

Abstract
Alzheimer's disease (AD) is an aging-related progressive neurodegenerative disorder. Previous studies suggested that various soluble Aβ species are neurotoxic and able to activate apoptosis and autophagy, the type I and type II programmed cell death, respectively. However, the sequential and functional relationships between these two cellular events remain elusive. Here we report that low molecular weight Aβ triggered cleavage of caspase 3 and poly (ADP-ribose) polymerase to cause neuronal apoptosis in rat cortical neurons. On the other hand, Aβ activated autophagy by inducing autophagic vesicle formation and autophagy related gene 12 (ATG12), and up-regulated the lysoso-mal machinery for the degradation of autophagosomes. Moreover, we demonstrated that activation of autophagy by Aβ preceded that of apoptosis, with death associated protein kinase phosphorylation as the potential molecular link. More importantly, under Aβ toxicity, neurons exhibiting high level of autophagosome formation were absent of apoptotic features, and inhibition of autophagy by 3-methylade-nine advanced neuronal apoptosis, suggesting that autophagy can protect neurons from Aβ-induced apoptosis.
Keywords: Alzheimer's disease, β-amyloid, autophagy, apoptosis, death associated protein kinase
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disorder related to aging. Among the pathological hallmarks, loss of synaptic communications and even neurons in hippocampus and cortex have been considered as the most immediate causes of cognitive impairment [1, 2]. Recent reports suggested that various soluble Aβ species can induce neurotoxicity [3–5], possibly through different types of programmed cell death (PCD) including apoptosis, the type I PCD, and autophagy, the type II PCD [6–8].
Apoptosis is a tightly regulated machinery to regulate cell death. It can be activated through intrinsic pathway (mitochondria and endoplasmic reticulum) and extrinsic death receptor-related pathway [7]. Upon activation, the apoptotic signal transduction is proceeded by a hierarchy of upstream initiator and downstream effector caspase proteases. The effector caspases then cleave a variety of protein substrates and cause cell death, by exhibiting condensation and fragmentation of nuclei. Apoptosis can also be triggered through caspase-independent pathway, in which apoptosis inducing factor (AIF), a pro-apoptotic factor, is released from mitochondria when the mitochondrial integrity is compromised. AIF can induce nuclear condensation without caspase activation [9].
Unlike apoptosis which is more clearly a cell killing event, autophagy has been accepted to provide a pro-survival role during cellular stress such as starvation [10] and organelle damage [11]. Autophagy mediates the lysosomal degradation of damaged organelles and protein aggregates [12]. During autophagy (referring to macroautophagy herein), cytosol components to be degraded are engulfed by limiting membrane to form an autophagosome, which then fuses with lysosomes for content breakdown. Useful substances will be recycled for energy and other biosynthetic processes. Although autophagy has shown cytoprotective properties, dysregulated activation of it causes irreversible cellular atrophy and thus leads to cell death [13].
Activation of both autophagy and apoptosis by Aβ has been reported in literature [8, 11, 14]. However, the sequential relationship between them has not been clearly elucidated. Crosstalk and potential functional relationship between these two cellular events also remains elusive. Here we report that low molecular weight (MW) Aβ triggered neuronal apoptosis through cleavage of caspase 3 and poly (ADP-ribose) poly-merase (PARP). On the other hand, Aβ activated autophagy by inducing autophagosome formation and autophagy related gene 12 (ATG12). Aβ also enhanced the lysosomal machinery in neurons. We demonstrated that activation of autophagy by low MW Aβ preceded that of apoptosis, with the potential molecular switch as death associated protein kinase (DAPK). More importantly, when stressed with Aβ, neurons exhibiting high numbers of autophagosomes were of negative immunoreactivity of cleaved caspase 3, and inhibition of autophagy advanced neu-ronal apoptosis, suggesting that autophagy can protect neurons from Aβ-induced apoptosis.
Materials and methods
Antibodies and chemicals
Cleaved caspase 3 (Asp 175) (1:1000 for Western blotting; 1:100 for immunocytochemistry), poly (ADP-ribose) polymerase (PARP) (1:1000), beclin-1 (BECN1) (1:100), ATG12 (1:100), AIF (1:100) antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Lysosome-associated membrane protein 2 (LAMP2) (1:100) antibody was from Abcam (Cambridge, MA, USA). Phospho-DAPK (p-DAPK) (1:2000) and β actin (1:10000) were from Sigma-Aldrich, Inc. (Saint Louis, MO, USA). Antibody against LC3 (1:1000) was from Medical and Biological Lab (Naka-ku Nagoya, Japan). Anti-Aβ 1–16 or APP antibody (6E10) was purchased from Signet (Covance, NJ, USA). 3-methyladenine (3-MA) was purchased from Sigma (St. Louis, MO, USA).
Preparation of oilgomeric Aβ
Aβ1–42 was purchased from Yale University. The Aβ1–42 peptide powder was first dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (Sigma) by vor-texing. A gentle stream of nitrogen gas was then used to dry the peptide solution, which was afterwards resuspended into anhydrous dimethyl sul-foxide (DMSO) (Sigma) to a final concentration of 2 mM. The peptide was incubated in a sonicator for 30 min. at room temperature. Aliquoted pep-tide was snap frozen in liquid nitrogen for storage. It was later diluted to working concentrations during experiments as specified.
