

Abstract
Exceptional mechanical and electrical properties of carbon nanotubes (CNT) have attracted neuroscientists and neural tissue engineers aiming to develop novel devices that interface with nervous tissues. In the central nervous system (CNS), the perinatal chloride shift represents a dynamic change that forms the basis for physiological actions of γ-aminobutyric acid (GABA) as inhibitory neurotransmitter, a process of fundamental relevance for normal functioning of the CNS. Low intra-neuronal chloride concentrations are maintained by chloride-extruding transporter, potassium chloride cotransporter 2 (KCC2). KCC2's increasing developmental expression underlies the chloride shift. In neural injury, repressed KCC2 expression plays a co-contributory role by corrupting inhibitory neurotransmission. Mechanisms of Kcc2 up-regulation are thus pertinent because of their medical relevance, yet they remain elusive. Here we show that primary CNS neurons originating from the cerebral cortex, cultured on highly-conductive few-walled-CNT (fwCNT) have a strikingly accelerated chloride shift caused by increased KCC2 expression. KCC2 upregulation is dependent on neuronal voltage-gated calcium channels (VGCC), furthermore on calcium/calmodulin-dependent kinase II, which is linked to VGCC-mediated calcium-influx. We also demonstrate accelerated Kcc2 transcription in brain-slices prepared from genetically-engineered reporter mice, in which Kcc2 promoter drives luciferase, when the cerebral cortex of these mice is exposed to fwCNT-coated devices. Based on these findings, we can now address whether fwCNT can enhance neural engineering devices for the benefit of neural injury conditions associated with elevated neuronal intracellular chloride concentration such as pain, epilepsy, traumatic neural injury and ischemia. Taken together, our novel insights illustrate how fwCNTs can promote low neuronal chloride in individual neurons and thus inhibitory transmission in neural circuits.
Keywords: few-walled, carbon, nanotube, neuronal, chloride
1. Introduction
In view of known nanoscopic size, extraordinary strength and electrical conductivity of carbon nanotubes (CNT),[1, 2] we addressed whether CNT exposure alters neurons’ gene expression so that their cell- and network physiological properties could be modified. One suitable and highly informative neuronal cell-physiological parameter is intraneuronal chloride, which dictates neurons’ transmission in response to GABA and glycine, known to be inhibitory in the mature brain and spinal cord, and excitatory in neural development.[3-7] The perinatal chloride shift is a profound developmental transformation of CNS neurons as they change from migratory phenotype to synaptic connectivity.[8] Developmentally earlier high intraneuronal chloride renders GABA excitatory, which is converted into the mature phenotype of low intraneuronal chloride by transcriptional upregulation of the chlorideextruding transporter KCC2.[4, 6] Low intraneuronal chloride sustains inhibitory GABA-ergic neurotransmission in neural circuits, a quintessential function of the vertebrate CNS. Aside from its basic relevance, a reversal of the developmental chloride shift has been demonstrated in the pathogenesis of diseases with a common feature of neural injury, namely epilepsy, pain, traumatic brain injury and cerebral ischemia.[5, 7]
Returning to the question of whether exposing primary CNS neurons to CNT matrices can alter their gene expression so that their cell- and network physiological properties could be enhanced, we set out to interrogate neuronal chloride regulation in response to CNT matrix.
2. Results
We chose to work with few-walled CNT (fwCNT) because simplified synthesis protocols of these CNTs have led to exceptional purity as compared to single-walled CNT.[9, 10] Compared with multi-walled CNT, fwCNT have lower defect density and higher electric conductivity. In order to improve aqueous solubility, biocompatibility with and adherence of neural cells and tissue, we suspended nanotubes in gum arabic (GA), a natural gum from hardened sap of Acacia senegal trees.[11] GA, a complex mixture of polysaccharides and glycoproteins has excellent biocompatibility as evidenced by its edibility. As a result of our efforts, we obtained high-quality/high-purity fwCNTs that showed an extraordinary intrinsic electrical conductivity of 3,000 S cm-1 (Figure 1a(i)). Moreover, stable suspensions of fwCNT in GA led to homogenous coating of various substrates as used in this study (Figure 1a(ii)). We found our preparations of fwCNT to be devoid of traces of metals except calcium (probably a contamination from de-ionized water used for washing), in particular no evidence for presence of cadmium, magnesium, cobalt and molybdenum (Supplementary Figure 1).
Figure 1. fwCNT preparation, culture of primary cortical neurons and ultrastructure.

a (i) Transmission electron micrograph of fwCNT demonstrating high-degree purity of fwCNT (arrows) against a support film on an EM grid (arrow-heads); (ii) Scanning electron micrograph of gum arabic (GA)-dispersed fwCNT, after spraying the fwCNT onto the matrix, showing a homogeneous carpet of non-aggregating fwCNT.
b Confocal micrographs (green fluorescence projected on bright-field images) of primary cortical neurons, plated on regular, poly-d-lysine (pDL)-coated matrix, vs. fwCNT-coated matrix. Note upregulation of neuronal differentiation marker ß3-tubulin and enlargement of the neurons (DIV2).
We found a suitable source of neural cells to address our question in primary neurons derived from the developing cerebral cortex of late embryonic rats.[6] Their culture on fwCNT, prepared with a final coating of poly-d-lysine, sustained vital primary cultures with robust formation of neural processes, enlarged somata, increased expression of neuronal marker ß3-tubulin, and notably the absence of any signs of cytotoxicity-neurotoxicity and biological non-compatibility in our cultures (Figure 1b). However, dedicated in-vivo neurotoxicity experiments of our fwCNT preparations will have to be conducted in future studies (see also ref.[12]). SEM confirmed enlarged soma size. Moreover, it showed the neurons in intimate proximity to the fwCNT matrix (Figure 2a). Direct proximity of the neuronal plasma membrane to matrix-bound fwCNT could be documented using TEM (Figure 2b-d). We interpreted enlarged soma size and increased expression of neuronal differentiation marker as signs of accelerated maturation. We further reasoned that fwCNT-cultured neurons show high-efficiency electrical coupling to their matrix because of their physical closeness and because of the high electrical conductivity of the preparation of fwCNT that we have provided here. Electrical coupling of neurons to CNT matrices has been mentioned previously,[1] but fwCNT as prepared for the present study show an electrical conductivity approximately 100x increased over regular CNT-preparations.
Figure 2. Ultrastructure of fwCNT-cultured primary cortical neurons.
