

Abstract
Aberrant telomere homeostasis is essential for cell immortality, enabling cells to evade telomere dependent senescence. Disruption of telomere structure and function in cancer cells is highly toxic as shown by detailed pre-clinical evaluation of telomerase inhibitors. Under telomerase inhibition, cells must divide sufficiently frequently to allow one or more telomeres to shorten to an unprotected length. Functioning telomeres are disguised from the DNA damage machinery by DNA remodelling and other activities of the telomere binding complex shelterin. Direct interference with shelterin has been shown to result in cell killing and small molecules directly targeting telomere DNA also have anti-tumour effects partially dependent on shelterin disruption. However, shelterin components have not generally been regarded as therapeutic targets in their own right. In this review, we explore the possibilities for therapeutic targeting of the shelterin complex.
Keywords: shelterin, telomere, telomerase, telosome, targeting, novel therapeutics, cancer, senescence
Introduction
In recent years, understanding of the molecular mechanisms underlying tumour cell biology has provided a variety of potential targets for drug development. One important current goal is to determine the best targets for therapeutic intervention given a particular molecular aetiology. For some targets it will be possible to develop highly specific and effective treatments, though it is likely that many pathways will prove refractory to targeting in the clinical setting. In particular, many leads with promising pre-clinical activity may ultimately prove effective only in specific patient subsets [1]. Therefore, agents with broad spectrum activity will presumably form the backbone of cancer therapy for some time to come.
Immortality is a near-universal phenotype of cancer cells that has attracted considerable interest recently in respect of therapeutic interventions [2]. Most normal somatic cells have a pre-defined maximum lifespan in vitro, bounded by the onset of replicative senescence. In contrast, tumour cells circumvent senescence and divide indefinitely. The complete network of signalling events that underlie senescence establishment and maintenance are poorly understood. However, the central role of telomere homeostasis is now firmly established [3].
Mammalian telomeric DNA comprises extended repeats of the sequence TTAGGG ending in a 3′ G-rich single stranded overhang on both termini. The natural end must be protected from recognition as damaged DNA to prevent cell cycle arrest in proliferating cells. In normal somatic cells the end replication problem, oxidative stress and processing by nucleases cause progressive telomere shortening during each round of cell division. Ultimately, compromised protection causes DNA damage signalling at critically shortened telomeres and telomere dependent senescence [3]. In contrast, most cancer cells aberrantly express telomerase to counteract telomere attrition, maintaining telomeres at a stable length [4].
Telomerase is present in the vast majority of human tumours and directly controls immortality, whereas its core RNA (hTR) and protein (hTERT) subunits are transcriptionally repressed in normal cells [2]. Thus, it is widely regarded as a highly attractive cancer target and has been the subject of intense drug development efforts. Thorough pre-clinical validation of multiple telomerase targeting approaches in cancer and normal cells has proven reproducibility of results across multiple in vitro and in vivo model systems in a large number of studies [2, 4]. Telomerase enzyme inhibition and targeting of hTR in cancer cells generally results in progressive telomere shortening and delayed onset senescence in a telomere length dependent manner, while a rapid growth inhibition and apoptosis induced by dysfunctional telomeres has been documented with hTERT targeting agents [5, 6]. In contrast, normal cells are usually unaffected. Encouragingly, several telomerase-directed therapies are now in clinical trial [2, 4].
Telomerase inhibition with the oligonucleotide enzyme inhibitor GRN163L provides indisputable pre-clinical proof of concept that induction of telomere dysfunction in cancer cells is an attractive therapeutic mechanism and there is good reason to be optimistic about its clinical prospects [2, 4]. However, evaluation is at an early stage and in a worst-case scenario that efficacy is not demonstrated, there are currently no alternative small molecule telomerase enzyme inhibitors scheduled for clinical trials.
A second class of agent directly targeting telomeric DNA secondary structure have also been investigated and found to cause toxicity in cancer cells (G-quadruplex (G4) targeting agents, GTAs). It was originally envisaged that these would block access of telomerase to the G-overhang. However, an emerging consensus is that GTAs elicit their effects at least in part by affecting the specialized telomere capping complex shelterin [7]. Recent studies comparing sensitivity of normal and cancer cells to GTAs combined with growing evidence of in vivo efficacy now lend support to the view that many of the agents in this class will display an acceptable therapeutic index in the pre-clinical setting. These findings suggest that targeting shelterin directly might also have acceptable specificity for cancer cells.
Targeting the telomere
Telomeric DNA is able to adopt a ‘basket-like’ secondary structure in vitro (G4 DNA) resulting from planar stacking of Hoogsteen bonded G-tetrads formed from guanine bases of adjacent telomere repeats. Evidence from direct labelling experiments suggests that telomeric G4 structure also exists in vivo where, like the t-loop, it may provide 3′ end protection. Telomere repeat binding factor 2 (TRF2) affects formation of telomeric G4 in vitro and, conversely, G4 DNA may affect the function of shelterin components in vivo, particularly protection of telomeres 1 (POT1) [8, 9]. The main attempt so far to directly target telomeres in cancer cells is through the use of agents that bind and stabilize G4 DNA (GTAs). Because G4 DNA must be resolved to allow telomerase access, GTAs were originally envisaged as inhibitors of telomerase primer extension, though they are now also known to cause more rapid toxic effects. Many chemically diverse GTAs have been described [7]. Our focus is on the development paths of three agents that have come close to clinical testing.
