

Abstract
Charcot-Marie-Tooth disease type 4B is caused by mutations in the genes encoding either the lipid phosphatase myotubularin-related protein-2 (MTMR2) or its regulatory binding partner MTMR13/SBF2. Mtmr2 dephosphorylates PI-3-P and PI-3,5-P2 to form phosphatidylinositol and PI-5-P, respectively, while Mtmr13/Sbf2 is an enzymatically inactive member of the myotubularin protein family. We have found altered levels of the critical signalling protein AKT in mouse mutants for Mtmr2 and Mtmr13/Sbf2. Thus, we analysed the influence of Mtmr2 and Mtmr13/Sbf2 on signalling processes. We found that overexpression of Mtmr2 prevents the degradation of the epidermal growth factor receptor (EGFR) and leads to sustained Akt activation whereas Erk activation is not affected. Mtmr13/Sbf2 counteracts the blockage of EGFR degradation without affecting prolonged Akt activation. Our data indicate that Mtmr2 and Mtmr13/Sbf2 play critical roles in the sorting and modulation of cellular signalling which are likely to be disturbed in CMT4B.
Keywords: Charcot-Marie-Tooth disease, neuropathy, myotubularin, EGFR, Akt
Introduction
The family of myotubularin-related proteins (Mtmrs) consists of 14 members in human beings and orthologues have been found in all eukaryotes but not in bacteria. Mutations in three family members are associated with human diseases. Mutations in myotubularin, the founding member of the Mtmr family, lead to X-linked myotubular myopathy [1]. X-linked myotubular myopathy is a severe muscle disease characterized by hypotonia and generalized muscle weakness in affected newborn males. Histopathological studies of the skeletal muscle in these patients reveal the presence of small rounded muscle fibres that contain centrally located nuclei. Since these fibres resemble foetal myotubes, it was proposed that the terminal differentiation of the muscle fibre is blocked [2]. Mutations in MTMR2 or MTMR13/SBF2 lead to Charcot-Marie-Tooth (CMT) diseases type 4B1 or CMT4B2, respectively. These recessive peripheral neuropathies are clinically indistinguishable and are characterized by focally folded myelin sheets and demyelination in peripheral nerves [3–5].
The Mtmr proteins are defined by a phosphoinositide-binding GRAM-pleckstrin homology domain (GRAM-PH), a phosphatase domain and a coiled coil. Active members dephosphorylate PI-3-P to phosphatidylinositol and PI-3,5-P2 to PI-5-P. Although initially described as a protein tyrosine/dual specificity phosphatase based on the primary sequence, no phosphoprotein has been identified as substrate so far [6]. Mtmr2, an active member of the family, is a 73 kD protein that exists as a dimer in eukaryotic cells. It contains a C-terminal Post-synaptic density protein-95, Drosophila disc large tumour suppressor, and Zonula occludens-1 protein (PDZ) binding motif in addition to the standard Mtmr domains. The GRAM-PH domain binds to several phosphoinositides and thereby mediates membrane binding [7]. Interestingly, six of the 14 members have substitutions in the catalytic site of the phosphatase domain rendering them inactive. Several inactive myotubularins interact with active Mtmrs [8]. Alternatively, inactive Mtmrs can also exist independently within cells [9, 10]. Mtmr13/Sbf2, an inactive member, is a 210 kD protein that interacts with Mtmr2 and enhances its catalytic activity. The formation of this complex is crucially important for the integrity of the peripheral nervous system since loss of either of the proteins leads to CMT4B. Mtmr2 contributes the disease-relevant catalytic activity to this complex while the important functions or domains of Mtmr13/Sbf2 have not yet been identified [11]. It is likely that Mtmr13/Sbf2 guides the catalytic activity to the appropriate place within the cell. Putative domains for this purpose are the N-terminal differentially expressed in neoplastic versus normal cells (DENN) domain which influences the proliferation of cells or the C-terminal PI-3,4,5-P3 binding pleckstrin homology domain [12]. Mtmr12/3-PAP, another inactive member, also interacts with Mtmr2 [13]. In contrast to Mtmr13/Sbf2, Mtmr12/3-PAP contains only the standard Mtmr domains indicating that the functional mechanisms are likely to be different from Mtmr13/Sbf2.
The correct metabolism of phosphoinositides is a critical issue for the integrity of peripheral nerves [14]. Several disease-associated proteins have phosphoinositide binding domains (MTMR2, MTMR13/SBF2, DYNAMIN2, N-myc downstream regulated gene 1 [NDRG1] and FRABIN) or dephosphorylate phosphoinositides as MTMR2 and the Sac type phosphatase FIG4 (pheromone-regulated or induced gene 4) [15–19]. Phosphoinositides are fixed to membranes by acyl chains and expose their phosphorylated head to the cytoplasm. Seven different phosphoinositides are generated by the reversible phosphorylation and dephosphorylation of the inositol ring of phosphatidylinositol at positions D3, D4 and D5. Particular phosphoinositides are enriched in certain subcellular compartments. The exposed head groups are anchor sites for phospho-inositide-binding domains which allow the assembly of multiprotein effector complexes with compartment-specific cellular functions [20, 21]. PI-3-P and PI-3,5-P2, the substrates of Mtmr2, localize mainly to the endosomal compartment pointing to a function in protein trafficking, sorting and degradation. PI-3,5-P2 is produced from PI-3-P by PIKfyve, an enzyme that is found on early and late endo-somes [22, 23]. It is transiently formed after the stimulation of cells with different treatments [24–26]. Several effector proteins including Mtmrs, sorting nexins (SNX), Svp1p and vps24 can bind to PI-3,5-P2 with low affinity and specificity [27–29]. Depending on the bound effector, the cargo of the corresponding vesicle is either directed to the lysosome or to the trans-Golgi network [30, 31].
This report is based on the observation that the overexpression of myotubularin blocks the degradation of the epidermal growth factor receptor (EGFR) [26]. We show that Mtmr2 has a comparable effect and that the adaptor unit Mtmr13/Sbf2 can counteract this outcome. In addition, we demonstrate that the blocked degradation leads to sustained Akt, but not Erk activation. Mtmr13/Sbf2 can counteract this effect if its C-terminal PI-3,4,5-P3 binding pleckstrin homology domain is removed. Active and inactive Mtmrs therefore integrate the PI-3-kinase-derived PI-3,4,5-P3 and the PIKfyve-derived PI-3,5-P2 signal to generate sustained Akt activation. After stimulation of cells, Mtmrs can colocalize with SNX in the endosomal compartment. Our results suggest that Mtmrs modulate the phosphoinositide content of endosomal membranes and thereby influence the sorting of EGFR-SNX complexes. The specificity in downstream signalling was also observed in Mtmr2 // Mtmr13/Sbf2 knockout animals.