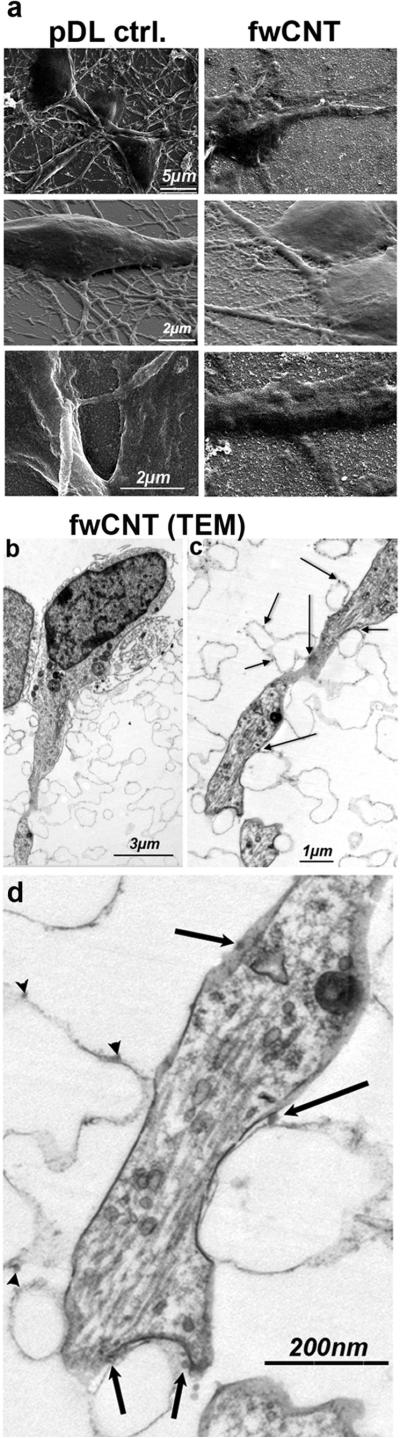
a Scanning electron micrographs of pDL-control vs. fwCNT matrix cultured primary neurons. Primary cortical neurons (DIV2) cultured on fwCNT show soma enlargement, and morphology highly suggestive of a very close interfacing with the fwCNT matrix. b Depicted is a low-power electron micrograph of a cultured neuron, note soma and dendrite. c Shown is a higher magnification of b. Arrows point to fwCNT bound to the cellulose acetate matrix, some of which in direct contact to the neuronal outer membrane, some in their vicinity. d Transmission electron micrograph of ultrathin sections of fwCNT-cultured primary cortical neuron, higher magnification. Process of a fwCNT-cultured primary cortical neuron shows very close interfacing and direct physical contact with fwCNT deposited on the cellulose-acetate matrix. Arrows point at fwCNT in direct contact with the neuron, arrowheads to fwCNT nearby. Note that ~60-90nm section thinness renders direct contact of nearby fwCNT highly likely in adjacent planes.
In our rat primary cortical neurons, we found the chloride shift robustly and strikingly accelerated (Figure 3a). Using directed expression of genetically-encoded chloride indicator, Clomeleon,[6, 13] we detected a reduction of neuronal chloride from 87.5 to 54.6mm (4 repeat experiments each comprising ≥50 neurons analyzed per group; all conducted on the days in vitro (DIV) 2). This was specific for fwCNT since a nanomaterial with similar nanostructure, yet lacking electrical conductivity, SiOx nanowires, did not affect the chloride shift (Supplementary Figure 2a). Of note, SiOx nanowires were GA-coated. Additionally, use of filtrate of fwCNT-GA coating did not lead to acceleration of the chloride shift either (Supplementary Figure 2b). Thus, fwCNT matrix, facilitated by GA-mediated solubilization of fwCNT, evoked a unique acceleration of the chloride shift in primary rat cortical neurons derived from late (the embryonic day 18 or E18) embryos. Our control experiments make co-contributory effects of contaminants of fwCNT synthesis highly unlikely. There was no positive effect of a matrix consisting of a non-carbon nanostructure showing greatly reduced electrical conductivity, SiOx, and of the fwCNT solubilization agent, GA.
Figure 3. Cl− down-regulation, KCC2 up-regulation in primary cortical neurons exposed to fwCNT.
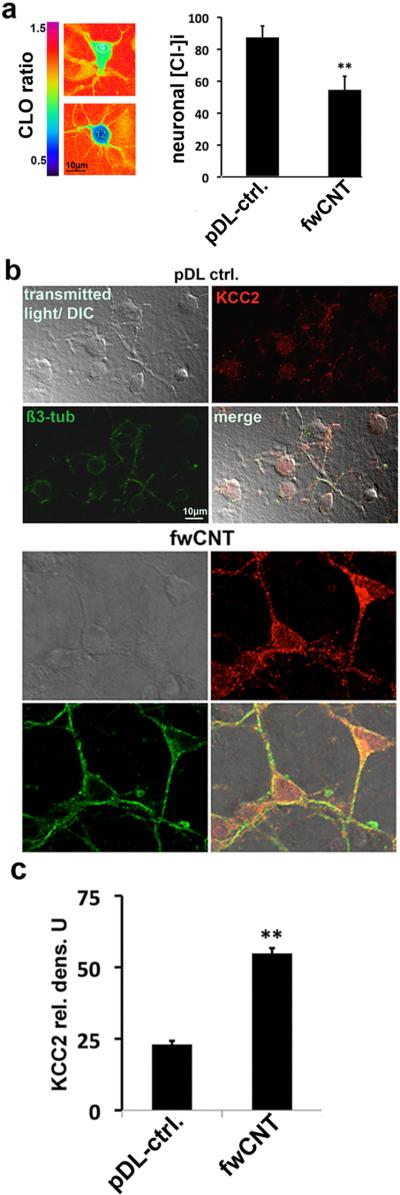
a Clomeleon-based ratiometric chloride imaging in primary cortical neurons.[6] The left-hand panel shows ratiometric pseudo-images of primary cortical neurons (DIV2) that express the genetically-encoded chloride indicator protein, clomeleon, pseudocolored according to their clomeleon emission ratios. The left-hand vertical bar shows the color-scheme from blue (ratio = 0.5) to purple (ratio = 1.5). Using calibration experiments,[6] these ratios were converted to chloride concentrations inside the neuron (note the inverse relationship of clomeleon emission ratio to intracellular chloride concentration ([Cl−]i): high ratio = low chloride, low ratio = high chloride). Results from four independent experiments (each n≥50 neurons/matrix) indicate a robust down-regulation of [Cl−]i in fwCNT-cultured primary neurons, shown in the bar-graph (right); ** = p<0.01.
b Confocal micrographs of primary cortical neurons. DIV2-neurons, grown on pDL conrol vs. fwCNT matrix are shown for bright-field, red channel (anti-KCC2 immunolabeling), green channel (anti-ß3-tubulin) and merged. Note increased KCC2 expression in soma and dendrite of fwCNT-exposed neurons, also increased expression of neuronal marker ß3-tubulin and increased soma size.
c Increased KCC2 expression of fwCNT-cultured neurons. KCC2 densitometry [6] is depicted, average of 4 independent cultures, n≥25 neurons/culture; ** = p<0.01 (t-test).