A diamidoanthraquinone was the first reported GTA, showing slight preference for binding G4 over duplex DNA [10]. Intensive structure-activity optimization led to development of the trisubsti-tuted acridine BRACO19 [11]. BRACO19 inhibits telomerase activity in cancer cells at sub-toxic concentrations and can reduce telomere length, though rapid telomere length independent growth suppression with increased telomere fusions has also been observed [12–14]. Growth suppression of uterine cancer and paclitaxel pre-treated vulval carcinoma xenografts was observed following delivery by the intraperitoneal route, though oral dosing was found to be inactive, presumably because the compound exhibits poor epithelial transport and is relatively unstable [12, 13, 15]. Derivative AS1410 was lead compound for a phase I trial planned by Antisoma Plc (London, UK). In 2005, the company indicated to investors that the trial would not commence because of compound toxicity unrelated to mechanism of action and that another lead would be selected (http://www.antisoma.com). In January 2008, collaboration was established with Argenta Discovery to deliver improved GTA candidates (http://www.argentadiscovery.com).
RHPS4 is a pentacyclic acridium salt with good specificity for binding triplex and G4 DNA over other conformations. It inhibits telomerase activity, has relatively low toxicity, is efficiently taken up into breast and lung cancer cells and is stable in culture medium [16]. Sub-toxic doses of RHPS4 inhibit telomerase in several cell lines but can also lead to rapid growth suppression and G1 arrest in the absence of significant telomere shortening [17]. However, analysis of the sensitivities of 36 tumour cell lines established that short telomere length does correlate with RHPS4 sensitivity [18].
The compound exhibits broad spectrum activity against a range of tumour cell lines in vitro and in xenograft models of melanoma and uterine, prostate, colorectal, breast and lung cancer [17–20]. Furthermore, it efficiently potentiates the activity of several other chemotherapy agents. However, context dependent effects have been observed: combination with paclitaxel was synergistic in MCF7 breast cancer cells but antagonistic in M14 melanoma cells [18, 19]. In 2006, Pharminox agreed in-licensing of rights to preclinical development of RHPS4 (http://www.pharminox.com). Two related acridinium salts were recently identified as potential backup leads on the basis of improved quadruplex binding specificity and low non-specific toxicity [21]. Additionally, a new and more flexible synthetic route has been described for RHPS4 and substituted derivatives [22].
Telomestatin, a natural macrocyclic pentaoxazole isolated from Streptomyces anulatus, is among the most efficient G4 stabilizing agents discovered [23]. Telomestatin inhibits telomerase activity and causes telomere shortening and apoptosis in a range of cancer cell lines in vitro and inhibits growth of leukaemia xenografts [24, 25]. Treatment also augmented apoptosis induced by daunorubicin, mitoxantrone and vincristine in human leukaemia cell lines and enhanced inhibition of colony formation by imatinib in primary chronic myeloid leukaemia (CML) cells [26]. in vivo evidence of telomestatin efficacy is currently limited, though suppression of human leukaemia cell xenografts has been shown [25].
The pharmaceutical company Sosei was to undertake collaborative pre-clinical development of telomestatin (GM-95/SOT-095) (http://www.sosei.com). However, in a 2005 pipeline review the company refocused on products in later phases of development. Low yield has presumably adversely affected the telomestatin development path: US patent 6613759 describes telomestatin purification yielding 3.2 mg from 84 L Streptomyces anulatus culture. Total synthesis is complex, low yield, and proved refractory to a variety of schemes [27, 28]. However, considerable interest surrounds chemistry of macrocyclic oxazoles in general. Synthetic routes for related compounds including telomestatin derivatives have been reported and these compounds are also under investigation as GTAs [29].
Though most GTAs do appear to inhibit telomerase activity, their effects are likely to be overestimated by the telomere repeat amplification protocol (TRAP) assay [7]. An emerging consensus is that telomerase inhibition reflects only part of the activities of these compounds. Generally speaking, high concentrations induce rapid cytotoxicity prior to the onset of telomere shortening accompanied by a telomere uncapping phenotype. Observed effects include telomeric fusions in the absence of significant telomere shortening, degradation of the 3_ overhang and loss of POT1 and/or TRF2 binding (reviewed in [6, 8]).
Interestingly GTAs also elicit a growth suppressive effect on cells utilizing the alternative recombination based mechanism of telomere maintenance, alternative lengthening of telomeres (ALT) [6]. ALT is active in around 10–15% of human tumours and prevalent in certain tumour types with poor outcome including those of mesenchymal origin [30]. These cells do not express telomerase activity so are generally refractory to telomerase inhibition, making the effective targeting of ALT cells an additional advantage to GTAs over other telomerase inhibitors. RHPS4 was shown to induce short-term growth arrest in the ALT cell line GM847 [17] in addition to interfering with telomere replication to induce telomere dysfunction and Ataxia Telangiectasia and Rad3 related (ATR)-dependent Ataxia Telangiectasia Mutated (ATM) signalling [31]. ALT cells are characterized by long heterogeneous telomeres, which provide a larger substrate for GTAs to bind. This may enhance the efficacy of G4 stabilization and prevent recombination required for telomere maintenance. Consistent with this telomestatin has been observed to block topoisomerase III recruitment to telomere G4 DNA in ALT cells resulting in telomere uncapping and DNA damage signalling [32].
