

Abstract
Airway inflammation is a common condition where glucocorticoids (GC) are a well-established therapy. It has been demonstrated that GC stimulate components of innate immunity. Specifically, GC up-regulate TLR2 expression and activation upon inflammatory stimuli; however, little is known about the signalling involved in this process. To determine the mechanism by which dexamethasone modulates TLR2-induced cytokine production this signalling pathway was monitored in a lung epithelial cell line exposed to the TLR2 synthetic agonist, Pam3-Cys-Ser-Lys4. These experiments demonstrate that phosphatidylinositol 3-kinase (PI3K) is critical for the TLR2 downstream effects of GC. Cells expressing a PI3K mutant (p85-dominant negative, DN; p85 Δ478–511) and exposed to Pam3-Cys-Ser-Lys4 in the presence or absence of dexamethasone, showed enhanced tumour necrosis factor (TNF)α expression while AP-1 and NF-κB transcriptional activity were repressed. We provide experimental evidence that PI3K physically interacts with the glucocorticoid receptor (GR) through two putative PI3K recruitment consensus YxxM binding motifs in the GR, suggesting that some functions regulated by this receptor might occur through kinase interaction. Mutations of two tyrosine residues in the GR, 598 and 663, to phenylalanine significantly reduced interaction with PI3K and the GC effects on TLR2-induced TNF-α expression. However, these mutations did not alter GR transcriptional activity nor affect cellular localization of the expressed mutant GR in COS-1 cells. Therefore, the PI3K-GR interaction may contribute to the effects of GC on the TLR2 pro-inflammatory signalling cascade, thus defining a novel signalling mechanism with a profound impact on innate immune responses.
Keywords: lung mucosa inflammation, TLR2, glucocorticoid receptor, phosphatidylinositol-3-kinase
Introduction
Airway inflammation, such as asthma, chronic obstructive pulmonary disease and infection, is a frequent condition whose incidence has increased. Therapeutic strategies currently in use often rely on the use of anti-inflammatory agents such as glucocorticoids (GC). GC have been shown to enhance innate immune responses, including those related to the toll-like receptors (TLRs) [1]. While the suppressive effects on inflammatory signalling are well characterized and are shown to involve protein–protein interactions between glucocorticoid receptor (GR) and transcription factors such as NF-κB and AP-1 [2], the pro-inflammatory effects of GC still remain mechanistically unresolved.
TLRs have been shown to be crucial in triggering epithelial innate immune response by recognizing pathogen-associated molecular patterns (PAMPs) and stimulating host immune cells against several microbial products [3]. To date, 13 distinct receptors have been reported which belong to the TLR family and are responsible for recognizing and triggering a response to microbial products such as LPS, peptidoglycan, flagellin, viral and bacterial CpG-DNA motifs, and single- and double-stranded viral RNAs [4–8]. TLR2, one of the members of the TLR family, is a pathogen recognition receptor (PRR) for gram-positive-derived stimuli or synthetic lipopeptides, such as Pam3-Cys-Ser-Lys4[6, 9–12]. Lung epithelial cells actively secrete and respond to inflammatory cytokines [1, 13] that are produced when the TLR2 signalling pathway is activated [6]. Once TLR2 is activated by gram-positive bacteria, MyD88 and TIRAP adaptor proteins are recruited to the receptor intracellular domain [14]. The serine/threonine kinase, IRAK, is subsequently recruited leading to an interaction of the TLR complex with the downstream signalling molecule TRAF6 which subsequently activates NF-κB [14], Jun amino terminal kinase [3], extra cellular signal related kinase and p38 MAP kinases [15]. The phosphatidylinositol 3-kinase (PI3K) and small GTPase Rac1 can also participate in the TLR2 signalling pathway activated by Staphylococcus aureus[9]. Rac1 controls NF-κB-dependent gene expression and transactivation through the alternative PI3K sig-nalling pathway after TLR2 [9] or TLR4 activation [16].
GC have been reported to increase tumour necrosis factor (TNF) α and Haemophilus influenza induced expression of TLR2 mRNA and protein. This action seems to be mediated by up-regulation of MAPK phosphatase 1 and negative cross-talk with p38 MAPK [1, 17]. Moreover, TLR2 and TLR4 mRNAs were shown to be induced in resting but not activated white cells [18, 19]. However, it remains unclear if the GC effects have an impact on TLR2 signalling by influencing the alternative PI3K pathway. In this study, we show that PI3K is critical for GC effects on TLR2-induced pro-inflammatory cytokine production and that PI3K-GR interaction occurs upon TLR2 and GR activation. Moreover, we demonstrate that impairment of putative PI3K interactive sites in human GR expressed in lung epithelial cells specifically counteracts TNF expression. These effects suggest that the PI3K-GR interaction represents a molecular mechanism with profound effects on innate immunity responses.
Material and methods
Reagents and antibodies
A synthetic bacterial lipopeptide Palmitoyl-Cys((RS)-2,3-(dipalmitoyloxy)-propyl)-OH (Pam3-Cys-Ser-Lys4) analogous to the N-Terminal region of gram-positive lipoproteins was purchased from Bachem Bioscience (King of Prussia, PA, USA), dexamethasone was purchased from Steraloids (Wilton, NH, USA). Mefipristone (RU486) was a gift from Roussel UCLAF (Romain Ville Cadex, France). Anti-Flag antibody was obtained from Sigma-Aldrich (St. Louis, MO, USA). The previously characterized antibody to the human GR Ab57 [20] was used in all studies. Anti-p85 and Anti-Akt antibodies were purchased from BD Bioscience (San Jose, CA, USA), Anti-phosphoAkt (Ser473) and Anti-phosphotyrosine was purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-TNF-α-FITC and TLR2-PE conjugated antibodies were purchased from eBioscience (San Diego, CA, USA). The peroxidase-labelled secondary antibodies and enhanced chemi-luminescence (ECL) reagents were purchased from Amersham Pharmacia Biotech (Piscataway, NJ, USA).
Cells, transient transfection, recombinant expression vectors and reporter gene assays
A549 cells were cultured in Dulbecco's modified Eagle's medium (DMEM)/F12 and COS-1 or Hek-293 cells were cultured in DMEM medium, all culture media were supplemented with 5% foetal calf serum, 100 IU/ml penicillin and 100 mg/ml streptomycin. Cell cultures were maintained in a 5% CO2 humidified incubator at 37°C and passaged every 3–4 days. All transfections were carried out in cell lines with Fugene reagent according to the manufacturer's protocol (Roche, Indianapolis, IN, USA). Fugene reagent (3 μl per μg of transfected plasmid) was added to OPTIMEM (Life Sciences, Inc., St. Petersburg, FL, USA) with the purified plasmid DNA and allowed to incubate for 40 min. at room temperature before being added to cells plated in OPTIMEM.