To address whether the accelerated chloride shift that was evoked by fwCNT matrix was caused by KCC2 up-regulation, we next determined KCC2 protein expression by immunocytochemistry. Robust KCC2 up-regulation was indeed detected (Figure 3b-c). Thus, culture of late embryonic mammalian cortical neurons on fwCNT vastly accelerated their chloride shift by significant upregulation of Kcc2, which leads to reduced chloride via KCC2's transporter function. This effect is specific for high-conductivity fwCNT, and was not apparent in any of our comprehensive controls.
We next investigated critical signaling mechanisms that underlie the neuronal chloride shift and upregulation of Kcc2. L-type voltage-gated calcium channels (VGCC) have been implied in the chloride shift.[4, 6] We therefore first verified their functional expression using a fluorescently-labeled compound that directly binds to VGCC, bodipy-DM-dihydropyridine, which has been used with benefit previously, also on primary neurons.[14] Interestingly, fwCNT-cultured primary cortical neurons expressed VGCC at dramatically increased levels of DHP-binding vs. control (Figure 4a, Supplementary Figure 3a-b). In order to discern the effects of a high-conductivity matrix, we used a gold (Au)-matrix, a material with known high electric conductance (30,000 S cm−1), yet with a homogenous, not particulate nanostructure compared with CNT. Increased functional expression of VGCC was also observed in Au-matrix cultured neurons, evidenced by increased DHP-binding (Figure 4b). Of note, DHP-binding patterns were qualitatively similar in appearance despite the vast differences in amount of binding, with the typical polar enrichment in one part of the soma and the more even decoration of the dendrite [14] (Supplementary Figure 3a-b). We found VGCC in cultured neurons to be of the Cav1.3 and 1.4 type, with additional detection of Cav1.1 in fwCNT-cultured neurons (Supplementary Figure 3c). Importantly, increased VGCC levels contributed critically towards the accelerated chloride shift since blocking them specifically with nifedipine abrogated both, chloride shift and KCC2 upregulation in fwCNT- and Au-matrix cultured neurons (Figure 4c). Even in pDL controls, chloride increased moderately in response to nifedipine, indicating the regular pace of the chloride shift during primary neuronal culture. Thus, high-conductivity culture matrices support increased DHP-binding indicative of functional VGCC expression, and blocking VGCC prevents KCC2 increase, which underlies the accelerated chloride shift.
Figure 4. Dependence of accelerated chloride shift on L-type VGCC and CaMKII in primary cortical neurons.

a Increased abundance of functional L-type VGCC expression in fwCNT-cultured cortical neurons. Confocal micrographs of DIV2 cultures are shown. Left-hand panel depicts binding of fluorescently-labeled dihydropyridine (Bodipy-DHP) to L-type VGCC in red; note fore-mostly the drastically-increased binding of Bodipy-DHP to fwCNT-cultured neurons, subcellular pattern in keeping with [14]. In particular, the delicate cellular detail, ubiquitous neuronal expression and a polar pattern with localized high-intensity decoration of a subsection of the soma can be appreciated; no counterstain shown in this micrograph. The right-hand panel, using identical acquisition settings, shows regular abundance of Bodipy-DHP binding sites in pDL control-cultured primary cortical neurons. Nuclear counterstain with DAPI is shown to better illustrate cellularity. See also Supplementary Figure 3a-b.
b Increased abundance of functional L-type VGCC expression in fwCNT-cultured cortical neurons, revealed by Bodipy-DHP binding, quantitative assessment. Left-hand panels show fluorescent micrographs depicting increased expression of L-type VGCC, verified by bindings of bodipy-DM-DHP, which yields green fluorescence in non-confocal microscopy, on fwCNT-cultured neurons, also on Au-cultured neurons, vs. control expression levels on pDL control-cultured cells (all cultures DIV2). Note the highly polar pattern present on all three matrices (as in [14]). Right-hand bar diagram shows significantly increased quantities of Bodipy-DHP binding for fwCNT and Au-cultured neurons vs. pDL control, for both areas of interest that were examined densitometrically. *** p<0.001, ** p<0.01, n≥25 neurons/condition.
c Specific block of L-type VGCC eliminates effects of fwCNT and Au-matrix on neuronal chloride. Bar diagrams illustrate the effect of blocking L-type VGCC, using 20μm nifedipine, on neuronal [Cl−]i and KCC2 abundance, depending on the culture matrix; same methods as in Figure 3. Left-hand bar diagram shows modest increase in [Cl−]i caused by nifefipine in PDL control-cultured neurons, yet a drastic increase of the robustly reduced [Cl−]i for fwCNT- and Au-cultured neurons (all cultures DIV2). KCC2 expression behaves inversely. *** p<0.001, ** p<0.01, * p<0.05; n≥40 neurons per condition.
d Specific block of CaMKII eliminates effects of fwCNT, not Au-matrix on neuronal chloride. Similar experiment as in Figure 4c, yet shown here are effects of specific antagonism of CaMKII with KN93 (1μM). Neuronal [Cl−]i measurements (left) indicate a specific and powerful effect of KN93 on fwCNT-cultured neurons, not on pDL control- and also not on Au-cultured neurons. Neuronal [Cl−]i findings are partially reflected by KCC2 abundance, particularly for fwCNT and pDL control groups. However, note modest reduction of KCC2 expression of Au-cultured neurons, suggesting moderate functional compensation by other mechanisms on Au-control matrix. *** p<0.001, n≥40 neurons per condition; see also Supplementary Figure 3d-e.
These findings prompt the question how a rise in intracellular calcium leads to increased expression of Kcc2. Based on known interactions in central neurons between increased intracellular calcium, calmodulin and calcium/calmodulin-dependent kinase II (CaMKII),[15] we tested whether CaMKII activity is required for the accelerated chloride shift and KCC2 upregulation when culturing on fwCNT matrix. Using specific inhibitors of CaMKII, we confirmed this novel concept, but remarkably only for fwCNT-, not for Au matrix-cultured neurons (Figure 4d, Supplementary Figure 3d-e). Thus, intracellular CaMKII-dependent signaling is specific for acceleration of the chloride shift in fwCNT cultures, and, in contrast, is dispensable for Au-matrix. This interesting difference could be due to the different conductivities of the two materials (Au matrix 10x higher than fwCNT), and also to the different structural properties of the materials: Au matrix is not a particulate material, fwCNT consists of nanotubes so that a neuron can be “wired” from the outside to itself and to its neighbors.