Despite these potentially non-selective effects on telomerase there is growing evidence that GTAs exhibit tumour specificity. BRACO19 caused acute toxicity in breast, prostate, colorectal, ovarian, lung and gastric cancer cell lines at lower concentrations than in two normal lung fibroblast strains [33]. Concentrations of telomestatin effective against leukaemia cells had little effect on normal CD34+ cells or normal fibroblasts while concentrations causing acute toxicity and telomere uncapping in cervical and breast cancer cells did not affect normal fibroblast or breast epithe-lial cells [25, 26, 34]. Similarly, RHPS4 treatment induced DNA damage signalling, telomere dysfunction and toxicity in melanoma cells but not in normal fibroblasts [20]. Alongside promising results from in vivo models, it is possible that many tumour cells may be ‘primed’ for telomere dependent death, suggesting that targeting shelterin directly may also have specificity.
Composition of the shelterin complex
The core reactions of telomere homeostasis are controlled by the six-member complex shelterin, comprising the proteins TRF1 and TRF2), POT1, TRF1 interacting protein 2 (TIN2), transcriptional repressor/activator protein 1 (RAP1) and POT1- and TIN2-organizing protein (TPP1 [PTOP/TINT1/PIP1]) [3]. Double stranded telomeric DNA is bound directly by homodimers of TRF1 and TRF2 which differentially recruit other proteins to the telosome, including shelterin proteins TIN2 and RAP1 (recruited by TRF1 and 2, respectively), in addition to accessory factors with roles in DNA damage signalling and repair such as Apollo [35]. The single stranded G-overhang is bound by the POT1/TPP1 complex, in which TPP1 stimulates POT1 recruitment to the telomere in vitro and in vivo[36]. Finally, the single strand and double strand binding subcomplexes are physically linked via interaction between TPP1 and TIN2 [3].
TRF1/2 share similar domain organization, though the N-terminus of TRF1 has an acidic region, whereas that of TRF2 is rich in Gly/Arg residues (basic region). The TRF2 N-terminus displays sequence non-specific DNA binding activity targeting to replication forks and Holliday junctions which may affect telomere replication and end protection [3, 37]. TRF1/2 perform a range of DNA remodelling activities: TRF1 bends DNA and promotes DNA looping and strand pairing by binding to non-adjacent half sites with conformational flexibility [3]. TRF2 binding to the junction of the G-overhang promotes formation of t-loops, which provide 3′ end protection by sequestration of the G-overhang within the telomere duplex. Recent observations point to a mechanism involving DNA melting mediated by super-coiling [3, 38].
Shelterin proteins diversely affect telomere length, with TRF1, TIN2 and RAP1 implicated in negative length regulation. One aspect of TIN2 activity appears to be suppression of TRF1 PARsylation by tankyrase, a telomere-associated PARP which targets TRF1 to the proteasome pathway [39]. TRF2 was also characterized as a negative regulator in overexpression experiments; however, TRF2 inhibition, like POT1, results in rapid telomere deprotection [3, 6, 40]. POT1 appears to have a dual role in length regulation: protection of the single stranded region may prevent telomerase access and in this context POT1 has been implicated as the effector of TRF1 dependent negative length regulation [41]. However, TPP1 directly interacts with telomerase, recruiting it to the telomere [42] and the POT1/TPP1 complex stimulates telomerase processivity [36, 43].
In addition to length regulation, shelterin proteins play essential roles in suppression of DNA damage signalling and inappropriate repair by homologous recombination and non-homologous end-joining. Disruption of these protective functions (telomere ‘uncapping’) occurs in ageing normal cells or can be achieved by dominant negative and knockdown approaches targeting TRF2 or POT1 which are essential for suppression of ATM and ATR, respectively [44]. TRF2 or POT1 presumably act in part by blocking access of the Mre11/Rad50/Nbs1 complex (MRN) and replication protein A (RPA) damage sensors to telomeric DNA [3]. Similar to the ultimate outcome of telomerase inhibition, though with far more rapid effects, such interventions induce highly fuso-genic telomeres and telomeric co-localization of DNA damage sig-nalling foci containing activated checkpoint factors including ATM/ATR, 53BP1 and phosphorylated histone H2AX (telomere dysfunction induced foci) [3, 6].
The roles of other shelterin proteins in cooperating with TRF2 and POT1 to suppress damage signalling and repair remain to be fully investigated, though targeting TPP1 or TIN2 can also be cytotoxic at least in some cells [45, 46]. Interestingly, recent observations point to the existence of specific shelterin subcomplexes with distinct roles in modulating telomere protection in vivo[46]. In particular, selective targeting of TIN2 subcomplexes containing POT1/TPP1 or TRF2 using a TIN2 deletion mutant specifically cooperated with p53 deficiency to induce cell death, whereas senescence was induced in p53 wild-type cells [46]. These observations suggest that appropriate pharmacological agents tailored to disrupt specific shelterin components or subcomplexes might elicit a variety of damage phenotypes, potentially with selectivity for different cancer genotypes.
Shelterin targeting approaches
Due to the complexity of drug development even in its early phases, in order to have confidence that targeting the telomere will be fully validated in the clinic it is appropriate to explore a range of targets. Therefore, it is timely to consider strategies for compound discovery with shelterin as the focus. However, the protein components of shelterin are not generally regarded as potential targets in their own right, with intervention studies frequently focused on functional rather than translational questions [6]. Because shelterin components may prove difficult to drug, particularly in the case of potentially targeting protein–protein interactions, novel discovery solutions may need to be applied to identify promising leads. Encouragingly, there is a growing list of small molecule inhibitors of similar unconventional targets. Development of the Bcl2 homology domain 3 (BH3)-mimetic Bcl2 inhibitor ABT-737 using ‘structure/activity relationship (SAR) by nuclear magnetic resonance (NMR)’ provides notable proof-of-concept that fragment based screening coupled with structure led design is an appropriate approach [47].
