

Abstract
Dyslipidemia is a well-established condition proved to accelerate the progression of chronic kidney disease leading to tubulo-interstitial injury. However, the molecular aspects of the dyslipidemia-induced renal damage have not been fully clarified and in particular the role played by low-density lipoproteins (LDLs). This study aimed to examine the effects of native non-oxidized LDL on cellular oxidative metabolism in cultured human proximal tubular cells. By means of confocal microscopy imaging combined to respirometric and enzymatic assays it is shown that purified native LDL caused a marked increase of cellular reactive oxygen species (ROS) production, which was mediated by activation of NADPH oxidase(s) and by mitochondrial dysfunction by means of a ROS-induced ROS release mechanism. The LDL-dependent mitochondrial alterations comprised inhibition of the respiratory chain activity, enhanced ROS production, uncoupling of the oxidative phosphorylation efficiency, collapse of the mtΔΨ, increased Ca2+ uptake and loss of cytochrome c. All the above LDL-induced effects were completely abrogated by chelating extracellular Ca2+ as well as by inhibition of the Ca2+-activated cytoplas-mic phospholipase A2, NADPH oxidase and mitochondrial permeability transition. We propose a mechanicistic model whereby the LDL-induced intracellular redox unbalance is triggered by a Ca2+ inward flux-dependent commencement of cPLA2 followed by activation of a lipid- and ROS-based cross-talking signalling pathway. This involves first oxidants production via the plasmamembrane NADPH oxidase and then propagates downstream to mitochondria eliciting redox- and Ca2+-dependent dysfunctions leading to cell-harming conditions. These findings may help to clarify the mechanism of dyslipidemia-induced renal damage and suggest new potential targets for specific therapeutic strategies to prevent oxidative stress implicated in kidney diseases.
Keywords: Low density lipoproteins, kidney proximal tubular cells, reactive oxygen species, mitochondria, NADPH oxidase, cytoplasmic phospholipase A2, chronic kidney disease, redox signalling, lipid signalling, ROS-induced ROS release
Introduction
Oxidized low-density lipoproteins (oxLDL) are proved to exert their role in the multi-steps atherogenetic process much more pronouncedly than native LDL (nLDL) [1, 2]. Although the chemical nature of the LDL oxidized products and their specific effects have not yet been identified, nevertheless the occurrence of different receptors for nLDL and oxLDL suggests a distinct mechanism of action [3]. Vascular endothelium is not the unique target of LDL pathogenetic properties. Indeed oxLDL have been recently shown to elicit inflammatory and fibrotic processes also in extra-vascular tissues such as renal mesangial and tubular epithelial cells [4, 5].
Under normal physiological conditions, the glomerular filtrate contains almost undetectable amount of lipoproteins. However, in chronic kidney disease (CKD), which is hallmarked by a progressive impairement of the glomerular barrier permselectivity, the renal tubule can be exposed to high molecular-weight molecules [6]. Hyperlipidemia has been proved to accelerate the progression of the CKD inducing a tubulo-interstitial injury [7, 8]. Evidence of oxLDL-induced oxidative stress has been provided supporting the idea that alteration of the reactive oxygen species (ROS) home-ostasis is an early event in the oxLDL-related diseases priming the subsequent tissue/organ disfunction [3, 4, 9, 10].
The overwhelming body of evidence supporting the deleterious effects of oxLDL has weakened the interest on unmodified nLDLs, which, however, still deserve attention as their interaction with extra-vascular tissues has been poorly characterized. On these grounds the present study investigated the effect of nLDL exposure on an in vitro model of proximal tubular epithe-lium with specific focus on the cellular oxidative metabolism. We demonstrate for the first time that non-oxidized nLDL elicit dysregulation of the cellular oxidation state by activating a redox signalling between different ROS-producing compartments in the cell. The role of altered ROS homeostasis in the development of LDL-related kidney damage and possible therapeutic interventions are discussed.
Materials and methods
Cell culture
HK-2 cells (ATCC, Manassas, VA, USA), which are normal proximal renal tubular epithelial cells immortalized by transduction with the human papilloma virus 16 E6/E7 genes, were cultured in DMEM/F12 (Sigma-Aldrich, Milan, Italy) medium supplemented with penicillin (50 U/ml) and streptomycin (50 mg/ml) and with 10% heat-inactivated foetal calf serum (FCS) (Sigma). Cultured cells were grown in monolayers at 37 °C in a humidified atmosphere containing 5% CO2.
Lipoprotein separation
LDL (d= 1.020–1.050 g/l) were separated from fasting normolipidemic plasma by sequential ultracentrifugation. The lipid (total and free cholesterol, triglycerides and phospholipid) content of lipoprotein fractions was measured by enzymatic techniques. The protein content was measured by the Lowry assay. Purified lipoprotein fractions were stored at 4°C in sodium bromide and dialyzed over night against saline colution solution (0.9% NaCl) before use.
Measurement of cell respiration and NADPH oxidase activity
Cultured cells were gently detached from the dish by trypsinization, washed in PBS, harvested by centrifugation and immediately assessed for O2 consumption by a Clark-type electrode (Hansatech) in a thermostated gas-tight chamber equipped with a stirring device. A total of 3 × 106 viable cells/ml were assayed in 50 mM KPi, 10 mM Hepes, 1 mM ethylenediaminetetraacetic acid (EDTA), pH 7.4 at 37 °C; after attainment of a stationary endogenous substrate-sustained respiratory rate, 2 μg/ml of oligomycin was added. The rates of O2 consumption were corrected for 5 mM KCN-insensitive respiration. The respiratory control ratio (RCR) was obtained dividing the rates of the oxygen consumption achieved before and after the addition of oligomycin. NADPH oxidase activity was assessed by following the reduction of extracellular acetylated-cytochrome c (SIGMA). Briefly, 20 μM ferri-cytochrome c was directly added to the cell cultures 60 min before the end of the nLDL-incubation times. At the desired time-points 100 μl of the culturing medium was transferred to a microcuvette and the reduction level of cytochrome c evaluated by the absorbance in the triple-wavelength mode (A549-(A540-A556)) using a Δɛ= 19.1 mM−1cm−1. In a pilot testing with a 24 hrs-nLDL-treated cell sample the absorbance of ferrocytochrome c increased linearly up to 90 min. The values attained were corrected for those obtained in parallel nLDL-treated cell samples but supplemented with superoxide dismutase (SOD) (500 U/ml) in the culturing medium.
Laser scanning confocal microscopy (LSCM) functional imaging of mitochondria in live cells.
Cells cultured at low density on fibronectin-coated 35 mm glass bottom dishes were incubated for 20 min at 37 °C with the following probes: 0.5 μM nonyl acridine orange (NAO) for the mitochondrial mass; 2 μM tetramethylrhodamine, ethyl ester (TMRE) for the mitochondrial membrane potential (mtΔΨ)); 0.5 μM MitoSOX or 10 μM 2′,7′-dichlorodihy-drofluorescein diacetate (H2DCF-DA) for mitochondrial O2− and cellular H2O2, respectively; 5 μM X-Rhod-1 AM for mitochondrial Ca2+. All the probes used were from Molecular Probes (Eugene, OR, USA). Stained cells were washed with PBS and examined by a Nikon TE 2000 microscope (images collected using a 60× objective (1.4 NA)) coupled to a Radiance 2100 dual laser (four-lines Argon–Krypton, single-line Helium–Neon) confocal laser scanning microscopy system (Biorad). Confocal planes (18–20) of 0.2 μm in thickness were examined along the z-axes, going from the top to the bottom of the cells. Acquisition, storage and analysis of data were performed with LaserSharp and LaserPix software (Biorad) or ImageJ (NIH, Bethesda, MD, USA). Quantification of the emitted fluorescent signal was achieved by averaging the pixel intensity values within the outline of single cells, as a function of each focal plane. Correction was made for minimal background in cell-free fields. The integrated value of the xz profile was taken as a measure of the fluorescence intensity and quantified in arbitrary units. At least 20 cells were randomly selected in each of 8–10 different optical fields under the indicated conditions and statistically analysed.