Vectors containing the NF-κB reporter (3XMHC-Luc) and the GRE reporter (MMTV-Luc) were previously described [21]. The AP-1 reporter (AP-1-Luc) (Stratagene, La Jolla, CA, USA) was transfected in combination with 1 μg of the Rous Sarcoma virus driven c-fos and c-jun expression plasmids which were provided by Drs. Bikiri and Yaniv, Institut Pasteur of Paris, France [22]. For promoter activity experiments, 1–2 μg of 3XMHC-Luc, MMTV-Luc or AP-1-Luc were transfected into cells in combination with 10 ng of pGL3-hRL (renilla) as a control [19]. Twenty-four hours after trans-fection, cells were treated with various stimuli for 18 hrs. Cells were then lysed in passive lysis buffer (Promega Corp., Madison, WI, USA) and the lysates were used to determine luciferase activity using the dual luciferase reporter assay system (Promega Corp.). The luciferase activity was measured using the 96-well plate format with an MLX automated microtitre plate luminometer from Dynex (Richfield, MN, USA). Luciferase activity was then normalized to the renilla activity and to the control. All of the luciferase assays shown in the current study represent three separate experiments assayed in duplicate. Representative results are shown for each experiment.
Site directed mutagenesis was used to generate three GR mutants containing specific mutations in two MxxY consensus sites which have been shown to be important for the p85 subunit of PI3K recruitment [23]. Tyrosines at position 598 and/or 663 were mutated to phenylalanine in the pCMV-hGRalpha construct (Y598F-hGR; Y663F-hGR and Y598/663F-hGR). A dominant negative construct of PI3K subunit p85 (Δ478–511), which lacks the p110-binding domain (p85-DN) was provided by Dr. L.C. Cantley (Division of Signal Transduction, Dept. of Cell Biology, Harvard Medical School, Boston, MA, USA) [24]. A wild-type (wt) p85 construct was kindly provided by Dr. J. Downward (Imperial Research Fund, London, UK) [25].
Immunoblots and immunoprecipitation
Following experimental treatment, cells were detached from the flasks using 1× trypsin/ethylenediaminetetraacetic acid (EDTA), pelleted and then re-suspended in low detergent buffer (LDB, 20 mM Tris-Cl, pH 7.5, 2 mM EDTA, 150 mM NaCl and 0.5% Triton X-100, protease and phos-phatase inhibitors) and homogenized. Total protein was measured using the Bio-Rad Protein Assay reagent (Bio-Rad Laboratories, Inc., Hercules, CA, USA) according to the manufacturer's protocol, and equivalent amounts of total protein were used for immunoprecipitation. Normal mouse IgG was initially used to reduce non-specific reactivity and total homogenate was incubated for 15 min. at 4°C with end-over-end rotation. Protein A/G agarose was then used to remove the non-specific binding for 1 hr, and the cleared supernatant was incubated with specific antibodies at 4°C over night. Finally, the antigen-antibody complex was pulled down with a second exposure to protein A/G agarose and the pellet was washed with LDB plus phosphatase and protease inhibitors. Sample buffer containing SDS and -mercaptoethanol was used to elute the immunoprecipitated protein, and samples were run on an 8% SDS-PAGE gel and then transferred to nitrocellulose membranes. Densitometric analysis of immunore-active bands was processed with the Gel Pro Analyzer 4 software (MediaCybernetics, Inc., Bethesda, MD, USA). For TLR2 tyrosine phospho-rylation, pull down TLR2-Flag was blotted with an anti-phosphotyyrosine antibody. For expressed-TLR2 interaction with the PI3K subunit p85, pull down TLR2-Flag was blotted with an anti-p85 antibody. For expressed-wt GR or F598Y-hGR; F663Y-hGR and Y598F/Y663F-hGR interaction with p85, pull down GR was blotted with the anti-p85 antibody.
Flow cytometry
To determine TNF-α content, cells were treated with different concentrations of Pam3-Cys-Ser-Lys4 and/or 100 nM of dexamethasone for 4 hrs with the addition of Brefeldin-A after the first 30 min. to stop cytokine secretion. Cells were fixed with 2% paraformaldehyde for 15 min. at 4°C, and then prepared in a solution of 0.05% saponin/1× PBS/1% bovine serum albumin. A fluorescein–isothiocyanate conjugated antibody raised against TNF-α (anti-TNF-α-FITC) was diluted with saponin solution and added to the cells for 30 min. at 4°C in the dark. Cells were washed and fixed again with 2% paraformaldehyde and stored at 4°C. For TLR2 surface determination, cells were treated with different concentrations of Pam3-Cys-Ser-Lys4 and/or 100 nM of dexamethasone for 24 hrs and a phycoery-thrin conjugated anti-TLR2 antibody (anti-TLR2-PE) was added to the cells for 30 min. at 4°C in the dark. Cells were washed and then fixed with 2% paraformaldehyde and stored at 4°C. When PI3K participation was assessed on these two molecular expression patterns, cells were trans-fected with 1 μg of p85-DN for 24 hrs before treatment as described above. Stained cells were analysed with a Becton Dickinson (Franklin Lakes, NJ, USA) FACSort using CellQuest or WinMD software.
Statistical analysis
All pair treated groups were compared with the ANOVA Tukey–Kramer analysis. Significant differences have a P < 0.05. Analysis was carried out with the JMP Software, Statistics Made Visual, SAS Institute, Inc. (Cary, NC, USA).
Results
Mutual inhibition of PI3K and GR activity in cells pulsed with Pam3-Cys-Ser-Lys4
GC have been shown to enhance innate immune response in clinical studies and animal models [26, 27]. Recently, we reported that GC cooperatively enhanced the expression of TLR2 induced by the pro-inflammatory cytokine TNF-α[1]. To determine if GC have an impact on TLR2-mediated signalling mechanisms activated by Pam3-Cys-Ser-Lys4, we first investigated the activation of PI3K. TLR2 has two tyrosine (Tyr) residues susceptible to phos-phorylation when the receptor is activated by gram-positive bacterial agents [9, 28]. In Hek-293 cells transiently expressing TLR2 and GR and treated briefly with Pam3-Cys-Ser-Lys4 and/or dexam-ethasone, tyrosine phosphorylation was enhanced (Fig. 1A). These results indicate that Pam3-Cys-Ser-Lys4 stimulates early TLR2 signalling events and that GC induce a cooperative effect on receptor activation.
Fig 1.