As an example of the application of this general strategy to shelterin, we submitted the structure of the TRF1/TIN2-peptide interaction ([35] (Protein Data Bank code 3BQO) for computational solvent mapping on the FTMAP server (http://ftmap.bu.edu). The FTMAP algorithm is a computational solution to detect candidate druggable sites on protein surfaces building on observations from NMR-screening and ‘multiple solvent crystal structures’ techniques that druggable ‘hotspots’ can be identified on the basis of their ability to bind many individual small molecule fragments/probes irrespective of affinity [48]. Computational docking of 16 small molecule probes into PDB structures is performed with a fast Fourier transform correlation approach to identify and rank likely clustering sites. The algorithm correctly identifies known drug-binding sites in several proteins [48].
Nine probe clusters were found with the top ranking cluster localized in a cleft adjacent to the TIN2 peptide binding site (Fig. 1A). Mutation either of TRF1-F142 or of TIN2-L260 is sufficient to ablate TRF1/TIN2 interaction in 293T cells [35]. The predicted site is in close juxtaposition with F142 and at the lip of the hydrophobic pocket which interacts with L260. The same analysis performed for the TRF2/TIN2 structure 3BU8 [35] also revealed a candidate drug-gable pocket in TRF2 bounded by the stacked helices 1 and 3 and the loop region connecting helices 8 and 9. This site is distal from the TIN2 probe, though might conceivably play a role in other TRF2 protein interactions. Similarly a structure was recently reported for an SCFFb_4-TRF1 interaction. Fb × 4, an F-box protein subunit of Skp, Cullin, F-box containing (SCF) ubiquitin E3 ligase, mediates ubiquitination and degradation of TRF1. This structure may represent an additional target for development of small molecule inhibitors [49]. Given the considerable structural understanding of several shelterin components [3], structure based design using similar algorithms seems a rational approach to identify suitable ligands.
Fig 1.
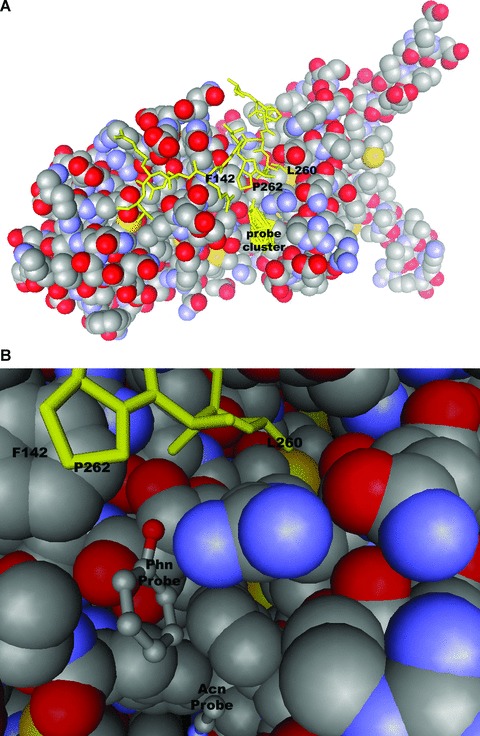
Computational solvent mapping of TRF1/TIN2 interactions. Probes were docked using fast Fourier transform correlation (http://ftmap.bu.edu). (A) shows location of the top ranking cluster in space filling model of TRF1. (B) Orientation of phenol probe adjacent to critical TRF1/TIN2 interacting residues. Stick representation of TIN2 peptide and probe clusters are highlighted in yellow.
Cell based assays could provide a second general approach to identify shelterin inhibitors [2]. Importantly, many interactions could be addressed without pre-existing knowledge of the pathway anatomy. We recently reported that promoter screening can identify inhibitors of hTERT expression [50]. Similar assays might be justified for some shelterin related targets such as TRF2, which is overexpressed in several human tumours and may play a role in drug resistance in some settings [6, 51, 52]. Similarly the telomere uncapping phenotype itself could be used in a screening approach to identify potential inhibitors of telomere dysfunction or telomerase itself. In addition, the availability of a good cellular model of telomere uncapping will enable further investigation of the signalling events underlying the telomere uncapping response and may identify new biomarkers of telomere dysfunction, which could aid future drug discovery strategies targeting the telomere (Fig. 2A). We designed a cell based screening assay, which utilizes an adenoviral vector expressing a form of hTR mutated in the template region for reverse transcription by the telomerase catalytic subunit hTERT (Ad-hTR-mut). When incorporated into telomeres this template synthesizes mutant telomere repeats of the sequence TATATATATAA [53], which are predicted to be incapable of binding key components of the shelterin complex, POT1, TRF1 and TRF2. Expression of this construct in cancer cell lines induces rapid telomere uncapping, DNA damage signalling and cyto-toxicity in an hTERT-dependent manner [53–57], thereby circumventing the problem of phenotypic lag which has hampered identification of telomerase inhibitors through screening assays in the past. In the context of this screening assay any inhibitors of telomere uncapping or the downstream signalling events will result in increased cell viability as measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetra-zolium bromide (MTT) assay, therefore any compounds inducing non-specific toxic effects will be removed in the initial screening process. With an estimated proportion of 30% of drugs failing in clinical trial due to non-specific toxicity [58] this is a tantalizing prospect from a drug development point of view.
Fig 2.