Materials and methods
Plasmids and cell lines
The previously described His-tagged Mtmr2 and HA-tagged Sbf2 cDNAs were transferred from pcDNA3.1 zeo(+) into pcDNA5/FRT [10]. These constructs were then used to generate stable cell lines with the Flp-In system according to manufacturer's recommendations (Invitrogen, Paisley, UK). Two independent lines were used for the experiments (except for Mtmr2 C417S). Transient transfection was not used since the cotransfec-tion efficiency with these long plasmids was too low. Cell lines expressing two Mtmrs were obtained by random integration of a second, pcDNA3. 1-based construct and selection with zeocin. Cell lines were checked by immunofluorescence for homogenous expression. More than 95% of the cells express the indicated Mtmrs (see Fig. S1)
EGFR degradation
Three 10 cm dishes were transfected with 2 μg pRC-hEGFR plasmid (kindly provided by Gordon Gill, San Diego) and 6 μl Fugene6. One day after transfection, cells were distributed in twelve 3.5 cm dishes for triplicates at four time-points. The transfection rate was between 10% and 20% and did not vary between cell lines. After an additional 24 hrs in culture, the cells were starved overnight in 1% bovine serum albumin (BSA)/ Dulbecco's modified Eagle medium (DMEM)/Penicillin/Streptomycin. Cells were stimulated with 50 ng/ml EGF for 0/20/40/180 min. and then lysed in 0.5% Triton-X100/ 100 mM NaCl/ 50 mM Tris-HCl pH 7.5. The supernatant was used for Western blot analysis after sonication and centrifugation. The following antibodies were used: 1:1000 rabbit anti-EGFR (Neomarkers, Fremont, CA, USA; RB-1417), 1:2000 mouse anti-actin (Sigma, Buchs, Switzerland; A5316), rabbit anti-Akt (Cell Signaling, Danvers, MA, USA; 9272), mouse anti-P-Akt (Cell Signaling; 4051), rabbit anti-Erk (Cell Signaling; 9102), mouse anti-P-Erk (Cell Signaling; 9106) . As secondary antibodies, alkaline phosphatase-coupled goat anti mouse and horse radish peroxidase (HRP)-coupled goat anti-rabbit were used, followed by chemiluminescence detection. Quantification was performed with ImageJ. The experiment was repeated three times for each cell line.
HRP uptake
FlpIn293 cells stably expressing Mtmrs were plated in triplicates in 3.5 cm dishes 2 days prior to the experiment. Cells were starved for 30 min. in starvation medium (1% BSA/DMEM) and then pulsed with 2 mg/ml HRP in starvation medium for the indicated time. Cells were immediately put on ice, washed two times with ice cold PBS/1% BSA followed by three washes with ice cold PBS and then scraped from the plate with PBS. Cells were lysed by sonification in 0.15% Triton-X100/PBS/protease inhibitors and centrifuged. HRP activity was measured with o-Dianisidine and H2O2 and normalized to the total protein content.
Immunofluorescence microscopy
COS cells were transfected in 3.5 cm dishes with Fugene6 or FugeneHD according manufacturers recommendations. Equal amounts of Mtmr plas-mids (His-Mtmr2 or HA-Sbf2) and myc-tagged SNX1/2/5 (kindly provided by Jo-Ann Trejo, Chapel Hill and Rohan Teasdale, St. Lucia) were taken for the transfections. The cells were fixed 24 or 36 hrs after transfection with 2% paraformaldehyde in PBS. Cells were blocked in blocking buffer (10% foetal calf serum/0.05% Saponin/PBS) for 30 min. prior to incubation with antibodies. Antibodies were diluted as follows in blocking buffer: rabbit anti-Mtmr2 (1:1000; [11]), rabbit anti-Mtmr13/Sbf2 (1:1000; [10]), mouse anti-myc 9E10 supernatant (1:10) and rabbit anti-EGFR (Neomarkers, RB-1417, 1:500). Appropriate Cy3 and ALEXA-488 secondary antibodies were used for visualization. Single optical sections with a thickness of 0.3 μm were taken with a confocal laser scanning microscope (Leica SP2 or SP5, Leica Microsystems, Wetzlar, Germany) for the analysis. Pictures of Figs 1 and S1 were taken with an Olympus IX81 microscope (Olympus Corporation, Tokyo, Japan).
Transgenic mice
Animal experiments were approved by the veterinary office of the Canton of Zurich. The generation and genotyping of Mtmr2 and Mtmr13/Sbf2 knockout mice was described previously [32, 33]. Sciatic nerves from three adult animals (P60) were pooled, powdered in liquid nitrogen, and then lysed in 0.5% Triton-X100, 100 mM NaCl, 50 mM Tris-HCl pH 7.5. Twenty micrograms protein was used for Western blot analysis as described above.
Results
Influence of Mtmr overexpression on receptor trafficking and fluid phase endocytosis
The degradation of the EGFR is a well-established paradigm for the switching off of activated receptors. A previous study revealed that overexpression of myotubularin blocks the degradation of the EGFR after ligand stimulation [26]. We were interested if Mtmr2 and Mtmr13/Sbf2 have a similar effect. We therefore generated FlpIn293 cell lines stably overexpressing Mtmr2 and/or Mtmr13/Sbf2 or mutants thereof to manipulate the intracellular levels and ratios of these enzymes. We did not use an RNAi approachbecause compensatory effects of the highly homologues and ubiquitously expressed proteins Mtmr1 and Mtmr5/Sbf1 may interferewith the effect and we used stable cell lines to overcome the problems of transient transfections (see ‘Materials and methods’ for details). The expression levels of Mtmr2 and its mutated forms were always higher than the expression level of Mtmr13/Sbf2 (based on Coomassie stained immunoprecipitation experiments and Western blotting, data not shown). Overexpression of Mtmr2 blocked the degradation of the EGFR in a similar way as previously described for myotubularin (Fig. 1A, B). Immunostaining of these cells revealed an enhanced surface staining of the EGFR indicating that the EGFR is either not internalized or more efficiently recycled (Fig. 1C). This effect depends on the phosphatase activity since an Mtmr2 mutant lacking the phos-phatase activity (C417S) has no effect. Similarly, overexpression of Mtmr13/Sbf2, a phosphatase-dead member of the myotubu-larin family, has also no effect on degradation. Interestingly, EGFR degradation is not blocked in cells overexpressing both Mtmr2 and Mtmr13/Sbf2 indicating that Mtmr13/Sbf2 acts as an antagonist of Mtmr2. This antagonistic function seems to be mediated by the N-terminal Mtmr2-binding part of Mtmr13/Sbf2, since the deletion of the C-terminal pleckstrin homology domain of Mtmr13/Sbf2 does not reverse this dominating effect. Mtmr2 has therefore the same effect on EGFR degradation as myotubularin and this effect is regulated by the adaptor unit Mtmr13/Sbf2.