Primary cell cultures of rat cortical neurons and transfection
Primary rat cortical neuronal cultures in Neurobasal medium (Gibco-BRL, Rockville, MO, USA) were prepared from the cerebral cortex of embryonic day 17 Sprague-Dawley rats (The Laboratory Animal Unit, The University of Hong Kong, accredited by Association for Assessment and Accreditation of Laboratory Animal Care International) as described before with modifications [15]. Briefly, cortices were mechanically dissociated in 1× PBS supplemented with glucose (18 mM). Neurons were later seeded onto 6-well culture plates, or with 10 mm glass cover slips, or MatTek glass-bottomed dishes (MatTek, Ashland, MA, USA) coated with poly-L-lysine (25 μg/ml) at 1 × 106, 3.5 × 105 and 3 × 105 cells per well, respectively. Neurons were cultured in Neurobasal medium supplemented with B-27 (Gibco-BRL), L-glutamine (2 mM) (Sigma), β-mercaptoethanol (10 μM) and penicillin/streptomycin (100 U and 100 μg/ml) for 7 days at 37°C in a humidified 5% CO2 incubator, with half of the growth media being refreshed on day 4. On day 5, neurons were transfected with specified constructs using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) for 1 hr. Neurons were treated with oligomeric Aβ 48 hrs after transfection. LC3-DsRed was a gift from Dr. N.S. Wong (The University of Hong Kong, Hong Kong). Mito-GFP plasmid was kindly given by Dr. S.K. Kong (School of Biomedical Science, The Chinese University of Hong Kong). The ER retention signal encoding plasmid (KDEL-GFP) was purchased from Invitrogen.
Immunocytochemical analysis and live cell imaging
Immunocytochemical staining was performed as described elsewhere with modifications [11, 16]. Briefly, neurons were cultured onto 6-well culture plates with 10 mm glass cover slips. After treatment, neurons were fixed with paraformaldehyde of 4% for 20 min. They were then permeabilized with Triton X-100 of 0.1% in TBS for 5 min. at room temperature, and washed with TBS twice. After blocking with 10% BSA for 1 hr, primary antibody incubation was carried out for 2 hrs at room temperature, or overnight at 4°C for ATG12. After TBS washing, secondary antibodies incubation with anti-rat (for LAMP2) or anti-rabbit Alexa-fluor 488 or 568 (Molecular Probes, Eugene, OR, USA) was carried out at 1:400 dilution for 1 hr at room temperature. Neurons were counterstained with DAPI for 5 min. at room temperature. Carl Zeiss LSM510-Meta system (Göttingen, Germany) was used to capture confocal images. Z-stack images were captured at 0.7 μm intervals. Signal intensity was determined by Image J (NIH, Bethesda, MD, USA).
For live cell imaging experiments, neurons culture was performed on MetTek glass-bottomed dishes coated with poly-L-lysine. After treatment, lysosomes were visualized after incubation with LysoTracker Green® (Molecular Probes) of 200 nM for 15 min. Carl Zeiss LSM510-Meta system (Göttingen, Germany) was used to capture confocal Z-stack images at 0.7 μm intervals.
Protein extraction and Western blot analysis
Protein extraction and Western blot analysis were performed as described elsewhere with modifications [17, 18]. Briefly, after treatment, cortical neurons were lysed to extract total protein lysates by ice-cold lysis buffer containing Tris (10 mM, pH 7.4), NaCl (100 mM), ethylenediaminetetraacetic acid (1 mM), ethylene glycol tetraacetic acid (EGTA) (1 mM), NaF (1 mM), Na4P2O7 (20 mM), Na3VO4 (2 mM) Triton X-100 (1%), glycerol (10%), SDS (0.1%), deoxycholate (0.5%), phenylmethylsulfonyl fluoride (1 mM), protease inhibitor cocktail (Sigma) and phosphatase inhibitor cocktail (Sigma). After brief sonication, lysates were centrifuged at 11,000 × g for 30 min. at 4°C. Protein contents of supernatants were determined by a protein assay kit (Bio-Rad, Hercules, CA, USA). Cytosol and membrane/organelle fraction lysates from cortical neurons were extracted using ProteoExtract® Subcellular Proteome Extraction Kit (Calbiochem, CA, USA) according to product protocols. Sample concentrations were determined by Bradford reagent (Bio-Rad).
Western blot analysis was performed by 12.5% SDS-PAGE. Proteins were then transferred onto polyvinylidene fluoride (PVDF) membrane (GE Healthcare, Little Chalfont, UK). Membranes were blocked with 5% non-fat milk in Tris-buffered saline (pH 7.4) containing Tween-20 of 0.1% for 1 hr at room temperature. Primary antibody incubation was carried out for 1 hr at room temperature for p-DAPK and β actin, 2 hr for LC3, overnight at 4°C for cleaved caspase 3 and PARP. Secondary antibody incubation with horseradish peroxidase-conjugated goat anti-rabbit and goat antimouse antibodies (DAKO, Glostrup, Denmark) was afterwards performed at 1:2000 dilution for 1 hr at room temperature. Bands were visualized with Amersham ECL Plus™ Western Blotting Detection Reagents (Amersham Bioscience, Piscataway, NJ, USA) on X-ray films (Amersham Bioscience).
Molecular size of low MW Aβ peptide prepared was detected by tris-tricine gel. Different concentrations of low MW Aβ peptides were mixed with 2× sample buffer for tris-tricine gel (1:1). After denaturing at 95°C for 5 min., Aβ peptides were loaded onto 16% tris-tricine gel for Western blot analysis. Gel electrophoresis was performed for 1.5 hrs at 100 V. After protein transfer and blocking, primary 6E10 antibody incubation at 1:1000 dilution was carried out at room temperature for 2 hrs. Secondary antibody incubation with horseradish peroxidase-conjugated goat antimouse antibody was then performed at 1:2000 dilution for 1 hr at room temperature. Bands were visualized on X-ray films.