These findings originated from cortical neurons maintained as primary cultures, which recapitulate the chloride shift as it happens in perinatal development of the vertebrate CNS. Despite cultured primary cortical neurons sharing this developmental hallmark, our present findings have been obtained in dissociated culture, in the absence of an organotypic context. We therefore examined properties of fwCNT matrices in brain slice cultures which maintain neurons in their natural surrounding of, for example, a layered cortical architecture with neural connectivity as a result of in-vivo development. For this purpose, we engineered mice so that a 2500 base-pair proximal promoter fragment of the Kcc2b gene drives a red-shifted luciferase (red LUC) reporter gene. This construct was genomically integrated, by homologous recombination in mouse embryonic stem cells, into the inert Rosa26 locus, giving rise to otherwise normal and fertile mice, which transmitted the engineered mutation (Figure 5a-b). Of note, these mice are Kcc2 wildtype. Importantly, when we cultured their late embryonic cortical neurons (E16.5), these neurons showed an increase in red LUC activity paralleling the known increase in Kcc2 expression (Figure 5c). First, this unambiguously demonstrates generation of functional reporter, luciferase, controlled by a 2.5kB DNA sequence of the proximal promoter fragment of the Kcc2 gene, in primary cortical neurons cultured from these mice. Second, it suggests common principles of transcriptional regulation shared by the 2.5kB fragment and the endogenous Kcc2b promoter. For exposure of cultured brain slices we generated semi-flexible devices made of poly-dimethyl-siloxane (PDMS, polysil), using molds with customized conical indentations of >200μm length, so that 250μm thin brain slices were exposed throughout their depth. The PDMS casts were coated with GA-solubilized fwCNT, finally with poly-d-lysine, and omitting fwCNT coating for controls (Figure 6a). Rectangular sections of the PDMS devices were positioned to indent and expose the parietal cortex of cortical brain slice cultures derived from newborn mice at the postnatal day 0 (P0) (Figure 6b). At 72h, we found red LUC activity increased by 22% for fwCNT-exposed brain vs. control, a statistically-significant difference. This increase appeared to involve areas not exposed to the fwCNT-coated indentation devices in both hemispheres, likely due to lack of resolution based on light scattering in the brain tissue, which had been exposed to fwCNT also in the depth, not only on the surface. To confirm and extend the finding of increased red LUC reporter activity in brain slices, we assessed gene regulation by quantitative RT-PCR and KCC2 immunolabeling for the part of the cortex that was exposed to fwCNT or control. qRT-PCR findings were consistent with red LUC activity in slice. We recorded similar significant increases for both endogenous gene (Kcc2) and red Luc reporter gene driven by Kcc2 promoter (Figure 6c), validating the key role of the proximal 2.5kB promoter in directing expression of Kcc2.[6] Based on this important conclusion, these findings also ratify the chosen approach of our Kcc2-red LUC Rosa26 knockin mouse. Furthermore, to verify increased gene expression at the protein level, slices were immunolabeled for KCC2, showing significant upregulation in fwCNT-exposed slices (Figure 6d). These findings suggest that 2.5kB of proximal Kcc2b promoter receive sufficient transcriptional drive in a brain slice, and that transcriptional drive is increased when the slice is exposed to fwCNT. Thus, in the organotypic context of the brain, exposure to fwCNT drives Kcc2b transcription, which leads to increased abundance of Kcc2 mRNA, which increases KCC2 protein expression.
Figure 5. Generation of Kcc2-red LUC reporter mice.

a This schematic illustrates how the genetic construct, 2.5kB of proximal Kcc2b promoter positioned 5’ of red Luc cDNA, was inserted into a plasmid vector specifically designed for targeting of the Rosa26 genomic locus. After homologous recombination in mouse embryonic stem cells, the engineered mutation went germline in mice.
b The panel shows DNA-genotyping results by PCR from tail-biopsies of stably transmitting mice.
c Recapitulation of the chloride-shift in primary cortical neurons derived from Kcc2 red LUC reporter mice. The panel shows red LUC activity over cortical neuronal development in primary neuronal culture, derived from Kcc2-red LUC reporter mice. Primary cortical neurons were generated at developmental stage E16.5 (DIV3 is the P0 (birth) equivalent). Note that functional reporter gene is generated by these neurons, and that red LUC reporter gene-activity recapitulates developmental Kcc2 upregulation.
Figure 6. Findings from brain slices derived from Kcc2-red LUC reporter mice are in keeping with primary cortical neuronal culture findings.

a fwCNT-coated PDMS devices for exposure of the cerebral cortex. The panel depicts scanning electron micrographs of the engineered flexible PDMS matrices, fwCNT-coated, so that these devices can expose the cerebral cortex of Kcc2 red LUC mice. Panels (i) to (ii) show lower and higher magnification of the customized conical poles and their relative density against the sheet-like basic structure of the PDMS matrix. Panels (iii) and (iv) show higher magnifications of (i) and (ii) so that the homogenous and non-aggregating nature of the fwCNT-coating can be appreciated.
b Increased reporter gene activity of brain slices from Kcc2 red LUC mice exposed to fwCNT-matrix. Upper panel schematically illustrates how cortical slices were exposed (slices generated at P0, exposition for 72h), the middle panel shows two slices at luminescence acquisition, and the lower panel their luminescent pseudoimages, clearly increased for fwCNT matrix. The right-hand bar diagram shows luminescence quantification, indicating a statistically significant increase from slices exposed to fwCNT-coated PDMS devices vs. control-coated PDMS devices. This increase reflects increased transcriptional drive on the Kcc2 promoter for fwCNT-coating; n=4/group, * p<0.05.
c Increased Kcc2 and Luc gene expression in cortex exposed to fwCNT. Bar diagrams show qRT-PCR quantification from slices, using exposed cortex as template. pDL control values for Kcc2 (left) and Red Luc (right) were set as “1”, indicating a significant upregulation for both, endogenous gene as well as Luc reporter when exposing brain slices of Kcc2 red LUC mice to fwCNT matrix; n=4-5 per group, ** p<0.01.
d Increased KCC2 protein expression in cortex exposed to fwCNT. Left-hand micrographs illustrate KCC2-immunolabeling of exposed cortex, and their respective quantification in the bar diagram on the right. Findings indicate a significant up-regulation of KCC2 protein for cortex exposed to fwCNT matrix. Note that in contrast to cultured neurons, immunolabeled KCC2 in organotypic preparations, similar to labeling in the native brain, presents as a diffuse staining of gray matter (such as cortical gray matter), rather not specifically highlighting individual neurons’ somata and/or processes. n=4-5 per group, * p<0.05, t-test.
