

Abstract
Anti-angiogenesis represents a promising therapeutic strategy for the treatment of various malignancies. Isthmin (ISM) is a gene highly expressed in the isthmus of the midbrain–hindbrain organizer in Xenopus with no known functions. It encodes a secreted 60 kD protein containing a thrombospondin type 1 repeat domain in the central region and an adhesion-associated domain in MUC4 and other proteins (AMOP) domain at the C-terminal. In this work, we demonstrate that ISM is a novel angiogenesis inhibitor. Recombinant mouse ISM inhibited endothelial cell (EC) capillary network formation on Matrigel through its C-terminal AMOP domain. It also suppressed vascular endothelial growth factor (VEGF)-basic fibroblast growth factor (bFGF) induced in vivo angiogenesis in mouse. It mitigated VEGF-stimulated EC proliferation without affecting EC migration. Furthermore, ISM induced EC apoptosis in the presence of VEGF through a caspase-dependent pathway. ISM binds to αvβ5 integrin on EC surface and supports EC adhesion. Overexpression of ISM significantly suppressed mouse B16 melanoma tumour growth through inhibition of tumour angiogenesis without affecting tumour cell proliferation. Knockdown of isthmin in zebrafish embryos using morpholino antisense oligonucleotides led to disorganized intersegmen-tal vessels in the trunk. Our results demonstrate that ISM is a novel endogenous angiogenesis inhibitor with functions likely in physiological as well as pathological angiogenesis.
Keywords: isthmin, angiogenesis, anti-angiogenesis, cancer
Introduction
Angiogenesis, the formation of new blood vessels from pre-existing vasculature, is fundamental for tumour growth, progression and metastasis [1]. Inhibition of angiogenesis is a promising therapeutic approach for cancer. Angiogenesis inhibitors are being investigated for applications in cancer therapies. Under normal physiological conditions, angiogenesis is believed to be regulated by a local balance of endogenous stimulators and inhibitors. Endogenous protein angiogenesis inhibitors include a few gene products such as thrombospondin-1 (TSP-1) and pigment epithe-lium-derived factor as well as a large number of protein proteolytic fragments such as angiostatin, endostatin and tumstatin [2]. Although angiogenic stimulators act on endothelial cells (ECs) to stimulate angiogenesis, angiogenic inhibitors tend to be pleiotropic in function and their expression is not necessarily related to angiogenesis regulation. Up to now, very few genes that encode proteins that specifically inhibit angiogenesis have been discovered and their physiological roles are divergent.
Isthmin (ISM) is a secreted protein first identified in Xenopus but its function is not yet known. During neuronal stage, ISM is highly expressed in the isthmus organizer, the signalling centre located at the midbrain–hindbrain boundary (MHB). Additional expression was detected in the paraxial mesoderm and neural folds in tail bud stage as well as in notochord in neuronal stage [3]. In zebrafish, isthmin expression could be up-regulated by overexpression of Wnt8 or down-regulated by Wnt/β-catenin inhibitor, indicating its involvement in Wnt signalling regulated processes during embryonic development [4].
Sequence analysis indicated that ISM contains a centrally localized thrombospondin type 1 repeat (TSR) and a C-terminal domain called adhesion-associated domain in MUC4 and other proteins (AMOP) (Fig. 1A) [3]. TSR was initially identified in the natural angiogenesis inhibitor TSP-1 and plays a critical role for the anti-angio-genic activity of TSP-1 [5, 6]. TSR has since been found in many secreted proteins or the extracellular portions of transmembrane proteins [7]. The functions of TSRs appear to be related to cell migration, communication and tissue remodelling. The other domain in ISM, AMOP, is a predicted protein domain containing eight invariant cysteine residues and is also found only in secreted proteins or the extracellular domains of transmembrane proteins. AMOP domain is speculated to be involved in cell adhesion due to its presence in cell adhesion molecules [8]. Besides the common cysteine residues that are shared by all AMOPs among different proteins, AMOP in ISM also has a ‘KGD’ motif, which is found in many antagonists of platelet aggregation and involved in integrin-dependent cell adhesion and tumour metastasis [9–11]. Therefore, the interaction between AMOP containing proteins and integrin may be involved in regulating angio-genesis. We therefore investigated the role of ISM in angiogenesis.
Fig 1.
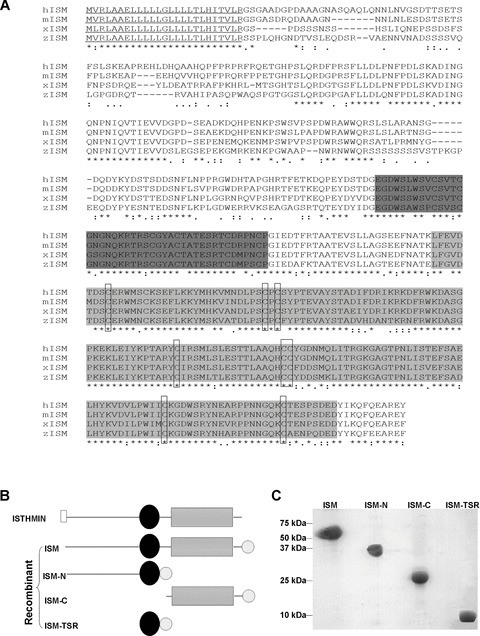
Sequence comparison, expression and purification of recombinant mouse ISM and its truncated fragments. (A) Amino acid alignment of ISM from mouse, human, Xenopus and zebrafish. The tentative signal peptide is underlined. Dark grey region represents TSR and light grey region indicates AMOP domain. The eight invariant cysteines in AMOP domain of different species are boxed. (B) Diagrams illustrating the domain organization of native ISM and its recombinant forms. Open rectangle represents signal peptide; black oval represents TSR domain; thin grey line represents N-terminal portion of ISM; dashed rectangle represents AMOP domain; dotted circle represent His-Tag. (C) SDS-PAGE gel showing purified recombinant ISM and its truncated fragments. Molecular weight marker is indicated on the left.
In this study, we demonstrate that ISM is a novel angiogenesis inhibitor. Recombinant mouse ISM inhibited angiogenesis in vitro and in vivo. Overexpression of ISM in B16 melanoma inhibited tumour growth and tumour angiogenesis in mice. Knockdown of isthmin in zebrafish embryos led to abnormal intersegmental vessel (ISV) formation in the trunk. ISM therefore plays a role in both physiological as well as pathological angiogenesis.
Materials and methods
In general, all experiments were repeated at least three times, with duplicates or triplicates within each experiment.
Cloning, expression and purification of recombinant full length and truncated isthmin in E. coli
The complete coding sequence of mouse ISM cDNA was acquired by assembling several EST clones. The full ORF was amplified by RT-PCR using primers Ism1F (ATGGTGCGCCTGGCTGC) and Ism1R (TTAG-TACTCTCTGGCTTCTTGG) and cloned into pGEM-T Easy Vector (Promega, Madison, WI, USA). The sequence was confirmed and is identical to the coding sequence of NM_001126490 in the NCBI Genbank. ISM-TSR, ISM-C, ISM-N and ISM (full-length) were cloned into pET-M vector (modified from pET32, Novagen, San Diego, CA, USA) using the following primers: IsmF1EcoRI: CGCGCGGAATTCAGGATGGTGCGCCTGGCTGC; IsmR1XhoI: CGCGCGCTCGAGGTACTCTCTGGCTTCTTGGAACTG; IsmAMOPF1EcoRI: CGCGCGGAATTCGAAGTGAGTCTGCTTGCGGG; IsmTSRR1XhoI: CGCGCG-CTCGAGCCCGCAAGCAGACTCACTTC. The sequence coding for N-terminal signal peptide in ISM and ISM-N were further removed by BamHI digestion. The recombinant proteins were expressed in E. coli (BL21DE3) and purified using Ni-NTA affinity chromatography in 6 M urea according to the manufactures’ instructions (Qiagen, Valencia, CA, USA). The proteins were then further purified by reverse-phase high-performance liquid chromatography (HPLC). Protein concentration was determined using BioRad Bradford assay reagent (Bio-Rad Laboratories, Hercules, CA, USA).
Cell culture
ECs used in this work are human umbilical vein endothelial cells (HUVECs). Fresh umbilical cords for HUVEC isolation were collected from consented maternal ward patients at the National University Hospital according to the protocol (DSRB C/00/553) which is approved by Singapore National Healthcare Group's Domain-Specific Review Board (DSRB) ethics approval committee. ECs were cultured in CSC complete medium (Cell System Corporation, Kirkland, WA, USA). Only cells of passages 3–6 were used in experiments. ECs used for all the assays were starved in basal CSC medium containing 2% fetal bovine serum (FBS) for 4 hrs before being treated with ISM recombinant protein. B16F10 cells were obtained from American Type Culture Collection (ATCC) and cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% FBS. Stable transfected B16 cell lines were maintained in DMEM containing 10% FBS supplemented with 400 μg/ml zeocin.
Capillary network formation assay
ECs (2 × 104) were pre-treated with different concentrations of ISM proteins for 30 min. before being plated onto the polymerized Matrigel (Chemicon International, Inc., Temecula, CA, USA) in 96-well plate. After 6 hrs, tube-like structure formation was documented using an inverted microscope (Zeiss Axiovert200, Carl Zeiss International, Singapore). Capillary length was quantified by measuring the length of branches from representative fields using NIH Image J 1.32 software. Recombinant endo-statin was obtained from Sigma (St. Louis, MO, USA). To investigate at which stage ISM interfered with in vitro angiogenesis, time-course analyses were conducted where ISM proteins were added into culture medium at time-points of 0, 2, 4 hrs after seeding ECs onto Matrigel.