Fig 1.
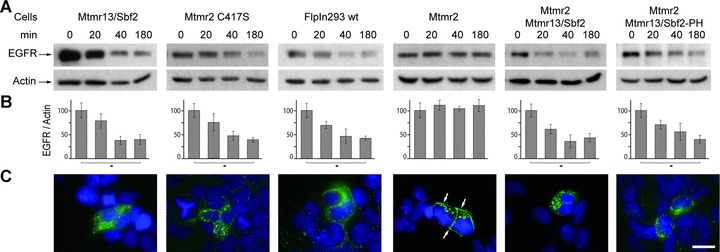
Influence of Mtmr overexpression on EGFR degradation. (A) FlpIn293 cells stably overexpressing the indicated Mtmr and transiently transfected with EGFR were stimulated with EGF. The EGFR level was determined after 0/20/40 and 180 min. (B) Quantification of the above blots (mean ± S.D., n= 3, lysates from independent transfections, *P < 0.05 [Student's t-test, unpaired, two-tailed]). (C) Immunostaining of EGFR transfected cells 180 min. after EGF stimulation. EGFR (green) was mainly seen at the plasma membrane of Mtmr2 overexpressing cells. In all other cell lines, the EGFR was mainly in intracellular vesicles. See Fig. S1 for the quantification of this effect. Cell nuclei were counterstained with DAPI. Scale bar: 20 μm. Mtmr2 C417S: Mtmr2 without phosphatase activity; Mtmr13/Sbf2-PH: Mtmr13/Sbf2 without C-terminal pleckstrin homology domain.
Next, we were interested whether Mtmrs influence only the sorting of membrane proteins or if fluid phase endocytosis is also affected. Fluid phase endocytosis was monitored by measuring the uptake of HRP, a marker that reaches the lysosomes by endocyto-sis. We did not observe significant differences in HRP uptake by overexpressing Mtmrs indicating that general vesicle trafficking is not affected (Fig. 2). This is in line with the finding that overexpres-sion of Mtmrs does not induce vacuoles under resting condition, a phenotype previously described for a dominant negative form of PIKfyve (K1831E), an inhibitor of fluid phase uptake [10, 34].
Fig 2.
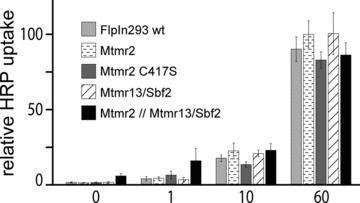
Mtmrs do not change fluid phase uptake. Stably transfected FlpIn293 cells were pulsed with HRP and its uptake was determined after 0/1/10/60 min. (mean ± S.D., n= 3).
Colocalization with sorting nexins
SNX are a family of proteins which bind to receptor tyrosine kinases and phosphoinositides [35]. Several members of this family regulate EGFR degradation in a positive or negative manner. They are therefore a putative link between the phosphoinositide-metabolizing Mtmrs and the observed block in EGFR degradation. Consequently, we tested if Mtmr2 and Mtmr13/Sbf2 colocalize with SNX. COS cells were cotransfected either with Mtmr2 or Mtmr13/Sbf2 and with SNX1, SNX2 or SNX5. The phosphoinosi-tide binding properties and the influence on EGFR degradation of these SNX are well established. Whereas overexpression of SNX1 and SNX2 promote EGFR degradation, overexpression of SNX5 plays an inhibitory role. The phosphoinositide binding specificities of these SNX are broad, but they bind to PI-3,5-P2, a substrate of Mtmr2. SNX5 also binds to PI-5-P, which can be produced by Mtmr2 [30, 36, 37]. Mtmrs and SNX show a diffuse cytoplasmic, vesicular or plasma membrane staining under resting conditions (Fig. S2). Cells were stimulated with EGF and analysed after 30 min. when the EGFR passes the endosomal compartment and when PI-3,5-P2 peaks [26]. The cells respond at this time-point with an enlargement of endosomes allowing an accurate colocal-ization analysis. Mtmrs are able to colocalize with SNX suggesting that they influence sorting of receptors by modulating the phos-phoinositide composition of membranes (Fig. 3).
Fig 3.
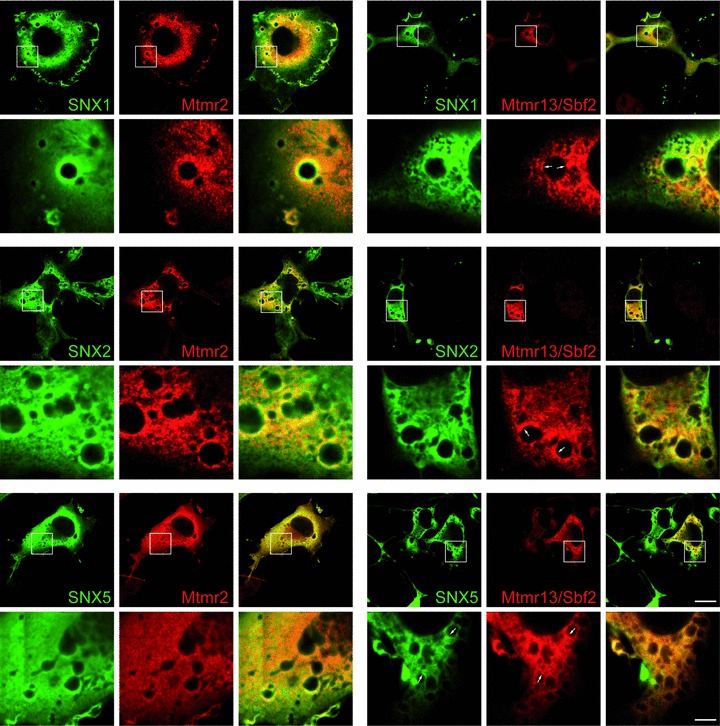
Colocalization of Mtmr2 or Mtmr13/Sbf2 with SNX1, SNX2 or SNX5. COS cells were cotransfected with the indicated plasmids, starved overnight 24 hrs later and then pulsed for 20 min. with EGF. Colocalization on vesicles was seen for all combinations of Mtmrs with SNX. Note that Mtmr13/Sbf2 localizes only to subdomains of vesicles (arrows). Scale bars: 20 μm (upper panel), 4 μm (lower panel).