Immunocytochemistry
HK-2 cells cultured at low density on fibronectin coated 35 mm glass bottom dishes were fixed (4% paraformaldehyde), permeabilized (0.2% Triton X-100), blocked (3% bovine serum albumin (BSA) in PBS) and then incubated 1 h at room temperature with 1:200 diluted 1 mouse mAb anti-cytochrome c (Promega). After two washes in PBS/BSA the sample was incubated for 1 h at room temperature with 10 μg/ml of FITC-labelled goat antimouse IgG (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The fluorescent signals emitted by the FITC conjugated Ab (λex, 490 nm; λem, 525 nm) of the labelled cells was imaged by LSCM as previously described. Direct treatment of the cell with the secondary FITC-Ab did not result in appreciable fluorescent staining.
Statistical analysis
Two tailed Student's t-test was applied with a P < 0.05 to evaluate the statistical significance of differences measured throughout the data-sets reported.
Results
Native LDL cause enhanced ROS production in HK-2 cell line
LDLs isolated from healthy donors were tested for their oxidation state by electrophoretic mobility shift assay and UV spectrophotometry (see supplementary material and Fig. S1A,B). The results obtained indicated that the isolated LDLs did not show detectable evidence of oxidative modifications and remained as such under the experimental settings of the present study. Indeed, incubation of HK2 cell line with native LDL, for different intervals up to 24 hrs, did not modify significantly their oxidation state. Therefore the effects described hereafter are bona fide referred as elicited by non-oxidized native LDL. Twenty-four hours treatment of HK-2 cell line with nLDL (100 μg protein/ml) did not alter their viability (assessed by Trypan-Blue assay) neither caused marked morphological changes. Conversely similar treatment with in vitro-oxidised LDL caused profound alterations in the cultured cells diagnostic of induced-distress (supplementary material, Fig. S2A). However when tested by the Annexin V/fluorescein diacetate assay the nLDL-treated HK2 cell line showed evidence of early signs of apoptosis as compared to untreated cells (supplementary material, Fig. S2B). As expected, incubation of HK2 with oxidised LDL revealed indication of late apoptosis/necrosis. The concentration of the nLDL used throughout this study (i.e. 100 μg/ml) was comparable with that used in studies aimed to assess the in vitro effect of oxLDL on endothelial cells and within the physio-pathological range in the glomerulo-filtrate of hyperlipidemic patients.
Figure 1A illustrates the effect of nLDL-treatment of HK-2 on ROS production as assessed by confocal microscopy performed with ROS-specific fluorescent probes. It is shown that 100 μg/ml of nLDL-treatment for 24 hrs caused a large increase of the dichlorofluorescein (DCF)-related fluorescence signal over the basal level, diagnostic of intracellular production of H2O2. This was fully prevented by N-acetyl cysteine (NAC) and not observed following treatment of HK-2 with 15 mg/ml of albumin. MitoSox, a probe sensing intramitochondrial O2− production [11], failed to detect evidence of increased ROS-production. However, reassessment of the nLDL-induced DCF fluorescence under different instrumental settings resulted in brighter spotting in sub-cellular compartments clearly resembling the mitochondrial network morphology (Fig. 1B).
Fig 1.
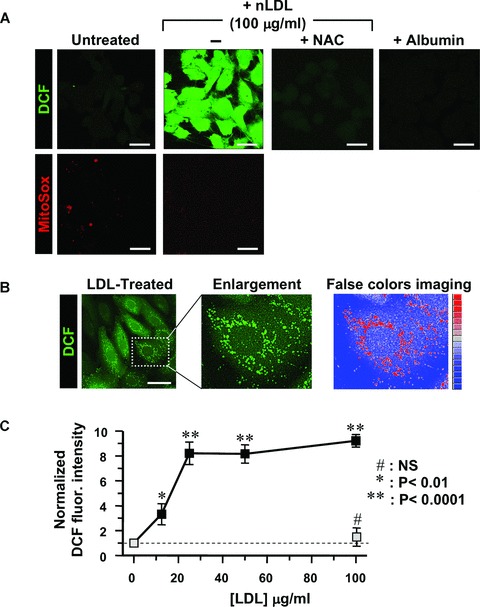
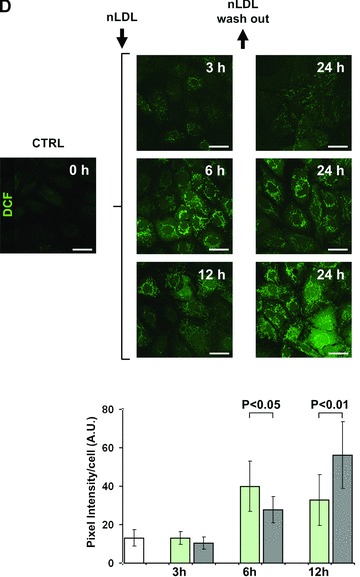
Treatment of HK-2 with nLDL results in unbalance of the cellular redox state. (A) LSCM for detection of H2O2 and mitochondrial O2− by the fluorescent probes DCF and MitoSox respectively. HK-2 cells were incubated for 24 hrs with 100 μg/ml nLDL alone or in the presence of 20 mM NAC or 15 mg/ml albumin. Untreated HK-2 cells were used as control. See Materials and Methods for details. Exciting Argon laser bean for DCF-related fluorescence was set at 5% of its maximal intensity and the PMT gain at 60%. Representative of at least four different preparations in each condition. (B) LSCM analysis of the DCF-related fluorescence and false-colours imaging. nLDL-treatment of HK-2 as in (A). The exciting Argon laser bean was set at 5% of its maximal intensity and the PMT gain at 30%. A false colors rendering of the enlarged detail was generated by ImageJ 1.38× (http://rsb.info.nih.gov/ij/). (C) Dose-dependence of DCF fluorescence. HK-2 cells were treated with the indicated concentrations of nLDL for 24 hrs and the DCF-related fluorescence recorded by LSCM as described in Materials and Methods. The fluorescence intensities of nLDL-treated HK-2 cells (black squares) were normalized to that of untreated cells and represent the average standard error of means (S.E.M.) of three independent experiments (n= 3) together with statistical analysis. The effect of NAC coincubation with 100 μg/ml of nLDL is also shown as light-grey square. (D) Effect of LDL wash-out on ROS production. HK-2 cells were treated with 100 μg/ml nLDL for 3, 6 and 12 hrs after that the cells were washed with a nLDL-free medium and maintained in culture for further 21, 18 and 12 hrs, respectively. Images of a representative experiment is presented showing the DCF-related fluorescence recorded before each nLDL wash-out and at 24 hrs from the beginning of the treatment. The histogram shows the quantitative analysis of the DCF fluorescence intensity. White bar, untreated cells; green bars, cell treated with nLDL for 3, 6 and 12 hrs; grey bars, cells incubated with nLDL for 3, 6 and 12 hrs, washed out and analysed after 24 hrs from the beginning of the treatment. Each bar is the average of three independent experiments ± S.E.M.; where indicated the statistical significance is shown. Bars inside all the micrographs: 30 μm.