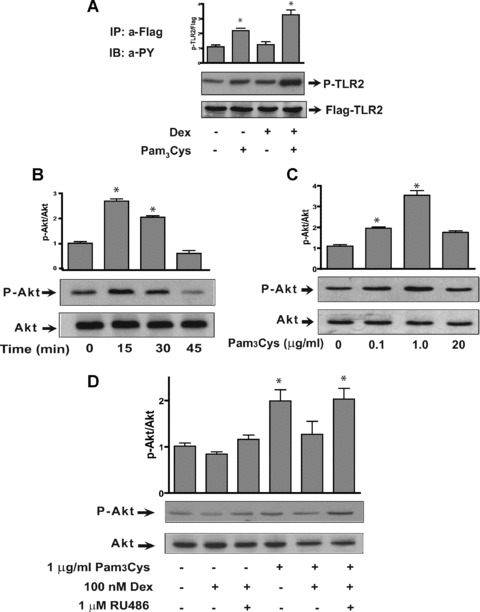
Pam3-Cys-Ser-Lys4 and dexamethasone regulate Akt phosphorylation. (A) Hek-293 cells were transiently transfected with TLR2 and GR and treated with 1 μg/ml Pam3-Cys-Ser-Lys4 and /or 100 nM dexamethasone for 5 min. and TLR2 was immunoprecipitated from Hek-293 cell homogenate using an antibody raised against Flag epitope, then solved in a SDS-PAGE and blotted with an anti-phophotyrosine antibody (n= 3; *P < 0.05 significant over the control). The bar graph represents the densitometric ratio between P-tyrosine/TLR2-Flag and over control as shown in the representative Western blot at the top of each graph (n= 3; *P < 0.05 significant over the control). (B) A549 cell extracts separated on SDS-PAGE and blotted with an anti-phospho Ser473 Akt (P-Akt) and anti-Akt, followed by ECL detection, after stimulation with 1 μg/ml Pam3-Cys-Ser-Lys4 for the indicated times. A transient but significant increase in P-Akt was seen after 15 min. of TLR2 activation. The bar graph represents the densitometric ratio between P-Akt/Akt and over control as shown in the representative Western blot at the top of each graph (n= 3; *P < 0.05 significant over the control). (C) A549 cell extracts were separated on SDS-PAGE and blotted with an anti-phospho Ser473 Akt (P-Akt) and anti-Akt, followed by ECL detection, after stimulation with different concentrations of Pam3-Cys-Ser-Lys4 for 15 min. A significant increase in P-Akt occurred with 1 μg/ml Pam3-Cys-Ser-Lys4. The bar graph represents the densitometric ratio between P-Akt/Akt and over control as shown in the representative Western blot at the top of each graph (n= 3; *P < 0.05 significant over the control). (D) A549 cells were treated with the GR antagonist RU486 (1 μM) and one hr later with 1 μg/ml Pam3-Cys-Ser-Lys4 and 100 nM dexamethasone. Treatment with dexamethasone counteracted Akt phosphory-lation induced by TLR2 activation, an effect that required dexamethasone binding to the GR as RU486 reversed this inhibition (n= 3; *P < 0.05 significant over the control).
Tyrosine phosphorylation induced by TLR2 activation has a strong impact on PI3K recruitment to the intracellular receptor domain, which in turn phosphorylates and activates the serine-threonine kinase Akt. A549 cells exposed to the TLR2 agonist Pam3-Cys-Ser-Lys4 showed a time and a dose-dependent increase in Akt phosphorylation (P-Akt) (Fig. 1B, C). Akt phosphorylation significantly increases after 15 min. treatment with 1 μg/ml of Pam3-Cys-Ser-Lys4 (2.5-fold) (Fig. 1B) and the maximum kinase activity was observed with 1 μg/ml (3.5-fold) (Fig. 1C). These data suggest rapid activation of the PI3K alternative pathway after exposure of epithelial cells to the TLR2 ligand.
To determine the effects of dexamethasone in the TLR2/PI3K/Akt signalling pathway, we examined Akt phosphoryla-tion in A549 cells treated with Pam3-Cys-Ser-Lys4 concomitantly with dexamethasone. Akt phosphorylation induced by Pam3-Cys-Ser-Lys4 was specifically inhibited by dexamethasone, as this inhibition was blocked by the GR antagonist RU486 (Fig. 1D). These results suggest that the GR may block PI3K activation induced by TLR2 stimulation and may play an important role in modulating the TLR2/PI3K/Akt signalling pathway.
Phosphatidylinositol 3-kinase sensitizes A549 cells to counteract TNF-α production induced by TLR2
To determine the role of the PI3K pathway on TLR2-mediated pro-inflammatory cytokine production, we analysed the effect of PI3K on the expression of TNF-α upon TLR2 activation in the presence or absence of dexamethasone. Expression of TNF-α was studied in A549 cells expressing a dominant negative construct of the PI3K subunit p85 (Δ478–511) which lacks the p110-binding domain (p85-DN) stimulated with Pam3-Cys-Ser-Lys4 alone or in combination with dexamethasone. PI3K intervention with the p85-DN induced a significant increase in TNF-α intracellular expression that was greatly enhanced with 20 μg/ml of Pam3-Cys-Ser-Lys4. Dexamethasone addition did not increase TNF-α when PI3K was abrogated (Fig. 2A).
Fig 2.
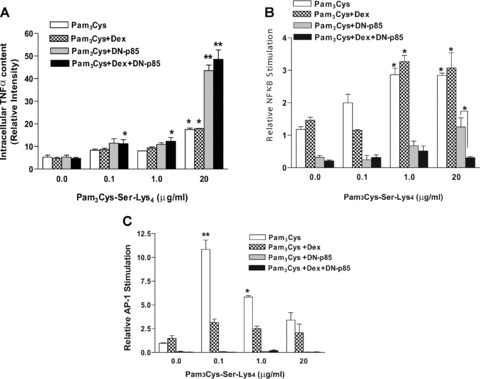
PI3K regulates the production of TNF-α and signalling events induced by Pam3-Cys-Ser-Lys4 and dexamethasone. (A) A549 cells transiently trans-fected with a p85 dominant negative mutant (p85-DN) as indicated, were exposed to Pam3-Cys-Ser-Lys4 (0.1 to 20 μg/ml) in the presence or absence of 100 nM dexamethasone for 4 hrs. Brefeldin-A was added 30 min. after exposure to the stimuli to stop cytokine secretion. Intracellular TNF-α production was determined by flow cytometry and normalized to untreated cells (n= 3; *P < 0.05, **P < 0.001 significant over the control). (B) A549 cells were co-transfected with the NF-κB reporter plasmid 3XMHC-Luc, pGL3-hRL and p85-DN mutant constructs indicated. Cells were stimulated for 18 hrs with different concentrations of Pam3-Cys-Ser-Lys4 (0.1–20 μg/ml) in the presence or absence of 100 nM dexamethasone and analysed for NF κ B activation through luciferase activity. (n= 3, *P < 0.05, **P < 0.01 significant over the vehicle treated control). (C) A549 cells were co-transfected with the reporter plasmids AP-1-Luc, pGL3-hRL and p85-DN mutant as indicated, together with vectors expressing c-fos and c-jun. Cells were left either untreated or stimulated for 18 hrs with different concentrations of Pam3-Cys-Ser-Lys4 (0.1–20 μg/ml) in the presence or absence of 100 nM dexamethasone. AP-1 transcriptional activity was assessed through luciferase activity. (n= 3, *P < 0.05, **P < 0.01 significant over the vehicle treated control.)