Influence on downstream signalling
Stimulation of the EGFR leads to a transient activation of the Akt and Erk pathways. We wanted to test if the blocked degradation has consequences on signal output by the EGFR. We observed a transient phosphorylation of Akt, Erk1 and Erk2 in wild-type FlpIn293 cells after 20 and 40 min. (Fig. 4). The phosphorylation decreases to basal levels after 180 min. In contrast, sustained activation of Akt was observed in FlpIn293 cells overexpressing Mtmr2. Sustained phosphorylation of Akt depends on phosphatase activity since the phosphatase-dead mutant of Mtmr2 (C417S) and Mtmr13/Sbf2 have no effect on signal output. The effect is specific for Akt since Phospho-Erk levels decrease to basal level as in wild-type cells. Interestingly, the sustained Akt activation was also observed in cells overexpressing Mtmr2 and Mtmr13/Sbf2 together which degrade the EGFR with normal efficiency (Fig. 1). These findings suggest that sustained Akt activity is not simply due to the lack of EGFR degradation but due to specific sorting. If Mtmr2 is overexpressed with a form of Mtmr13/Sbf2 which lacks the C-terminal PI-3,4,5-P3 binding pleckstrin homology domain, phospho-Akt returns to basal level after 180 min. similar to wild-type cells. We conclude that sustained Akt activation is only achieved when Mtmr2 phosphatase activity and the PI-3,4,5-P3-binding pleckstrin homology domain of Mtmr13/Sbf2 are available.
Fig 4.
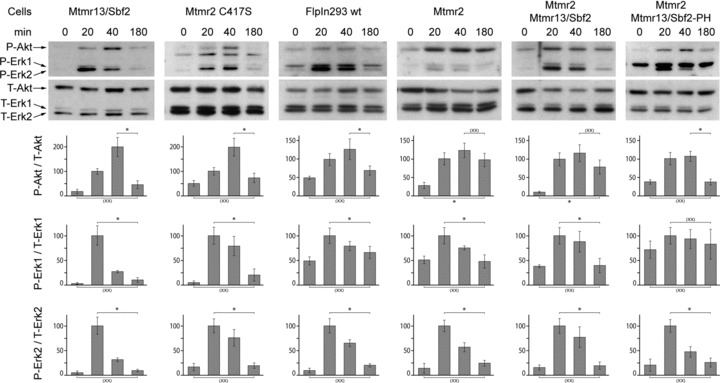
Influence of Mtmr overexpression on EGFR downstream signalling. FlpIn293 cells stably overexpressing the indicated Mtmr and transiently trans-fected with EGFR were stimulated with EGF. The levels of phospho-Akt, phospho-Erk1 (p44) and phospho-Erk2 (p42) were determined after 0/20/40 and 180 min. and normalized to the total amount of the corresponding protein (mean ± S.D., n= 3, lysates from independent transfections). P-Akt levels in cells overexpressing Mtmr2 and Mtmr2 // Mtmr13/Sbf2 do not return to basal levels after 3 hrs (P < 0.05) and the activation level after 3 hrs is statistically not different from the maximal activation at 40 min. (P < 0.05). P-values (Student's t-test, unpaired, two-tailed) are indicated with a single asterisk if a statistical significant difference was observed (P < 0.05) or with (xx) if no difference was observed (P > 0.05). Mtmr13/Sbf2-PH: Mtmr13/Sbf2 without C-terminal pleckstrin homology domain; Mtmr2 C417S: Mtmr2 without phosphatase activity.
Signalling in knockout animals
We were also interested if the observed in vitro influence of Mtmr2 overexpression on Akt signalling is relevant in vivo. For this purpose, we used our previously described Mtmr2 and Mtmr13/Sbf2 knockout animals that, in contrast to the acute stimulation experiments described so far, reflect a steady state condition. These animals show a similar but milder pathology than human beings affected with CMT 4B1 or CMT 4B2 [32, 33]. The sciatic nerves of these animals were analysed because the peripheral nervous system is the only affected tissue identified so far. We expected a down-regulation of phospho-Akt in mice lacking both Mtmr2 and Mtmr13/Sbf2 since Mtmr2 overexpression leads to sustained Akt activation. Indeed a reduction of the steady state ratio of phospho Akt to total Akt was observed in the mutant. The total level of phospho-Akt in mutant animals, however, is comparable to the levels in wild-type animals while the level of total Akt is significantly increased (Fig. 5). Mutant mice therefore appear to compensate for the inefficient phosphorylation of Akt by up-regulating the total level of this kinase. Neither total-Erk nor phospho-Erk differed between wild-type and mutant mice. Akt is a complex regulator of peripheral nerve development and function [38]. How our data integrate with the critical role of Akt in this tissue remains to be elucidated.
Fig 5.
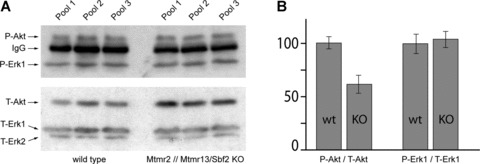
Phosphorylation of Akt and Erk in 60 day old Mtmr2 // Mtmr13/Sbf2 double knockout mice. (A) Western blot analysis of sciatic nerve lysate from wild-type and Mtmr2 // Mtmr13/Sbf2 double knockout animals. Endogenous mouse IgGs (arrow, upper panel) were also recognized by the secondary alkaline phosphatase coupled goat anti-mouse antibody. (B) Quantification of the blot. Knockout animals express higher levels of Akt to reach the same phosphorylation level. The ratio of phosphorylation of Erk1 is identical in wild-type and knocks out animals.