3. Discussion
Results presented here document a dramatic acceleration of a fundamental neural maturation mechanism by a fwCNT-matrix. Findings imply that this process is driven by gene regulatory changes, namely upregulation of Kcc2. We speculate that this might have involved transcriptional de-repression, as demonstrated for physiological in-vitro development.[6] Mechanistically, we could verify a direct contact of fwCNT matrix to the neuronal outer plasma membrane in primary neuronal culture. fwCNT as generated for our study are highly conductive, so that “external wiring” of single neurons will enhance their electrical activity cell-autonomously. Extraordinarily high electrical conductivity of the fwCNT culture matrix and direct interfacing of the neuronal plasma membrane with fwCNT will also promote neuron-to-neuron connections, in addition to biological neuron-to-neuron connectivity based on direct proximity of somata and processes of individual neurons. This indicates that the observed novel effect of fwCNT can be regarded as based on two major principles. One is the increased electrical conductivity of the matrix – similar in principle to the Au-matrix that we have used as control (even though conductivity of Au is substantially increased over that of the used fwCNT). In addition, different from Au, is the “external wiring” of the nerve cell, cell-autonomously and neuron-to-neuron, by an electrically conductive nanomaterial. The joint effect will be increased electrical activity of the neurons. Increased activity translates into increased functional expression of VGCC, which promotes Kcc2 expression via calcium influx. We are providing here the novel concept of CaMKII as involved in the accelerated chloride shift, whereby CaMKII provides the link between calcium influx via VGCC and subsequent transcriptional activation of Kcc2. How increased electrical connectivity causes increased functional expression of VGCC will be the subject of future studies, which will also do justice to the 4 different VGCC isoforms, 3 of which were expressed in fwCNT-cultured neurons. More relevant for the present study, VGCC activity is critical for the accelerated chloride shift, as critical as CaMKII, since their respective selective block eliminated the accelerated chloride shift (concept summarized in Figure 7).
Figure 7. Conceptual representation of our findings.

Exposure of cortical neurons to fwCNT matrix leads to strikingly increased functional expression of L-type VGCC, which will increase voltage change-mediated calcium influx, in particular on high-conductance fwCNT matrix. This in turn will accelerate transcriptional activation of the Kcc2 gene, in a VGCC-calcium dependent manner, as shown previously,[4, 6] and in a CaMKII dependent manner, as now shown here. Increased transcription of Kcc2 will lead to increased expression and function of KCC2, which, via its chloride-extruding transporter function, will lower neuronal chloride. This in turn will render activation of GABAA-receptors and glycine-receptors hyperpolarizing, which translates into inhibitory neurotransmission, and thus attenuation of excitation in neural circuits.[24]
The mechanism outlined for primary neuronal cell culture could also occur in brain slices, yet in this platform awaits more in-depth study and definitive proof in future studies.
The documented acceleration of the chloride shift via interfacing with fwCNT could form the basis for their use as an advantageous and innovative tool to precisely control cell-physiological properties of CNS neurons, which in turn dictate neural circuits’ functions. KCC2 has recently been characterized as neuroprotective.[7] This means that approaches that can upregulate KCC2, such as direct exposure to fwCNT, will have a neuroprotective effect. Future use of fwCNT-coated devices can now be envisioned in conditions of neural injury that have been associated with Kcc2 down-regulation and increased neuronal chloride. Based on our findings here, the question can now be addressed in future experiments whether use of fwCNT in neural tissue engineering and neuroprosthetics will promote re-normalization of inhibitory neurotransmission that has been corrupted by injury, by evoking increased Kcc2 expression.
4. Conclusions
Primary cortical neurons, cultured on highly-conductive few-walled-CNT (fwCNT) show an accelerated chloride shift caused by increased KCC2 expression. Our results illustrate how fwCNTs can promote low intraneuronal chloride concentrations through enhanced chloride extrusion in individual central neurons and therefore inhibitory transmission in neural circuits.
5. Experimental Section
5.1. Carbon Nanotubes and Silicon Oxide Nanowires (SiOx)
Growth of Carbon Nanotubes and Silicon Oxide Nanowires
Few-walled carbon nanotubes (fwCNTs) were grown using a catalytic chemical vapor deposition (CVD) method. Co/Mo catalyst supported on porous MgO powder was used as the catalyst for nanotube growth and carbon monoxide (CO) was used as the carbon precursor.[9] In a typical synthesis procedure, the as-prepared catalyst powder was placed in a horizontal tubular furnace (3 inches) and heated at 950 °C. Then CO was infused to the growth chamber for 30 minutes. The as-grown CNT product was then oxidized at 570 °C in air-argon mixture to remove amorphous carbon impurities produced during growth. Next, the material was boiled in HCl (5 m) to remove metal catalysts and MgO support. Purified nanotubes were obtained after filtration and washing with deionized water. Conductivity of the purified CNT thin film (~3,000 S cm−1) was determined by four-probe measurement.
Silicone oxide nanowires were grown by the CVD method following a previous protocol from our group.[16]
Purity analysis of fwCNT preparation with X-ray photoelectron spectroscopy (XPS)
To analyze purity of our fwCNT preparation, we used XPS, a spectroscopy surface chemical analysis technique that can quantitatively acquire the elemental composition, chemical and electronic state of the elements contained in a material. This method is characterized by great sensitivity. We measured as-prepared fwCNT films in an X-ray photoelectron spectrometer, model Axis Ultra (Kratos Analytical, Manchester, UK), following the instructions of the manufacturer, followed by processing of the primary spectra with CasaXPS software (Casa Software).
Preparation of gum arabic (GA) solution, GA-coating and thin films
GA aqueous solution was prepared by adding GA (5 mg; Laboratory Grade, Fisher Scientific) in deionized water (100 mL) and stirring for 20 min. Dried-pure fwCNT (0.7 mg) was added to the GA solution and sonicated for 1 h. The fwCNT-GA solution was then centrifugated (7200 rpm or 4400 G) in an IEC Centra MP4 centrifuge for 2h to remove aggregates. The weight ratio of fwCNT to GA was 0.2. The same procedure was applied for solubilizing SiOx. Filtration of the fwCNT-GA solution through a polycarbonate membrane filter (pore size: 0.4 μm; Millipore, HTTP02500) yielded the GA-filtrate control reagent.
