliability of C4d staining has not been formally evaluated. Lastly, we used an arbitrary MFI threshold to define DSA positivity. However, there is currently no widely accepted definition of DSA positivity and different labs use different cutoffs. Future studies will be necessary to address these limitations.
Despite the initial clinical improvement in many patients, long-term outcomes were poor because of a high incidence of CLAD development after the initial recovery. We used various antibody-directed regimens but cannot make firm conclusions about their relative efficacy because the sample sizes were small and the decision to use a particular treatment was individualized and based on the clinical course. Clearly, additional studies are necessary to identify the optimal treatment, but an accepted and validated definition of acute AMR is necessary before treatment trials can be conducted. In summary, we propose that acute AMR after lung transplantation can be defined by the following criteria: allograft dysfunction, DSA, pathology of acute lung injury, and capillary endothelial C4d deposition. A multicenter study is necessary to refine and validate this definition.
Acknowledgments
Disclosure statement: This study was supported in part by HL092514 (TM, RRH), HL105412 (JPG, RDY, TM, RRH), and The BJC Foundation (TM).
Footnotes
None of the authors has a financial relationship with a commercial entity that has an interest in the subject of the presented manuscript or other conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Thabut G, Christie JD, Kremers WK, Fournier M, Halpern SD. Survival differences following lung transplantation among US transplant centers. JAMA. 2010;304:53–60. doi: 10.1001/jama.2010.885. [DOI] [PubMed] [Google Scholar]
- 2.Christie JD, Edwards LB, Kucheryavaya AY, et al. The Registry of the International Society for Heart and Lung Transplantation: 29th adult lung and heart-lung transplant report-2012. J Heart Lung Transplant. 2012;31:1073–86. doi: 10.1016/j.healun.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 3.Bhorade SM, Stern E. Immunosuppression for lung transplantation. Proc Am Thorac Soc. 2009;6:47–53. doi: 10.1513/pats.200808-096GO. [DOI] [PubMed] [Google Scholar]
- 4.Durrbach A, Francois H, Beaudreuil S, Jacquet A, Charpentier B. Advances in immunosuppression for renal transplantation. Nature reviews Nephrology. 2010;6:160–7. doi: 10.1038/nrneph.2009.233. [DOI] [PubMed] [Google Scholar]
- 5.Jeannet M, Pinn VW, Flax MH, Winn HJ, Russell PS. Humoral antibodies in renal allotransplantation in man. N Engl J Med. 1970;282:111–7. doi: 10.1056/NEJM197001152820301. [DOI] [PubMed] [Google Scholar]
- 6.Jaramillo A, Smith MA, Phelan D, et al. Development of ELISA-detected anti-HLA antibodies precedes the development of bronchiolitis obliterans syndrome and correlates with progressive decline in pulmonary function after lung transplantation. Transplantation. 1999;67:1155–61. doi: 10.1097/00007890-199904270-00012. [DOI] [PubMed] [Google Scholar]
- 7.McKenna RM, Takemoto SK, Terasaki PI. Anti-HLA antibodies after solid organ transplantation. Transplantation. 2000;69:319–26. doi: 10.1097/00007890-200002150-00001. [DOI] [PubMed] [Google Scholar]
- 8.Girnita AL, McCurry KR, Iacono AT, et al. HLA-specific antibodies are associated with high-grade and persistent-recurrent lung allograft acute rejection. J Heart Lung Transplant. 2004;23:1135–41. doi: 10.1016/j.healun.2003.08.030. [DOI] [PubMed] [Google Scholar]
- 9.Girnita AL, Duquesnoy R, Yousem SA, et al. HLA-specific antibodies are risk factors for lymphocytic bronchiolitis and chronic lung allograft dysfunction. Am J Transplant. 2005;5:131–8. doi: 10.1111/j.1600-6143.2004.00650.x. [DOI] [PubMed] [Google Scholar]
- 10.Moll S, Pascual M. Humoral rejection of organ allografts. Am J Transplant. 2005;5:2611–8. doi: 10.1111/j.1600-6143.2005.01086.x. [DOI] [PubMed] [Google Scholar]
- 11.Glanville AR. Antibody-mediated rejection in lung transplantation: myth or reality? J Heart Lung Transplant. 2010;29:395–400. doi: 10.1016/j.healun.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 12.Stewart S, Fishbein MC, Snell GI, et al. Revision of the 1996 working formulation for the standardization of nomenclature in the diagnosis of lung rejection. J Heart Lung Transplant. 2007;26:1229–42. doi: 10.1016/j.healun.2007.10.017. [DOI] [PubMed] [Google Scholar]
- 13.Frost AE, Jammal CT, Cagle PT. Hyperacute rejection following lung transplantation. Chest. 1996;110:559–62. doi: 10.1378/chest.110.2.559. [DOI] [PubMed] [Google Scholar]
- 14.Bittner HB, Dunitz J, Hertz M, Bolman MR, 3rd, Park SJ. Hyperacute rejection in single lung transplantation--case report of successful management by means of plasmapheresis and antithymocyte globulin treatment. Transplantation. 2001;71:649–51. doi: 10.1097/00007890-200103150-00012. [DOI] [PubMed] [Google Scholar]