Cell attachment and spreading assay
ECs (5000) were pre-treated with different concentration of ISM proteins for 30 min. before being plated onto a fibronectin or diluted Matrigel-coated 96-well plate and incubated for 30 min. at 37°C. Attached cells were then fixed and stained with Giemsa (Sigma). The number of cells attached was counted under light microscopy. For cell attachment and spreading on ISMs-coated surface, 96-well-plate was coated with ISM proteins at concentrations of 100 nM, 500 nM and 1 μM at 37°C for 2 hrs. Excess ISM proteins then were removed and the wells were blocked with 3% bovine serum albumin for 2 hrs. Cells were seeded on the wells and incubated for 2 hrs at 37°C. The attached cells were photographed under light microscopy.
Cell migration assay
Cell chemotaxis migration assay was performed with 8 μm Falcon cell culture inserts as previously described [12]. Briefly, ECs were starved for 4 hrs in CSC medium and 2% FBS. ISM proteins at concentrations of 100 nM, 500 nM and 1 μM with 20,000 cells were seeded on gelatin-coated cell culture insert. VEGF at 15 ng/ml (R&D Systems, St. Paul, MN, USA) with CSC medium supplemented with 2% FBS were placed on the lower chamber. After 8 hrs incubation, cells on the upper surface of the insert were removed with a cotton swab. Migrated cells on the lower surface of inserts were fixed, and stained with Giemsa (Sigma). The migrated cells were counted and quantified using light microscopy. Similar experiments were also performed in the presence of 15 ng/ml bFGF or 10% FBS.
For chemokinesis cell migration assay, the chemoattractant was present in both the bottom and upper wells at the same concentration. All other procedures were the same as chemotaxis described above.
Cell proliferation assay
ECs (2 × 104 per well) were cultured overnight in a coated 96-well plate in CSC complete medium at 37°C. Cells were starved the following day for 3 hrs in CSC basal medium and ISM proteins were then added to the medium together with 15 ng/ml VEGF (R&D Systems). After 24 hrs of incubation, EC proliferation was determined using BrdU cell proliferation kit (Chemicon International, Inc.) according to manufacturer's instruction. Briefly, BrdU was added into the culture media and incubated for 2 hrs. ECs were then fixed and anti-BrdU antibody was used to stain BrdU+ cells. Relative proliferation is represented by the amount of BrdU in cells measured by absorption at 450 nm using a microplate reader. Similar experiments were also performed in the presence of 15 ng/ml bFGF or 10% FBS.
For non-ECs (Swiss3T3, NIH3T3, B16 or HepG2), 10,000 cells were cultured overnight in 96-well plate in DMEM supplemented with 10% FBS at 37°C. Cells were then starved for 24 hrs with DMEM. Subsequently, ISM was added to culture medium with or without 10% FBS. After additional 24 hrs incubation, cell proliferation was determined using the same method described above.
Cell apoptosis determination
ECs (3 × 105 per well) were cultured in coated 6-well plate in CSC complete medium overnight at 37°C. ISM proteins (100 nM, 500 nM and 1 μM), VEGF (15 ng/ml) and z-VAD-fmk (10 μM) (Calbiochem, Inc., Darmstadt, Germany) were added to the culture medium after 3 hrs of cell starvation and incubated for 24 hrs prior to apoptosis detection. Apoptosis was determined by measuring cytosolic oligonucleosome-bound DNA using a Cell Death ELISA kit (Roche Diagnostics GmbH, Penzberg, Germany). Activated caspase 3 was detected by Western blot using an antibody specific for active caspase 3 (R&D Systems). For non-ECs (Swiss3T3, NIH3T3, B16 or HepG2 cells), 2 × 105 cells per well were cultured in 6-well plate in DMEM supplemented with 10% FBS at 37°C overnight. Cells were then starved in DMEM for 24 hrs. Subsequently, ISM were added to the culture medium and incubated for an additional 24 hrs prior to apoptosis detection.
Generation of anti-ISM antibody
Recombinant mouse ISM purified from E. coli using the same method described above was used for generation of anti-ISM antibody. Anti-serum were raised in rabbits and purified by Biogenes GmbH (Berlin, Germany) using standard procedures.
Immunoprecipitation and immunoblot
HUVECs membrane extract was prepared using subconfluent HUVECs. After washing the cells twice with ice-cold PBS, HUVECs were resuspended in 1 ml ice-cold hypotonic buffer [10 mM HEPES, pH 7.9, 0.5 mM dithio-threitol, 0.5 mM phenylmethylsulfonyl fluoride, and one tablet of protease inhibitor cocktail (Roche, Penzberg, Germany)]. The cells were disrupted with 50 strokes of a tight-fitting Dounce homogenizer. The homogenate was checked under phase contrast microscope, and no intact cell could be observed. Nuclei and mitochondria were removed from the homogenate by centrifugation at 8000 ×g for 10 min. at 4°C. The supernatant was then centrifuged at 100,000 ×g for 30 min. at 4°C. The membrane fraction, obtained as the pellet, was dissolved in 2 ml hypotonic buffer. Membrane proteins were released by treating with 1% Triton X-100 for 1 hr at 4°C.
Purified recombinant ISM protein was incubated with HUVECs membrane extract for 2 hrs at 4°C. Subsequently, the anti-αvβ3, anti-αvβ5, control IgG and protein A/G Sepharose beads (25 μl, Santa Cruz, Inc., Santa Cruz, CA, USA) were added into the co-immunoprecipitation reaction, and incubated for a further 2 hrs at 4°C. The precipitated proteins were resolved by SDS-PAGE, and blotted with anti-His antibody. Also, the anti-ISM antibody, control IgG and protein A/G Sepharose beads were added into the co-immunoprecipitation reaction and incubated for 2 hrs at 4°C. The precipitated proteins were resolved by SDS-PAGE and blotted with anti-αv, β1, β3 and β5 antibody, respectively.
Directed in vivo angiogenesis assay in mice
The role of ISM in in vivo angiogenesis in Matrigel plug was determined using the directed in vivo angiogenesis assay kit (Trevigen, Inc., Gaithersburg, MD, USA) according to manufacturer's instructions [13]. Briefly, the angioreactors were either filled with basement membrane extracts alone, in combination with VEGF or with VEGF supplemented with 500 nM or 1 μM ISM proteins and incubated at 37°C for 1 hr to allow gelling. The angioreactors were then implanted into the dorsal flank of 7–8-week-old female nude mice. After 2 weeks of incubation, the angioreactors were harvested. The invaded ECs were isolated, and labelled with fluorescein isothio-cyanate (FITC)-lectin at 4°C overnight. The fluorescence was measured in 96-well plates using a SPECTRAmax microplate spectrofluorometer (excitation 485 nm, emission 510 nm; Molecular Devices, Downingtown, PA, USA).
Establishment of stable ISM over-expressing B16 cell lines
Mouse ISM cDNAs containing full-length (without its natural signal pep-tide) or truncated fragments were cloned into mammalian secretive expression vector pSecTag2B (Invitrogen, Carlsbad, CA, USA). B16F10 cells were transfected with 1 μg of ISM expression plasmids or control empty vectors using pSecTag2B according to manufacturer's instructions. Zeocin (Invitrogen) was supplemented with the culture medium at a final concentration of 400 μg/ml to select resistant cells. Zeocin-resistant colonies appeared after 2–3 weeks. Five colonies (B16/ISMa, b, c, d, e) and B16/Vec were selected. Conditioned culture medium from each individual clone was collected and concentrated by Microcon filter unit (Millipore, Billerica, MA, USA). Western blot were performed with anti-His antibody to detect exogenously introduced ISM (Santa Cruz, Inc.).
Xenograft mouse tumour model and immunohistochemistry
One million tumour cells of the control group (vector transfected B16) and experimental group (B16 overexpressing ISM) in 100 μl PBS were injected subcutaneously into the left and right flank of the same C57BL/6J mouse. Tumour growth was monitored every other day with a digital calliper. Tumour volume was calculated using the formula 0.52 × length × (width2) [14]. Tumours were harvested 14 days after inoculation and fixed with 4% paraformaldehyde for 72 hrs. Paraffin tumour tissue sections were made according to standard methodology. For immunohistochemistry analysis of tumour blood vessels, tumour tissue sections were stained with anti-CD31 antibody followed by Alexa-fluor 568-conjugated secondary antibody (Molecular Probes, Carlsbad, CA, USA). The nuclei were counter stained blue with 4′,6-diamidino-2-phenylindole (DAPI). Microvessel density was determined according to previously described methodology [15]. All CD31+ lumen and non-lumen structures (such as cell clusters and spots) were counted as vessels. Consistent with general practice, only angiogenesis hot spot areas of a tumour section were selected for vessel analyses. For vessel quantification, the average number of vessels per microscopic field, from three microscopic fields per tumour section, three tumour sections per tumour (representing the upper, middle and lower portion of the tumour) and four tumours for each experimental group (B16/Vec, B16/ISMa or B16/ISMb) were analysed. Apoptotic cells were determined by the ApoAlert TUNEL Assay Kit (Clonetech, Mountain View, CA, USA) and stained green by FITC-conjugated antibody. The average number of apoptotic cells per microscopic field was determined by analysing three microscopic fields per tumour section, three tumour sections per tumour and three tumours in each experimental group. For immunohistochemistry analysis of β5 integrin, B16 tumour sections were stained with a rabbit polyclonal anti-β5 integrin antibody (Santa Cruz, Inc.) and horseradish peroxidase (HRP)-conjugated secondary antibody. The sections were then developed with diaminobenzi-dene (DAB, DAKO) and counterstained with haematoxylin.