Discussion
Mtmrs play a crucial role in human health and disease. Active family members dephosphorylate PI-3-P and PI-3,5-P2 in vitro and in vivo, but the consequences for the phosphoinositide-regulated cellular processes are poorly understood. We have overex-pressed Mtmr2 to negatively regulate the levels of these two phos-phoinositides in vivo. A more specific regulation is not possible since there is no mutant Mtmr2 available which specifically dephosphorylates only one of the two substrates. Thus, whether PI-3-P or PI-3,5-P2 or both are the relevant substrates in vivo cannot be distinguished. Whereas the level of PI-3-P is high within cells, the level of PI-3,5-P2 is low under resting conditions and increases transiently after stimulation of cells [24–26, 39]. The observation that Mtmr2 relocalizes within cells under conditions with elevated PI-3,5-P2 levels, e.g. under hypoosmotic conditions or after EGFR stimulation, point to PI-3,5-P2 as an important in vivo substrate [10, 26]. In addition, one might speculate that the binding pocket for the D5 phosphate would have been closed during evolution if the activity towards PI-3,5-P2 is not necessary in vivo. This has been partially achieved in vitro with the artificial mutation H357N in Mtmr2 which reduces the activity towards PI-3,5-P2 but not towards PI-3-P [40].
Mtmr2 and Mtmr13/Sbf2 can acquire three different structural states in vitro and in vivo which affect the signalling of cell in different ways: (i) as a Mtmr2 dimer, (ii) as a Mtmr13/Sbf2 dimer and (iii) as a Mtmr2// Mtmr13/Sbf2 heterotetramer. Our results indicate, that the concentration, ratios and localization of these two Mtmrs have an important impact in which compartments the receptors are guided. Overexpression of Mtmr2 blocks the degradation of EGFR. Since this effect depends on phosphatase activity, it can either result from (i) the dephosphorylation of PI-3-P to phosphatidylinositol (ii) the dephosphorylation of PI-3,5-P2 or (iii) the production of PI-5-P. PI-3,5-P2 is produced in the endosomal compartment and assembles protein complexes for the trafficking into other subcellular locations of the cell [41, 42]. Vps24 is part of the ESCRT-III complex and one of these adaptors [29]. Previous studies revealed that the knockdown of vps24 with RNAi or the overex-pression of dominant-negative forms of vps24 block the degradation of the EGFR [43, 44]. PI-3,5-P2 is therefore an important metabolite for the degradation of the EGFR and PI-3,5-P2 dephosphorylation by Mtmr2 is one of the ways to interfere with EGFR degradation. PI-5-P is the least characterized phosphoinositide. SNX5 has a PX domain that binds PI-5-P. Interestingly, overexpres-sion of SNX5 prevents the degradation of the EGFR indicating that it guides the EGFR away from lysosomes [37]. Consequently, reduction of PI-3,5-P2 levels and the production of PI-5-P are both possible explanations for the observed block in EGFR degradation. It is therefore reasonable to propose that the phosphatase activity of Mtmrs block downstream pathways depending on PI-3,5-P2 and redirects receptors to pathways depending on PI-5-P (Fig. 6). Overexpression of Mtmr13/Sbf2 can counteract this function of Mtmr2. We have previously described that Mtmr13/Sbf2 exists in double transfected cells in a complex with Mtmr2 or separately [10]. It is therefore not clear if Mtmr13/Sbf2 fulfils this counteracting function in a complex with Mtmr2 or independently.
Fig 6.
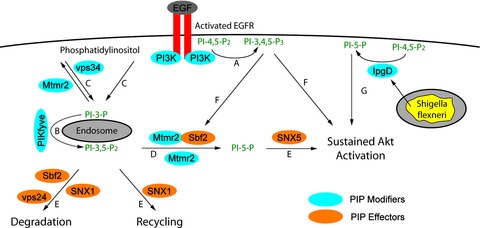
Model for sustained Akt activation. (A) Stimulation of the EGFR leads to the immediate production of PI-3,4,5-P3 which allows transient activation of Akt. The EGFR is internalized during this process and binds to phosphoinositide-binding SNX1 and SNX5 (not depicted in the picture). (B) PI-3,5-P2 is produced in a secondary, delayed process by PIKfyve. (C) It is not clear, if the necessary PI-3-P is produced by EGFR-linked PI-3-kinase or vps34. (D) Cells overex-pressing Mtmr2 or Mtmr2 // Mtmr13/Sbf2 increase the dephospho-rylation of PI-3,5-P2 to PI-5-P. (E) The dephosphorylation of PI-3,5-P2 prevents the effects of PI-3,5-P2 effectors (SNX1, vps24) and allows the function of PI-5-P effectors (SNX5). (F) The Mtmr2 // Mtmr13/Sbf2 complex leads only to sustained activation of Akt if the C-terminal pleckstrin homology domain of Mtmr2/Sbf2 is present indicating that the initially produced PI-3,4,5-P3 is important. (A, E, F) Sustained Akt phosphorylation is therefore only possible if PI-3,4,5-P3 and PI-5-P are available. (G) Shigella flexneri which resides in endo-somes secretes IpgD into the cytoplasm. IpgD dephosphorylates PI-4,5-P2 to PI-5-P which also leads to sustained Akt activation. PIP, phosphoinositide.
Overexpression of Mtmr2 leads to sustained Akt activation. Surprisingly, this effect on downstream signalling of the EGFR is limited to Akt whereas Erk shows the normal transient activation observed in growth factor stimulated cells. The same specific activation pattern was observed after infection of HeLa cells with Shigella flexneri. Phosphorylation of Akt depends in this case on IpgD, a phosphatidylinositol-4,5-bisphosphate-4-phosphatases which is secreted into the cytoplasm of host cells leading to increased PI-5-P levels [45]. The generation of PI-5-P from PI-4,5-P2 seems to be a widespread mechanism of pathogenic bacteria to activate the Akt pathway to increase survival of host cells [46]. Bacteria probably use PI-4,5-P2 as a substrate because its level is constitutively high within cells, whereas PI-3,5-P2 levels are low and only available transiently. Enhanced PI-5-P levels were also observed when myotubularin was overexpressed in Jurkat cells [47]. Mtmr13/Sbf2 appears to play a different role in downstream signalling than in receptor degradation. Although Mtmr13/Sbf2 counteracts the protective function of Mtmr2 in EGFR degradation, it does not interfere with sustained Akt activation. Sustained Akt activation is therefore due to different trafficking of the receptor and not due to increased levels of the EGFR. The permissive function on sustained MTMR2-induced Akt activation of Mtmr13/Sbf2 is lost, when its C-terminal pleckstrin homology domains is deleted. This suggests that the sustained activation of Akt by the Mtmr2 // Mtmr13/Sbf2 complex is only possible when PI-3,4,5-P3-binding pleckstrin homology domain of Mtmr13/Sbf2 is present. PI-3,4,5-P3 as well as PI-3,5-P2 are available after stimulation of the EGFR [26, 48]. Mtmr2 and Mtmr13/Sbf2 recognize both lipids with their GRAM-PH and pleckstrin homology domains and are able to modulate the PI-3,5-P2 level in the endosomal compartment (Fig. 6). Receptors may recognize the phosphoinositide composition of membranes indirectly via their bound SNX and are then transported to distinct compartments where they fulfil their function. Mtmr13/Sbf2 is a catalytically inactive Mtmr. Dead Mtmrs emerged only in higher eukaryotes and do not exist in yeast [6]. PI-3,4,5-P3 and Akt signalling are also absent in yeast suggesting that the function of dead Mtmrs might have coevolved with PI3-kinase signalling in higher eukaryotes.