Total RNA extraction, cDNA synthesis and quantitative real-time PCR (Q-PCR)
After treatment, total RNA was extracted from neurons with Trizol reagent (Invitrogen). Cell homogenates were mixed vigorously with chloroform and then centrifuged at 11,000 ×g for 15 min. at 4°C. The upper aqueous layer which contained RNA was mixed with pre-cooled isopropanol to precipitate the RNA. Mixtures were centrifuged at 11,000 ×g for 30 min. at 4°C afterwards. After ethanol wash, RNA pellets were resuspended in DEPC-treated H2O. Concentration of RNA samples was determined by GeneQuantII spec-trophotometer (Pharmacia Biotech, Stockholm, Sweden).
Total RNA isolated from neurons was first reverse-transcribed to cDNA by using SuperScript™ III First-Strand Synthesis System for RT-PCR (Invitrogen), in GeneAmp R PCR System 9700 thermal cycler (PE Biosytems, Foster City, CA, USA). Expression levels of target genes were then determined by using Platinum® SYBR® Green qPCR SuperMix-UDG (Invitrogen). Diluted cDNA samples were mixed with specific primers and SYBR Green mastermix and then incubated in iCycler™ (Bio-Rad) for 40 cycles of 95°C denaturation for 15 sec., 58.7°C annealing for 15 sec. and 72°C extension for 15 sec. Specific gene amplification was confirmed by the melt-curve function on the real-time instrument and 1.5% (w/v) agarose gel electrophoresis. Gene expression was analysed by iCycler™ IQ Optical System Software (Bio-Rad) and the Pfaffl method. Primer sequences used were 5′-AAACGGCTACCACATCCAAG-3′ and 5′-CAATTACAGGGCCTCGAAAG-3′ for 18S ribosomal RNA, 5′-AACTCTGGAGGTCTCGCTCT-3′ and 5′-CCTTAGACCCCTCCATTCTT-3′ for BECN1, 5′-TGGCCTACTGTTCGATCTTC-3′ and 5′-AAGTGAGCCTCAACTGCATC-3′ for ATG5, 5′-GGTATCCGTTCGGAGAGTGA-3′ and 5′-CGTAGTCCTCCCCTTCAACA-3′ for AIF.
Statistical analysis
The results are expressed in means ± S.E.M.. Quantitative data was analysed by one-way ANOVA followed by Student–Newman–Keul test according to the statistical program SigmaStat® (Jandel Scientific, San Jose, CA, USA) to compare the level significance. A P-value less than 0.05 was regarded as significant, at *P < 0.05.
Result
Induction of apoptosis in neurons by low MW Aβ
The present project aimed to study Aβ-induced PCD events, to start with, we at first characterized the molecular size of the low MW Aβ prepared for subsequent experiments. By 16% tris-tricine gel elec-trophoresis followed by 6E10 antibody detection, we found that low MW Aβ1–42 contained Aβ species from monomeric to tetrameric form, with the monomer as the majority of the preparation (Fig. 1A). Incubation in culture media induced the formation of Aβ species of higher MW from 4 hrs, as denoted by asterisks in Fig. 1B.
Fig 1.

Western blotting of low MW Aβ peptide. Sixteen percent tris-tricine SDS-PAGE showing the molecular weight of low MW Aβ (A) of indicated amount loaded, and (B) after incubation in culture media of indicated time. 6E10 antibody was used for the detection of Aβ peptide.
To examine the toxicity of low MW Aβ in causing neuronal cell death, we demonstrated that low MW Aβ induced apoptosis in neurons, but not DMSO (vehicle)-treated control. By Western blot analysis, we showed that low MW Aβ induced cleavage of caspase 3 and PARP, which are typical apoptotic features [7]. Cleavage of these two proteins was shown to be dose- and time-dependent. It first occurred at 18 hrs of Aβ treatment and became prominent from 24 hrs onwards (Fig. 2). We also examined the effect of Aβ on AIF expression and intracellular localization. Q-PCR did not show significant induction of AIF mRNA levels upon Aβ treatment from 4 to 30 hrs (Fig. 3). Immunofluorescent analysis of AIF showed that, in vehicle-treated control, AIF signals co-localized with Mito-GFP at 8 and 24 hrs (Fig. 4). When neurons were treated with Aβ, there was no significant induction of AIF immunoreactiv-ity and the localization of AIF was comparable to the vehicle-treated control at both time-points.
Fig 2.

Low MW Aβ induced cleavage of caspase 3 and PARP. Western blotting of cleaved caspase 3 (Asp 175) and PARP in primary cortical neurons treated with low MW Aβ1–42 of indicated concentrations or DMSO (vehicle) for 4 to 30 hrs. β-actin was used as an internal control. Figure shows the representative result from three independent experiments.
Fig 3.

Low MW Aβ did not significantly affect AIF gene expression. AIF mRNA level in neurons treated with low MW Aβ1–42 of indicated concentrations or DMSO (vehicle) for 4 to 30 hrs was measured by Q-PCR with 18S ribosomal RNA as internal control. Relative gene expression level was analysed with Pfaffl method. Bar chart plots the means ± S.E.M. of a triplicate from three independent experiments. Empty bars indicate the control at each time-point.
Fig 4.