Zebrafish studies
Zebrafish were maintained at standard conditions [16]. Embryos were staged according to the standard morphologic criteria [17]. Embryos derived from wide-type and Tg(fli-1:EGFP) fish were used. The ISM mor-pholino antisense oligonucleotides (MOs) were purchased from Gene Tools (Philomath, OR, USA), and have the following sequences:
spl MO, 5′-TGTGAGCATCTACCTGTATATTGGG-3′; mis MO 5′-TGTcAGgATCTACCTcTATtTTcGG-3′;
ismATG MO, 5′-CTCCGCCGCCAGACGCACCATCCTC-3′;
ATGmis MO, 5′-CTgCGCCcCCAGAgGCACgATCgTC-3′. MOs were reconstituted in nuclease-free water to a stock concentration of 1 mM. Microinjections of one-cell stage zebrafish embryos with MO were carried out as described [18]. Zebrafish isthmin full length sequence (NM_001012376) were cloned into pGEM-T easy vector by RT-PCR using primers zismf (TGGCGGCGGAGCTGCTGCTGCTTT) and zismr (GTTTATAG-TAGTCCTCATCCTGAGGG). The pGEMT-easy/zism plasmid was linearized using NcoI, and SP6 polymerase was used for DIG-labelled antisense RNA synthesis. DIG-labelled RNA probe was synthesized using the in vitro DIG labelling kit (Roche). Zebrafish in situ hybridization procedures were performed as previously described [19].
Statistical analysis
Statistical analysis was performed with paired Student's t-test as indicated. A P-value of less than 0.05 was considered significant in all cases (*P < 0.05; **P < 0.01).
Results
Cloning and expression of recombinant mouse isthmin and its truncated fragments in E. coli
To study the function of ISM, we cloned the full-length mouse and zebrafish isthmin cDNAs and compared the deduced amino acid sequences with ISM of human and Xenopus. The TSR domains are highly conserved with 98% identity between mouse and human, 87–88% identity between mouse and zebrafish or mouse and Xenopus. The C-terminal AMOP domains are also highly conserved with 99% identity between mouse and human; 91% identity between mouse and Xenopus and 85% identity between mouse and zebrafish. The eight invariant cysteines in the AMOP domain are all conserved cross species and they have been pre-dicted to be involved in disulfide bond formation [8]. The signal peptide is also highly conserved among all four species. In comparison, the N-terminal region outside the TSR is relatively more diverged with 85% identity between mouse and human and 62% between mouse and Xenopus, respectively. No known protein domains are identified in the N-terminal region.
To examine the function of ISM protein and its domains in angio-genesis, we expressed and purified His-Tagged full-length mouse ISM protein (ISM) as well as three truncated forms (ISM-TSR, ISM-C and ISM-N) containing TSR, AMOP and TSR plus N-terminal region (Fig. 1B). The recombinant proteins were purified by Ni-NTA affinity chromatography followed by further reverse-phase HPLC purification (Fig. 1C). The EC cytotoxicity and endotoxin level of these purified recombinant proteins were determined. No acute cytotoxic-ity to ECs was observed up to 1 μM although the endotoxin levels of the recombinant proteins were below 0.5 EU/mg (data not shown).
ISM inhibits in vitro capillary network formation through its C-terminal AMOP domain
ECs can rapidly align and form tubular structures within 6–8 hrs when cultured on Matrigel [20]. We premixed ECs with different concentrations of ISM, ISM-C, ISM-N or ISM-TSR and plated them onto Matrigel and monitored the extent of capillary network formation. All capillary network formations were documented at 6 hrs after ECs were plated onto Matrigel. As shown in Fig. 2A and B, ISM and ISM-C both inhibited EC capillary network formation in a dose-dependent manner. Recombinant endostatin, a known endogenous angiogenesis inhibitor, inhibited EC tube formation in similar fashion (Fig. 2A) [21]. In contrast, ISM-N and ISM-TSR had no such activity. Moreover, the ED50 of ISM (318 ± 61 nM) is comparable to the ED50 of ISM-C (334 ± 65 nM), suggesting that the anti-capillary network formation function of ISM is largely mediated through its C-terminal AMOP domain.
Fig 2.
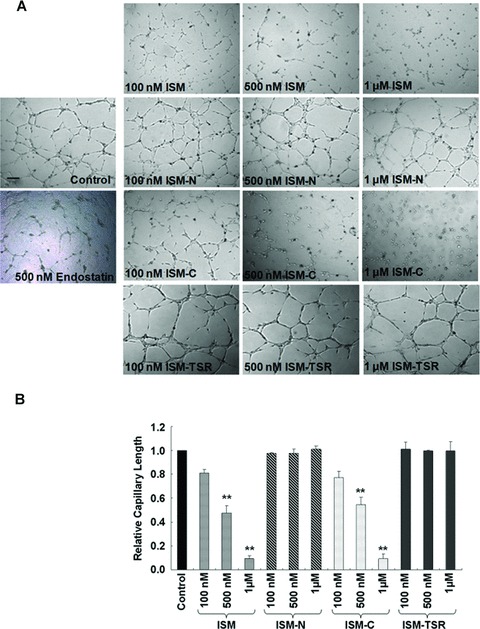
ISM and its C-terminal AMOP domain inhibit EC capillary network formation in a dose-dependent manner. (A) ISM and ISM-C dose-dependently inhibited EC tube formation although ISM-N and ISM-TSR have no such activity. Magnification 50 ×n= 4. The angiogene-sis inhibitor endostatin was used as a positive control. (B) Quantification of capillary length in different concentrations of ISM and truncates. All capillary network formation was documented at 6 hrs after EC plating onto Matrigel. The scale bar represents 200 μm.
ISM disrupts capillary network formation in a time-dependent manner
The in vitro formation of EC capillary network is a multi-step and dynamic process including cell attachment to matrix, cell migration, cell spreading, cell–cell adhesion, morphogenesis as well as apoptosis [22]. After plating ECs onto Matrigel, we observed that cells attached to Matrigel and migrated during the 0–1 hr period; cells then spread and elongated to form cell–cell alignment during 1–2 hrs; between 2 and 3 hrs, some short cell–cell connections have been formed; capillary tubes appeared by 4 hrs and finally extensive cellular network was fully formed by 6 hrs (data not shown).
To investigate at which stage ISM interfered with in vitro capillary network formation, recombinant ISM and ISM-C (both at 1 μM) were added to the assay culture media at 0, 1, 2 and 4 hrs, respectively, after ECs were plated onto Matrigel. When ISM or ISM-C is added together with ECs to Matrigel (0 hr), hardly any tubular network was formed at the end of the experiment (6 hrs after EC plating onto Matrigel) (Fig. 3). There was a gradual loss of angio-genesis inhibition observed at 6 hrs when ISM or ISM-C was added into the angiogenesis assay at later time-points. After 2 hrs of EC plating onto Matrigel, ISM could no longer disrupt capillary network formation. These results indicate that ISM and ISM-C inhibited EC capillary network formation mainly by interfering with the early stages of in vitro angiogenesis on Matrigel. Possibly, cell-matrix attachment, cell migration, cell–cell adhesion or even apop-tosis could be affected.
Fig 3.

ISM and ISM-C inhibit EC capillary network formation in a time-dependent manner. ISM and ISM-C (1 μM) are required to be present from the early stages of in vitro angiogene-sis assay (0 hr, the time when ECs were plated onto Matrigel) in order to prevent capillary network formation. All capillary network formation was documented at 6 hrs after EC plating onto Matrigel. Magnification 50×, n= 4. The time at which ISM or ISM-C was added to the culture media after ECs have been plated onto Matrigel are indicated in the panel. The scale bar represents 200 μm.
ISM inhibited VEGF-, bFGF- or serum-induced EC proliferation without affecting EC migration
To analyse the mechanisms of ISM function, we examined the effects of ISM and its truncated fragments on the various aspects of in vitro angiogenesis. VEGF is the most important endothelial-specific angiogenic growth factor, stimulating multiple aspects of angiogenesis [23]. ISM or its fragments has no effect on VEGF-stimulated chemotactic (directional) EC migration (Fig. 4A and more details in Fig. S1). ISM also did not influence EC chemokine-sis (non-directional migration) in the presence or absence of VEGF (Fig. S2A and B).
Fig 4.