- 15.Masson E, Stern M, Chabod J, et al. Hyperacute rejection after lung transplantation caused by undetected low-titer anti-HLA antibodies. J Heart Lung Transplant. 2007;26:642–5. doi: 10.1016/j.healun.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 16.Astor TL, Weill D, Cool C, Teitelbaum I, Schwarz MI, Zamora MR. Pulmonary capillaritis in lung transplant recipients: treatment and effect on allograft function. J Heart Lung Transplant. 2005;24:2091–7. doi: 10.1016/j.healun.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 17.Girnita AL, McCurry KR, Yousem SA, Pilewski J, Zeevi A. Antibody-mediated rejection in lung transplantation: case reports. Clin Transpl. 2006:508–10. [PubMed] [Google Scholar]
- 18.Astor TL, Galantowicz M, Phillips A, Palafox J, Baker P. Pulmonary capillaritis as a manifestation of acute humoral allograft rejection following infant lung transplantation. Am J Transplant. 2009;9:409–12. doi: 10.1111/j.1600-6143.2008.02467.x. [DOI] [PubMed] [Google Scholar]
- 19.Morrell MR, Patterson GA, Trulock EP, Hachem RR. Acute antibody-mediated rejection after lung transplantation. J Heart Lung Transplant. 2009;28:96–100. doi: 10.1016/j.healun.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 20.Daoud AH, Betensley AD. Diagnosis and treatment of antibody mediated rejection in lung transplantation: A retrospective case series. Transpl Immunol. 2013;28:1–5. doi: 10.1016/j.trim.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 21.Takemoto SK, Zeevi A, Feng S, et al. National conference to assess antibody-mediated rejection in solid organ transplantation. Am J Transplant. 2004;4:1033–41. doi: 10.1111/j.1600-6143.2004.00500.x. [DOI] [PubMed] [Google Scholar]
- 22.Berry G, Burke M, Andersen C, et al. Pathology of pulmonary antibody-mediated rejection: 2012 update from the Pathology Council of the ISHLT. J Heart Lung Transplant. 2013;32:14–21. doi: 10.1016/j.healun.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 23.Hachem RR, Yusen RD, Meyers BF, et al. Anti-human leukocyte antigen antibodies and preemptive antibody-directed therapy after lung transplantation. J Heart Lung Transplant. 2010;29:973–80. doi: 10.1016/j.healun.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yousem SA, Zeevi A. The histopathology of lung allograft dysfunction associated with the development of donor-specific HLA alloantibodies. Am J Surg Pathol. 2012;36:987–92. doi: 10.1097/PAS.0b013e31825197ae. [DOI] [PubMed] [Google Scholar]
- 25.Denicola MM, Weigt SS, Belperio JA, Reed EF, Ross DJ, Wallace WD. Pathologic findings in lung allografts with anti-HLA antibodies. J Heart Lung Transplant. 2013;32:326–32. doi: 10.1016/j.healun.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sis B, Jhangri GS, Bunnag S, Allanach K, Kaplan B, Halloran PF. Endothelial gene expression in kidney transplants with alloantibody indicates antibody-mediated damage despite lack of C4d staining. Am J Transplant. 2009;9:2312–23. doi: 10.1111/j.1600-6143.2009.02761.x. [DOI] [PubMed] [Google Scholar]
- 27.Campos EF, Tedesco-Silva H, Machado PG, Franco M, Medina-Pestana JO, Gerbase-DeLima M. Post-transplant anti-HLA class II antibodies as risk factor for late kidney allograft failure. Am J Transplant. 2006;6:2316–20. doi: 10.1111/j.1600-6143.2006.01503.x. [DOI] [PubMed] [Google Scholar]
- 28.Langan LL, Park LP, Hughes TL, et al. Post-transplant HLA class II antibodies and high soluble CD30 levels are independently associated with poor kidney graft survival. Am J Transplant. 2007;7:847–56. doi: 10.1111/j.1600-6143.2006.01691.x. [DOI] [PubMed] [Google Scholar]
- 29.Flechner SM, Fatica R, Askar M, et al. The role of proteasome inhibition with bortezomib in the treatment of antibody-mediated rejection after kidney-only or kidney-combined organ transplantation. Transplantation. 2010;90:1486–92. doi: 10.1097/TP.0b013e3181fdd9b0. [DOI] [PubMed] [Google Scholar]
- 30.DeVos JM, Gaber AO, Knight RJ, et al. Donor-specific HLA-DQ antibodies may contribute to poor graft outcome after renal transplantation. Kidney Int. 2012;82:598–604. doi: 10.1038/ki.2012.190. [DOI] [PubMed] [Google Scholar]
- 31.Cunningham AC, Zhang JG, Moy JV, Ali S, Kirby JA. A comparison of the antigen-presenting capabilities of class II MHC-expressing human lung epithelial and endothelial cells. Immunology. 1997;91:458–63. doi: 10.1046/j.1365-2567.1997.d01-2249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shreeniwas R, Schulman LL, Narasimhan M, McGregor CC, Marboe CC. Adhesion molecules (E-selectin and ICAM-1) in pulmonary allograft rejection. Chest. 1996;110:1143–9. doi: 10.1378/chest.110.5.1143. [DOI] [PubMed] [Google Scholar]