Low MW Aβ did not induce mito-chondrial release and nuclear entry of AIF. Neurons transfected with Mito-GFP were treated with low MW Aβ1–42 of indicated concentrations or DMSO (vehicle) for 8 or 24 hrs. The neurons were stained with anti-AIF antibody. Representative Z-stack confocal images from two independent experiments are shown. Scale bar, 10 μM.
Induction of autophagy in neurons by low MW Aβ
We next sought to examine the effect of Aβ on the activation of autophagy, which is also known as the type II PCD. We trans-fected microtubule-associated protein 1 light chain 3 (LC3)-DsRed construct into neurons and examined its expression upon Aβ challenge. LC3 is a microtubule-associated protein binding protein. It can be post-translationally processed from LC3-I to LC3-II during autophagy and attached to the inner and outer membranes of autophagosomes [19]. Therefore, it is used as a marker for autophagosome formation. By transient expression of LC3-DsRed in neurons, we showed that the florescence intensity in vehicle-treated control was low. However, upon Aβ treatment, the expression of the protein increased with the formation of vesicular LC3 from 8 hrs onwards, in both soma (Fig. 5) and dentritic processes (Fig. 12). The size and number of vesicles increased with time (Fig. 5). An increase in the autophagosomes formation induced by Aβ treatment could also be observed in live neurons (Fig. 10B).
Fig 5.

Low MW Aβ induced the formation of autophagic vesicles. Primary cortical neurons transfected with LC3-DsRed were treated with low MW Aβ1–42 of 10 μM or DMSO (vehicle) for indicated time. Representative Z-stack confocal images from three independent experiments are shown. Scale bar, 10 μM.
Fig 12.

Autophagy protects neurons from Aβ-induced apoptosis. (A) Neurons trans-fected with LC3-DsRed were treated with low MW Aβ1–42 of indicated concentrations or DMSO (vehicle) for 18 hrs. Neurons were then stained with anti-cleaved caspase 3 (Asp 175) antibody. Representative Z-stack con-focal images from two independent experiments are shown. Scale bar, 10 μM. (B) Percentage of LC3-trans-fected cells showing positive immunoreactivity for cleaved caspase 3. Bar chart plots the means ± S.E.M. of three independent experiments. *P < 0.05, represents significant difference from the reference value. Empty bars indicate the control at each time-point.
Fig 10.

Low MW Aβ induced the lysosomal machinery. Neurons were treated with low MW Aβ1–42 of indicated concentrations or DMSO (vehicle) for 24 hrs. (A) Neurons were stained with anti-LAMP2 antibody. Scale bar, 20 μM. (B) Neurons transfected with LC3-DsRed prior treatment were stained with 200 nM LysoTracker Green®. Representative live cell Z-stack confocal images from two independent experiments are shown. Scale bar, 10 μM.
Apart from LC3, we also examined the expression of other genes involved in the autophagy process. By Q-PCR, we found that low MW Aβ did not significantly affect the mRNA level of BECN1 (Fig. 6) after treatment from 4 to 30 hrs, which has been suggested to play a role in the early induction of autophagy [20]. The protein level of BECN1 also showed no significant change after Aβ treatment of 10 μM (0.98 ± 0.03 at 8 hrs) and 20 μM (1.09 ± 0.12 at 8 hrs), as revealed by immunocytochemical analysis (Fig. 7). Low MW Aβ showed no obvious effect on the gene expression level of ATG5 (Fig. 8), which facilitates autophagosome expansion and completion [21]. Although Aβ did not significant up-regulate the total protein level of ATG12 (1.07 ± 0.26 at 10 μM, 1.07 ± 0.13 at 20 μM) (Fig. 9A, B), which also plays a role in autophagosomes expansion [21], it induced ATG12 puncta formation which co-localized with autophagosomes vesicles (Fig. 9C).
Fig 6.

Low MW Aβ did not significantly affect BECN1 gene expression. mRNA levels of BECN1 in neurons treated with low MW Aβ1–42 of indicated concentrations or DMSO (vehicle) for 4 to 30 hrs were measured by Q-PCR with 18S ribosomal RNA as internal control. Relative gene expression level was analysed with Pfaffl method. Bar chart plots the means ± S.E.M. of a triplicate from three independent experiments. Empty bars indicate the control at each time-point.
Fig 7.

Low MW Aβ did not significantly induce BECN1. (A) Neurons were treated with low MW Aβ1–42 of indicated concentrations or DMSO (vehicle) for 4 to 8 hrs. The neurons were stained with anti-BECN1 antibody. Representative Z-stack con-focal images from two independent experiments are shown. Scale bar, 20 μM. (B) Relative intensity of BECN1 staining. Intensity of signals were measured by Image J. Bar chart plots the means ± S.E.M. of at least three images. *P < 0.05, represents significant difference from the reference value. Empty bars indicate the control at each time-point.
Fig 8.
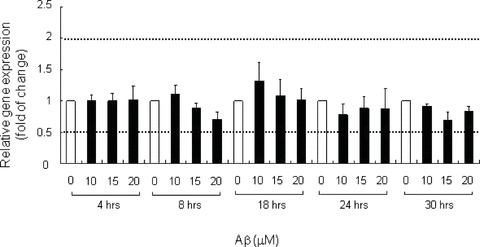
Low MW Aβ did not significantly affect ATG5 gene expression. mRNA levels of ATG5 in neurons treated with low MW Aβ1–42 of indicated concentrations or DMSO (vehicle) for 4 to 30 hrs were measured by Q-PCR with 18S ribosomal RNA as internal control. Relative gene expression level was analysed with Pfaffl method. Bar chart plots the means ± S.E.M. of a triplicate from three independent experiments. Empty bars indicate the control at each time-point.