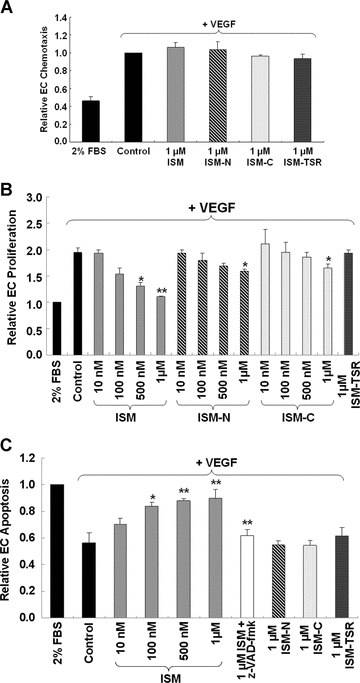
The effects of ISM and its various domains on various aspects of in vitro angiogenesis. (A) ISM does not influence VEGF-induced chemotactic EC migration. The concentrations of ISM, ISM-N, ISM-C and ISM-TSR tested were from 1 nM to 1 μM. Only results of 1 μM were shown. (B) ISM suppressed VEGF-induced EC proliferation. Both ISM-N and ISM-C have a weaker activity comparing to ISM, whereas ISM-TSR is not active. Only 1 μM result was shown for ISM-TSR. (C) ISM induced EC apoptosis in the presence of VEGF in a dose-dependent manner. The ISM-induced EC apop-tosis was abolished when pan-caspase inhibitor z-VAD-fmk was added. None of the ISM truncated fragments (at concentrations from 10 nM to 1μM) showed such activity (only 1 μM result was shown) *P < 0.05, **P < 0.01, n= 4. VEGF used was 15 ng/ml in all experiments.
However, ISM significantly inhibited VEGF-induced EC proliferation in a dose-dependent manner (Fig. 4B). Both ISM-N and ISM-C inhibited EC proliferation, although each was much less effective comparing to the full-length protein whereas ISM-TSR had no such activity. Similarly, ISM also inhibited bFGF (another potent angiogenic growth factor) or serum-stimulated EC proliferation without affecting EC migration induced by these growth factors (Figs S3 and S4).
To determine whether ISM preferentially affects EC proliferation, we also examined the effect of ISM on serum-stimulated proliferation of fibroblasts and tumour cells including NIH3T3 fibroblasts, Swiss 3T3 fibroblasts, B16 melanoma cells and HepG2 hepatocellular carcinoma cells. The results showed that ISM only mildly inhibited serum-stimulated fibroblasts proliferation (Fig. S5A and B) and has no effect on serum-stimulated tumour cell proliferation (Fig. S5C and D).
ISM induced EC apoptosis in the presence of VEGF, bFGF or serum
ISM dose-dependently induced EC apoptosis in the presence of VEGF (Fig. 4C and more details in Fig. S6A). The extent of apopto-sis induced by 1 μM ISM in the presence of VEGF is similar to apoptosis induced by serum withdrawal (control in Fig. 4C). This apoptosis induction appears to be caspase dependent because the Pan-caspase inhibitor Z-VAD-fmk effectively abolished this function (Fig. 4C). Furthermore, ISM dose-dependently induced the activation of caspase 3, a key downstream caspase in the extrinsic as well as intrinsic apoptosis pathways (Fig. S6B). None of the ISM truncated fragments could induce apoptosis under the same condition. Similarly, ISM also induced EC apoptosis in the presence of bFGF or serum (Fig. S7).
To test if ISM can also induce apoptosis of non-ECs, we investigated whether ISM can induce apoptosis of NIH3T3 fibroblasts, Swiss 3T3 fibroblasts, B16 melanoma cells and HepG2 hepatocellular carcinoma cells. As shown in Fig. S8, ISM marginally induced fibroblast apoptosis in the presence of serum, but did not induce apoptosis of tumour cells. It therefore seems that ISM may have a preferential effect on ECs in proliferation and apoptosis.
ISM supports EC adhesion through its C-terminal AMOP domain without affecting EC attachment to matrix
Although ISM seems to suppress the early stages of EC capillary network formation on Matrigel, neither ISM nor its truncated fragments had any effect on EC attachment to gelatin-, fibronectin- or diluted Matrigel-coated surface (Fig. 5A and data not shown). Similarly, ISM also did not influence EC spreading on these matrix molecules (Fig. S9). On the other hand, ECs can attach and spread onto ISM-coated surface in a similar fashion compared to gelatin-coated surface (Fig. 5B). Furthermore, ISM-C but not ISM-N could support this attachment and spreading (Fig. 5B). These attachment results are in line with results of Fig. 2 above which indicated that only ISM-C inhibited EC capillary network formation similar to full-length ISM. It therefore seems that ISM interacts with ECs through its C-terminal AMOP domain and that ISM most likely interacts with EC surface molecules distinct from receptors for gelatin, fibronectin or the major component of Matrigel such as laminin and collagen IV.
Fig 5.
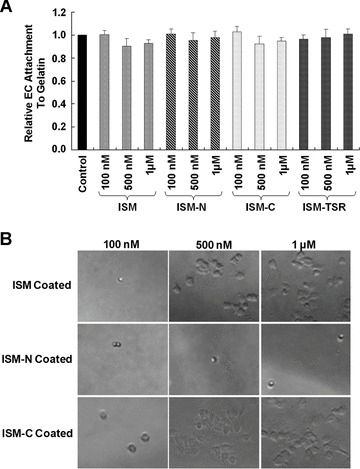
Effects of ISM and its domains on EC attachment and spreading. (A) ISM does not interfere with EC attachment to gelatin-coated surface. Neither ISM nor its truncated fragments at various concentrations have any effect on EC attachment to gelatin-coated surface. (B) ECs can attach and spread onto ISM coated surface. ISM-C support EC attachment and spreading as efficient as ISM whereas ISM-N cannot support this function.
ISM binds to ECs through αvβ5 integrin
Integrins are the major cell surface receptors mediating cell adhesion to matrix molecules and play important roles in angiogenesis [24]. We therefore investigated the interaction of ISM with inte-grins expressed in ECs. We isolated the plasma membrane extract of cultured ECs and incubated with recombinant ISM in vitro. ECs are known to express αvβ3, αvβ5 and a series of β1 integrins. Co-immunoprecipitation experiments using either anti-His antibody (to detect recombinant ISM) or anti-integrin antibody (to detect a particular integrin subunit or heterodimer) were carried out. As shown in Fig. 6, only integrin αvβ5 was co-immunoprecipitated by anti-His antibody (Fig. 6A and B). On the other hand, only anti-αvβ5, anti-αv or anti-β5 antibodies could co-precipitate ISM. Furthermore, ISM-C interacted with αvβ5 but not ISM-N (Fig. 6, part G and data not shown). These results suggest that ISM could interact with ECs through αvβ5 integrin.
Fig 6.
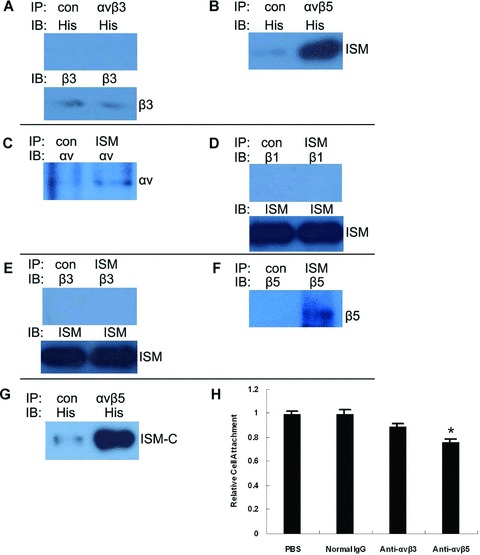
ISM binds to αvβ5 inte-grin on ECs. Recombinant His-tagged ISM was incubated with membrane protein extract of ECs and subjected to immunoprecipitation (IP) followed by immunoblot. (A) and (B) show results of immuno-precipitation using anti-αvβ5 or control IgG and immunoblot with anti-His and anti-β3 antibody. (C)–(F) show results of immunoprecipitation using anti-ISM antibody followed by immunoblot with antibodies against integrin αv and β1, β3, β5 and ISM. Only αv and β5 are co-immunoprecipated by anti-ISM antibody. (G) shows the results of immunoprecipi-tation using anti-αvβ5 or control IgG and immunoblot with anti-ISM antibody. (H) presents the effect of αvβ5 neutralizing antibody in blocking EC attachment to ISM-coated surface in comparison to normal IgG and αvβ3 neutralizing antibody.
To further confirm this, anti-αvβ5 neutralizing antibody was pre-incubated with ECs before cells were plated onto ISM-coated surface. As shown in Fig. 6, anti-αvβ5 antibody partially but significantly blocked EC attachment to ISM-coated surface (Fig. 6, part H). Under the same condition, normal mouse IgG or anti-αvβ3 antibody did not present significant blocking of EC attachment to ISM-coated surface. These results demonstrate that ISM interacts with ECs at least partly through αvβ5 integrin.
ISM inhibits angiogenesis in vivo
To further examining if ISM could inhibit angiogenesis in vivo, we used a modified Matrigel plug angiogenesis assay in which the Matrigel is retained in a silicon tube angioreactor. In each mouse, a control and experimental angioreactor was implanted subcutaneously into each side of the dorsal flank to reduce variations between individual mice. The amount of angiogenesis in each angioreactor was quantified by measuring the number of ECs using fluorescently labelled EC-binding lectin. As shown in Fig. 7, a mixture of VEGF and bFGF induced potent angiogenesis in mice compared to control (Matrigel alone). When ISM was added together with VEGF/bFGF, it potently suppressed VEGF/bFGF induced angiogenesis at 0.5 μM and 1 μM, concentrations where it is also effective in inhibiting angiogenesis in vitro. These results demonstrate that ISM functions as an angiogenesis inhibitor in vivo. Surprisingly, when ISM-C was tested under the same experimental condition, it did not suppress in vivo angiogenesis at 0.5 μM or 1 μM (Fig. 7 and data not shown).
Fig 7.