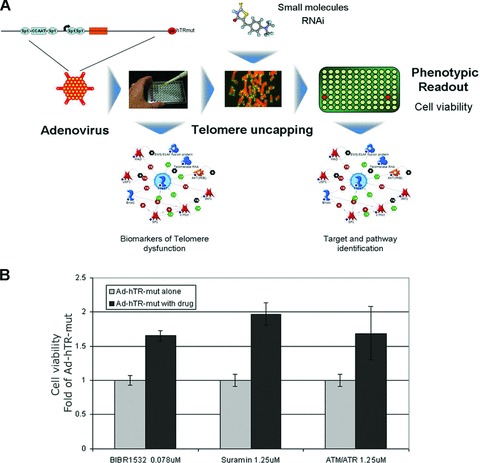
Cell based screening for inhibitors of telomere uncapping. (A) Infection with an adenoviral vector expressing mutant hTR (Ad-hTR-mut) induces rapid telom-ere uncapping that can be used as a model system to investigate novel bio-markers or signalling events downstream of telomere dysfunction, or as a screening approach for inhibitors of these pathways. (B) Colorectal carcinoma cell line HCT116 was infected with Ad-hTR-mut. One day after infection cells were drugged with 1.25 μM Suramin or ATM/ATR inhibitor CGK733 (Both Merck, Darmstadt, Germany) or 0.078 μM BIBR1532 (Tocris Bioscience, Ellisville, MO, USA). Five days after infection cell viability was assessed by MTT cytotoxicity assay. Viability of treated cells was expressed as fold of virus alone.
Three validated reagents, known telomerase inhibitors BIBR1532 [59, 60] and suramin [61] and commercially available general inhibitor of ATM/ATR kinase, postulated to rescue viral cytotoxicity by blocking incorporation of mutant telomere repeats through inhibition of telomerase in the case of BIBR1532 and Suramin [60, 61], or by blocking the downstream DNA damage signalling that ensues from telomere uncapping in the case of the ATM/ATR inhibitor [62], increased cell viability compared to Ad-hTR-mut alone (Fig. 2B). These proof-of-concept studies show the power of such a model system in combination with well-defined reagents including small molecules or siRNA, as a screening approach and as a tool to probe signalling pathways downstream of telomere uncapping. More detailed investigation of shelterin expression profiles is clearly needed and well validated and standardized biomarker assays would be an advantage in this respect.
Inhibiting the enzymes that post-translationally modify shelterin could also be an attractive therapeutic mechanism, as has previously been suggested for telomerase. Tankyrase-1 is widely considered a good target in this regard, while a new candidate is the Pin1 prolyl isomerase which also contributes to TRF1 instability and might provide an alternative or complementary target [39, 63]. Other known modifications of TRF1 include phosphorylation by Plk1 which promotes DNA binding [64]. Therefore, blocking negative regulators of Plk1 might similarly enhance TRF1 telomere association. Because regulation of the stability and/or localization of shelterin components is of major functional importance for telomere biology, existing functional fluorescent tagged shelterin expression constructs could rapidly be put to use as screening reagents to interrogate this class of pathways [65, 66]. Importantly, screening could further enhance understanding of telomere homeostasis even if not all interactions constitute valid therapeutic targets.
Conclusion
To identify potential new anti-cancer agents, knowledge from mechanistic studies of tumour cell biology must be employed to evaluate potential targets in the translational setting. Increasing evidence suggests that directly targeting telomeric DNA has acceptable specificity for cancer cells. Therefore, agents directed against the shelterin complex may also have anti-tumour activity. A growing list of examples suggests that unconventional targets should not be ignored in drug discovery. Approaches such as structure-led design and cell-based screening might provide pharmacological access to ‘undruggable’ proteins in the shelterin complex. The most effective telomere directed therapies will be identified by exploration of all targets using suitable assays based on mechanistic understanding of telomere homeostasis and the unique biology of each candidate. However, well-designed drug discovery assays could generate novel small molecule tools to further probe telomere biology even if high quality clinical leads are not identified.