Fig 9.
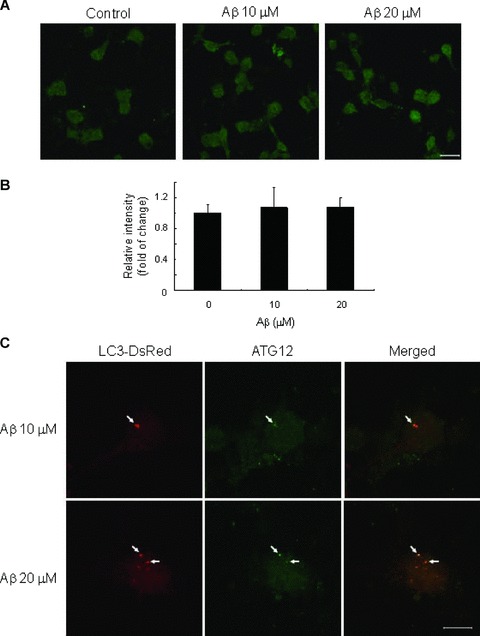
Low MW Aβ induced ATG12. Neurons were treated with low MW Aβ1–42 of indicated concentrations or DMSO (vehicle) for 24 hrs. (A) Neurons were stained with anti-ATG12 antibody. Representative confocal images from two independent experiments are shown. Scale bar, 20 μM. (B) Relative intensity of ATG12 staining. Intensity of signals were measured by Image J. Bar chart plots the means ± S.E.M. of three images. (C) Neurons trans-fected with LC3-DsRed prior treatment were stained with anti-ATG12 antibody. Representative Z-stack confocal images are shown. Scale bar, 10 μM.
Induction of lysosomal machinery in neurons by low MW Aβ
As lysosomes are required for the degradation of the content of autophagosomes, we further examined if Aβ treatment would enhance the lysosomal machinery in neurons. After Aβ treatment for 24 hrs, by immunohistochemical analysis, we demonstrated that the immunoreactivity of LAMP2 was greatly enhanced in the treatment group, but not in the vehicle-treated control (Fig. 10A). LAMP2 signals were mainly localized in soma and aggregates could also be observed in neurons treated with a higher dose (20 μM) of Aβ. Apart from that, we also visualized the acidic lysosomes with LysoTracker Green® in live neurons. Autophagosomes formation was revealed by expressing LC3-DsRed in neurons. In untransfected and vehicle-treated neurons, the lysosomes appeared in round small puncta (Fig. 10B). However, after 24 hrs of Aβ treatment, the size and number of lysosomes increased, which mainly localized in the soma. Lysosomes co-localized with autophagosomes, concurs with the concept that autophagosomes are delivered to lysosomes for degradation [22].
Phosphorylation of death-associated protein kinase in Aβ-induced autophagy and apoptosis
After establishing that low MW Aβ could induce both autophagy and neuronal apoptosis in cortical neurons, we next sought to investigate the potential switch between these two cellular events. Increasing lines of evidence suggested that DAPK plays an important role in regulating autophagy, apoptosis, development and cancer formation [23–26]. Therefore, we assessed the potential role of DAPK in the switching between Aβ-induced autophagy and apoptosis. DAPK is a calcium/calmodulin serine/threonine kinase which can be activated through dephosphorylation at serine 308 to cause cell death [27]. Upon Aβ treatment, we showed that the phosphorylation state of DAPK at serine 308 reduced from 24 hrs (Fig. 11), which agrees with the time-point of prominent neuronal apoptosis observed in our system (Fig. 2). Phosphorylation of DAPK continued to down-regulate at 30 hrs and became undetectable at 20 μM of Aβ treatment.
Fig 11.

Low MW Aβ reduced phosphorylation of DAPK. Western blotting of p-DAPK (S308) in neurons treated with low MW Aβ1–42 of indicated concentrations or DMSO (vehicle) for 4 to 30 hrs. β-actin was used as an internal control. Figure shows the representative result from two independent experiments.
Autophagy protects neurons from Aβ-induced apoptosis
Previous reports suggested that autophagy and apoptosis crosstalk to one another [28–32], so that the cellular responses during specific stress can be tightly regulated. In view of this, we were interested in examining whether there is a functional relationship between autophagy and apoptosis under low MW Aβ challenge and how these two events orchestrate to determine the fate of a neuron. After transfecting the neurons with LC3-DsRed plas-mid to reveal autophagosome production, we treated the neurons with Aβ and then assessed the occurrence of apoptosis by cleaved caspase 3 immunoreactivity. In vehicle-treated control, we observed that the neuron with minimal autophagosomes formation was immunoreactive negative for cleaved caspase 3 (Fig. 12A). Confirmed by DAPI staining to reveal normal nuclear morphology, we demonstrated that vehicle treatment did not cause significant cytotoxicity in neurons, which agrees with our cleaved caspase 3 Western blot analysis (Fig. 2). However, when neurons were treated with Aβ for 18 hrs, we found that neurons exhibiting high levels of vesicular LC3 were also immunoreactive negative for cleaved caspase 3 and displayed normal nuclear morphology (Fig. 12A). This phenomenon was also demonstrated in neurons with 24 hrs Aβ treatment (data not shown). We counted the number of neurons showing positive staining of cleaved caspase 3 and they made up for only 13%± 1.2, 14%± 4.5 and 17%± 1.7 of the population of cells expressing vesicular LC3 after 18 hrs treatment of vehicle, Aβ of 10 μM and 20 μM, respectively (Fig. 12B). Although the percentage increased with time, none of the treatment groups showed a cleaved caspase 3-immunoreactivity positive subpopulation exceeding 35% at 24 hrs (18%± 3.4 at 10 μM, 23%± 2.5 at 20 μM) and 30 hrs (28%± 1.2 at 10 μM, 31%± 1.4 at 20 μM), indicating that the majority (65%) of the LC3-transfected population did not show apoptotic features. Together with the Western blot analysis (Fig. 2), our results suggested that a subpopulation of neurons might be protected from Aβ-induced apoptosis by activating autophagy.