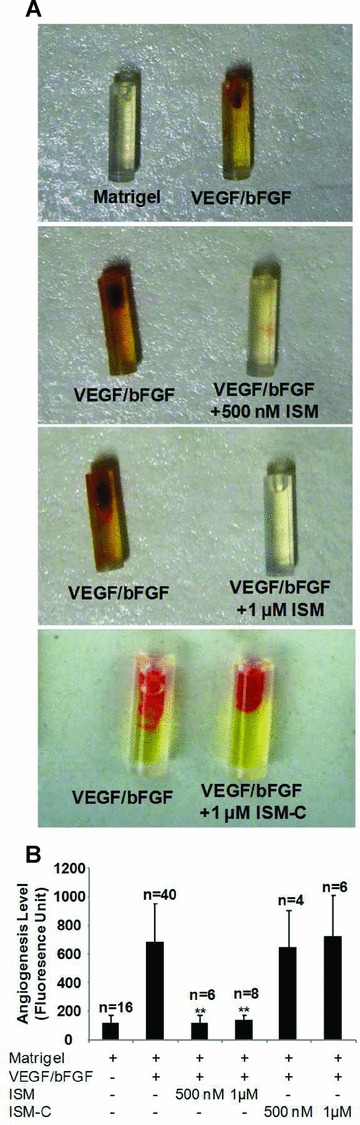
ISM suppresses angiogenesis in vivo. Effect of ISM on in vivo angiogenesis was examined using the directed in vivo angiogenesis assay by implanting a Matrigel based angioreactor in mice (Trevigen, Inc.). (A) ISM potently suppressed VEGF/bFGF induced angiogenesis in the angioreactor. Control (Matrigel alone) only showed minimum angio-genesis. ISM-C failed to suppress VEGF/bFGF induced angiogenesis. Representative photographs are presented. (B) Quantitative measurement of angiogenesis in the angioreactor. ECs inside the angioreactor were quantified using FITC-lectin. **P < 0.01 when compared with VEGF/bFGF sample. Number of samples in each category is indicated on top of the bar.
Overexpression of ISM in B16F10 melanoma cells suppressed tumour angiogenesis and tumour growth in mice
Solid tumour growth is known to be angiogenesis dependent. To investigate if ISM could suppress tumour angiogenesis and tumour progression, we established several mouse B16F10 melanoma stable cell lines that overexpress ISM as a secreted protein (Fig. 8A, B16/ISMa and B16/ISMb). B16F10 cells express very low levels of endogenous ISM (data not shown). No differences of cell proliferation and apoptosis in culture were observed among the ISM over-expressing stable lines compared to the vector modified or parental B16 cells (data not shown). In each C57BL mouse, cells of B16/ISMa or B16/ISMb (1 million) were injected subcutaneously into one side of the dorsal flank while the same number of B16/Vec cells was injected into the opposite side of the dorsal flank in the same mouse. Representative sets of tumours from B16/ISMa and B16/Vec of each individual mouse at day 14 are shown in Fig. 8B. Tumour growths were significantly reduced in tumours formed from B16/ISMa and B16/ISMb compared to those of B16/Vec (Fig. 8B and C). At the end of the experiment (14 days after tumour cell inoculation), the tumour size and tumour weight from B16/ISMa or B16/ISMb group was reduced up to 60% comparing to B16/Vec (Fig. 8C and D; P < 0.01).
Fig 8.
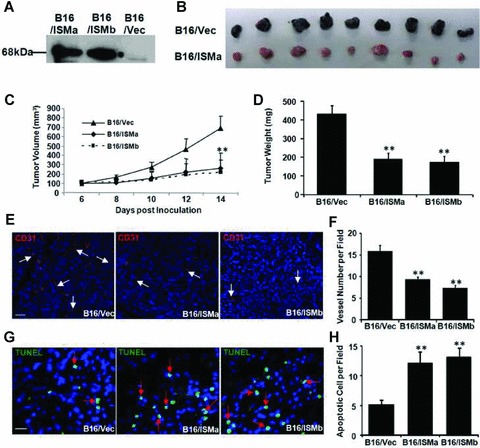
ISM inhibits B16 tumour growth in vivo. (A) Establishment of stable B16 melanoma cell lines overex-pressing mouse ISM. Expression of ISM in B16 stable lines were detected by Western blotting using anti-His antibody. Lines B16/ISMa and B16/ISMb showed strong expression of ISM. Vector transfected B16 (B16/Vec) is used as control. B16 cells express very low level of endogenous ISM. (B) Overexpression of ISM inhibits B16 tumour growth. Each pair of tumours (B16/ISMa and B16/Vec) was extracted from the same mouse. (C) Tumour growth curve in mice. X-axis represents the days after tumour cell inoculation. ISM suppressed tumour volume up to 70% at 14 days after inoculation. (D) Tumour weight at the end of the experiment (14 days after tumour cell inoculation). ISM led to tumour weight reduction of more than 50%. **P < 0.01, n× 4. (E) B16/ISM tumours show a reduced vascularization compared to controls. Tumour vessels were visualized by CD31 staining (in red, indicated by white arrows). Representative pictures are shown. Blue staining by DAPI indicates cell nucleus. Magnification 200 ×, n= 4. The scale bar is 50 μm. (F) Relative vessel densities of tumours from B16/Vec and B16/ISM. **P < 0.01, n = 4. (G) B16/ISM tumours showed increased apoptosis. Apoptotic cells were stained by TUNEL assays indicated in green (red arrows). (H) Relative apoptosis of B16/Vec and B16/ISMs. **P < 0.01, n = 4. The scale bar is 20 μm.
To investigate whether the decreased tumour growth in B16/ISMa and B16/ISMb was attributable to decreased tumour angiogenesis, we evaluated blood vessel density and morphology of tissue sections of various tumours. Immunofluorescence staining of tumour tissue sections with anti-CD31 antibody (which stains tumour ECs) revealed a decreased tumour vascularization compared with B16/Vec tumours (Fig. 8E, tumour vessels in red, indicated by white arrows). The average tumour vascular density of each tumour group, which included all lumen and non-lumen CD31+ structures or cells, was decreased by more than 40% in B16/ISMa and B16/ISMb tumours (P < 0.01, Fig. 8F). A significant increase in cell apoptosis was also observed in B16/ISMa and B16/ISM6 tumour tissue sections as determined by terminal deoxynucleotidyl trans-ferase dUTP nick end labeling (TUNEL) staining (Fig. 8G, apoptotic cells in green, indicated by red arrows). The average apoptotic indexes quantified by analysing multiple tumour samples in each tumour group were shown in Fig. 8H. Increased apoptosis would also contribute to the slow growth of these tumours.
Overexpression of VEGF has been documented in most type of cancers [23]. VEGF is also highly expressed in xenograph B16F10 tumour in mice [25, 26]. Because ISM potently inhibits VEGF-induced angiogenesis in vitro, it is possible that ISM also suppressed VEGF induced angiogenesis in B16 tumour. Interestingly, overexpression of ISM also led to reduction of melanin production in B16F10 melanoma cells (Fig. 8B). However, it is unclear at this point if the reduction of melanin production is linked to the reduced tumour progression in mice. An earlier report has shown that melanin production is not linked to B16 melanoma cell invasiveness in vitro[27].
Knockdown of isthmin in zebrafish embryos disrupted trunk intersegmental vessel formation
To study the role of isthmin in physiological angiogenesis, we used zebrafish embryos as a model to investigate embryonic angiogenesis. Expression of ism during zebrafish embryogenesis was studied by whole mount in situ hybridization. ism is expressed from the late gastrulation/early segmentation stage in the MHB and in the posterior trunk region (Fig. 9A and B). Subsequently, its expression is restricted in the tail bud region and notochord. At 22 hrs after fertilization (hpf), high level expression was observed in the notochord, MHB (Fig. 9D). The tail bud expression declined by 30 hpf (Fig. 9E). Notochord expression declined at 48 hpf and disappeared by 72 hpf (Fig. 9F and G) although low level MHB expression remained until 72 hpf.
Fig 9.
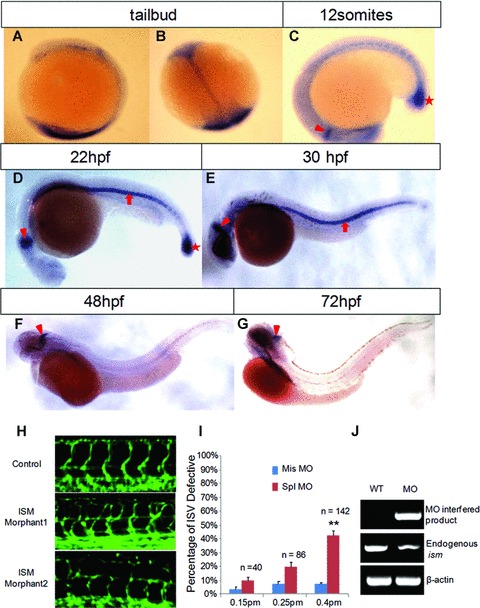
Knockdown of isth-min in zebrafish embryos disrupted trunk ISV formation. (A–G) Expression of ism during zebrafish embryogenesis. Notochord is indicated by arrow, MHB by arrowhead, and tail bud by star. (H) Knockdown of ism by MOs led to disrupted ISV formation. Abnormal ISVs in morphants are indicated by white arrows. (I) Quantitation of ism mor-phants with defective ISV formation. (J) Efficacy of spl MO in knockdown endoge-nous ism mRNA expression in morphants. The specific splicing-interfering MO (spl MO) interfered mRNA product is only detected in mor-phants while endogenous mRNA level was down about 70%. β-actin is used as a control.