What are the potential implications for human diseases? CMT4B is a severe demyelinating peripheral neuropathy with myelin out-foldings as the pathological hallmark [49]. The overall structure and thickness of the myelin sheath appear normal in patients and in animal models pointing to an increased synthesis or impaired membrane degradation in regions of active myelin turnover. Here we show that Mtmrs have an influence on protein turnover in vitro. In the peripheral nerve, Mtmrs may either be involved in the turnover of structural myelin proteins or indirectly influence the signal output of an unidentified receptor. Pharmacological targeting of Mtmr2 or Mtmr13/Sbf2 might be promising since curcumin, an inhibitor of Pikfyve, shows a positive effect in some animal models of peripheral neuropathies. Curcumin prevents the accumulation of mutant peripheral myelin protein 22 (PMP22) or protein zero (P0) in the endoplasmic reticulum in vitro suggesting that the levels of PI-3-P and/or PI-3,5-P2 play a general role in the development of peripheral neuropathies [50–52]. Future studies may elucidate which disease-associated proteins play a role in this pathway leading to peripheral neuropathies (reviewed in [53, 54]).
Acknowledgments
We thank Dr. Matthias Wymann for helpful discussions, Dr. Ned Mantei for critically reading the manuscript and Drs. Gordon Gill (San Diego), Jo-Ann Trejo (Chapel Hill) and Rohan Teasdale (St. Lucia) for plasmids. This work has been supported by the Swiss National Science Foundation and the National Center for Competence in Research (NCCR) Neural Plasticity and Repair.
Supporting Information
Characterization of the cell lines overexpressingHis-tagged Mtmr2 and/or HA-tagged Mtmr13/Sbf2. Cells were plated onglass cover slips and fixed after 3 days. Mtmr2 was detected with amouse anti-His antibody (Qiagen, Hilden, Germany) and Mtmr13 with arabbit anti-Mtmr13/Sbf2 antibody. Nuclei of the cells were stainedwith 4′,6-Diamidino-2-phenylindol (DAPI). Note that more than95% of the cells express the indicated transgenicprotein. Scale bar. 20 μm.
Mtmrs and sorting nexins in unstimulated COScells Colocalization of Mtmr2 or Mtmr13/Sbf2 with SNX1, SNX2 orSNX5 in unstimulated cells. COS cells were cotransfacted with theindicated plasmids and then fixed after 28 hrs (Mtmr13/Sbf2transfected cells) or 42 hrs (Mtmr2 transfected cells). Allproteins show a diffuse cytoplasmic or vesicular staining. Scalebars: 20 μm (upper panel), 5 μm (lower panel).
References
- 1.Laporte J, Hu LJ, Kretz C, et al. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat Genet. 1996;13:175–82. doi: 10.1038/ng0696-175. [DOI] [PubMed] [Google Scholar]
- 2.Sarnat HB. Myotubular myopathy: arrest of morphogenesis of myofibres associated with persistence of fetal vimentin and desmin. Four cases compared with fetal and neonatal muscle. Can J Neurol Sci. 1990;17:109–23. doi: 10.1017/s0317167100030304. [DOI] [PubMed] [Google Scholar]
- 3.Bolino A, Muglia M, Conforti FL, et al. Charcot-Marie-Tooth type 4B is caused by mutations in the gene encoding myotubu-larin-related protein-2. Nat Genet. 2000;25:17–9. doi: 10.1038/75542. [DOI] [PubMed] [Google Scholar]
- 4.Senderek J, Bergmann C, Weber S, et al. Mutation of the SBF2 gene, encoding a novel member of the myotubularin family, in Charcot-Marie-Tooth neuropathy type 4B2/11p15. Hum Mol Genet. 2003;12:349–56. doi: 10.1093/hmg/ddg030. [DOI] [PubMed] [Google Scholar]
- 5.Azzedine H, Bolino A, Taieb T, et al. Mutations in MTMR13, a new pseudophos-phatase homologue of MTMR2 and Sbf1, in two families with an autosomal recessive demyelinating form of Charcot-Marie-Tooth disease associated with early-onset glaucoma. Am J Hum Genet. 2003;72:1141–53. doi: 10.1086/375034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robinson FL, Dixon JE. Myotubularin phosphatases: policing 3-phosphoinosi-tides. Trends Cell Biol. 2006;16:403–12. doi: 10.1016/j.tcb.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 7.Berger P, Schaffitzel C, Berger I, et al. Membrane association of myotubularin-related protein 2 is mediated by a pleck-strin homology-GRAM domain and a coiled-coil dimerization module. Proc Natl Acad Sci USA. 2003;100:12177–82. doi: 10.1073/pnas.2132732100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lorenzo O, Urbe S, Clague MJ. Systematic analysis of myotubularins: heteromeric interactions, subcellular local-isation and endosome related functions. J Cell Sci. 2006;119:2953–9. doi: 10.1242/jcs.03040. [DOI] [PubMed] [Google Scholar]
- 9.Cui X, De Vivo I, Slany R, et al. Association of SET domain and myotubu-larin-related proteins modulates growth control. Nat Genet. 1998;18:331–7. doi: 10.1038/ng0498-331. [DOI] [PubMed] [Google Scholar]
- 10.Berger P, Berger I, Schaffitzel C, et al. Multi-level regulation of myotubularin-related protein-2 phosphatase activity by myotubularin-related protein-13/set-binding factor-2. Hum Mol Genet. 2006;15:569–79. doi: 10.1093/hmg/ddi473. [DOI] [PubMed] [Google Scholar]
- 11.Berger P, Bonneick S, Willi S, et al. Loss of phosphatase activity in myotubularin-related protein 2 is associated with Charcot-Marie-Tooth disease type 4B1. Hum Mol Genet. 2002;11:1569–79. doi: 10.1093/hmg/11.13.1569. [DOI] [PubMed] [Google Scholar]
- 12.Firestein R, Cleary ML. Pseudo-phos-phatase Sbf1 contains an N-terminal GEF homology domain that modulates its growth regulatory properties. J Cell Sci. 2001;114:2921–7. doi: 10.1242/jcs.114.16.2921. [DOI] [PubMed] [Google Scholar]