The LDL-induced ROS production was dose-dependent (Fig. 1C) with a 3.5 fold increase over the basal level already detectable at a concentration as low as 12.5 μg nLDL/ml, which levelled off at values of eight to ninefold at 25–100 μg LDL/ml. The nLDL-induced ROS overproduction was not due to down-regulation of the main anti-oxidant enzymes (SOD1, SOD2, catalase, GPX1, GPX4; supplementary material, Fig. S3).
Six and twelve hours of nLDL-incubation caused oxidative changes that did not restore after removal of nLDL from the culturing medium and persisted or even increased at 24 hrs. On the other hand, 3 hrs of exposition to nLDL did not cause apparently any actual or delayed effect on ROS production (Fig. 1D).
nLDL-Treatment of HK-2 cells caused mitochondrial dysfunction
Because MitoSox accumulates into the mitochondria by a trans membrane potential (mtΔΨ)-driven process [11], we tested the possibility that nLDL-treatment affected the mtΔΨ. To this aim we used tetramethylrhodamine ethyl ester (TMRE) a sensitive mtΔΨ-probe. Figure 2A shows that although the nLDL-treatment did not change significantly the mitochondrial mass and its overall morphology (assessed by the cardiolipin dye 10-N-NAO), nevertheless it caused a dramatic decrease of the mtΔΨ. This was further verified by high-resolution respirometry on intact cells. Figure 2B shows that nLDL-treatment resulted in a slight, although significant, decrease of the endogenous resting oxygen consumption rate. This, in the presence of oligomycin, a specific inhibitor of the mtΔΨ-driven H+-FoF1 ATP-synthase, was much higher in nLDL-treated than in untreated HK-2 cells. As a consequence a drop in the respiratory control coupling ratio (RCR) [12] was observed in treated cells.
Fig 2.
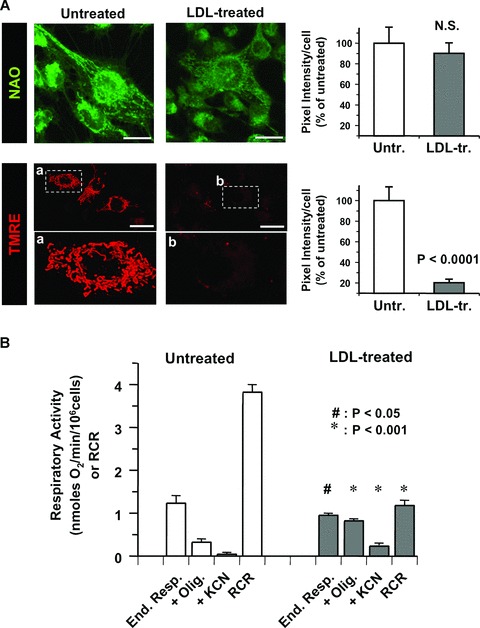
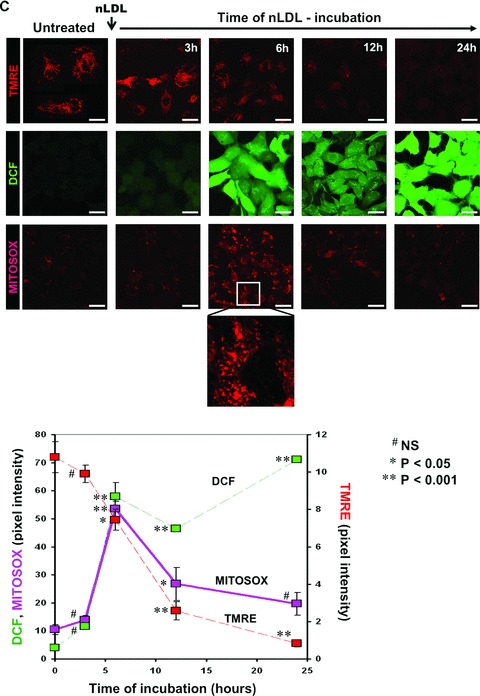
Treatment of HK-2 with nLDL causes mitochondrial dysfunction. (A) LSCM analysis of mito-chondrial mass and mtΔΨ by the fluorescent probes NAO and TMRE respectively. HK-2 cell line were incubated for 24 hrs with 100 μg/ml of nLDL. For the TMRE stained samples enlarged details of the picture, indicated by the rectangles a and b are shown. On the right the quantitative and statistical analysis of the NAO- and TMRE-related fluorescence is shown with white and grey bars indicating the average ± S.E.M. (n= 3) in untreated and nLDL-treated cells respectively. (B) Respirometric analysis. Untreated and nLDL-treated intact HK-2 cells were assayed for oxygen consumption as detailed under Material and Methods. White and grey bars represent the average ± S.E.M (n= 5) of the following conditions: resting endogenous respiration (End. resp); in the presence of oligomycin (+Olig.). The values were corrected for the KCN-insensitive respiration which is also reported (+KCN). The RCR was obtained dividing the resting respiration by that attained in the presence of oligomycin. The statistical significance of each figure between untreated and nLDL-treated cells is also shown. (C) Time-course of the effect of nLDL on mtΔΨ, and H2O2 and mitochondrial O2•− production. HK-2 cells were treated with 100 μg/ml of nLDL for the indicated incubation times, then stained with the fluorescent probes and analysed by LSCM as described in Materials and Methods. An enlargement of the micrograph relative to the MitoSox-treated cells incubated for 6 hrs with nLDL is also shown. The lower graph shows the quantitative analysis of the fluorescence intensity for each probe as a function of the nLDL-time of incubation; the values are the averages ± S.E.M. (n= 3 for each condition). The statistical significance with respect to untreated HK-2 cells is also reported. Bars inside all the micrographs: 30 μm.
The time-course of the effect of nLDL on the ΔΨ decline showed that it slightly progressed within 3–6 hrs to accelerate thereafter (Fig. 2C). Parallel detection of ROS formation displayed a significant DCF-fluorescence signal occurring after 6 hrs of nLDL-incubation. Importantly, when mitochondrial O2− generation was assessed by MitoSox a clear spotted fluorescence signal was evident at 6 hrs which progressively disappeared at longer time of nLDL-treatment. This observation indicated that at relatively short time of nLDL-incubation the mtΔΨ, although reduced, still drove the electrophoretic MitoSox probe accumulation allowing to detect formation of mitochondrial O2·−. A severe collapse of the mtΔΨ, as that attained after 12–24 hrs of nLDL-treatment, impaired the mitochondrial accumulation of MitoSox. Mitochondrial ROS-generation was, however, still occurring as displayed by the mtΔΨ-independent DCF probe. Thus this result supported the direct involvement of mitochondria as source of superoxide and peroxide because the early steps of the nLDL-induced change in the cellular redox-state.
nLDL-Induced ROS production in HK-2 cells was prevented by inhibition of NAD(P)H oxidase
Besides mitochondria, the membrane-bound NADPH oxidase (NOX) constitutes an additional cellular source of ROS [13] and its activation has been extensively reported for endothelial cells upon exposure to oxLDL [14]. To ascertain the possible involvement of NOX to the observed nLDL-induced ROS production we tested the effect of pharmacological inhibitors. Figure 3A shows that co-incubation of nLDL with diphenyleneiodinium (DPI), a widely used pan-inhibitor of flavo-enzymes [15] or with apocynin, a more selective inhibitor of NOX [16] resulted in the complete abrogation of the ROS-linked DCF signal. Consistently, measurement of the SOD-inhibitable superoxide generation by the externally added cytochrome c reduction assay resulted in a progressive increase starting at 6 hrs of nLDL-incubation. Importantly, the extra-production of external superoxide over the basal level was either DPI-and apocyin-sensitive (Fig. 3B), thus supporting their attribution to NADPH oxidase activity.