The spray coating method was used to prepare uniform thin films of fwCTN and SiOx nanowires on cover slips and cell culture dishes. For additional control experiments, the same cell-culture substrates were coated with gold films (100 nm) by E-beam evaporator (CHA Industries Solution). Finally, for all substrates, poly-d-lysine (pDL) coating was applied before neuronal culture.
fwCNT PDMS device preparation
Polydimethylsiloxane (PDMS) substrates with pillars were fabricated by the standard molding procedures used previously in soft lithography for fabrication of PDMS stamps.[17] A sheet of aluminum foil (0.25 mm) punched with over-the-counter quilting needles (size 7) was used as the replica mold. The elastomer and curing agent mixture (Sylgard 184 Silicone Elastomer Kit, Dow Corning) was cast on the mold. After degassing and curing, the pillared PDMS stamp was removed from the mold surface. The PDMS substrate was irradiated in a plasma sterilizer for 5min to render the surface hydrophilic before fwCNT coating. Then the fwCNT-GA solution was spray-coated on the PDMS substrate. SEM images of the as-prepared PDMS stamps with pillars protruding from the surface were acquired at 30° tilt of the sample stage (Figure 5a). SEM micrographs documented a uniform CNT film on both flat surface and cone-shaped pillars of the PDMS substrate.
5.2. Primary Cortical Neurons
Culture
The preparation of primary cortical neurons was adapted from a previous protocol.[6] Briefly, cortices were microdissected from embryonic rats (E18) or mice (E16.5). The tissue was dissociated using papain, followed by mechanical dissociation. Cytosine arabinoside (2.5 μm) was added to cultures to inhibit the proliferation of non-neuronal cells. Cell suspension was plated at a density of 106 cells ml−1 onto tissue-culture dishes. Matrix-covered glass coverslips (diameter: 12 mm) were contained in tissue culture dishes, typically n=3. Cortical neuronal culture prepared by this method yielded a majority population of neuronal cells, with negligible glial contamination, as evidenced by the absence of astrocytic protein, GFAP by Western blotting.[6] Neuronal viability and differentiation were ascertained microscopically before, during and after experiments, and we did not obtain evidence of neurotoxicity of our fwCNT preparation.
All animal procedures leading to primary cells and organotypic cultures, as used in this study, were performed with approval of the Duke University Animal Care and Use Committee under a valid institutional animal protocol, and in observance of National Institutes of Health guidelines.
Chloride imaging
Clomeleon-based ratiometric chloride imaging was conducted as described previously,[6, 13] taking advantage of the ratiometric fluorescent chloride indicator protein, clomeleon, after appropriate calibration reactions in primary cortical neurons.
VGCC receptor binding studies
Fluorescence imaging of dihydropyridine binding to L-type VGCC was conducted following previous reference.[14] Briefly, Bodipy-DM-DHP (1 μm Invitrogen) was applied to primary cortical neuronal cultures on DIV2 for 1h (37°C), cells were washed and fixed in paraformaldehyde (4 %) for 20min, mounted on glass-slides using fluoromount, and imaged, using either green or red filter settings. Quantitative assessment was conducted following a previous reference,[6] using ImageJ.
Immunocytochemistry
Confocal fluorescence imaging was conducted after immunolabeling for ß3-tubulin (mouse monoclonal antibody; Iowa hybridoma bank; 1:200), and KCC2 (rabbit polyclonal antibody; Abcam; 1:200).[6] Fluorescently labeled sections were visualized in a Zeiss LSM710 confocal imaging suite with lasers tuned to the emission spectra of the secondary fluorescent antibodies (coupled to Alexa-488 and Alexa-595 dyes), as described previously.[18]
Wide-field fluorescence microscopy was conducted after immunolabeling for VGCC isoforms Cav1.1, 1.3 and 1.4. Labeling for Cav1.2 was not conducted because preliminary testing revealed absence of a PCR product for Cav1.2, whereas all other isoforms could be detected in fwCNT-cultured neurons (primers and conditions available upon request). The following primary antibodies were used: mouse anti-Cav1.1 (Thermo Scientific Cat# MA3-920; 1:100), mouse anti-Cav1.3 (NeuroMab Clone# N38/8; 1:100), and rabbit anti-Cav1.4 (LS Biosciences, Cat#: LS-C94032; 1:100). Secondary fluorescent detection antibodies as described above “Confocal Imaging”. Micrographs were acquired on an Olympus BX61 upright microscope using a 40x Olympus objective, connected to Roper high-resolution CCD-camera with ISEE software.
Scanning electron microscopy
SEM was conducted according to previous reference.[19] In short, the samples were dehydrated through a graded ethanol series (30%, 50%, 70%, 90%, 100%) and then dried with hexa-methyl-disilazane. The samples were sputter-coated with gold using a Denton Desk IV system. Samples were imaged using a FEI XL30 FE-SEM.
Transmission electron microscopy
TEM was conducted according to previous reference,[20] with the following modification to accommodate culture of primary cortical neurons on poly-d-lysine, and fwCNT-coated matrices. Cellulose acetate was used as tissue culture matrix (pore size: 0.45 μm; Millipore, PIHA 03050), coated with fwCNT and finally with poly-d-lysine. This matrix can be readily processed for TEM including ultra-microtome sectioning (60-90 nm).
Morphometry for confocal and light-microscopy acquired images was conducted as previously described.[6]
5.3. Brain Slices
Mouse gene targeting of the Rosa26 locus with Kcc2-promoter LUC reporter construct
We first constructed a plasmid, pKcc2-red Luc that contains a mouse 2.5kb Kcc2b (fore-brain specific Kcc2 isoform) promoter DNA fragment downstream of a polyA+ cassette to protect against cis-acting promoter/enhancer interference of the Rosa26 locus.[21] This cassette containing the polyA+-Kcc2-promoter DNA was subcloned upstream of the red luciferase coding region in a vector called pBasicRedLuc, harboring a codon-optimized Italian firefly (Luciola italica) luciferase (Genetargeting Systems). PacI and AscI restriction sites were added at the 5' and 3' ends of the final construct to enable cloning into a modified Rosa26 targeting vector, pROSA26Am1.[22] The resulting construct was linearized with MfeI, purified and electroporated into embryonic stem (ES) cells, strain R1. PCR screening of G418-resistant ES-cell colonies was used to identify homologous recombination into the Rosa26 locus. For that purpose one primer was designed upstream of the short homology arm of the Rosa26 targeting vector. This primer (R26F) had the sequence : 5'-CCTAAAGAAGAGGCTGTGCTTTGG-3'.