- 13.Nandurkar HH, Layton M, Laporte J, et al. Identification of myotubularin as the lipid phosphatase catalytic subunit associated with the 3-phosphatase adapter protein, 3-PAP. Proc Natl Acad Sci USA. 2003;100:8660–5. doi: 10.1073/pnas.1033097100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suter U. Phosphoinositides and Charcot-Marie-tooth disease: new keys to old questions. Cell Mol Life Sci. 2007;64:3261–5. doi: 10.1007/s00018-007-7381-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zuchner S, Noureddine M, Kennerson M, et al. Mutations in the pleckstrin homology domain of dynamin 2 cause dominant intermediate Charcot-Marie-Tooth disease. Nat Genet. 2005;37:289–94. doi: 10.1038/ng1514. [DOI] [PubMed] [Google Scholar]
- 16.Kachhap SK, Faith D, Qian DZ, et al. The N-Myc down regulated Gene1 (NDRG1) Is a Rab4a effector involved in vesicular recycling of E-cadherin. PLoS ONE. 2007;2:1–11. doi: 10.1371/journal.pone.0000844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delague V, Jacquier A, Hamadouche T, et al. Mutations in FGD4 encoding the Rho GDP/GTP exchange factor FRABIN cause autosomal recessive Charcot-Marie-Tooth type 4H. Am J Hum Genet. 2007;81:1–16. doi: 10.1086/518428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stendel C, Roos A, Deconinck T, et al. Peripheral nerve demyelination caused by a mutant Rho GTPase guanine nucleotide exchange factor, frabin/FGD4. Am J Hum Genet. 2007;81:158–64. doi: 10.1086/518770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chow CY, Zhang Y, Dowling JJ, et al. Mutation of FIG4 causes neurodegenera-tion in the pale tremor mouse and patients with CMT4J. Nature. 2007;448:68–72. doi: 10.1038/nature05876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–7. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- 21.Balla T. Inositol-lipid binding motifs: signal integrators through protein-lipid and protein-protein interactions. J Cell Sci. 2005;118:2093–104. doi: 10.1242/jcs.02387. [DOI] [PubMed] [Google Scholar]
- 22.Sbrissa D, Ikonomov OC, Shisheva A. Phosphatidylinositol 3-phosphate-interacting domains in PIKfyve. Binding specificity and role in PIKfyve. Endomenbrane localization. J Biol Chem. 2002;277:6073–9. doi: 10.1074/jbc.M110194200. [DOI] [PubMed] [Google Scholar]
- 23.Rutherford AC, Traer C, Wassmer T, et al. The mammalian phosphatidylinositol 3-phosphate 5-kinase (PIKfyve) regulates endosome-to-TGN retrograde transport. J Cell Sci. 2006;119:3944–57. doi: 10.1242/jcs.03153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dove SK, Cooke FT, Douglas MR, et al. Osmotic stress activates phosphatidylinos-itol-3,5-bisphosphate synthesis. Nature. 1997;390:187–92. doi: 10.1038/36613. [DOI] [PubMed] [Google Scholar]
- 25.Jones DR, Gonzalez-Garcia A, Diez E, et al. The identification of phosphatidyli-nositol 3,5-bisphosphate in T- lymphocytes and its regulation by interleukin-2. J Biol Chem. 1999;274:18407–13. doi: 10.1074/jbc.274.26.18407. [DOI] [PubMed] [Google Scholar]
- 26.Tsujita K, Itoh T, Ijuin T, et al. Myotubularin regulates the function of the late endosome through the GRAM domain-phosphatidylinositol 3,5-bisphosphate interaction. J Biol Chem. 2004;279:13817–24. doi: 10.1074/jbc.M312294200. [DOI] [PubMed] [Google Scholar]
- 27.Seet LF, Hong W. The Phox (PX) domain proteins and membrane traffic. Biochim Biophys Acta. 2006;1761:878–96. doi: 10.1016/j.bbalip.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 28.Dove SK, Piper RC, McEwen RK, et al. Svp1p defines a family of phosphatidyli-nositol 3,5-bisphosphate effectors. EMBO J. 2004;23:1922–33. doi: 10.1038/sj.emboj.7600203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whitley P, Reaves BJ, Hashimoto M, et al. Identification of mammalian Vps24p as an effector of phosphatidylinositol 3,5-bisphosphate-dependent endosome compartmentalization. J Biol Chem. 2003;278:38786–95. doi: 10.1074/jbc.M306864200. [DOI] [PubMed] [Google Scholar]
- 30.Kurten RC, Cadena DL, Gill GN. Enhanced degradation of EGF receptors by a sorting nexin, SNX1. Science. 1996;272:1008–10. doi: 10.1126/science.272.5264.1008. [DOI] [PubMed] [Google Scholar]
- 31.Carlton J, Bujny M, Peter BJ, et al. Sorting nexin-1 mediates tubular endosome-to-TGN transport through coincidence sensing of high- curvature membranes and 3-phosphoinositides. Curr Biol. 2004;14:1791–800. doi: 10.1016/j.cub.2004.09.077. [DOI] [PubMed] [Google Scholar]
- 32.Bonneick S, Boentert M, Berger P, et al. An animal model for Charcot-Marie-Tooth disease type 4B1. Hum Mol Genet. 2005;14:3685–95. doi: 10.1093/hmg/ddi400. [DOI] [PubMed] [Google Scholar]
- 33.Tersar K, Boentert M, Berger P, et al. Mtmr13/Sbf2-deficient mice: an animal model for CMT4B2. Hum Mol Genet. 2007;16:2991–3001. doi: 10.1093/hmg/ddm257. [DOI] [PubMed] [Google Scholar]
- 34.Ikonomov OC, Sbrissa D, Foti M, et al. PIKfyve controls fluid phase endocytosis but not recycling/degradation of endocy-tosed receptors or sorting of procathepsin D by regulating multivesicular body mor-phogenesis. Mol Biol Cell. 2003;14:4581–91. doi: 10.1091/mbc.E03-04-0222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Worby CA, Dixon JE. Sorting out the cellular functions of sorting nexins. Nat Rev Mol Cell Biol. 2002;3:919–31. doi: 10.1038/nrm974. [DOI] [PubMed] [Google Scholar]