Fig 3.
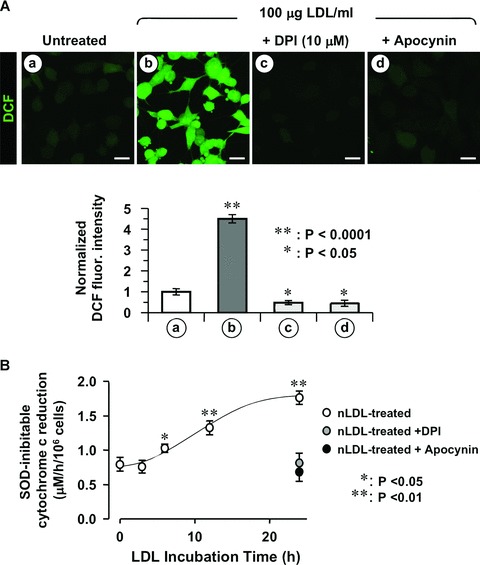
The nLDL-induced ROS production is sensitive to NOX inhibition. HK-2 cells were incubated for 24 hrs with 100 μg/ml nLDL alone or in the presence of either 10 μM DPI or 100 μM apocynin. (A) Upper panel: representative LSCM analysis of the effect of NOX inhibitors on nLDL-induced ROS production probed by the fluorescent probe DCF. Lower panel: quantitative analysis of the DCF fluorescence; each bar indicate the averages ± S.E.M. of n= 3. Statistical evaluation of the values measured in treated vs untreated cells is also shown. Bars inside all the micrographs: 30 μm. (B) NADPH oxidase activity assessed by the SOD-inhibitable cytochrome c reduction assay. See Materials and Methods for experimental details. The values are means ± S.E.M. of n= 3. Statistical evaluation of the values versus untreated cells, when significant, is also shown.
NOX isoforms have a distinct cellular localization in the kidney [17]. RT-PCR analysis unveiled in HK-2 major expression of the NOX1 and NOX4 isoforms and differential absorbance spec-trophotometry allowed to assess the presence of a catalytically active NOX-related b-type cytochrome (supplementary material, Fig. S4). Twenty-four hours treatment of HK-2 with nLDLs resulted neither in changes of the NOXs expression level nor in NOX-linked b-type cytochrome amount (data not shown).
LDL-linked mitochondrial ROS production in HK-2 cells is induced by NOX-related ROS signaling
Because NOXs release O2− in the extracellular space and the probes used detected LDL-linked intracellular ROS production we pondered that ROS released by NOX acted as messengers triggering mitochondrial ROS generation. To test this hypothesis the HK-2 cells were co-incubated with nLDL and the membrane-impermeant ROS scavengers superoxide dismutase (SOD) and catalase (CAT). As shown in Figure 4A SOD and CAT prevented completely the nLDL-induced ROS generation when added together and largely when tested separately. This indicated that the effects of O2− and of its dismutation product H2O2 were largely interchangeable.
Fig 4.
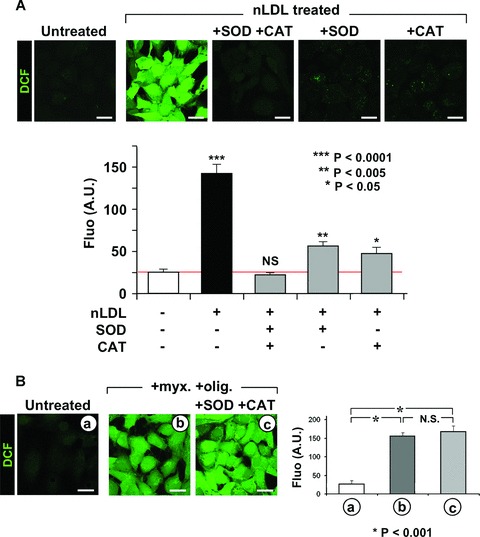
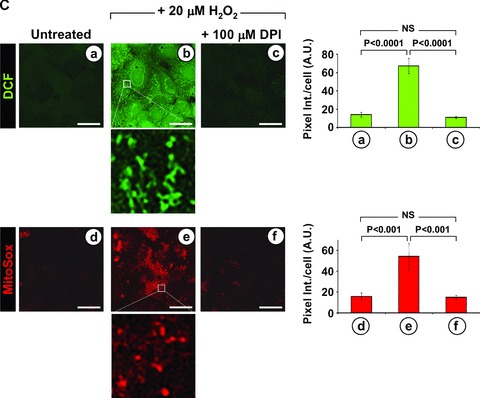
Extracellular ROS generation induces mitochondrial ROS production. (A) Effect of extra-cellular ROS scavengers on the nLDL-induced ROS production. HK-2 cells were incubated for 24 hrs with 100 μg/ml nLDL alone or supplemented with 500 U/ml SOD and/or 200 μg/ml catalase (CAT). Upper panel: representative LSCM analysis of the effect of SOD and/or CAT on the nLDL-induced ROS production probed by the fluorescent probe DCF (see Figure 1A and Materials and Methods). Lower panel: quantitative analysis of the DCF fluorescence; each bar indicate the averages ± SEM of n= 3. Statistical evaluation of the values measured in treated versus untreated cells is shown. (B) Effect of extra-cellular ROS scavengers on the mitochondrial ROS production induced by inhibition of the respiratory chain. HK-2 cells were treated for 2 hrs with 10 μM myxothiazol (myx) plus 1 μg/ml oligomycicn (olig) in the absence or in the presence of SOD + CAT (same concentrations as in panel (A)). ROS production probed by DCF is shown for a representative LSCM assay along with the statistical evaluation of the fluorescence intensity averaged ± S.E.M. from n= 3 for each condition. (C) Effect of externally added H2O2 on the intracellular ROS production. HK-2 cell were treated for 6 h with 20 μM H2O2 either alone or with 100 μM DPI. The intracellular H2O2 and intramitochondrial O2•− production was assessed by LSCM by DCF and MitoSox as described under Materials and Methods. Representative images of the LSCM analysis are show along with the statistical evaluation of the fluorescence intensity averaged ± S.E.M. from n= 3 for each condition. Enlargments showing subcellular compartmentalization of the DCF and MitoSox fluorescence are also shown for the H2O2-treated cells. Bars inside all the micrographs: 30 μm.
As proof of principle we induced a mitochondrial oxidative stress by incubating HK-2 with myxothiazol plus oligomycin a condition forcing the respiratory chain to release ROS [18] and tested on it the effect of extracellular anti-oxidant scavengers. Figure 4B shows that under this condition SOD and CAT were unable to prevent the endogenous mitochondrial ROS production. Moreover, we tested the effect of externally added sub-cytotoxic concentration of H2O2 to untreated HK-2 on peroxide and mito-chondrial superoxide production. As shown in Figure 4C, 6 hrs of incubation with 20 μM of the membrane permeant H2O2 resulted in enhanced generation of both peroxide and mitochondrial super-oxide which was fully prevented by inhibition of the respiratory chain complexes. This shows that the DCF fluorescent signal was not trivially due to diffusion into the cell of the externally added H2O2 and that the mitochondrial respiratory chain was the main target and source of the observed H2O2-induced ROS release.