Knockdown of ism expression in Tg(fli-1:EGFP) transgenic zebrafish embryos were carried out by microinjecting antisense MO into fertilized eggs. In this transgenic line, EGFP is expressed in all ECs under the control of the endothelial specific fli-1 gene promoter from as early as 3-somite stage of embryogenesis [28]. As shown in Fig. 9, a splicing-interference MO (spl MO) dose-dependently disrupted ISV formation in the trunk during the second day of embryogenesis, the period when ISV forms through angio-genesis by branching from the dorsal aorta and axial vein (Fig. 9). In ism morphants which showed no gross trunk morphological defects, ISVs were often mis-joined (Fig. 9H, morphant 1). In ism morphants which showed a curved body, severe ISV defects were observed, with some ISVs failed to form (Fig. 9H, morphant 2). In contrast, the dorsal aorta and axial vein were formed normally through vasculogenesis in these morphants. Semi-quantitative RT-PCR showed that the endogenous ism mRNA was effectively reduced about 70% in the morphants (Fig. 9). Similar morphant phenotypes were also obtained using an ATG blocking MO (data not shown). The trunk ISV defects in ism morphants correlate with the high level ism expression in the notochord during the second day of embryogenesis. No obvious vessel defects were observed in the head region from 12–24 hpf despite the high level ism expression in the MHB during this period.
Discussion
Angiogenesis inhibitors play important roles in regulating angio-genesis in physiology and pathology. Over the years, very few genes have been discovered which directly code for anti-angio-genic proteins. Although ISM was discovered as a secreted protein highly expressed in the isthmus of the brain [3], its function has remained unknown. In this work, we demonstrated for the first time that ISM is a novel endogenous angiogenesis inhibitor.
Recombinant ISM inhibits angiogenesis in vitro and in vivo
Recombinant ISM inhibited multiple aspects of angiogenesis in vitro (Table 1). It suppressed EC capillary network formation on Matrigel in a dose-dependent manner (Fig. 2). Addition of ISM at earlier times (0–2 hrs) after EC plating inhibits capillary formation (Fig. 3), suggesting that ISM most likely influences the early stages of the process. Recombinant ISM also inhibited VEGF/bFGF induced in vivo angiogenesis in implanted Matrigel plug in mice (Fig. 7).
Table 1.
Summary of the function of ISM and its various domains in in vitro angiogenesis
| Protein | Capillary network formation | Chemotaxis migration | Proliferation | Apoptosis | Attachment/spreading on matrix | Support EC attachment |
|---|---|---|---|---|---|---|
| ISM | ↓↓ | - | ↓↓ | ↑↑ | - | ↑↑ |
| ISM-N- | - | - | ↓ | - | - | - |
| ISM-C | ↓↓ | - | ↓ | - | - | ↑↑ |
| ISM-TSR | - | - | - | - | - | - |
-, no effect; ↓↓, strong inhibitory effect; ↓, inhibitory effect; ↑↑, strong stimulatory effect.
ISM inhibits VEGF-, bFGF- and serum-stimulated EC proliferation in a dose-dependent manner (Figs 4B and S3). It also induced EC apoptosis in the presence of VEGF (or bFGF or serum) through a caspase-dependent pathway (Figs 4C, S6 and S7). Interestingly, only the full-length ISM protein has this apoptosis-inducing activity. EC survival and apoptosis are known to play key roles in angio-genesis [29, 30]. However, it has no effect on tumour cell apopto-sis and only induced mild apoptosis of fibroblasts in culture (Figs 4, S3, S5–S8). The specific effect on ECs could be an advantage for therapeutic drug development.
A recent report has shown that although VEGF stimulates EC chemotaxis, it reduces EC chemokinesis [31], indicating that chemotaxis is the main mode of cell migration during VEGF-induced angiogenesis. ISM neither interferes with EC chemotaxis induced by VEGF, bFGF or serum (Figs 4 and S4) nor does it affect EC chemoki-nesis in the presence of VEGF or 2% serum (Fig. S2). Furthermore, our preliminary study indicated that ISM also has no influence on EC haptotaxis (migration towards a gradient of solid ECM) to collagen I or fibronectin in the presence of VEGF (data not shown).
ISM may inhibit angiogenesis through αvβ5 integrin
ISM also does not influence EC attachment and spreading to several matrix molecule- coated surfaces such as fibronectin, gelatin (collagen) and diluted Matrigel. In spite of this, ISM supports EC attachment and spreading in similar fashion as gelatin or fibronectin (Figs 5 and S8). We further demonstrated that ISM selectively interacts with αvβ5 integrin on EC surface (Fig. 6). Anti-αvβ5 neutralizing antibody partially blocked EC adhesion and spreading to ISM-coated surface, further supported that ISM may interact with EC and influence angiogenesis through this integrin (Fig. 6). Because ISM has no effect on EC attachment and spreading to matrix molecules such as fibronectin, gelatin (collagen) or diluted Matrigel whose major components are laminin and collagen IV (Figs 5A and S9). This is consistent with the fact that inte-grin αvβ5 is not the receptor for collagen, fibronectin or laminin.
Other integrins expressed in ECs such as αvβ3, and various β1 integrins do not seem to interact with ISM. Integrin αvβ5 is known to be highly expressed in ECs in tumour, ECs in active angiogene-sis as well as in some tumour cells [24]. It is often co-targeted with αvβ3 integrin by pharmacological molecules to suppress tumour angiogenesis and tumour progression [24]. However, the presence of αvβ5 integrin in B16F10 melanoma cells has not been reported. Although low level αv integrin subunit is detected in B16F10 melanoma cells in culture, the dominant integrin subunits in this tumour cell are α4 and α5 [32]. In addition, the α1 integrin has been documented to be main β subunit in this cell whereas β5 integrin expression has not been documented. Because ISM did not affect B16 tumour cell proliferation or apoptosis in vitro (Figs S5 and S8), we propose that ISM mainly affected ECs in B16 tumours and suppressed tumour growth through suppressing angiogenesis.
ISM inhibits tumour angiogenesis in mice
B16 melanomas that stably overexpressed ISM showed markedly reduced xenograft tumour growth along with decreased tumour vascular density (Fig. 8 and data not shown). Two independent ISM-overexpressing cell lines showed similar and consistent effect in mice. This result strongly suggests that continued presence of high level ISM in the tumour milieu inhibits tumour angio-genesis and hence tumour growth in mice. Whether ISM could inhibit the growth and angiogenesis of pre-established tumour in mice when delivered systematically will need further investigation. Several angiogenesis inhibitor proteins or their truncated fragments have been shown to suppress pre-established tumours in mice, making them candidates for anticancer drug development [21, 33, 34].
We found that ISM is expressed in many human tumours at various levels (in some cases higher than normal tissue whereas in other cases it was lower; data not shown). Endogenous ISM is also secreted into the media of certain cultured human tumour cells and ECs (data not shown). However, VEGF treatment (up to 6 hrs) did not significantly increase or decrease ISM secretion in cultured human ECs (data not shown). It would be interesting to examine the role of ISM in human tumours.
ism influences physiological angiogenesis in zebrafish embryonic development
When ism gene was knocked down in zebrafish embryos by MO, an obvious disruption of trunk ISVs was observed. ISVs are formed by angiogenesis by sprouting from the dorsal aorta and axial vein during the second day of zebrafish embryogenesis. In contrast, early blood vessel formation through vasculogenesis such as dorsal aorta and axial vein were not affected by ism knockdown, suggesting ism may specifically influence certain angiogenesis process. Interestingly, ism is expressed at very high levels in notochord during the second day of zebrafish embryogenesis. Notochord is known to play an important role in zebrafish development including vascular development possibly by secreting various morphogens and regulatory factors such as sonic hedgehog [35]. How the expression of ism in notochord might influence ISV formation through angiogenesis needs further investigation.
Similar to Xenopus, ism is expressed at high levels in the developing MHB in zebrafish (Fig. 9). In 2–3-week-old mouse, Ism is expressed at very high levels in lung and brain compared to other tissues (data not shown). In addition, ISM is expressed in many human tissues including brain and lung (data not shown). It would be interesting to study whether Ism has additional physiological functions.
AMOP domain plays a key role in ISM's function
ISM has two recognizable protein domains, a centrally localized TSR and a C-terminal AMOP. Although TSR has been linked to the anti-angiogenic activity of TSP-1 [5], deletion mutant studies indicate that the TSR domain of ISM has no anti-angiogenic activity (Fig. 2). Instead, the inhibition of EC capillary network formation is mainly mediated through the C-terminal AMOP domain, correlating with the fact that ECs attach to ISM-coated surface through this domain (Figs 2 and 5, and Table 1). Although AMOP domain was demonstrated to be involved in cell adhesion based on its presence in MUC4 and other adhesion molecules [8], its function in cell adhesion has not been experimentally validated. Our work here provides the first experimental evidence that AMOP domain in ISM mediates EC adhesion to ISM and plays an important role in the inhibition of capillary network formation (Figs 2 and 5).