- 36.Gullapalli A, Garrett TA, Paing MM, et al. A role for sorting nexin 2 in epidermal growth factor receptor down-regulation: evidence for distinct functions of sorting nexin 1 and 2 in protein trafficking. Mol Biol Cell. 2004;15:2143–55. doi: 10.1091/mbc.E03-09-0711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu H, Liu ZQ, Chen CX, et al. Inhibitory regulation of EGF receptor degradation by sorting nexin 5. Biochem Biophys Res Commun. 2006;342:537–46. doi: 10.1016/j.bbrc.2006.01.179. [DOI] [PubMed] [Google Scholar]
- 38.Ogata T, Iijima S, Hoshikawa S, et al. Opposing extracellular signal-regulated kinase and Akt pathways control Schwann cell myelination. J Neurosci. 2004;24:6724–32. doi: 10.1523/JNEUROSCI.5520-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cao C, Backer JM, Laporte J, et al. Sequential actions of myotubularin lipid phosphatases regulate endosomal PI(3)P and growth factor receptor trafficking. Mol Biol Cell. 2008;19:3334–46. doi: 10.1091/mbc.E08-04-0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Begley MJ, Taylor GS, Kim SA, et al. Crystal structure of a phosphoinositide phosphatase, MTMR2: insights into myotubular myopathy and Charcot-Marie-Tooth syndrome. Mol Cell. 2003;12:1391–402. doi: 10.1016/s1097-2765(03)00486-6. [DOI] [PubMed] [Google Scholar]
- 41.Shisheva A, Rusin B, Ikonomov OC, et al. Localization and insulin-regulated relocation of phosphoinositide 5- kinase PIKfyve in 3T3-L1 adipocytes. J Biol Chem. 2001;276:11859–69. doi: 10.1074/jbc.M008437200. [DOI] [PubMed] [Google Scholar]
- 42.Cabezas A, Pattni K, Stenmark H. Cloning and subcellular localization of a human phosphatidylinositol 3-phosphate 5-kinase, PIKfyve/Fab1. Gene. 2006;371:34–41. doi: 10.1016/j.gene.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 43.Bache KG, Stuffers S, Malerod L, et al. The ESCRT-III subunit hVps24 is required for degradation but not silencing of the epidermal growth factor receptor. Mol Biol Cell. 2006;17:2513–23. doi: 10.1091/mbc.E05-10-0915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yan Q, Hunt PR, Frelin L, et al. mVps24p functions in EGF receptor sorting/trafficking from the early endosome. Exp Cell Res. 2005;304:265–73. doi: 10.1016/j.yexcr.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 45.Pendaries C, Tronchere H, Arbibe L, et al. PtdIns5P activates the host cell PI3-kinase/Akt pathway during Shigella flexneri infection. EMBO J. 2006;25:1024–34. doi: 10.1038/sj.emboj.7601001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Knodler LA, Finlay BB, Steele-Mortimer O. The Salmonella effector protein SopB protects epithelial cells from apoptosis by sustained activation of Akt. J Biol Chem. 2005;280:9058–64. doi: 10.1074/jbc.M412588200. [DOI] [PubMed] [Google Scholar]
- 47.Tronchere H, Laporte J, Pendaries C, et al. Production of phosphatidylinositol 5-phosphate by the phosphoinositide 3-phosphatase myotubularin in mammalian cells. J Biol Chem. 2004;279:7304–12. doi: 10.1074/jbc.M311071200. [DOI] [PubMed] [Google Scholar]
- 48.Bjorge JD, Chan TO, Antczak M, et al. Activated type I phosphatidylinositol kinase is associated with the epidermal growth factor (EGF) receptor following EGF stimulation. Proc Natl Acad Sci USA. 1990;87:3816–20. doi: 10.1073/pnas.87.10.3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quattrone A, Gambardella A, Bono F, et al. Autosomal recessive hereditary motor and sensory neuropathy with focally folded myelin sheaths: clinical, electrophysio-logic, and genetic aspects of a large family. Neurology. 1996;46:1318–24. doi: 10.1212/wnl.46.5.1318. [DOI] [PubMed] [Google Scholar]
- 50.Ikonomov OC, Sbrissa D, Mlak K, et al. Requirement for PIKfyve enzymatic activity in acute and long-term insulin cellular effects. Endocrinology. 2002;143:4742–54. doi: 10.1210/en.2002-220615. [DOI] [PubMed] [Google Scholar]
- 51.Khajavi M, Inoue K, Wiszniewski W, et al. Curcumin treatment abrogates endo-plasmic reticulum retention and aggregation-induced apoptosis associated with neuropathy-causing myelin protein zero-truncating mutants. Am J Hum Genet. 2005;77:841–50. doi: 10.1086/497541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khajavi M, Shiga K, Wiszniewski W, et al. Oral curcumin mitigates the clinical and neuropathologic phenotype of the Trembler-J mouse: a potential therapy for inherited neuropathy. Am J Hum Genet. 2007;81:438–53. doi: 10.1086/519926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Berger P, Niemann A, Suter U. Schwann cells and the pathogenesis of inherited motor and sensory neuropathies (Charcot-Marie-Tooth disease) Glia. 2006;54:243–57. doi: 10.1002/glia.20386. [DOI] [PubMed] [Google Scholar]
- 54.Niemann A, Berger P, Suter U. Pathomechanisms of mutant proteins in Charcot-Marie-Tooth disease. Neuromolecular Med. 2006;8:217–42. doi: 10.1385/nmm:8:1-2:217. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Characterization of the cell lines overexpressingHis-tagged Mtmr2 and/or HA-tagged Mtmr13/Sbf2. Cells were plated onglass cover slips and fixed after 3 days. Mtmr2 was detected with amouse anti-His antibody (Qiagen, Hilden, Germany) and Mtmr13 with arabbit anti-Mtmr13/Sbf2 antibody. Nuclei of the cells were stainedwith 4′,6-Diamidino-2-phenylindol (DAPI). Note that more than95% of the cells express the indicated transgenicprotein. Scale bar. 20 μm.
Mtmrs and sorting nexins in unstimulated COScells Colocalization of Mtmr2 or Mtmr13/Sbf2 with SNX1, SNX2 orSNX5 in unstimulated cells. COS cells were cotransfacted with theindicated plasmids and then fixed after 28 hrs (Mtmr13/Sbf2transfected cells) or 42 hrs (Mtmr2 transfected cells). Allproteins show a diffuse cytoplasmic or vesicular staining. Scalebars: 20 μm (upper panel), 5 μm (lower panel).