To further evaluate the role of PI3K on the TLR2-induced classical signalling pathway, we analysed NFκB transcriptional activity in cells where PI3K is inhibited by expression of p85-DN. Cells in which PI3K was transiently inhibited or deficient showed a diminished basal NFκB transcriptional activity that followed a similar trend as control cells exposed to Pam3-Cys-Ser-Lys4 (Fig. 2B). However, dexamethasone significantly antagonized NFκB induction under conditions where PI3K activity was abolished and high concentrations of Pam3-Cys-Ser-Lys4 (20 μg/ml) were added. The concentration of p85-DN used in these experiments (1 μg/ml) also significantly antagonized the induction of NFκB in the absence of Pam3-Cys-Ser-Lys4 (data not shown) suggesting that, under these conditions, it might counteract the endogenous kinase. Together, these results indicate that PI3K positively regulates NFκB activity induced by Pam3-Cys-Ser-Lys4 in lung epithelial cells and further contributes to the effect of GC on TNF-α production when a high dose of TLR2-agonist is used.
To determine the specificity of this process, we examined the role of PI3K on AP-1 transcriptional activity upon TLR2 activation in the presence or absence of dexamethasone. Cells transiently expressing the p85-DN construct did not show an AP-1-driven transcriptional activity when treated with Pam3-Cys-Ser-Lys4 either in the presence or absence of dexamethasone (Fig. 2C), suggesting that PI3K also positively regulates AP-1-mediated transcriptional upon TLR2 activation.
Physical interaction between PI3K and GR
The TLR2 intracellular domain as well as the GR steroid binding domain contain tyrosine residues vulnerable to phosphorylation when activated by microbial products or steroid agonists, respectively. This molecular event may promote PI3K subunit p85 recruitment to phospho-tyrosine residues. To evaluate the role of GR on TLR2 signalling activation, co-immunoprecipitation assays were conducted on cells overexpressing TLR2-Flag, p85 and GR recombinant vectors. The diagram in Fig. 3A shows the location of potential p85 subunit recruitment consensus motifs (YxxM) in the amino acid sequences of TLR2 and GR. (These results obtained using the DS Gene v1.5 Accelrys software, Inc., San Diego, CA, USA) The TLR2 intracellular domain and the GR steroid-binding domain, both located at the carboxyl-terminus of their molecules, contain one and two YxxM motifs, respectively. This observation is supported by the evidence that the p85 subunit co-immunopre-cipitates with GR and this interaction is enhanced by dexametha-sone in vivo[29]. Cells treated with dexamethasone induced a 2-fold increase in the GR/PI3K immunoprecipitated product as compared to untreated cells (Fig. 3B, lanes 1 and 3). Treatment with dexamethasone in the presence of 1 μg/ml Pam3-Cys-Ser-Lys4 did not change dexamethasone-induced GR/p85 co-immunoprecipitation (Fig. 3B, lanes 2 and 4). These data suggest that activated GR is needed to enhance GR/PI3K interaction. In addition, treatment with dexamethasone did not alter TLR2/p85 co-immunoprecipitation (Fig. 3C) suggesting that PI3K recruitment to the TLR2 intracellular domain is not modulated by GC. Moreover, TLR2 agonist did not alter TLR2/p85 co-immunoprecipitation. Experiments were also conducted to assess the impact of PI3K on GR-mediated transcriptional activation in cells transiently co-expressing the MMTV-Luc and p85-DN constructs. When the PI3K dominant negative was expressed (p85-DN), a significant inhibition of GC-induced transcription was observed (Fig. 3D) indicating that p85 recruitment to GR is important for GR-mediated gene transcription. These finding indicate that there is a cross-talk between GR and PI3K that results in mutual inhibitory function.
Fig 3.
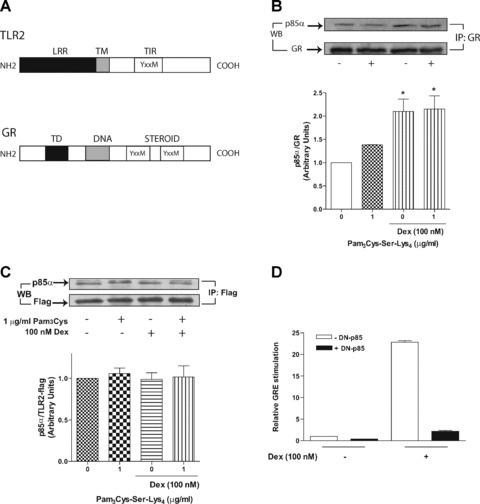
Interaction between p85/GR and p85/TLR2. (A) Schematic drawing of the YxxM motifs in the TLR2 and GR amino acid sequences. (B) GR and p85 co-immunoprecipitation assay. A549 cells overexpressing p85-wt and wt-GR constructs were exposed to 1 μg/ml Pam3-Cys-Ser-Lys4 in the presence or absence of 100 nM dexamethasone for 12 min. (the Western blot is representative of three independent experiments, * represents the fold of the densitometric ratio between p85/GR as compared 0 nM control). (C) A549 cells overexpressing p85-wt and TLR2-Flag constructs were exposed to 1 μg/ml Pam3-Cys-Ser-Lys4 in the presence or absence of 100 nM dexamethasone for 12 min. (the Western blot is representative of three independent experiments). (D) PI3K regulates GRE transcriptional activity induced by dexamethasone. A549 cells were co-transfected with the reporter plasmids MMTV-Luc, pGL3-hRL and the p85-DN mutant as indicated. Cells were stimulated for 18 hrs with 100 nM dexamethasone and analysed for GRE-driven transcriptional activation through luciferase activity (n= 3).
Functional analysis of GR, and YxxM-GR mutants
We next evaluated the molecular mechanism underlying the interaction between PI3K and GR. Experiments were conducted to assess the impact of PI3K as well the GR tyrosine mutation on GR-mediated transcriptional activation in cells transiently co-expressing the MMTV-Luc and p85-DN constructs or YxxM-hGR mutants (Y598F-hGR, Y663F-hGR or Y598/663F-hGR), respectively. When both tyrosine residues in the YxxM-GR motif are mutated, a slight reduction in GC-mediated transcription was seen indicating that phosphorylation of these amino acids has some role in receptor-mediated gene transcription (Fig. 4A). Experiments were then completed to show if the interaction between GR and PI3K is functionally involved in regulating TLR2-induced TNF-α expression. COS-1 cells transiently expressing the Y/F-hGR mutant recombinant vectors were treated with dexam-ethasone in the presence or absence of 10 μg/ml Pam3-Cys-Ser-Lys4 and intracellular TNF-α expression was determined by flow cytometry. Pam3-Cys-Ser-Lys4 induced a rise in TNF-α expression that is synergized by co-treatment with dexamethasone in cells transiently transfected with empty vector (control) or wt-GR (Fig. 4B). TLR2 activation in cells expressing the Y/F hGR mutants also showed an increase in intracellular TNF-α; however, dexamethasone co-treatment repressed the Pam3-Cys-Ser-Lys4 effect on cytokine expression (Fig. 4B). Dexamethasone treatment alone increased intracellular TNF-α in cells expressing the Y/F GR mutants suggesting that GR is a platform for TLR2 signalling pathway regulation which may require intact phosphorylation sites [30, 31]. These data indicate that a PI3K dominant negative effect is enhanced in cells where the YxxM p85 recruitment motifs in GR are altered and in the presence of TLR2 and GR agonists. Interaction between p85 and GR was studied in COS-1 cells expressing the wt-GR and YxxM-hGR mutant recombinant vectors: Y599F-hGR, Y663F-hGR or Y598/663F-hGR, together with p85. Upon dexamethasone treatment p85/GR interaction was exclusively detected in cells expressing the wt-GR construct but not with any of the YxxM-hGR mutants (Fig. 4C). These data indicate that activated GR is needed to enhance GR/p85 interaction and that both tyrosine residues, present in the steroid-binding region of GR, are required for p85 recruitment to GR.