To further provide evidence that mitochondrial ROS production was triggered by NOX activation, HK-2 cells were incubated for 6 hrs with nLDL and the ensuing enhanced DCF staining was image processed. The intracellular fluorescence was thresholded in order to eliminate the low-intensity cellular signal. Following this procedure a networked high-intensity fluorescence signal emerged that could be clearly attributable to the mitochondrial subcellular compartment (Fig. 5A). The co-incubation of nLDL-treated HK-2 cells with either apocynin or SOD or the membrane-permeant anti-oxidant Tempol resulted in a practically complete prevention of the mitochondria-linked DCF fluorescence signal when the images were processed under identical conditions. Interestingly, co-incubation of HK-2 with nLDL and catalase alone resulted in residual mitochondrial DCF signal over the basal level. This suggests that the extracellular superoxide was somehow more efficient than peroxide in mediating the ROS-induced ROS production.
Fig 5.
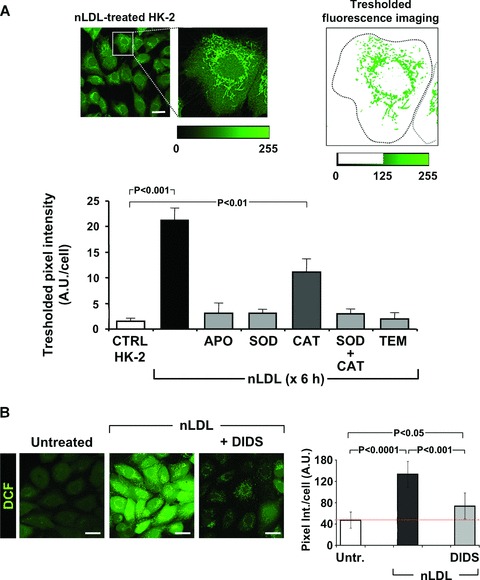
ROS generated by NOX(s) are the primary triggers of the ROS-dependent mitochondrial ROS production. (A) Processing of LSCM image. HK-2 cells were treated with nLDL for 6 h and stained for hydroperoxide detection with DCF as described in Figure 1B and Materials and Methods. An enlargement of a representative optical field pinpointing a selected cell is shown along with the pixel intensity scale (from 0 to 255 a.u.) of the green channel. The image on the right shows the result obtained removing the pixels with intensity values below 125. The thresholded pixel texture was quantitated dividing its overall intensity by the cell surface. This procedure was applied to process at least 80–100 single cells from different fields per each experimental condition. The histogram shows the effect of the co-incubation of nLDL with each of the followings: 100 μM apocynin, 500 U/ml SOD, 200 μg/ml CAT, 500 U/ml SOD + 200 μg/ml CAT, 5 mM Tempol. Each bar is the average of three independent experiments ± S.E.M.; where indicated the statistical significance is shown. (B) Effect of DIDS on the intracellular ROS production. HK-2 cells were treated for 24 hrs with 100 μg/ml of nLDL either alone and in the presence of 200 μM DIDS after that the intracellular H2O2 production was assesses by LSCM by DCF. Representative images are shown along with the statistical evaluation of the fluorescence intensity averaged ± S.E.M. from n= 3 for each condition. Bars inside all the micrographs: 30 μm.
A recent report showed that O2− can be transported by members of the chloride channel (ClC) [19]. As the renal proximal tubular cells express several members of the ClC family [20], we tested the effect of 4,4-diisothiocyanatostilbene-2,2-disulfonic acid (DIDS), a commonly used inhibitor of the ClCs [17]. Figure 5B shows that DIDS largely prevented the LDL-linked ROS production thus suggesting a role of ClCs in mediating the ROS-induced ROS release.
nLDL-Induced ROS production was dependent on extracellular Ca2+ and activation of cPLA2
Stimulation of the Ca2+-dependent cytoplasmic phospholipase A2 (cPLA2) with release of arachidonic acid (AA) is a process known to activate NOX [13, 21]. Therefore, we tested the effect of arachidonyl trifluoromethyl ketone (AACOCF3), an inhibitor of cPLA2, on the nLDL-induced ROS overproduction. Figure 6A clearly shows that AACOCF3 abrogated completely the DCF-fluorescence in nLDL-treated HK-2. Moreover, either chelation of the external Ca2+ by EGTA and treatment with the Ca2+-channel blocker vera-pamil caused, similarly, a depression of the nLDL-induced ROS production. Importantly AACOCF3 prevented the nLDL-dependent extra-cellular production of superoxide (Fig. 6B) supporting the occurrence of NOX activation. These results indicated that the pro-oxidant effect of nLDL on HK-2 was largely mediated by the reaction products of the cPLA2 whose activation was likely linked to stimulation of the inward current of external Ca2+ into the cell.
Fig 6.
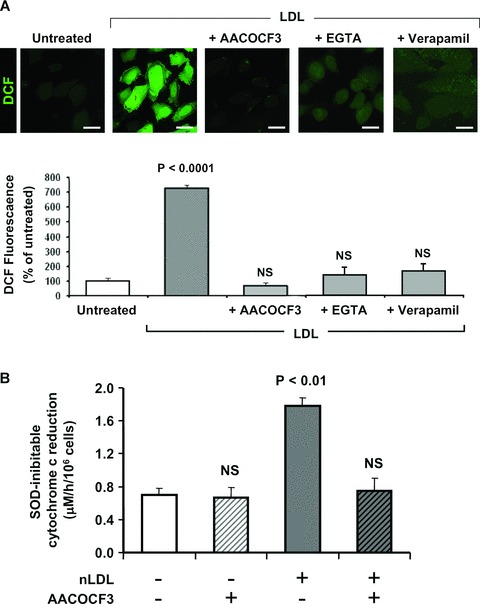
Extracellular Ca2+ mediates the nLDL-induced cPLA2-linked ROS production. (A) HK-2 cells were incubated for 24 hrs with 100 μg/ml nLDL either alone or in the presence of 20 μM AACOCF3 or 0.5 mM EGTA or 30 μM verapamil and after that assessed for intracellular H2O2 production by DCF. Representative images of the LSCM analysis are show along with the statistical evaluation of the fluorescence intensity averaged ± S.E.M. from n= 3 for each condition. Bars inside all the micrographs: 30 μm. (B) Effect of AACOCF3 on the nLDL-mediated activation of the NADPH oxidase. The SOD inhibitable cytochrome c reduction values are averages ± S.E.M. from n= 3 for each condition; the statistical differences versus untreated HK-2 cells is also indicated when significant.
nLDL Caused intramitochondrial Ca2+ accumulation
Mitochondria are known to provide a buffering power toward intracellular calcium rise [22]. Thus, we monitored the effect of nLDL on the intramitocondrial calcium (mtCa2+) level using the specific probe Rhod-1 [23]. Figure 7A shows that nLDL-treatment caused a significant increase of the Rhod1-linked fluorescence signal. The mtCa2+ load was linked to the collapse of the mtΔΨ. Indeed, the mitochondrial calcium uniporter inhibitor ruthenium red (RR) abrogated the nLDL-induced mitochondrial Ca2+ uptake and prevented at the same time the nLDL-induced mtΔΨ collapse (Fig. 7A). Dantrolene (an inhibitor of the ryanodine calcium channel) was ineffective whereas EGTA prevented both the mtCa2+ load and the mtΔΨ decrease in nLDL-treated HK-2 (Fig. 7B).
Fig 7.