We tried to confirm this hypothesis by manipulating autophagy by the chemical inhibitor 3-MA [33] which inhibits PI3K. Without 3-MA, we demonstrated that low MW Aβ significantly increased the levels of both LC3-I and LC3-II at 24 hrs, which indicated the activation of autophagy, when compared to the vehicle-treated control (Fig. 13). When neurons were co-treated with 10 mM of 3-MA for 24 hrs, the levels of LC3-I and II of the 10 μM Aβ treated group were comparable to the vehicle-treated control, indicating that autophagy was inhibited by 3-MA. LC3 levels after 20 μM Aβ treatment were lower in the 3-MA group than that without 3-MA but higher than the vehicle control with 3-MA. This indicated that 10 mM 3-MA did not completely abolish autophagy induced by 20 μM Aβ. When we examined the effect of autophagy inhibition on apoptosis, we first demonstrated that the levels of cleaved cas-pase 3 in vehicle-treated control with or without 10 mM 3-MA were comparable (Fig. 13), suggesting 3-MA did not cause a cytotoxic effect in neurons. When neurons were treated with Aβ, cleaved caspase 3 levels increased with dose, which agrees with our Western blot analysis in Fig. 2. However, when the neurons were co-treated with 10 mM of 3-MA, the levels of cleaved cas-pase 3 markedly increased in all doses of Aβ treatment, suggesting that inhibition of autophagy advanced apoptosis. This further supports our hypothesis that neurons can be protected from Aβ-induced apoptosis by activating autophagy.
Fig 13.

Inhibition of autophagy advanced Aβ-induced apopto-sis. Western blotting of (A) LC3, and (B) cleaved caspase 3 (Asp 175) in neurons treated with low MW Aβ1–42 of indicated concentrations or DMSO (vehicle) with or without 3-MA of 10 mM for 24 hrs. β-actin was used as an internal control. Figure shows the representative result from three independent experiments.
Discussion
In the present study, we demonstrated that low MW Aβ triggered neuronal apoptosis through cleavage of caspase 3 and PARP, as early as 18 hrs. This agrees with the common concept that soluble Aβ species are neurotoxic [3–5]. However, we found that low MW Aβ did not induce both the gene and protein expression levels of AIF, which can activate apoptosis independent of caspases [9]. Movsesyan et al. previously reported that AIF was released from mitochondria when cortical neurons were stressed with Aβ25–35[34]. However, we did not observe significant mitochondrial release and nuclear entry of AIF upon Aβ challenge, further supporting that low MW Aβ triggers apoptosis mainly via a caspase-dependent manner, which differs from the mechanism of cytotoxicity of Aβ25–35.
Apart from apoptosis, low MW Aβ also induced autophagy. This can be characterized by increased autophagosome formation in cortical neurons as early as 8 hrs. This observation is comparable with our previous report in the primary culture of hip-pocampal neurons [11], suggesting that the toxicity of Aβ to activate autophagy is shared among different regions of the brain. Low MW Aβ also up-regulated the lysosomal machinery by showing increased number and size of lysosomes and LAMP2 immunoreactivity. Co-localization between lysosomes and autophagosomes implicates that Aβ induced autophagosomes are delivered to lysosomes for degradation, which agrees with our previous observations [11].
BECN1 regulates autophagy by modulating its binding partner mammalian class III phosphatidylinositol 3-kinase (PI3K) Vps34 and recruiting membranes to form autophagosomes [35]. A study by Pickford et al. reported that BECN1 was down-regulated in brains of AD patients, and heterozygous deletion of BECN1 in mice decreased neuronal autophagy [36]. On the contrary, in our study, we did not observe significant changes in BECN1 expression at both mRNA and protein levels when neurons were exposed to extracellular low MW Aβ. This discrepancy may partly be explained by different cell composition of neurons and glial cells in the brain and in the primary cell culture. It also raises the possibility that low MW Aβ activates autophagy via a BECN1-independent method [37]. Other than BECN1, low MW Aβ did not affect the expression of ATG5 and ATG12 which play a role in the proper development of autophagic isolation membranes [19]. This could imply that neurons display a quick response to Aβ toxicity via recruitment of essential proteins to the site of autophagy rather than de novo gene expression and protein synthesis. In fact, we found that Aβ induced ATG12 in puncta, but only some of which co-localized with autophagosomes. This suggests that ATG12 is involved in autophagosomematuration [38] through a transient process which is difficult to detect by immunocytochemical methods.