Interestingly, only full-length ISM induced EC apoptosis although neither ISM-N nor ISM-C (AMOP domain) has such activity (Figs 4C and S6). On the other hand, ISM inhibited VEGF-induced EC proliferation much more potently than either ISM-N or ISM-C, although both fragments have weak anti-proliferative activities (Fig. 4B). As the anti-proliferation activity measured by BrdU incorporation (cells in active DNA synthesis) may actually be the combined effect of proliferation inhibition and apoptosis induction, the weak anti-proliferation effect of ISM-N or ISM-C may be due to their lack of pro-apoptotic activities. It therefore seems that the anti-proliferative and pro-apoptotic activity of ISM require different functional domains with the pro-apoptotic activity requiring the context of the full-length ISM protein.
However, ISM-C (AMOP) failed to inhibit in vivo angiogenesis of implanted Matrigel plug. In this experiment, ISM-C (similar to ISM) is pre-mixed with Matrigel before implantation under the mouse skin (a single dose) and angiogenesis are determined 2 weeks later. The stability of ISM-C in vivo under this assay condition may be a factor. Future studies involving repeated application of ISM-C during the in vivo assay or an evaluation of the in vivo stability of ISM-C are needed to clarify if ISM-C could inhibit angiogenesis in vivo.
Possible mechanisms of action of ISM
ISM may inhibit angiogenesis through mechanisms such as EC apoptosis induction, suppression of EC proliferation as well as inhibition of EC morphogenesis. Although ISM affects neither EC attachment to certain matrix molecules (such as collagen, fibronectin and laminin) nor EC migration, it may still influence EC interaction with other matrix molecules that preferentially interact with αvβ5 integrin such as vitronectin. Further studies on the roles of ISM-αvβ5 integrin interaction in the anti-angiogenic function of ISM are needed in order to thoroughly understand its molecular mechanisms of action.
Conclusion
We report here for the first time that ISM is a novel secreted angiogenesis inhibitor that inhibited angiogenesis in vitro and in vivo. It suppressed mouse melanoma tumour growth through inhibiting tumour angiogenesis. It also plays a role in physiological angiogenesis in zebrafish embryogenesis. The C-terminal AMOP domain plays an important role in the anti-angiogenic function of ISM by mediating ISM's interaction with αvβ5 integrin on EC surface. This is the first report of a biological function of the previously hypothetical AMOP domain in proteins. Further understanding of the molecular mechanisms of ISM as well as knowledge of its anti-angiogenic potency through various delivery methods in vivo will help to determine the therapeutic potential of this protein for cancer and other angiogenesis-related diseases.
Acknowledgments
This work is supported by grants 01/1/21/18/074 and 07/1/21/19/493 from the Singapore Biomedical Research Council (BMRC) to R.G. We thank Dr. Rong Gao for critically reading the manuscript.
Supporting Information
Dose analyses of the effects of ISM and itsvarious domains on EC migration. ISM does not influenceVEGF-induced chemotactic EC migration. The concentrations of ISM,ISM-N, ISM-C and ISM-TSR tested were from 1 nM to 1 μM.
ISM did not affect EC chemokinesis in thepresence or absence of VEGF. ISM concentration tested is 100 nM to1 μM and VEGF used is at 15 ng/ml. (A) ISM has noinfluence on EC chemokinesis in the absence of VEGF (2% FBS only).(B) ISM has no influence on EC chemokinesis in the presenceof VEGF.
ISM suppressed bFGF or serum-induced ECproliferation. (A) ISM inhibited bFGF-stimulated ECproliferation in a dose-dependent manner. bFGF is at 15 ng/ml.(B) ISM inhibited 10% FBS-stimulated EC proliferation in adose-dependent manner.*:P < 0.05, **: P < 0.01,n = 3.
ISM did not influence bFGF or serum-stimulatedchemo-tactic EC migration. (A) EC chemotactic migrationstimulated by 15 ng/ml bFGF. ISM did not influence EC chemotacticmigration up to 1 μM. (B) EC migration stimulated by 10%FBS. ISM did not influence EC migration up to 1 μM.
ISM inhibited serum-stimulated proliferation offibroblast cells but not tumour cells. (A) ISM mildlyinhibited 10% FBS-stimulated Swiss3T3 cell proliferation at 1μM. (B) ISM mildly inhibited 10% FBS-stimulated NIH3T3cell proliferation at 1 μM. (C) ISM did not influence 10%FBS-stimulated B16 cell proliferation up to 1 μM. (D) ISMdid not influence 10% FBS-stimulated HepG2 cell proliferation up to1 μM. *: P < 0.05, n = 3.
ISM induced EC apoptosis through acaspase-dependent pathway. (A) ISM induced EC apoptosis inthe presence of VEGF in a dose-dependent manner. The ISM-induced ECapoptosis was abolished when pan-caspase inhibitor z-VAD-fmk wasadded. None of the ISM truncated fragments (at concentrations from10 nM to 1μM) showed such activity. *: P < 0.05, **:P < 0.01, n = 4. VEGF used was 15 ng/ml in allexperiments. (B) ISM induced the activation of caspase 3 inthe presence of VEGF. The activated form of caspase 3 (17 kD) wasdetected by Western blot using an antibody that stains foractivated caspase 3 (detailed in 'Materials and methods'). Controlis 2% FBS without VEGF. ECs were treated with ISM for 8 hrs beforeharvested for this experiment.
ISM induced EC apoptosis in the presence of bFGFor serum. (A) ISM dose-dependently induced EC apoptosis inthe presence of 15 ng/ml bFGF. (B) ISM dose-dependentlyinduced EC apoptosis in the presence of 10% FBS. *: P <0.05, **: P < 0.01, n = 3.
ISM induced apoptosis of fibroblast cells butnot tumour cells in the presence of serum. (A) ISM mildlyinduced Swiss3T3 fibroblast cell apoptosis. Significant effects ofSwiss3T3 apoptosis were observed at 1 μM. (B) ISM mildlyinduced NIH3T3 cell apop-tosis at 1 μM. (C) ISM did notinfluence B16 cell apoptosis up to 1 μM. (D) ISM did notinfluence HepG2 cell apoptosis up to 1 μM. *: P <0.05, n = 3.
ISM does not interfere with EC spreading ontogelatin. Time course of EC spreading showed that ECs normally takeabout 2 hrs to spread onto gelatin-coated surface to form aflattened and extended morphology. The presence of ISM at 100 nM,500 nM and 1 μM did not interfere with this spreading. Controlis 2% FBS only.
References
- 1.Folkman J. What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst. 1990;82:4–6. doi: 10.1093/jnci/82.1.4. [DOI] [PubMed] [Google Scholar]
- 2.Sato Y. Update on endogenous inhibitors of angiogenesis. Endothelium. 2006;13:147–55. doi: 10.1080/1062332060069110. [DOI] [PubMed] [Google Scholar]
- 3.Pera EM, Kim JI, Martinez SL, et al. Isthmin is a novel secreted protein expressed as part of the Fgf-8 synexpression group in the Xenopus midbrain-hindbrain organizer. Mech Dev. 2002;116:169–72. doi: 10.1016/s0925-4773(02)00123-5. [DOI] [PubMed] [Google Scholar]
- 4.Weidinger G, Thorpe CJ, Wuennenberg-Stapleton K, et al. The Sp1-related transcription factors sp5 and sp5-like act downstream of Wnt/beta-catenin signaling in mesoderm and neuroecto-derm patterning. Curr Biol. 2005;15:489–500. doi: 10.1016/j.cub.2005.01.041. [DOI] [PubMed] [Google Scholar]
- 5.Iruela-Arispe ML, Lombardo M, Krutzsch HC, et al. Inhibition of angiogenesis by thrombospondin-1 is mediated by 2 independent regions within the type 1 repeats. Circulation. 1999;100:1423–31. doi: 10.1161/01.cir.100.13.1423. [DOI] [PubMed] [Google Scholar]
- 6.Guo NH, Krutzsch HC, Inman JK, et al. Antiproliferative and antitumor activities of D-reverse peptides derived from the sec-ond type-1 repeat of thrombospondin-1. J Pept Res. 1997;50:210–21. doi: 10.1111/j.1399-3011.1997.tb01187.x. [DOI] [PubMed] [Google Scholar]
- 7.Tucker RP. The thrombospondin type 1 repeat superfamily. Int J Biochem Cell Biol. 2004;36:969–74. doi: 10.1016/j.biocel.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 8.Ciccarelli FD, Doerks T, Bork P. AMOP, a protein module alternatively spliced in cancer cells. Trends Biochem Sci. 2002;27:113–5. doi: 10.1016/s0968-0004(01)02049-7. [DOI] [PubMed] [Google Scholar]
- 9.Nykvist P, Tasanen K, Viitasalo T, et al. The cell adhesion domain of type XVII collagen promotes integrin-mediated cell spreading by a novel mechanism. J Biol Chem. 2001;276:38673–9. doi: 10.1074/jbc.M102589200. [DOI] [PubMed] [Google Scholar]
- 10.Johansson MW. Cell adhesion molecules in invertebrate immunity. Dev Comp Immunol. 1999;23:303–15. doi: 10.1016/s0145-305x(99)00013-0. [DOI] [PubMed] [Google Scholar]