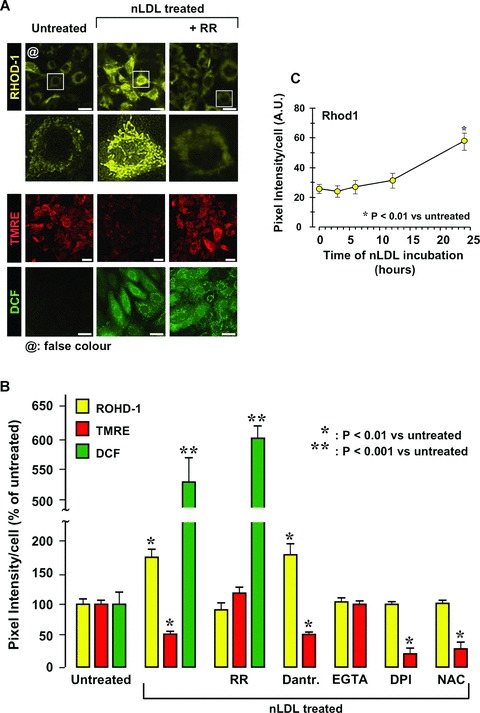
Treatment of HK-2 with nLDL causes increase of mitochondrial Ca2+ uptake. (A) Effect of nLDL on the intramito-chondrial level of Ca2+. HK-2 cells were treated for 24 hrs with 100 μg/ml of nLDL either alone or in combination with 5 μM ruthenium red (RR). After that the intramitochondrial Ca2+, mtΔΨ and H2O2 production were assessed by the specific probes Rhod-1, TMRE and DCF respectively as detailed under Materials and Methods. The panel shows a representative LSCM imaging. To note, in order to improve visually the data presented, the original red fluorescence of Rhod-1 was digitally rendered in yellow (by ImageJ 1.38×, NIH, USA, http://rsb.info.nih.gov/ij/) without altering the pixel intensity scale. Enlargments of single cell imaging for Rhod-1-probed of untreated and nLDL treated samples are also shown. (B) Quantitative analysis of the mtCa2+-, mtΔΨ- and H2O2-probe fluorescence intensities from LSCM imaging. In addition to the experimental setting shown in (A) the effect of coincu-bation of nLDL with 10 μM dantrolene, 0.5 mM EGTA, 10 mM DPI, 20 mM NAC is also reported for mtCa2+-and mtΔΨ-changes. Each bar represents the fluorescence intensity/cell averaged ± S.E.M. from at least n= 3. The statistical differences versus untreated HK-2 cells is also indicated when significant. (C) Time-dependence of the mtCa2+ changes as a function of the nLDL-incubation time. The Rhod-1-related fluorescence intensity for each time-point was measured as in (A, B) and refers to the average ± S.E.M. of n= 3. Bars inside all the micrographs: 30 μm.
These observations confirmed that the mtCa2+ load was mostly linked to the intracellular entry of external Ca2+ with negligible if any involvement of the intracellular ER calcium stores. Noteworthy RR, although impairing the calcium entry in the mitochondria, had no effect on the nLDL-induced ROS production (Fig. 7A,B). Consistent with this result is the observation that the nLDL-induced decrease of mtΔΨ was not prevented by DPI or NAC (Fig. 7B). Conversely, both DPI and NAC soppressed the LDL-induced mtCa2+ overload. Thus, the LDL-linked deregulations of the mtCa2+, ROS and mtΔΨ homeostasis interplayed by a complex modality and partly occurred independently rather than sequentially. This conclusion was further supported by the time-course of the nLDL-linked mtCa2+ changes showing that the mtCa2+ overload was significantly delayed (Fig. 7C) with respect to the mtΔΨ and ROS changes (Fig. 2C).
MtCa2+ overload is a condition known to open the mitochondrial permeability transition pore (MPTP) thereby collapsing mtΔΨ[22] and leading, in addition, to release of pro-apoptotic factors (such as cytochrome c) from the mitochondrial intermembrane space [24]. To test this possibility HK-2 were co-incubated with nLDL and cyclosporine A (CsA), an inhibitor of the MPTP. Figure 8 shows that CsA preserved efficiently the mtΔΨ without significant effect on the nLDL-induced mtCa2+ load. As proof of principle the re-equilibration of calcium gradients by the Ca2+-inonophore A23187 proved to prevent completely both the mtCa2+ and the mtΔΨ LDL-induced changes. Further it was shown that whilst untreated HK-2 presented, as expected, a punctuate appearance of the intramitochondrial cytochrome c-linked immunofluorescence, treatment with nLDL resulted in blurring of the fluorescence suggesting dilution of cytochrome c in the cytoplasm. Both CsA and A23187 preserved the brilliant punctuate appearance of the signal.
Fig 8.
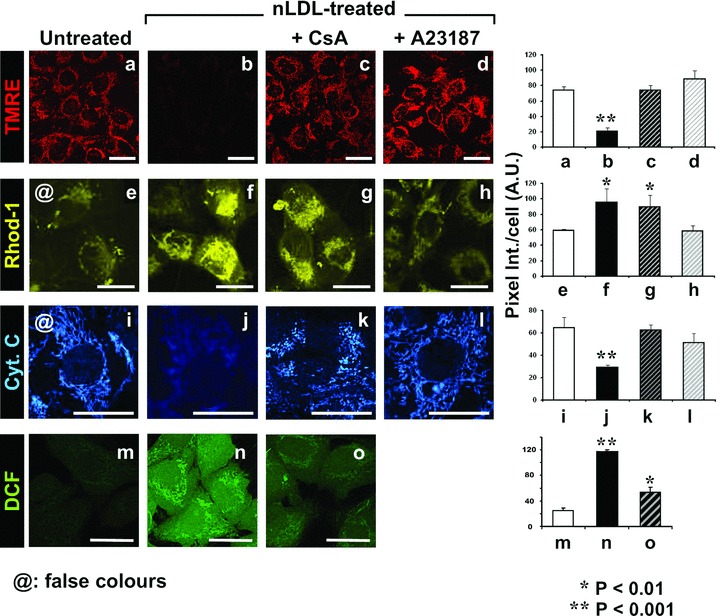
Treatment of HK-2 with nLDL induces opening of the mitochondrial PTP. HK-2 cells were treated for 24 hrs with 100 μg/ml of nLDL either alone or in combination with 1 μM cyclosporin A (CsA) or 5 μM of the Ca2+ ionophore A23187. After that the mtΔΨ, the intramitochondrial Ca2+ and the peroxide production were assessed by the specific probes TMRE, Rhod-1 and DCF respectively. In addition cytochrome c was detected by immunofluorescence as detailed under Materials and Methods. To improve visually the data presented, the original fluorescence of Rhod-1 and of the fluorescein isothiocyanate (FITC)-conjugated secondary mAb was digitally rendered in yellow and blue, respectively (by ImageJ 1.38×, NIH, USA, http://rsb.info.nih.gov/ij/) without altering the original pixel intensity scale. A representative LSCM imaging for each probe with a side-by-side statistical evaluation of the fluorescence intensity averaged ± S.E.M. from n= 3 for each condition is shown. Bars inside all the micrographs: 30 μm.
Next we tested the effect of MPTP blocking on the nLDL-dependent ROS production. Figure 8 shows that CsA-treatment resulted in a large reduction of the nLDL-induced DCF fluorescence. Similar results were obtained by using the more specific MPTP-blocker Debio 025 (data not shown).
Taken together these observations support the conclusion that the nLDL-linked collapse of mtΔΨ was due to calcium-induced MPTP opening.