Acknowledgments
Work in the authors laboratory is supported by Cancer Research UK, European Community grant Health-F2–2007-200950 and Glasgow University.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Gazdar AF. Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene. 2009;28:S24–31. doi: 10.1038/onc.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Keith WN, Bilsland AE. Targeting telom-erase: therapeutic options for cancer treatment. In: Rudolph KL, editor. Telomeres and telomerase in ageing, disease, and cancer. Berlin: Springer-Verlag; 2008. pp. 247–283. [Google Scholar]
- 3.Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–34. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- 4.Harley CB. Telomerase and cancer therapeutics. Nat Rev Cancer. 2008;8:167–79. doi: 10.1038/nrc2275. [DOI] [PubMed] [Google Scholar]
- 5.Folini M, Brambilla C, Villa R, et al. Antisense oligonucleotide-mediated inhibition of hTERT, but not hTERC, induces rapid cell growth decline and apoptosis in the absence of telomere shortening in human prostate cancer cells. Eur J Cancer. 2005;41:624–34. doi: 10.1016/j.ejca.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 6.Folini M, Gandellini P, Zaffaroni N. Targeting the telosome: therapeutic implications. Biochim Biophys Acta. 2009;1792:309–16. doi: 10.1016/j.bbadis.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 7.De Cian A, Lacroix L, Douarre C, et al. argeting telomeres and telomerase. Biochimie. 2008;90:131–55. doi: 10.1016/j.biochi.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 8.Lipps HJ, Rhodes D. G-quadruplex structures: in vivo evidence and function. Trends Cell Biol. 2009;19:414–22. doi: 10.1016/j.tcb.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 9.Pedroso IM, Hayward W, Fletcher TM. The effect of the TRF2 N-terminal and TRFH regions on telomeric G-quadruplex structures. Nucleic Acids Res. 2009;37:1541–54. doi: 10.1093/nar/gkn1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun D, Thompson B, Cathers BE, et al. Inhibition of human telomerase by a G-quadruplex-interactive compound. J Med Chem. 1997;40:2113–6. doi: 10.1021/jm970199z. [DOI] [PubMed] [Google Scholar]
- 11.Harrison RJ, Cuesta J, Chessari G, et al. Trisubstituted acridine derivatives as potent and selective telomerase inhibitors. J Med Chem. 2003;46:4463–76. doi: 10.1021/jm0308693. [DOI] [PubMed] [Google Scholar]
- 12.Burger AM, Dai F, Schultes CM, et al. The G-quadruplex-interactive molecule BRACO-19 inhibits tumor growth, consistent with telomere targeting and interference with telomerase function. Cancer Res. 2005;65:1489–96. doi: 10.1158/0008-5472.CAN-04-2910. [DOI] [PubMed] [Google Scholar]
- 13.Gowan SM, Harrison JR, Patterson L, et al. A G-quadruplex-interactive potent small-molecule inhibitor of telomerase exhibiting in vitro and in vivo antitumor activity. Mol Pharmacol. 2002;61:1154–62. doi: 10.1124/mol.61.5.1154. [DOI] [PubMed] [Google Scholar]
- 14.Incles CM, Schultes CM, Kempski H, et al. A G-quadruplex telomere targeting agent produces p16-associated senescence and chromosomal fusions in human prostate cancer cells. Mol Cancer Ther. 2004;3:1201–6. [PubMed] [Google Scholar]
- 15.Taetz S, Murdter TE, Zapp J, et al. Decomposition of the telomere-targeting agent BRACO19 in physiological media results in products with decreased inhibitory potential. Int J Pharm. 2008;357:6–14. doi: 10.1016/j.ijpharm.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 16.Cookson JC, Heald RA, Stevens MF. Antitumor polycyclic acridines. 17. Synthesis and pharmaceutical profiles of pentacyclic acridinium salts designed to destabilize telomeric integrity. J Med Chem. 2005;48:7198–207. doi: 10.1021/jm058031y. [DOI] [PubMed] [Google Scholar]
- 17.Gowan SM, Heald R, Stevens MF, et al. Potent inhibition of telomerase by small-molecule pentacyclic acridines capable of interacting with G-quadruplexes. Mol Pharmacol. 2001;60:981–8. doi: 10.1124/mol.60.5.981. [DOI] [PubMed] [Google Scholar]
- 18.Cookson JC, Dai F, Smith V, et al. Pharmacodynamics of the G-quadruplex-stabilizing telomerase inhibitor 3,11-diflu-oro-6, 8, 13-trimethyl-8H-quino[4, 3, 2-kl]acridinium methosulfate (RHPS4) in vitro: activity in human tumor cells correlates with telomere length and can be enhanced, or antagonized, with cytotoxic agents. Mol Pharmacol. 2005;68:1551–8. doi: 10.1124/mol.105.013300. [DOI] [PubMed] [Google Scholar]
- 19.Leonetti C, Scarsella M, Riggio G, et al. G-quadruplex ligand RHPS4 potentiates the antitumor activity of camptothecins in preclinical models of solid tumors. Clin Cancer Res. 2008;14:7284–91. doi: 10.1158/1078-0432.CCR-08-0941. [DOI] [PubMed] [Google Scholar]
- 20.Salvati E, Leonetti C, Rizzo A, et al. Telomere damage induced by the G-quadruplex ligand RHPS4 has an antitumor effect. J Clin Invest. 2007;117:3236–47. doi: 10.1172/JCI32461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng MK, Modi C, Cookson JC, et al. Antitumor polycyclic acridines. 20. Search for DNA quadruplex binding selectivity in a series of 8,13-dimethylquino[4,3,2-kl] acridinium salts: telomere-targeted agents. J Med Chem. 2008;51:963–75. doi: 10.1021/jm070587t. [DOI] [PubMed] [Google Scholar]
- 22.Kristensen JL. Synthesis of RHPS4 via an anionic ring closing cascade. Tetrahedron Letters. 2008;49:2351–2354. [Google Scholar]