In view of the cytoprotective properties exerted by autophagy during starvation through catabolizing cellular components for energy [10], and during pathological conditions through removal of toxic protein aggregates [12], autophagy is believed to occur before apoptosis. In the current study, we demonstrated that activation of autophagy by low MW Aβ occurred at 8 hrs, followed by apoptosis at 18 hrs. This result clearly indicates that autophagy precedes apoptosis under Aβ toxicity. As higher MW species of Aβ started to form after 4 hrs of incubation in culture media, temporal discrepancy in the induction of the two events was, therefore, not due to a change in the oligomeric state of Aβ, but more likely a regulated cellular response. Apart from the sequential relationship, interlacing and sometimes common upstream signals of autophagy and apoptosis implicate a potential functional relationship between the events. Studies have proposed different molecular links between autophagic and apoptotic responses, including ATG5 [29], Bcl-2 proteins [31], p53 [30], DAPK [24, 25]. Among them, DAPK is a calcium/calmodulin serine/threonine kinase which functions as a tumour suppressor. Its activity has been shown necessary for both autophagic and apoptotic cell death [24, 26]. The ability to regulate both processes suggests DAPK as an integrator in the crosstalk. In our present study, we have demonstrated that Aβ activated DAPK via removal of the inhibitory autophosphorylation at serine 308 from 24 hrs onwards. This phenomenon coincided with the occurrence of prominent neuronal apoptosis observed in our system, suggesting that low MW Aβ first induces autophagy in neurons and later activates DAPK to cause apoptosis. Further investigation of the role of phosphoryla-tion of DAPK in the switching between autophagy and apoptosis will be carried out in our next study. Nonetheless, our results revealed that the majority of the cell population undergoing autophagy did not show activated signals for apoptosis at later time-points. Inhibition of autophagy by suppressing PI3K activity further exaggerated the cleavage of caspase 3. These observations support our hypothesis that autophagy can, at least to a certain extent, protect neurons from apoptotic cell death exerted by Aβ. Similar implications have been obtained in other systems under nutrient depletion and neurodegenerative conditions [10, 12, 32]. However, a recent study by Maycotte et al. reported a delayed appearance of apoptotic features after autophagic inhibition in rat cerebellar granule neurons under potassium deprivation and stau-rosporine treatment [33], revealing a more complicated crosstalk between autophagy and apoptosis that may result in different cellular responses and modes of cell death when cells are exposed to different stresses.
In summary, our report provided evidence to show that low MW Aβ triggered neuronal apoptosis, autophagy and enhanced lysosomal machinery in rat cortical neurons. We also demonstrated that activation of autophagy by low MW Aβ preceded the appearance of apoptosis, with DAPK phosphorylation potentially involved in the molecular link between the two events. More importantly, this is the first report showing that apoptotic features were absent in cortical neurons exhibiting high level of autophago-some formation under Aβ toxicity; while on the other hand, inhibition of autophagy advanced neuronal apoptosis, suggesting that autophagy can protect neurons from Aβ-induced apoptosis. More studies have to be performed to understand how autophagic and apoptotic responses by Aβ orchestrate to contribute to the neu-rodegeneration in AD.
Acknowledgments
This work was supported by Research Grant Council GRF (7552/06M, 7616/09M), N_HKU (707/07M), HKU Seed Funding for Basic Research (200811159082) and HKU Alzheimer's Disease Research Network. YTC, NQZ, CHLH and CSWL are supported by the Graduate School and MSY is supported by Postdoctoral Fellowship from The University of Hong Kong. Confocal live-cell imaging was supported by Faculty Core Facility.
References
- 1.Mattson MP, Magnus T. Ageing and neu-ronal vulnerability. Nat Rev Neurosci. 2006;7:278–94. doi: 10.1038/nrn1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blennow K, de Leon MJ, Zetterberg H. Alzheimer's disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- 3.Lambert MP, Barlow AK, Chromy BA, et al. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–53. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lesne S, Koh MT, Kotilinek L, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–7. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 5.Nguyen HD, Hall CK. Molecular dynamics simulations of spontaneous fibril formation by random-coil peptides. Proc Natl Acad Sci USA. 2004;101:16180–5. doi: 10.1073/pnas.0407273101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bredesen DE, Rao RV, Mehlen P. Cell death in the nervous system. Nature. 2006;443:796–802. doi: 10.1038/nature05293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuan J, Lipinski M, Degterev A. Diversity in the mechanisms of neuronal cell death. Neuron. 2003;40:401–13. doi: 10.1016/s0896-6273(03)00601-9. [DOI] [PubMed] [Google Scholar]