Inhibition of cPLA2 prevents nLDL-linked mtCa2+ and mtΔΨ deregulation
Figure 9 shows that treatment of HK-2 with AACOCF3 resulted in complete prevention of the nLDL-induced mtΔΨ decrease and mtCa2+ load. In keeping with the reported effect of AACOCF3 in abrogating the nLDL-induced ROS formation (Fig. 6) this result suggested that the activation of cPLA2 and the formation of its reaction products were largely responsible of the observed mitochondrial dysfunction. However, the conflicting results of the effect of DPI- and NAC-treatment on the mtΔΨ and mtCa2+, in spite of their ability to fully prevent the oxidative unbalance, would indicate that the cPLA2-products exert multiple independent actions.
Fig 9.
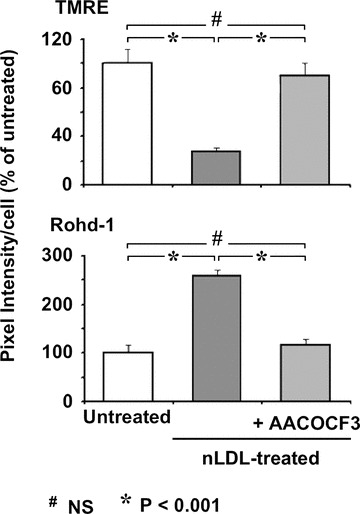
Inibition of cPLA2 causes abrogation of the nLDL-induced changes of mtΔΨ and mtCa2+. HK-2 cells were treated for 24 hrs with 100 μg/ml of nLDL either alone or in combination with 20 μM AACOCF3. After that the mtΔΨ and the intramitochondrial Ca2+ were assessed by the specific probes TMRE and Rhod-1, respectively, and assayed by LSCM as in Figure 7. The histograms represent the statistical evaluation of the fluorescence intensity averaged ± S.E.M. from n= 3 for each condition.
Discussion
Our primary finding in this study shows nLDL causing unbalance of the oxidation state in human proximal tubular cells. Oxidative modification of LDL components has long been considered a main factor promoting in the vasculature development of atherosclerotic lesions [3]. This led investigators to focus on oxLDL effects exerted on extra vascular tissue neglecting those of nLDL so much as to consider the lack of significant effects of nLDL as a negative control to emphasize the alterations caused by oxLDL. However in most of these reports the timing of nLDL in vitro treatment is too short to let the effects being detectable. Our results show, indeed, that enhanced ROS formation became reliable after 6 hrs of exposition of HK-2 to nLDL. Interestingly, in a comparative study evaluating the influence of native and hypochlorite-modified LDL on gene expression in HK-2 cells nLDL up-regulated the transcription of genes involved in ROS metabolism and cellular stress so as hypochlorite-oxidized LDL but following longer incubations [25].
In the attempt to characterize the mechanism underlying the nLDL-induced changes in the HK-2 cell redox homeostasis, a number of interconnected signalling pathways were found to be involved. The scheme drawn in Figure 10 convenes the evidence presented in this study with others reported in literature and provides a verifiable mechanicistic model of nLDL-induced cellular damage. The first step following the interaction of the nLDL with the HK-2 cells (Fig. 10, step 1) is the activation of an inward current of Ca2+ (Fig. 10, step 2). This interaction is most likely mediated by binding of nLDL to its specific receptor (LDL-R). HK-2 express several members of the LDL receptor family [26] as well as Ca2+ channel subtypes [27]. In addition to drive endocytotic LDL transportation the LDL-R is known to trigger activation of a number of adaptive signalling pathways [28]. Whatever is the mechanism of action, the nLDL-induced increase of the Ca2+ inward current is the earliest step required for all the subsequent events. Indeed chelation of external Ca2+ or inhibition of the L-type Ca2+ channel abolished completely the responsiveness of HK-2 to nLDL (Fig. 6A). Of note blockade of calcium influx through L-type calcium channel proved to attenuate apoptogenic insults occurring in hypoxic renal tubular cells [29].
Fig 10.
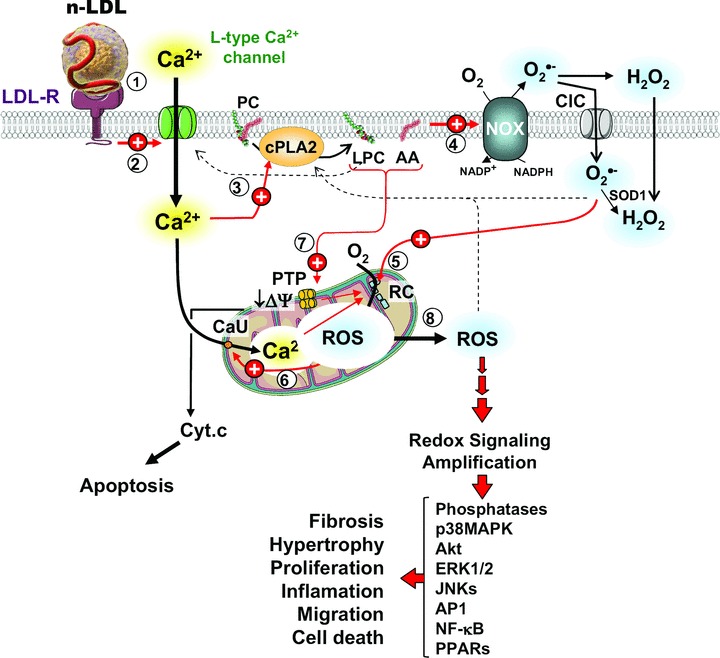
Schematic model of the effect of nLDL on HK-2. A sequence of events triggered by the interaction of nLDL with HK-2 and leading to alterations of the cell redox homeostasis is pictorially shown. The encircled numbers indicate the temporally cascade whereby different cell components are involved. Red arrows stand for stimulatory effects as suggested by the results presented in this study; dashed arrows stand for stimulatory effects reported in literature; black arrows indicate metabolite fluxes and enzymatic transformations. LDL-R, low density lipoprotein receptor; PC, phosphatidyl choline; cPLA2, Cytosolic phospholipase A2; LPC, lysophosphatidyl choline; AA, arachidonic acid; NOX, NADPH oxidase; ClC, chloride channel; SOD1, superoxide dismutase; PTP, mitochondrial permeability transition pore; RC, mitochondrial respiratory chain; CaU, calcium uniporter. See Discussion for explanation.
The enhanced nLDL-induced Ca2+ entry into the cell activates the cytoplasmic Ca2+-sensitive isoform of the phospholypase cPLA2 (Fig. 10, step 3). The central role of cPLA2 in mediating the nLDL-linked cellular alterations is directly demonstrated by the preventing effect of its specific inhibitor AACOCF3 (Figs. 6 and 9). cPLA2 hydrolizes the C-2 ester bond of phospholipids with high affinity to phosphatidylcholine (PC) containing arachidonic acid (AA) bound to C-2 thereby releasing lisophosphatidylcholine (LPC) and the free fatty acid, both with a documented signal transducing activity [30]. AA is a powerful activator of the plasma-membrane NOX(s) [31] and in this study we clearly show the involvement of NOX in the nLDL-linked oxidative imbalance (Fig. 10, step 4). Indeed, specific NOX inhibitors or externally added ROS-scavenging enzyme prevented completely the nLDL-dependent cell redox alterations (Figs. 3, 4A). Isoforms of the macrophagic NOX, unrelated to the host defence, exert regulatory functions by production of ROS acting as signalling molecules [13, 17]. A number of cell functions turns out to be regulated by redox sensitive signalling pathways and their molecular determinants are being now partly disclosing [32, 33].
Besides NOXs, the mitochondrial respiratory chain constitutes the main source of ROS in the cell [18, 34]. An ever-growing mass of evidence highlights the involvement of dysfunctioning mitochondria in the onset/development of many human pathologies [35] including atherosclerosis [36]. Of interest, specific blockade of mitochondrial ATP production was reported to promote proximal tubular cholesterol loading [37].