Another primer (KCC2R) was designed within the 5' end of the Kcc2 promoter and had the sequence : 5'-CTTATCCTTGAGAGACGTACTAGTCC-3'.
The 1.3kB PCR product amplified from genomic DNA of homologously recombined ES-cell clones was identified for 11 clones of a total of 48 clones screened, indicating correct, orthotopic targeting. Three such clones were expanded and verified again. Recombinant ES-cells from these clones were used for microinjection into blastocysts to generate chimeric mice. Breeding of these chimeras to wildtype C57Bl6 mice established germline transmission of the knockin and established the mouse line as used here.
Organotypic culture
Cortical slices were cut and then cultured from neonatal mice using a method described previously.[23] In brief, brains were removed from euthanized Kcc2 red LUC mice into complete HBSS. The brains were then embedded in low-gelling temperature agarose (2–3 % w/v). Coronal slices (300 μm) were cut from the agarose block with a Leica VT1000S vibratome. The slices were then transferred onto cell culture inserts (PICM0RG50; Millipore, Billerica, MA) in culture dishes (35 mm) filled with growth medium (BMEM, 10 % bovine calf serum, 1 mm l-glutamine, 50 U penicillin, 50 μg ml−1 streptomycin, 35 mm d-glucose). Small strips of PDMS sheets, as described above, with or without fwCNT-coating were placed facing-down onto cortical regions of the slices (illustrated in Figure 6a-b). Gentle pressure was applied to assure proper insertion of pillars/cones into the brain slices. They were maintained in a tissue culture incubator until the bioluminescence assays, with daily change of media.
After 3 days in culture, bioluminescence from cultured slices was determined with a cooled CCD camera (IVIS100; Xenogen, Alameda, CA). After removing PDMS sheets and obtaining a baseline, luciferin (20 μl; 500 μm) was dropped onto each slice and bioluminescence was measured afterwards. Bioluminescence reached a plateau within 5 min. Images were acquired at time-point 10 min after luciferin application with 5min exposure time.
For qRT-PCR assays, cortical regions exposed to PDMS sheets were dissected on the cell culture inserts, transferred into centrifuge tubes, then quick-frozen for subsequent total RNA extraction.
For KCC2 immunolabeling, brain slices were immersion-fixed in paraformaldehyde (2 %) for 2 h, then immunolabeled with KCC2-specific antibody as described under Immunocytochemistry.
qRT-PCR
Total RNA from cortical neurons was extracted and quantified as previously described.[18] Prior to reverse transcription, total RNA was subjected to DNaseI treatment (Invitrogen) to eliminate genomic DNA. DNaseI-treated total RNA (1 μg) was then subjected to first-strand cDNA synthesis with Superscript-III reverse transcriptase (Invitrogen). qPCR was performed using a ABI 7900 RT-PCR platform. First-strand cDNA (~100 ng or 2 μl of a 20 μl RT reaction) was processed using SYBR-Green PCR Mastermix (Qiagen). Each reaction was performed in triplicates. The following primers (mouse sequence) were used in qRT-PCR:
Forward Kcc2 5’-CTTCACCCGAAACAATGTCACAGAG-3’
Reverse Kcc2 5’-GCAGGGTGAAGTAGGAGGTCATATCAC-3’
Forward red Luc 5’-GAAGCCACCAGAGAAACTATTGA-3’
Reverse red Luc 5’-GGAACCCCGGCCACACCAGCATC-3’
Forward ß3-tubulin 5’-CCCCTTCATTGACCTCAACTACATGG-3’
Reverse ß3-tubulin 5’-GGCCATGCCAGTGAGCTTCCCGTTC-3’
The relative increase in reporter fluorescent dye emission was monitored in an ABI quantitative real-time thermocycler platform. The level of Kcc2 or red Luc mRNA, relative to ß3-tubulin, was calculated using the ΔΔCt method, where Ct was defined as the number of the cycle in which emission exceeded a pre-set threshold.
5.4. Chemicals
Chemical reagents and compounds were purchased from Sigma-Aldrich and Tocris.
5.5. Statistical Assessment
All results were expressed as mean ± SEM. Two-tailed student's t-test or one-way ANOVA with post-hoc Tukey analysis were applied to ascertain statistical significance with p<0.05 indicating significant differences.
Supplementary Material
Acknowledgements
The authors wish to thank Dr. Sidney Simon, Duke University, for critical reading of the manuscript, and the late Dr. Timothy Olliver, also Duke University, for assistance with confocal microscopy. This study was supported by funding from Duke University, the Klingenstein Fund, New York, NY, and the NIH (R21NS066307) to WL, and by the Center for the Environmental Implications of NanoTechnology, CEINT (maintained by the NSF and the EPA; NSF Cooperative Agreement Number EF-0830093) to JL. In regards to the latter, any opinions, findings, conclusions or recommendations expressed in this article are those of the authors and do not necessarily reflect the views of the NSF or the EPA. This work has not been subjected to EPA review and no official endorsement should be inferred. HZ was partially supported by a contractual agreement between Unidym, Inc. (Sunnyvale CA) and Duke University (PI JL).
Footnotes
Supporting Information is available on the WWW under http://www.small-journal.com or from the author.