- 8.Wei Z, Song MS, MacTavish D, et al. Role of calpain and caspase in beta-amy-loid-induced cell death in rat primary sep-tal cultured neurons. Neuropharmacology. 2008;54:721–33. doi: 10.1016/j.neuropharm.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 9.Yu SW, Wang H, Poitras MF, et al. Mediation of poly(ADP-ribose) poly-merase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–63. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- 10.Young JE, Martinez RA, La Spada AR. Nutrient deprivation induces neuronal autophagy and implicates reduced insulin signaling in neuroprotective autophagy activation. J Biol Chem. 2009;284:2363–73. doi: 10.1074/jbc.M806088200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lai CS, Preisler J, Baum L, et al. Low molecular weight Abeta induces collapse of endoplasmic reticulum. Mol Cell Neurosci. 2009;41:32–43. doi: 10.1016/j.mcn.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 12.Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polygluta-mine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11:1107–17. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- 13.Nixon RA. Autophagy in neurodegenera-tive disease: friend, foe or turncoat? Trends Neurosci. 2006;29:528–35. doi: 10.1016/j.tins.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 14.Ling D, Song HJ, Garza D, et al. Abeta42-induced neurodegeneration via an age-dependent autophagic-lysosomal injury in Drosophila. PLoS One. 2009;4:e4201. doi: 10.1371/journal.pone.0004201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ho YS, Yu MS, Yik SY, et al. Polysaccharides from wolfberry antagonizes glutamate excitotoxicity in rat cortical neurons. Cell Mol Neurobiol. 2009;29:1233–44. doi: 10.1007/s10571-009-9419-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheung YT, Lau WK, Yu MS, et al. Effects of all-trans-retinoic acid on human SH-SY5Y neuroblastoma as in vitro model in neurotoxicity research. Neurotoxicology. 2009;30:127–35. doi: 10.1016/j.neuro.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 17.Lai CS, Yu MS, Yuen WH, et al. Antagonizing beta-amyloid peptide neuro-toxicity of the anti-aging fungus Ganoderma lucidum. Brain Res. 2008;1190:215–24. doi: 10.1016/j.brainres.2007.10.103. [DOI] [PubMed] [Google Scholar]
- 18.Yu MS, Lai SW, Lin KF, et al. Characterization of polysaccharides from the flowers of Nerium indicum and their neuroprotective effects. Int J Mol Med. 2004;14:917–24. [PubMed] [Google Scholar]
- 19.Sou YS, Waguri S, Iwata J, et al. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol Biol Cell. 2008;19:4762–75. doi: 10.1091/mbc.E08-03-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suzuki K, Kirisako T, Kamada Y, et al. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 2001;20:5971–81. doi: 10.1093/emboj/20.21.5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mizushima N, Yamamoto A, Hatano M, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152:657–68. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lang T, Schaeffeler E, Bernreuther D, et al. Aut2p and Aut7p, two novel micro-tubule-associated proteins are essential for delivery of autophagic vesicles to the vacuole. EMBO J. 1998;17:3597–607. doi: 10.1093/emboj/17.13.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raveh T, Droguett G, Horwitz MS, et al. DAP kinase activates a p19ARF/p53-mediated apoptotic checkpoint to suppress oncogenic transformation. Nat Cell Biol. 2001;3:1–7. doi: 10.1038/35050500. [DOI] [PubMed] [Google Scholar]
- 24.Fujita Y, Taniguchi J, Uchikawa M, et al. Neogenin regulates neuronal survival through DAP kinase. Cell Death Differ. 2008;15:1593–608. doi: 10.1038/cdd.2008.92. [DOI] [PubMed] [Google Scholar]
- 25.Zalckvar E, Berissi H, Mizrachy L, et al. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009;10:285–92. doi: 10.1038/embor.2008.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Inbal B, Bialik S, Sabanay I, et al. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J Cell Biol. 2002;157:455–68. doi: 10.1083/jcb.200109094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shohat G, Spivak-Kroizman T, Cohen O, et al. The pro-apoptotic function of death-associated protein kinase is controlled by a unique inhibitory autophosphorylation-based mechanism. J Biol Chem. 2001;276:47460–7. doi: 10.1074/jbc.M105133200. [DOI] [PubMed] [Google Scholar]
- 28.Betin VM, Lane JD. Caspase cleavage of Atg4D stimulates GABARAP-L1 processing and triggers mitochondrial targeting and apoptosis. J Cell Sci. 2009;122:2554–66. doi: 10.1242/jcs.046250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yousefi S, Perozzo R, Schmid I, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–32. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- 30.Tasdemir E, Maiuri MC, Galluzzi L, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10:676–87. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heath-Engel HM, Chang NC, Shore GC. The endoplasmic reticulum in apoptosis and autophagy: role of the BCL-2 protein family. Oncogene. 2008;27:6419–33. doi: 10.1038/onc.2008.309. [DOI] [PubMed] [Google Scholar]
- 32.Yang DS, Kumar A, Stavrides P, et al. Neuronal apoptosis and autophagy cross talk in aging PS/APP mice, a model of Alzheimer's disease. Am J Pathol. 2008;173:665–81. doi: 10.2353/ajpath.2008.071176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maycotte P, Guemez-Gamboa A, Moran J. Apoptosis and autophagy in rat cerebel-lar granule neuron death: role of reactive oxygen species. J Neurosci Res. 2009 doi: 10.1002/jnr.22168. [DOI] [PubMed] [Google Scholar]
- 34.Movsesyan VA, Stoica BA, Faden AI. MGLuR5 activation reduces beta-amyloid-induced cell death in primary neuronal cultures and attenuates translocation of cytochrome c and apoptosis-inducing factor. J Neurochem. 2004;89:1528–36. doi: 10.1111/j.1471-4159.2004.02451.x. [DOI] [PubMed] [Google Scholar]
- 35.Zeng X, Overmeyer JH, Maltese WA. Functional specificity of the mammalian Beclin-Vps34 PI 3-kinase complex in macroautophagy versus endocytosis and lysosomal enzyme trafficking. J Cell Sci. 2006;119:259–70. doi: 10.1242/jcs.02735. [DOI] [PubMed] [Google Scholar]
- 36.Pickford F, Masliah E, Britschgi M, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118:2190–9. doi: 10.1172/JCI33585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chu CT, Zhu J, Dagda R. Beclin 1-independent pathway of damage-induced mitophagy and autophagic stress: implications for neurodegeneration and cell death. Autophagy. 2007;3:663–6. doi: 10.4161/auto.4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hanada T, Noda NN, Satomi Y, et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282:37298–302. doi: 10.1074/jbc.C700195200. [DOI] [PubMed] [Google Scholar]