Fig 4.
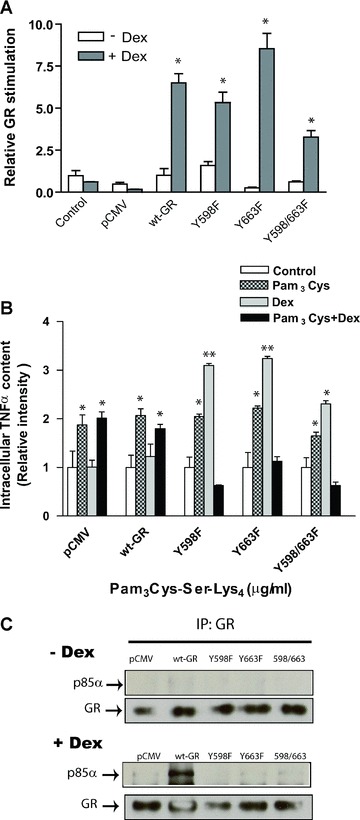
Functional implications of YxxM motifs present in hGR. (A) The hGR mutant proteins were analysed for transactivation potential following transient transfection into GR-deficient COS-1 cells. Cells were co-transfected with no DNA (control), the empty vector (pCMV) or the indicated GR expression vectors and a glucocorticoid-responsive MMTV-Luc reporter gene. Hormone response was measured as the fold induction of Luciferase activity in response to 100 nM dexamethasone treatment (+Dex, grey bars) over that of untreated cells (–Dex, white bars). Data shown are an average for four to seven experiments with the indicated standard error of the mean (*P < 0.05 significant over the control). (B) TNF-α expression regulation by YxxM motifs present in hGR. Cells expressing empty vector (control), wt-GR or Y598/663F-hGR mutant were exposed to Pam3-Cys-Ser-Lys4 (1 μg/ml) in the presence or absence of 100 nM dexamethasone for 4 hrs. Brefeldin-A was added 30 min. after exposure to the stimuli to stop cytokine secretion. Intracellular TNF-α production was determined by flow cytometry and normalized to untreated cells (n= 3, *P < 0.05, **P < 0.001 significant over the control). (C) GR and p85 co-immunoprecipitation assay in COS-1cells. COS-1 cells overexpressing p85-wt and hGR mutant constructs were exposed to 100 nM dexamethasone for 12 min. and immunoprecip-itated with antibody raised against GR was, then solved in a SDS-PAGE and blotted with an anti-p85 antibody (the Western blots shown are representative of three independent experiments).
Discussion
In the present study, we address the role of PI3K and GC as immunomodulatory molecules of the TLR2 signalling pathway in lung epithelial cells. These experiments demonstrate that PI3K plays a negative regulatory role in the pathways leading to TNF-α expression induced by TLR2 activation in the presence or absence of GC. Moreover, GR activity is critically controlled by PI3K. In particular, in A549 cells, stimulation through TLR2 in the presence or absence of GC resulted in increased intracellular TNF-α expression and NF-κB activation. In contrast, Akt phosphorylation and AP-1 activation upon TLR2 induction were significantly inhibited by dexametha-sone. PI3K co-immunoprecipitated with TLR2 and GR, suggesting that the negative regulation of PI3K on the pro-inflammatory TLR2 pathway might require protein–protein interactions.
PI3K participation in the TLR2 signalling pathway is controversial; however, the present results demonstrate a central role for PI3K in epithelial responses to TLR2-GR crosstalk. Whether PI3K has similar pro-inflammatory effects in other settings of epithelial activation remains to be determined. PI3K phosphorylates 3-phospholipids that in turn activate a series of cellular intermediates including tyrosine kinases, small G proteins and serine/threonine kinases such as protein kinase C (PKC-ζ) and Akt [32].
Recent studies have found that, in neutrophils and HEK293 cells, PI3K has a positive rather than a negative regulatory role on pro-inflammatory cytokine production [33]. However, PI3K had a negative regulatory effect in dendritic cells from PI3K deficient mice (PI3K−/−) or normal cells cultured in the presence of a TLR2 or TLR4 agonist [34, 35]. In this context, dendritic cells from deficient PI3K mice demonstrated increased release of IL-12 upon TLR2 activation. Our studies show that inhibition of PI3K results in a significant increase in TNF-α production but decreased NF-κB activation in TLR2-stimulated epithelial cells. These data are consistent with other reports [33] and confirm that signalling pathways involving PI3K and Akt modulate NF-κB activation and pro-inflammatory cytokine production in epithelial cells after TLR2 engagement. Even though we and others have shown a positive regulatory role for PI3K/Akt in NF-κB activation [9, 33, 36], this is not a universal finding in terms of NF-κB mediated cytokine production. Several studies in other cell types have reported a negative relationship between TLR4-activated NF-κB and PI3K/Akt [37–39]. Together these findings would suggest that the role of PI3K and Akt in modulating NF-κB activation is likely a cell type and stimulus dependent.
Although translocation of NF-κB dimers into the nucleus is important for NF-κB dependent transcription, phosphorylation of the p65 subunit at serine residues is required for maximal transcriptional activity [40, 41]. The molecular mechanism by which PI3K activates NF-κB does not appear to be involved in the nuclear translocation of NF-κB but does increase the duration and magnitude of the phosphorylation state [42–44]. This evidence suggests a mechanism by which PI3K is affecting the production of cytokines, such as TNF-α, in a different way than is shown here for TLR2-induced TNF-α expression.
Airway epithelium is one of the first lines of defence against invasion by airborne gram-positive bacterial pathogens. Bacterial PAMPs are recognized by TLR2, and its activation triggers pro-inflammatory reactions that induce the production of pro-inflammatory cytokines that stimulate the hypothalamic–pituitary–adrenal axis and induce the synthesis and release of antiinflammatory GC. This feedback loop affords a tuned homeostatic control over inflammation and excludes an over-response to inflammatory signals. Similarly, pathogenic endotoxins are also known to increase the expression of GR, which sensitizes the cells to GC thereby counteracting the effect of pro-inflammatory molecules. The interaction between GC and cytokines is often cell type specific and depends on the physiologic context of the cell. The antagonizing effect of GC on TNF-α-induced pro-inflammatory reactions involves the interaction between the GR with pro-inflammatory transcription factors bound to the promoter region of different cytokine and pro-inflammatory genes. However, cooperative regulation of TLR2 gene at both the mRNA and the protein levels in response to TNF-α and dexamethasone was demonstrated previously (1) and represents a novel signalling mechanism that involves recruitment of NF-κB and STAT transcription factors as well as the GR.