- 23.Kim MY, Vankayalapati H, Shin-Ya K, et al. Telomestatin, a potent telomerase inhibitor that interacts quite specifically with the human telomeric intramolecular g-quadruplex. J Am Chem Soc. 2002;124:2098–9. doi: 10.1021/ja017308q. [DOI] [PubMed] [Google Scholar]
- 24.Binz N, Shalaby T, Rivera P, et al. Telomerase inhibition, telomere shortening, cell growth suppression and induction of apoptosis by telomestatin in childhood neuroblastoma cells. Eur J Cancer. 2005;41:2873–81. doi: 10.1016/j.ejca.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 25.Tauchi T, Shin-ya K, Sashida G, et al. Telomerase inhibition with a novel G-quadruplex-interactive agent, telomestatin: in vitro and in vivo studies in acute leukemia. Oncogene. 2006;25:5719–25. doi: 10.1038/sj.onc.1209577. [DOI] [PubMed] [Google Scholar]
- 26.Tauchi T, Shin-Ya K, Sashida G, et al. Activity of a novel G-quadruplex-interactive telomerase inhibitor, telomestatin (SOT-095), against human leukemia cells: involvement of ATM-dependent DNA damage response pathways. Oncogene. 2003;22:5338–47. doi: 10.1038/sj.onc.1206833. [DOI] [PubMed] [Google Scholar]
- 27.Deeley J, Bertram A, Pattenden G. Novel polyoxazole-based cyclopeptides from Streptomyces sp Total synthesis of the cyclopeptide YM-216391 and synthetic studies towards telomestatin. Org Biomol Chem. 2008;6:1994–2010. doi: 10.1039/b802477d. [DOI] [PubMed] [Google Scholar]
- 28.Doi T, Yoshida M, Shin-ya K, et al. Total synthesis of (R)-telomestatin. Org Lett. 2006;8:4165–7. doi: 10.1021/ol061793i. [DOI] [PubMed] [Google Scholar]
- 29.Tera M, Lida K, Ishizuka H, et al. Synthesis of a potent G-quadruplex-binding macrocyclic heptaoxazole. Chembiochem. 2009;10:431–5. doi: 10.1002/cbic.200800563. [DOI] [PubMed] [Google Scholar]
- 30.Henson JD, Neumann AA, Yeager TR, et al. Alternative lengthening of telomeres in mammalian cells. Oncogene. 2002;21:598–610. doi: 10.1038/sj.onc.1205058. [DOI] [PubMed] [Google Scholar]
- 31.Rizzo A, Salvati E, Porru M, et al. Stabilization of quadruplex DNA perturbs telomere replication leading to the activation of an ATR-dependent ATM signaling pathway. Nucleic Acids Res. 2009;37:5353–64. doi: 10.1093/nar/gkp582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Temime-Smaali N, Guittat L, Sidibe A, et al. The G-quadruplex ligand telomestatin impairs binding of topoisomerase IIIalpha to G-quadruplex-forming oligonucleotides and uncaps telomeres in ALT cells. PLoS ONE. 2009;4:e6919. doi: 10.1371/journal.pone.0006919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gunaratnam M, Greciano O, Martins C, et al. Mechanism of acridine-based telom-erase inhibition and telomere shortening. Biochem Pharmacol. 2007;74:679–89. doi: 10.1016/j.bcp.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 34.Tahara H, Shin-Ya K, Seimiya H, et al. G-Quadruplex stabilization by telomestatin induces TRF2 protein dissociation from telomeres and anaphase bridge formation accompanied by loss of the 3′ telomeric overhang in cancer cells. Oncogene. 2006;25:1955–66. doi: 10.1038/sj.onc.1209217. [DOI] [PubMed] [Google Scholar]
- 35.Chen Y, Yang Y, van Overbeek M, et al. A shared docking motif in TRF1 and TRF2 used for differential recruitment of telomeric proteins. Science. 2008;319:1092–6. doi: 10.1126/science.1151804. [DOI] [PubMed] [Google Scholar]
- 36.Xin H, Liu D, Wan M, et al. TPP1 is a homologue of ciliate TEBP-beta and interacts with POT1 to recruit telomerase. Nature. 2007;445:559–62. doi: 10.1038/nature05469. [DOI] [PubMed] [Google Scholar]
- 37.Poulet A, Buisson R, Faivre-Moskalenko C, et al. TRF2 promotes, remodels and protects telomeric Holliday junctions. EMBO J. 2009;28:641–51. doi: 10.1038/emboj.2009.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amiard S, Doudeau M, Pinte S, et al. A topological mechanism for TRF2-enhanced strand invasion. Nat Struct Mol Biol. 2007;14:147–54. doi: 10.1038/nsmb1192. [DOI] [PubMed] [Google Scholar]
- 39.Seimiya H, Muramatsu Y, Ohishi T, et al. Tankyrase 1 as a target for telomere-directed molecular cancer therapeutics. Cancer Cell. 2005;7:25–37. doi: 10.1016/j.ccr.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 40.Smogorzewska A, van Steensel B, Bianchi A, et al. Control of human telomere length by TRF1 and TRF2. Mol Cell Biol. 2000;20:1659–68. doi: 10.1128/mcb.20.5.1659-1668.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Loayza D, De Lange T. POT1 as a terminal transducer of TRF1 telomere length control. Nature. 2003;423:1013–8. doi: 10.1038/nature01688. [DOI] [PubMed] [Google Scholar]
- 42.Abreu E, Aritonovska E, Reichenbach P, et al. TIN2-tethered TPP1 recruits human telomerase to telomeres in vivo. Mol Cell Biol. 2010;30:2971–82. doi: 10.1128/MCB.00240-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang F, Podell ER, Zaug AJ, et al. The POT1-TPP1 telomere complex is a telomerase processivity factor. Nature. 2007;445:506–10. doi: 10.1038/nature05454. [DOI] [PubMed] [Google Scholar]
- 44.Denchi EL, de Lange T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature. 2007;448:1068–71. doi: 10.1038/nature06065. [DOI] [PubMed] [Google Scholar]