References
- 1.Lee W, Parpura V. Prog Brain Res. 2009;180:110. doi: 10.1016/S0079-6123(08)80006-4. [DOI] [PubMed] [Google Scholar]
- 2.Odom TW, Huang JL, Lieber CM. Ann N Y Acad Sci. 2002;960:203. [PubMed] [Google Scholar]; b Lu J, Yuan D, Liu J, Leng W, Kopley TE. Nano Lett. 2008;8:3325. doi: 10.1021/nl801744z. [DOI] [PubMed] [Google Scholar]
- 3.Delpire E, Mount DB. Annu Rev Physiol. 2002;64:803. doi: 10.1146/annurev.physiol.64.081501.155847. [DOI] [PubMed] [Google Scholar]; b Boulenguez P, Liabeuf S, Bos R, Bras H, Jean-Xavier C, Brocard C, Stil A, Darbon P, Cattaert D, Delpire E, Marsala M, Vinay L. Nat Med. 2010;16:302. doi: 10.1038/nm.2107. [DOI] [PubMed] [Google Scholar]; b Coull JA, Boudreau D, Bachand K, Prescott SA, Nault F, Sik A, De Koninck P, De Koninck Y. Nature. 2003;424:938. doi: 10.1038/nature01868. [DOI] [PubMed] [Google Scholar]; b Fiumelli H, Cancedda L, Poo MM. Neuron. 2005;48:773. doi: 10.1016/j.neuron.2005.10.025. [DOI] [PubMed] [Google Scholar]; b Malek SA, Coderre E, Stys PK. J Neurosci. 2003;23:3826. doi: 10.1523/JNEUROSCI.23-09-03826.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Woo NS, Lu J, England R, McClellan R, Dufour S, Mount DB, Deutch AY, Lovinger DM, Delpire E. Hippocampus. 2002;12:258. doi: 10.1002/hipo.10014. [DOI] [PubMed] [Google Scholar]; b Zhu L, Polley N, Mathews GC, Delpire E. Epilepsy Res. 2008;79:201. doi: 10.1016/j.eplepsyres.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hubner CA, Stein V, Hermans-Borgmeyer I, Meyer T, Ballanyi K, Jentsch TJ. Neuron. 2001;30:515. doi: 10.1016/s0896-6273(01)00297-5. [DOI] [PubMed] [Google Scholar]; b Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. Nature. 1999;397:251. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- 4.Ganguly K, Schinder AF, Wong ST, Poo M. Cell. 2001;105:521. doi: 10.1016/s0092-8674(01)00341-5. [DOI] [PubMed] [Google Scholar]
- 5.Price TJ, Cervero F, de Koninck Y. Curr Top Med Chem. 2005;5:547. doi: 10.2174/1568026054367629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yeo M, Berglund K, Augustine G, Liedtke W. J Neurosci. 2009;29:14652. doi: 10.1523/JNEUROSCI.2934-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pellegrino C, Gubkina O, Schaefer M, Becq H, Ludwig A, Mukhtarov M, Chudotvorova I, Corby S, Salyha Y, Salozhin S, Bregestovski P, Medina I. J Physiol. 2011;589:2475. doi: 10.1113/jphysiol.2010.203703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bortone D, Polleux F. Neuron. 2009;62:53. doi: 10.1016/j.neuron.2009.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qi H, Qian C, Liu J. Chemistry of Materials. 2006;18:5691. [Google Scholar]
- 10.Feng Y, Zhang H, Hou Y, McNicholas TP, Yuan D, Yang S, Ding L, Feng W, Liu J. ACS Nano. 2008;2:1634. doi: 10.1021/nn800388g. [DOI] [PubMed] [Google Scholar]; b Hou Y, Tang J, Zhang H, Qian C, Feng Y, Liu J. ACS Nano. 2009;3:1057. doi: 10.1021/nn9000512. [DOI] [PubMed] [Google Scholar]; b Qian C, Qi H, Gao B, Cheng Y, Qiu Q, Qin LC, Zhou O, Liu J. J Nanosci Nanotechnol. 2006;6:1346. doi: 10.1166/jnn.2006.140. [DOI] [PubMed] [Google Scholar]
- 11.Bandyopadhyaya R, Nativ-Roth E, Regev O, Yerushalmi-Rozen R. Nano Lett. 2002;2:25. [Google Scholar]
- 12.Moon SU, Kim J, Bokara KK, Kim JY, Khang D, Webster TJ, Lee JE. Int J Nanomedicine. 2012;7:2751. doi: 10.2147/IJN.S30273. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Al-Jamal KT, Gherardini L, Bardi G, Nunes A, Guo C, Bussy C, Herrero MA, Bianco A, Prato M, Kostarelos K, Pizzorusso T. Proc Natl Acad Sci U S A. 2011;108:10952. doi: 10.1073/pnas.1100930108. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Higgins P, Dawson J, Walters M. Nat Nanotechnol. 2011;6:83. doi: 10.1038/nnano.2011.5. [DOI] [PubMed] [Google Scholar]; b Lee HJ, Park J, Yoon OJ, Kim HW, Lee do Y, Kim do H, Lee WB, Lee NE, Bonventre JV, Kim SS. Nat Nanotechnol. 2011;6:121. doi: 10.1038/nnano.2010.281. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gilmore JL, Yi X, Quan L, Kabanov AV. J Neuroimmune Pharmacol. 2008;3:83. doi: 10.1007/s11481-007-9099-6. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ciofani G, Raffa V, Vittorio O, Cuschieri A, Pizzorusso T, Costa M, Bardi G. Methods Mol Biol. 2010;625:67. doi: 10.1007/978-1-60761-579-8_7. [DOI] [PubMed] [Google Scholar]
- 13.Kuner T, Augustine GJ. Neuron. 2000;27:447. doi: 10.1016/s0896-6273(00)00056-8. [DOI] [PubMed] [Google Scholar]
- 14.Schild D, Geiling H, Bischofberger J. J Neurosci Methods. 1995;59:183. doi: 10.1016/0165-0270(94)00181-f. [DOI] [PubMed] [Google Scholar]
- 15.Wayman GA, Lee YS, Tokumitsu H, Silva AJ, Soderling TR. Neuron. 2008;59:914. doi: 10.1016/j.neuron.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng B, Wu Y, Yang P, Liu J. Advanced Materials. 2002;14:122. [Google Scholar]
- 17.Kumar A, Whitesides GM. Applied Physics Letters. 1993;63:2002. [Google Scholar]
- 18.Li J, Kanju P, Patterson M, Chew WL, Cho SH, Gilmour I, Oliver T, Yasuda R, Ghio A, Simon SA, Liedtke W. Environ Health Perspect. 2011;119:784. doi: 10.1289/ehp.1002807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rak K, Wasielewski N, Radeloff A, Scherzed A, Jablonka S, Hagen R, Mlynski R. J Biomed Mater Res A. 2011;97:158. doi: 10.1002/jbm.a.33042. [DOI] [PubMed] [Google Scholar]
- 20.Kesty NC, Mason KM, Reedy M, Miller SE, Kuehn MJ. Embo J. 2004;23:4538. doi: 10.1038/sj.emboj.7600471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eggermont J, Proudfoot NJ. Embo J. 1993;12:2539. doi: 10.1002/j.1460-2075.1993.tb05909.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pond BB, Berglund K, Kuner T, Feng G, Augustine GJ, Schwartz-Bloom RD. J Neurosci. 2006;26:1396. doi: 10.1523/JNEUROSCI.1421-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fiumelli H, Woodin MA. Curr Opin Neurobiol. 2007;17:81. doi: 10.1016/j.conb.2007.01.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.