Understanding the PI3K role in TLR2 activation by Pam3-Cys-Ser-Lys4 in the presence of dexamethasone will provide additional information about the cooperative effect observed after the combined treatment with pro- and anti-inflammatory molecules. One recent study suggests that the GR physically associates with PI3K and decreases the Akt activity in mouse skin cells that overexpress GR as well as in lens epithelial cells [29, 45]. Here, we demonstrated that epithelial cells exposed to Pam3-Cys-Ser-Lys4 in the presence of dexamethasone significantly increased TLR2 phosphorylation. This is the first evidence reporting the effect of GC on TLR2 early signalling events. Downstream of TLR2 activation, GC treatment repressed Akt phosphorylation as shown here and previously [29] indicating that GC effects on the PI3K/Akt signalling pathway is not exclusive to this cell type. The decrease in Akt phosphorylation might be related to a diminished phosphatidyli-nositol-3,4,5 triphosphate (PIP3) generation as it has been associated with Akt phosphorylation status. Moreover, these very rapid effects of GC require the physical interaction between GR and p85. Indeed, GR contains two PI3K recruitment motifs that directly contribute to the interaction between both molecules. However, transactivation experiments showed that these PI3K motifs are not relevant for GR transcriptional activity or translocation into the nucleus upon dexamethasone exposure.
Our docking simulations show that dexamethasone docks into the binding pocket located towards the intracellular end of the Y598/663F GR mutant receptor in the same manner as has been shown in the crystal structure for wt-GR (Fig. S1C). The close agreement between our mutagenesis and functional studies, as well as the simulated docking results, indicate that the tyrosine to phenylalanine mutations do not affect the binding interaction of GR with dexamethasone due to the distance of these amino acids from the binding pocket. However, mutational modification of GR has a functional impact on TLR2-induced TNF-α expression, and nuclear translocation of these GR mutants requires a higher dose of GC (Fig. S2A and B). Nonetheless, GR tyrosine mutations impair PI3K recruitment to the GR with a concomitant impact on TLR2-mediated pro-inflammatory effects.
Although the use of an epithelial cell line does not necessarily represent the situation in lung inflammation, these results clearly show that PI3K contributes to GR intervention in TLR2 pro-inflammatory responses (see diagram in Fig. 5). Taken together these results suggest that GC play a crucial role in the regulation of innate immune responses triggered by bacterial agents and that PI3K activation is differentially regulated by TLR2 activation both in the presence and absence of GC. Understanding the molecular regulation of TLRs that initiate the innate immune response and inflammation may be beneficial in the development and manage treatment of allergic as well as inflammatory disorders.
Fig 5.
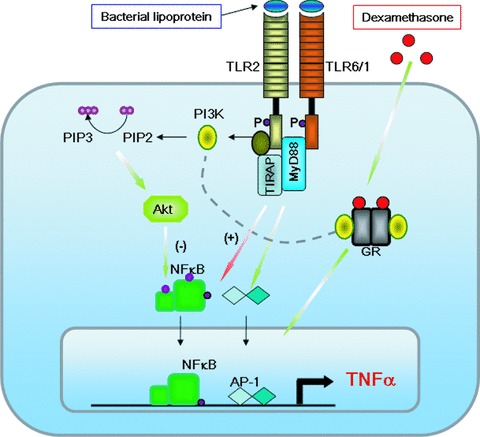
The diagram represents the molecular cross-talk between TLR2 heterodimers with TLR6/TLR1 and GR through the participation of PI3K. Upon TLR2 agonist sensing, TIR domain phosphorylation in Tyrosine (P) recruits PI3K activating an atypical signalling pathway that promotes Akt activation through membrane phospholipids. Akt seems to repress NF-κB and AP-1 related genes that are classically induced by the MyD88-dependent TLR pathway. Upon dexamethasone treatment Tyr phosphorylation in position 598 and 663 promotes also PI3K recruitment through p85 subunit. Both receptors might compete the binding of the kinase (see dotted line) resulting in activation or repression of inflammatory genes, such as TNF-α.
Acknowledgments
Funding support from FONDECYT 1080290, 1050451 and NIEHS/NIH, and to P. Silva and C. Beltrán for technical support and to Dr. F. Salazar for providing cytokines and cell line supplies.
Supporting Information
Molecular views of the wt-GR-dexamethasone(A) and Y598/663F-GR-dexamethasone complex (B)showing the position of dexamethasone (in light-blue as ball andstick model). The mutated residues Y598 and Y663 are shown in red.(C) Close-up view of the GR pocket of the model in B. Inblue, amino acid residues that participate in the interaction.Figures were generated with PyMOL (12).
GR translocates into the nucleus of transfectedcells in response to dexamethasone. (A) The subcellulardistribution of wt-GR, Y598F-GR, Y663F-GR and Y598/663F-GR mutantswas measured in the absence and presence of a range ofdexamethasone doses (0--100 nM). A representative image for eachcondition is shown. COS-1 cells were transiently transfected withplasmids expressing wt-GR, Y598F-GR, Y663F-GR or Y598/663F-GR andtreated with vehicle or treated for 3 hrs with 1, 10, 100 nMdexam-ethasone before being processed for immunocytochemistry andexamined by microscopy. Dexamethasone treatment caused nucleartranslocation of all GR constructs; only wt-GR translocated intothe nucleus with the lower dexamethasone doses. (B)Frequency histograms of the resulting localization scores areplotted (n ≥ 130). Black bars indicate receptor localization with vehicle treatment; striped bars indicate 1 to 100 nM dexamethasone. The localization of GR mutants in response to steroid treatment was quantified by determining the ratio of the fluorescence intensity in an area of the nucleus divided by the fluorescence intensity in a similarly sized area of the cytoplasm. The data were quantified by ranking cell staining according to their predominantly cytosolic (CC = 5), or equivalent (C = N = 3) distribution. The numbers on the Y-axis represent the percentages of cells counted which fell into the indicated rank. This analysis confirmed that wt-GR more efficiently translocated into the nucleus when cells were treated with dexamethasone. Full nuclear translocation of wt-GR, first became evident with 10 nM dexamethasone. Different ligand doses caused statistically significant changes in the frequency histogram of GR Y/F mutants reflecting its nuclear translocation: the percentage of cells scored as 5 is higher for the dexamethasone treatment than for the vehicle treatment, while there are more vehicle-treated cells scoring at 4 or below. In contrast, only wt-GR was able to nuclear translocate upon treatment with 1 nM dexamethasone.