- 11.Lu X, Davies J, Lu D, et al. The effect of the single substitution of arginine within the RGD tripeptide motif of a modified neurotoxin dendroaspin on its activity of platelet aggregation and cell adhesion. Cell Commun Adhes. 2006;13:171–83. doi: 10.1080/15419060600726183. [DOI] [PubMed] [Google Scholar]
- 12.Sulochana KN, Fan H, Jois S, et al. Peptides derived from human decorin leucine-rich repeat 5 inhibit angiogenesis. J Biol Chem. 2005;280:27935–48. doi: 10.1074/jbc.M414320200. [DOI] [PubMed] [Google Scholar]
- 13.Guedez L, Rivera AM, Salloum R, et al. Quantitative assessment of angiogenic responses by the directed in vivo angiogen-esis assay. Am J Pathol. 2003;162:1431–9. doi: 10.1016/S0002-9440(10)64276-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aoka Y, Johnson FL, Penta K, et al. The embryonic angiogenic factor Del1 accelerates tumor growth by enhancing vascular formation. Microvasc Res. 2002;64:148–61. doi: 10.1006/mvre.2002.2414. [DOI] [PubMed] [Google Scholar]
- 15.Weidner N, Semple JP, Welch WR, et al. Tumor angiogenesis and metastasis–correlation in invasive breast carcinoma. N Engl J Med. 1991;324:1–8. doi: 10.1056/NEJM199101033240101. [DOI] [PubMed] [Google Scholar]
- 16.Westerfield M, Doerry E, Kirkpatrick AE, et al. Zebrafish informatics and the ZFIN database. Methods Cell Biol. 1999;60:339–55. doi: 10.1016/s0091-679x(08)61909-3. [DOI] [PubMed] [Google Scholar]
- 17.Kimmel CB, Ballard WW, Kimmel SR, et al. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- 18.Nasevicius A, Ekker SC. Effective targeted gene ‘knockdown’ in zebrafish. Nat Genet. 2000;26:216–20. doi: 10.1038/79951. [DOI] [PubMed] [Google Scholar]
- 19.Jowett T. Analysis of protein and gene expression. Methods Cell Biol. 1999;59:63–85. doi: 10.1016/s0091-679x(08)61821-x. [DOI] [PubMed] [Google Scholar]
- 20.Madri JA, Pratt BM. Endothelial cell-matrix interactions: in vitro models of angiogenesis. J Histochem Cytochem. 1986;34:85–91. doi: 10.1177/34.1.2416801. [DOI] [PubMed] [Google Scholar]
- 21.O’Reilly MS, Boehm T, Shing Y, et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–85. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 22.Davis GE, Saunders WB. Molecular balance of capillary tube formation versus regression in wound repair: role of matrix metallo-proteinases and their inhibitors. J Investig Dermatol Symp Proc. 2006;11:44–56. doi: 10.1038/sj.jidsymp.5650008. [DOI] [PubMed] [Google Scholar]
- 23.Ferrara N. The role of VEGF in the regulation of physiological and pathological angiogenesis. EXS. 2005:209–31. doi: 10.1007/3-7643-7311-3_15. [DOI] [PubMed] [Google Scholar]
- 24.Silva R, D’Amico G, Hodivala-Dilke KM, et al. Integrins: the keys to unlocking angiogenesis. Arterioscler Thromb Vasc Biol. 2008;28:1703–13. doi: 10.1161/ATVBAHA.108.172015. [DOI] [PubMed] [Google Scholar]
- 25.Zhang S, Li M, Gu Y, et al. Thalidomide influences growth and vasculogenic mimicry channel formation in melanoma. J Exp Clin Cancer Res. 2008;27:60. doi: 10.1186/1756-9966-27-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Culp WD, Neal R, Massey R, et al. Proteomic analysis of tumor establishment and growth in the B16-F10 mouse melanoma model. J Proteome Res. 2006;5:1332–43. doi: 10.1021/pr060059q. [DOI] [PubMed] [Google Scholar]
- 27.Zhao W, Liu H, Xu S, et al. Migration and metalloproteinases determine the invasive potential of mouse melanoma cells, but not melanin and telomerase. Cancer Lett. 2001;162:S49–55. doi: 10.1016/s0304-3835(00)00656-x. [DOI] [PubMed] [Google Scholar]
- 28.Lawson ND, Weinstein BM. in vivo imaging of embryonic vascular development using transgenic zebrafish. Dev Biol. 2002;248:307–18. doi: 10.1006/dbio.2002.0711. [DOI] [PubMed] [Google Scholar]
- 29.Nor JE, Polverini PJ. Role of endothelial cell survival and death signals in angiogen-esis. Angiogenesis. 1999;3:101–16. doi: 10.1023/A:1009053411094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peters K, Troyer D, Kummer S, et al. Apoptosis causes lumen formation during angiogenesis in vitro. Microvasc Res. 2002;64:334–8. doi: 10.1006/mvre.2002.2438. [DOI] [PubMed] [Google Scholar]
- 31.Barkefors I, Le Jan S, Jakobsson L, et al. Endothelial cell migration in stable gradients of vascular endothelial growth factor A and fibroblast growth factor 2: effects on chemotaxis and chemokinesis. J Biol Chem. 2008;283:13905–12. doi: 10.1074/jbc.M704917200. [DOI] [PubMed] [Google Scholar]
- 32.Qian F, Zhang ZC, Wu XF, et al. Interaction between integrin alpha(5) and fibronectin is required for metastasis of B16F10 melanoma cells. Biochem Biophys Res Commun. 2005;333:1269–75. doi: 10.1016/j.bbrc.2005.06.039. [DOI] [PubMed] [Google Scholar]
- 33.Miao WM, Seng WL, Duquette M, et al. Thrombospondin-1 type 1 repeat recombinant proteins inhibit tumor growth through transforming growth factor-beta-dependent and -independent mechanisms. Cancer Res. 2001;61:7830–9. [PubMed] [Google Scholar]
- 34.O’Reilly MS, Holmgren L, Shing Y, et al. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metas-tases by a Lewis lung carcinoma. Cell. 1994;79:315–28. doi: 10.1016/0092-8674(94)90200-3. [DOI] [PubMed] [Google Scholar]
- 35.Stemple DL. Structure and function of the notochord: an essential organ for chordate development. Development. 2005;132:2503–12. doi: 10.1242/dev.01812. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dose analyses of the effects of ISM and itsvarious domains on EC migration. ISM does not influenceVEGF-induced chemotactic EC migration. The concentrations of ISM,ISM-N, ISM-C and ISM-TSR tested were from 1 nM to 1 μM.
ISM did not affect EC chemokinesis in thepresence or absence of VEGF. ISM concentration tested is 100 nM to1 μM and VEGF used is at 15 ng/ml. (A) ISM has noinfluence on EC chemokinesis in the absence of VEGF (2% FBS only).(B) ISM has no influence on EC chemokinesis in the presenceof VEGF.
ISM suppressed bFGF or serum-induced ECproliferation. (A) ISM inhibited bFGF-stimulated ECproliferation in a dose-dependent manner. bFGF is at 15 ng/ml.(B) ISM inhibited 10% FBS-stimulated EC proliferation in adose-dependent manner.*:P < 0.05, **: P < 0.01,n = 3.
ISM did not influence bFGF or serum-stimulatedchemo-tactic EC migration. (A) EC chemotactic migrationstimulated by 15 ng/ml bFGF. ISM did not influence EC chemotacticmigration up to 1 μM. (B) EC migration stimulated by 10%FBS. ISM did not influence EC migration up to 1 μM.
ISM inhibited serum-stimulated proliferation offibroblast cells but not tumour cells. (A) ISM mildlyinhibited 10% FBS-stimulated Swiss3T3 cell proliferation at 1μM. (B) ISM mildly inhibited 10% FBS-stimulated NIH3T3cell proliferation at 1 μM. (C) ISM did not influence 10%FBS-stimulated B16 cell proliferation up to 1 μM. (D) ISMdid not influence 10% FBS-stimulated HepG2 cell proliferation up to1 μM. *: P < 0.05, n = 3.
ISM induced EC apoptosis through acaspase-dependent pathway. (A) ISM induced EC apoptosis inthe presence of VEGF in a dose-dependent manner. The ISM-induced ECapoptosis was abolished when pan-caspase inhibitor z-VAD-fmk wasadded. None of the ISM truncated fragments (at concentrations from10 nM to 1μM) showed such activity. *: P < 0.05, **:P < 0.01, n = 4. VEGF used was 15 ng/ml in allexperiments. (B) ISM induced the activation of caspase 3 inthe presence of VEGF. The activated form of caspase 3 (17 kD) wasdetected by Western blot using an antibody that stains foractivated caspase 3 (detailed in 'Materials and methods'). Controlis 2% FBS without VEGF. ECs were treated with ISM for 8 hrs beforeharvested for this experiment.
ISM induced EC apoptosis in the presence of bFGFor serum. (A) ISM dose-dependently induced EC apoptosis inthe presence of 15 ng/ml bFGF. (B) ISM dose-dependentlyinduced EC apoptosis in the presence of 10% FBS. *: P <0.05, **: P < 0.01, n = 3.
ISM induced apoptosis of fibroblast cells butnot tumour cells in the presence of serum. (A) ISM mildlyinduced Swiss3T3 fibroblast cell apoptosis. Significant effects ofSwiss3T3 apoptosis were observed at 1 μM. (B) ISM mildlyinduced NIH3T3 cell apop-tosis at 1 μM. (C) ISM did notinfluence B16 cell apoptosis up to 1 μM. (D) ISM did notinfluence HepG2 cell apoptosis up to 1 μM. *: P <0.05, n = 3.
ISM does not interfere with EC spreading ontogelatin. Time course of EC spreading showed that ECs normally takeabout 2 hrs to spread onto gelatin-coated surface to form aflattened and extended morphology. The presence of ISM at 100 nM,500 nM and 1 μM did not interfere with this spreading. Controlis 2% FBS only.