- 45.Guo X, Deng Y, Lin Y, et al. Dysfunctional telomeres activate an ATM-ATR-dependent DNA damage response to suppress tumorigenesis. EMBO J. 2007;26:4709–19. doi: 10.1038/sj.emboj.7601893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim SH, Davalos AR, Heo SJ, et al. Telomere dysfunction and cell survival: roles for distinct TIN2-containing complexes. J Cell Biol. 2008;181:447–60. doi: 10.1083/jcb.200710028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chonghaile TN, Letai A. Mimicking the BH3 domain to kill cancer cells. Oncogene. 2008;27:S149–57. doi: 10.1038/onc.2009.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brenke R, Kozakov D, Chuang GY, et al. Fragment-based identification of druggable ‘hot spots’ of proteins using Fourier domain correlation techniques. Bioinformatics. 2009;25:621–7. doi: 10.1093/bioinformatics/btp036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zeng Z, Wang W, Yang Y, et al. Structural basis of selective ubiquitination of TRF1 by SCFFbx4. Dev Cell. 2010;18:214–25. doi: 10.1016/j.devcel.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bilsland AE, Hoare S, Stevenson K, et al. Dynamic telomerase gene suppression via network effects of GSK3 inhibition. PLoS ONE. 2009;4:e6459. doi: 10.1371/journal.pone.0006459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dong W, Wang L, Chen X, et al. Upregulation and CpG Island hypomethy-lation of the TRF2 gene in human gastric cancer. Dig Dis Sci. 2010;55:997–1003. doi: 10.1007/s10620-009-0810-8. [DOI] [PubMed] [Google Scholar]
- 52.Ning H, Li T, Zhao L, et al. TRF2 promotes multidrug resistance in gastric cancer cells. Cancer Biol Ther. 2006;5:950–6. doi: 10.4161/cbt.5.8.2877. [DOI] [PubMed] [Google Scholar]
- 53.Kim MM, Rivera MA, Botchkina IL, et al. A low threshold level of expression of mutant-template telomerase RNA inhibits human tumor cell proliferation. Proc Natl Acad Sci USA. 2001;98:7982–7. doi: 10.1073/pnas.131211098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goldkorn A, Blackburn EH. Assembly of mutant-template telomerase RNA into catalytically active telomerase ribonucleoprotein that can act on telomeres is required for apoptosis and cell cycle arrest in human cancer cells. Cancer Res. 2006;66:5763–71. doi: 10.1158/0008-5472.CAN-05-3782. [DOI] [PubMed] [Google Scholar]
- 55.Guiducci C, Cerone MA, Bacchetti S. Expression of mutant telomerase in immortal telomerase-negative human cells results in cell cycle deregulation, nuclear and chromosomal abnormalities and rapid loss of viability. Oncogene. 2001;20:714–25. doi: 10.1038/sj.onc.1204145. [DOI] [PubMed] [Google Scholar]
- 56.Li S, Rosenberg JE, Donjacour AA, et al. Rapid inhibition of cancer cell growth induced by lentiviral delivery and expression of mutant-template telomerase RNA and anti-telomerase short-interfering RNA. Cancer Res. 2004;64:4833–40. doi: 10.1158/0008-5472.CAN-04-0953. [DOI] [PubMed] [Google Scholar]
- 57.Marusic L, Anton M, Tidy A, et al. Reprogramming of telomerase by expression of mutant telomerase RNA template in human cells leads to altered telomeres that correlate with reduced cell viability. Mol Cell Biol. 1997;17:6394–401. doi: 10.1128/mcb.17.11.6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carden CP, Sarker D, Postel-Vinay S, et al. Can molecular biomarker-based patient selection in Phase I trials accelerate anticancer drug development? Drug Discov Today. 2010;15:88–97. doi: 10.1016/j.drudis.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 59.Damm K, Hemmann U, Garin-Chesa P, et al. A highly selective telomerase inhibitor limiting human cancer cell proliferation. EMBO J. 2001;20:6958–68. doi: 10.1093/emboj/20.24.6958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pascolo E, Wenz C, Lingner J, et al. Mechanism of human telomerase inhibition by BIBR1532, a synthetic, non-nucle-osidic drug candidate. J Biol Chem. 2002;277:15566–72. doi: 10.1074/jbc.M201266200. [DOI] [PubMed] [Google Scholar]
- 61.Cristofari G, Reichenbach P, Regamey PO, et al. Low- to high-throughput analysis of telomerase modulators with Telospot. Nat Methods. 2007;4:851–3. doi: 10.1038/nmeth1099. [DOI] [PubMed] [Google Scholar]
- 62.Stohr BA, Blackburn EH. ATM mediates cytotoxicity of a mutant telomerase RNA in human cancer cells. Cancer Res. 2008;68:5309–17. doi: 10.1158/0008-5472.CAN-08-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee TH, Tun-Kyi A, Shi R, et al. Essential role of Pin1 in the regulation of TRF1 stability and telomere maintenance. Nat Cell Biol. 2009;11:97–105. doi: 10.1038/ncb1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu ZQ, Yang X, Weber G, et al. Plk1 phosphorylation of TRF1 is essential for its binding to telomeres. J Biol Chem. 2008;283:25503–13. doi: 10.1074/jbc.M803304200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gomez D, O’Donohue MF, Wenner T, et al. The G-quadruplex ligand telomestatin inhibits POT1 binding to telomeric sequences in vitro and induces GFP-POT1 dissociation from telomeres in human cells. Cancer Res. 2006;66:6908–12. doi: 10.1158/0008-5472.CAN-06-1581. [DOI] [PubMed] [Google Scholar]
- 66.Krutilina RI, Oei S, Buchlow G, et al. A negative regulator of telomere-length protein trf1 is associated with interstitial (TTAGGG)n blocks in immortal Chinese hamster ovary cells. Biochem Biophys Res Commun. 2001;280:471–5. doi: 10.1006/bbrc.2000.4143. [DOI] [PubMed] [Google Scholar]