References
- 1.Hermoso MA, Matsuguchi T, Smoak K, et al. Glucocorticoids and tumor necrosis factor alpha cooperatively regulate toll-like receptor 2 gene expression. Mol Cell Biol. 2004;24:4743–56. doi: 10.1128/MCB.24.11.4743-4756.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McKay LI, Cidlowski JA. Cross-talk between nuclear factor-kappa b and the steroid hormone receptors: mechanisms of mutual antagonism. Mol Endocrinol. 1998;12:45–56. doi: 10.1210/mend.12.1.0044. [DOI] [PubMed] [Google Scholar]
- 3.Medzhitov R, Janeway CA., Jr Innate immunity: impact on the adaptive immune response. Curr Opin Immunol. 1997;9:4–9. doi: 10.1016/s0952-7915(97)80152-5. [DOI] [PubMed] [Google Scholar]
- 4.Medzhitov R, Janeway CA., Jr Innate immunity: the virtues of a nonclonal system of recognition. Cell. 1997;91:295–8. doi: 10.1016/s0092-8674(00)80412-2. [DOI] [PubMed] [Google Scholar]
- 5.Rock FL, Hardiman G, Timans JC, et al. A family of human receptors structurally related to drosophila toll. Proc Natl Acad Sci USA. 1998;95:588–93. doi: 10.1073/pnas.95.2.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoshino K, Takeuchi O, Kawai T, et al. Cutting edge: toll-like receptor 4 (tlr4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for tlr4 as the lps gene product. J Immunol. 1999;162:3749–52. [PubMed] [Google Scholar]
- 7.Hemmi H, Takeuchi O, Kawai T, et al. A toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–5. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 8.Chuang T, Ulevitch RJ. Identification of htlr10: a novel human toll-like receptor preferentially expressed in immune cells. Biochim Biophys Acta. 2001;1518:157–61. doi: 10.1016/s0167-4781(00)00289-x. [DOI] [PubMed] [Google Scholar]
- 9.Arbibe L, Mira JP, Teusch N, et al. Toll-like receptor 2-mediated nf-kappa b activation requires a rac1-dependent pathway. Nat Immunol. 2000;1:533–40. doi: 10.1038/82797. [DOI] [PubMed] [Google Scholar]
- 10.Means TK, Wang S, Lien E, et al. Human toll-like receptors mediate cellular activation by mycobacterium tuberculosis. J Immunol. 1999;163:3920–7. [PubMed] [Google Scholar]
- 11.Flo TH, Halaas O, Torp S, et al. Differential expression of toll-like receptor 2 in human cells. J Leukoc Biol. 2001;69:474–81. [PubMed] [Google Scholar]
- 12.Aliprantis AO, Yang RB, Mark MR, et al. Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science. 1999;285:736–9. doi: 10.1126/science.285.5428.736. [DOI] [PubMed] [Google Scholar]
- 13.Nissen RM, Yamamoto KR. The glucocorticoid receptor inhibits nfkappab by interfering with serine-2 phosphorylation of the rna polymerase ii carboxy-terminal domain. Genes Dev. 2000;14:2314–29. doi: 10.1101/gad.827900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Q, Dziarski R, Kirschning CJ, et al. Micrococci and peptidoglycan activate tlr2– myd88–irak–traf–nik–ikk–nf-kappab signal transduction pathway that induces transcription of interleukin-8. Infect Immun. 2001;69:2270–6. doi: 10.1128/IAI.69.4.2270-2276.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.An H, Yu Y, Zhang M, et al. Involvement of erk, p38 and nf-kappab signal transduction in regulation of tlr2, tlr4 and tlr9 gene expression induced by lipopolysaccharide in mouse dendritic cells. Immunology. 2002;106:38–45. doi: 10.1046/j.1365-2567.2002.01401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang TY, Daynes RA. Glucocorticoid conditioning of myeloid progenitors enhances tlr4 signaling via negative regulation of the phosphatidylinositol 3-kinase-akt pathway. J Immunol. 2007;178:2517–26. doi: 10.4049/jimmunol.178.4.2517. [DOI] [PubMed] [Google Scholar]
- 17.Imasato A, Desbois-Mouthon C, Han J, et al. Inhibition of p38 mapk by glucocor-ticoids via induction of mapk phosphatase-1 enhances nontypeable haemophilus influenzae-induced expression of toll-like receptor 2. J Biol Chem. 2002;277:47444–50. doi: 10.1074/jbc.M208140200. [DOI] [PubMed] [Google Scholar]
- 18.Galon J, Franchimont D, Hiroi N, et al. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. FASEB J. 2002;16:61–71. doi: 10.1096/fj.01-0245com. [DOI] [PubMed] [Google Scholar]
- 19.Lu NZ, Cidlowski JA. Translational regulatory mechanisms generate n-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol Cell. 2005;18:331–42. doi: 10.1016/j.molcel.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 20.Cidlowski JA, Bellingham DL, Powell-Oliver FE, et al. Novel antipeptide antibodies to the human glucocorticoid receptor: recognition of multiple receptor forms in vitro and distinct localization of cytoplasmic and nuclear receptors. Mol Endocrinol. 1990;4:1427–37. doi: 10.1210/mend-4-10-1427. [DOI] [PubMed] [Google Scholar]
- 21.Yudt MR, Jewell CM, Bienstock RJ, et al. Molecular origins for the dominant negative function of human glucocorticoid receptor beta. Mol Cell Biol. 2003;23:4319–30. doi: 10.1128/MCB.23.12.4319-4330.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delerive P, Martin-Nizard F, Chinetti G, et al. Peroxisome proliferator-activated receptor activators inhibit thrombin-induced endothelin-1 production in human vascular endothelial cells by inhibiting the activator protein-1 signaling pathway. Circ Res. 1999;85:394–402. doi: 10.1161/01.res.85.5.394. [DOI] [PubMed] [Google Scholar]
- 23.Cantley LC, Songyang Z. Specificity in recognition of phosphopeptides by src-homology 2 domains. Journal of cell science. 1994;18:121–6. doi: 10.1242/jcs.1994.supplement_18.18. [DOI] [PubMed] [Google Scholar]
- 24.Yaffe MB, Rittinger K, Volinia S, et al. The structural basis for 14-3-3:Phosphopeptide binding specificity. Cell. 1997;91:961–71. doi: 10.1016/s0092-8674(00)80487-0. [DOI] [PubMed] [Google Scholar]
- 25.Wennstrom S, Downward J. Role of phosphoinositide 3-kinase in activation of ras and mitogen-activated protein kinase by epidermal growth factor. Mol Cell Biol. 1999;19:4279–88. doi: 10.1128/mcb.19.6.4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barber AE, Coyle SM, Marano MA, et al. Glucocorticoid therapy alters hormonal and cytokine responses to endotoxin in man. J Immunol. 1993;150:1999–2006. [PubMed] [Google Scholar]
- 27.Fantuzzi G, Galli G, Zinetti M, et al. The upregulating effect of dexamethasone on tumor necrosis factor production is mediated by a nitric oxide-producing cytochrome p450. Cell Immunol. 1995;160:305–8. doi: 10.1016/0008-8749(95)80042-h. [DOI] [PubMed] [Google Scholar]
- 28.Henneke P, Morath S, Uematsu S, et al. Role of lipoteichoic acid in the phagocyte response to group b streptococcus. J Immunol. 2005;174:6449–55. doi: 10.4049/jimmunol.174.10.6449. [DOI] [PubMed] [Google Scholar]
- 29.Leis H, Page A, Ramirez A, et al. Glucocorticoid receptor counteracts tumorigenic activity of akt in skin through interference with the phosphatidylinositol 3-kinase signaling pathway. Mol Endocrinol. 2004;18:303–11. doi: 10.1210/me.2003-0350. [DOI] [PubMed] [Google Scholar]
- 30.Galliher-Beckley AJ, Williams JG, Collins JB, et al. Glycogen synthase kinase 3beta-mediated serine phosphorylation of the human glucocorticoid receptor redirects gene expression profiles. Mol Cell Biol. 2008;28:7309–22. doi: 10.1128/MCB.00808-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen W, Dang T, Blind RD, et al. Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol Endocrinol. 2008;22:1754–66. doi: 10.1210/me.2007-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carricaburu V, Lamia KA, Lo E, et al. The phosphatidylinositol (pi)-5-phosphate 4-kinase type ii enzyme controls insulin signaling by regulating pi-3,4,5-trisphosphate degradation. Proc Natl Acad Sci USA. 2003;100:9867–72. doi: 10.1073/pnas.1734038100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strassheim D, Asehnoune K, Park JS, et al. Phosphoinositide 3-kinase and akt occupy central roles in inflammatory responses of toll-like receptor 2-stimulated neutrophils. J Immunol. 2004;172:5727–33. doi: 10.4049/jimmunol.172.9.5727. [DOI] [PubMed] [Google Scholar]
- 34.Fukao T, Tanabe M, Terauchi Y, et al. Pi3k-mediated negative feedback regulation of il-12 production in dcs. Nat Immunol. 2002;3:875–81. doi: 10.1038/ni825. [DOI] [PubMed] [Google Scholar]
- 35.Kamda JD, Singer SM. Phosphoinositide 3-kinase-dependent inhibition of dendritic cell interleukin-12 production by giardia lamblia. Infect Immun. 2009;77:685–93. doi: 10.1128/IAI.00718-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gustin JA, Ozes ON, Akca H, et al. Cell type-specific expression of the ikappab kinases determines the significance of phosphatidylinositol 3-kinase/akt signaling to nf-kappa b activation. J Biol Chem. 2004;279:1615–20. doi: 10.1074/jbc.M306976200. [DOI] [PubMed] [Google Scholar]
- 37.Guha M, Mackman N. The phosphatidylinositol 3-kinase-akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem. 2002;277:32124–32. doi: 10.1074/jbc.M203298200. [DOI] [PubMed] [Google Scholar]
- 38.Cao X, Wei G, Fang H, et al. The inositol 3-phosphatase pten negatively regulates fc gamma receptor signaling, but supports toll-like receptor 4 signaling in murine peritoneal macrophages. J Immunol. 2004;172:4851–7. doi: 10.4049/jimmunol.172.8.4851. [DOI] [PubMed] [Google Scholar]
- 39.Fang H, Pengal RA, Cao X, et al. Lipopolysaccharide-induced macrophage inflammatory response is regulated by ship. J Immunol. 2004;173:360–6. doi: 10.4049/jimmunol.173.1.360. [DOI] [PubMed] [Google Scholar]
- 40.Vermeulen L, De Wilde G, Van Damme P, et al. Transcriptional activation of the nf-kappab p65 subunit by mitogen- and stress-activated protein kinase-1 (msk1) EMBO J. 2003;22:1313–24. doi: 10.1093/emboj/cdg139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haller D, Russo MP, Sartor RB, et al. Ikk beta and phosphatidylinositol 3-kinase/akt participate in non-pathogenic gram-negative enteric bacteria-induced rela phosphorylation and nf-kappa b activation in both primary and intestinal epithelial cell lines. J Biol Chem. 2002;277:38168–78. doi: 10.1074/jbc.M205737200. [DOI] [PubMed] [Google Scholar]
- 42.Madrid LV, Wang CY, Guttridge DC, et al. Akt suppresses apoptosis by stimulating the transactivation potential of the rela/p65 subunit of nf-kappab. Mol Cell Biol. 2000;20:1626–38. doi: 10.1128/mcb.20.5.1626-1638.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mayo MW, Madrid LV, Westerheide SD, et al. Pten blocks tumor necrosis factor-induced nf-kappa b-dependent transcription by inhibiting the transactivation potential of the p65 subunit. J Biol Chem. 2002;277:11116–25. doi: 10.1074/jbc.M108670200. [DOI] [PubMed] [Google Scholar]
- 44.Sizemore N, Lerner N, Dombrowski N, et al. Distinct roles of the ikappa b kinase alpha and beta subunits in liberating nuclear factor kappa b (nf-kappa b) from ikappa b and in phosphorylating the p65 subunit of nf-kappa b. J Biol Chem. 2002;277:3863–9. doi: 10.1074/jbc.M110572200. [DOI] [PubMed] [Google Scholar]
- 45.Gupta V, Awasthi N, Wagner BJ. Specific activation of the glucocorticoid receptor and modulation of signal transduction pathways in human lens epithelial cells. Investigative ophthalmology & visual science. 2007;48:1724–34. doi: 10.1167/iovs.06-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Molecular views of the wt-GR-dexamethasone(A) and Y598/663F-GR-dexamethasone complex (B)showing the position of dexamethasone (in light-blue as ball andstick model). The mutated residues Y598 and Y663 are shown in red.(C) Close-up view of the GR pocket of the model in B. Inblue, amino acid residues that participate in the interaction.Figures were generated with PyMOL (12).
GR translocates into the nucleus of transfectedcells in response to dexamethasone. (A) The subcellulardistribution of wt-GR, Y598F-GR, Y663F-GR and Y598/663F-GR mutantswas measured in the absence and presence of a range ofdexamethasone doses (0--100 nM). A representative image for eachcondition is shown. COS-1 cells were transiently transfected withplasmids expressing wt-GR, Y598F-GR, Y663F-GR or Y598/663F-GR andtreated with vehicle or treated for 3 hrs with 1, 10, 100 nMdexam-ethasone before being processed for immunocytochemistry andexamined by microscopy. Dexamethasone treatment caused nucleartranslocation of all GR constructs; only wt-GR translocated intothe nucleus with the lower dexamethasone doses. (B)Frequency histograms of the resulting localization scores areplotted (n ≥ 130). Black bars indicate receptor localization with vehicle treatment; striped bars indicate 1 to 100 nM dexamethasone. The localization of GR mutants in response to steroid treatment was quantified by determining the ratio of the fluorescence intensity in an area of the nucleus divided by the fluorescence intensity in a similarly sized area of the cytoplasm. The data were quantified by ranking cell staining according to their predominantly cytosolic (CC = 5), or equivalent (C = N = 3) distribution. The numbers on the Y-axis represent the percentages of cells counted which fell into the indicated rank. This analysis confirmed that wt-GR more efficiently translocated into the nucleus when cells were treated with dexamethasone. Full nuclear translocation of wt-GR, first became evident with 10 nM dexamethasone. Different ligand doses caused statistically significant changes in the frequency histogram of GR Y/F mutants reflecting its nuclear translocation: the percentage of cells scored as 5 is higher for the dexamethasone treatment than for the vehicle treatment, while there are more vehicle-treated cells scoring at 4 or below. In contrast, only wt-GR was able to nuclear translocate upon treatment with 1 nM dexamethasone.