In this study we provide hints suggesting that NOX-generated ROS may stimulate further ROS production by mitochondria (Fig. 10, step 5). This is proved by the complete ablation of the intracellular mitochondrial ROS production following NOX-inhibition or scavenging extracellular ROS (Fig. 5A). It has been suggested that a cross-talk between NOX activation and the mitochondrial respiratory chain occurs whereby extracellular ROS can function as signalling molecules by a paracrine/autocrine mechanism (Fig. 4C). The effect of DIDS, an inhibitor of the plasma membrane chloride channel, would suggest that ROS-induced ROS production is partly mediated by transmembrane transport of the superoxide anion (Fig. 5B). Once into the cell, O2− and H2O2 trigger ROS production by mitochondria. We cannot at the moment detail whether the activation is exerted directly by ROS or mediated by oxidized molecules acquiring signalling features neither the specific mitochondrial target. However, some clues are offered in the literature to get insights into the mechanism. Indeed reported propagation of an oxidative wave in the mitochondrial network was shown to be sensitive to inhibitors of either the MPTP or the inner mitochondrial anion channel [38–40]. It is worth mentioning that evidence indicating the occurrence of a cross-talk between mitochondria and NOXs have been recently reported [41, 42]. However, in those studies mitochondria appear to be upstream of a signalling path leading to enhanced expression and activation of NOX. Thus, our finding highlights a further different modality of cross-talk in the signalling axe between the two major ROS-generating sources in the cell. The nLDL-linked enhanced mito-chondrial ROS production induces further changes. Of note, DPI or NAC prevented the nLDL-induced mtCa2+ load but not the mtΔΨ decrease whereas blockade of the mtCa2+ uniporter by RR largely preserved the LDL-induced mtΔΨ collapse but was ineffective on the ROS over-production (Fig. 7). These puzzling observations can be explained assuming the occurrence of distinct pathways converging on the same target. In particular, it is proposed that ROS can favour the mtCa2+ overload by activating the Ca2+ uniport (Fig. 10, step 6) as shown for other redox modulated Ca2+-transporting systems [43]. This would place the activation of the inward mtCa2+ transport down-stream of the redox sig-nalling pathway explaining the effects of anti-oxidants and RR. The nLDL-induced collapse of the mtΔΨ seems to be dependent on both the mtCa2+ level and on products of the cPLA2 catalysis. Indeed both the AACOCF3-mediated inhibition of the cPLA2 and the RR-mediated suppression of the mitochondrial Ca2+ uptake preserved the nLDL-linked decrease of the mtΔΨ (Figs. 6, 9). The effect of CsA in preventing the nLDL-induced mtΔΨ- collapse as well as ROS production (Fig. 8) would indicate the causative involvement of the MPTP (Fig. 10, step 7).
In this study we observed that collapse of the mtΔΨ although fully prevented by RR-treatment precedes mt-Ca2+ accumulation (cf. Fig. 2CversusFig. 7C) implying that the two events are apparently distinct and that mitochondrial depolarization may occur independently of mt-Ca2+ uptake. Intramitochondrial Ca2+ overload is a key inducer of MPTP opening leading to loss of mtΔΨ[22]. However, it must be pointed out that activation of the MPTP by mt-Ca2+ has been described as occurring first by enhancing the intrinsic flickering of the pore [44]. The transient opening of the pore causes a rapid ionic re-equilibration between the intra- and extra-mitochondrial milieu leading to partial collapse of the mtΔΨ as well as to mt-Ca2+ exit. Therefore, mtΔΨ lowering would not necessarily require a high steady-state mt-Ca2+. At high concentration of mt-Ca2+ the MPTP is permanently opened and the mtΔΨ fully released. Another possibility is that the mtΔΨ decrease is due to accumulation of the cPLA2 products. Indeed a large body of evidence reported in the literature shows that LPC and AA are all inducers of the MPTP opening [45–47]. In addition, AA has been reported to inhibit the OXPHOS acting as mild uncoupler as well as inhibitor of the ETC [48] (consistent with the respirometric results reported in Fig. 2B). Of course all the aforementioned mechanisms are not mutually exclusive but may act at once.
A further point emerged from this study is the apparent irreversibility of the nLDL induced oxidative unbalance. Indeed removal of nLDL after 6–12 hrs of incubation (under our experimental protocol) did not result in resetting of the original cellular redox state but instead induced persistent alterations. This observation would suggest the occurrence of a forward feed back mechanism enabling self-maintenance of the oxidative insult once a critical threshold of effectors has been reached. The ROS-induced ROS production here proposed may well account for such a positive loop. Indeed release of ROS, in the form of the freely diffusible H2O2, from a mitochondrial subset could solicit ROS generation in other mitochondrial clusters thus propagating and self-maintaining the oxidative insult. It is worth mentioning that ROS signalling has been implicated in the activation of the plasma membrane L-type Ca2+ channels, the cPLA2 and the MPTP [38, 49–51]. On the other hand, mtCa2+ enhancement, release of lyso-PC and AA, opening of the MPTP are all conditions proved to cause mitochondrial ROS generation [22, 52, 53]. In addition, lyso-PC and AA were shown to activate directly the L-type Ca2+ channel [54, 55]. All these observations are fully consistent with a self-fuelling mechanism.
The here-reported nLDL-induced alterations in HK-2 were largely reproducible in a different cell phenotype (i.e. podocytes; C. Piccoli et al., unpublished data). This observation, in addition to its clinical relevance with respect to the role of hyperlipidemia in the progression of the CKD [7], would argue that the here-reported nLDL-induced alteration might be of more general interest. Progressive chronic renal disease of all types is characterized by tubular (atrophy, hypertrophy, hyperplasia) and interstitial (inflammatory cell infiltration, fibrosis) pathological changes, the severity of which correlates well with the degree of proteinuria and the decline in glomerular filtration. In response to proteinuria and additional factors [4–10], tubular cells are activated to produce a large number of chemoattractants, proinflammatory and profi-brotic cytokines and matrix proteins, which cause, at least in part, interstitial inflammation and fibrosis [8–10]. The clinical relevance of our observations results evident in keeping with the potential role of redox signalling in activating pro-inflamatory and fibro-genic pathways (Fig. 10, step 8) [8–10]. Dyslipidemia is a well-established factor contributing to exacerbate chronic renal disease independently of the priming noxious glomerular events [4–7]. This notion has led to the successful utilization of statin-based therapy to slow down or even halt the progression of CKD [56]. It is worth noting that statins, in addition to their hypocholes-terolemic capacity, exhibit additional pleiotropic effects including modulation of inflammatory processes. The anti-inflammatory actions of statins likely arise from their ability to prevent the synthesis of isoprenoid intermediates that are responsible for the post-translational-isoprenylation on the C-terminus of a variety of proteins including small GTPases such as Rac [57]. Because Rac-1/2 are critically involved in the activation of NOXs [14], it is tempting to speculate that the efficacy of statins may partly result from impairment of plasma membrane-translocation of Rac-1 necessary for NOX activation.
In conclusion the interplay between changes in the intracellular redox ‘tone’ and the proinflamatory and profibrogenic cytokines- and growth factors-mediated stimuli offers a mechanicistic basis to understand the role and effect of dyslipi-demia in the renal chronic disease. The nLDL-related activation of the ROS generating network disclosed in this study suggests novel pharmacological targets to be potentially exploited in the development of combined therapeutic strategies.
Acknowledgments
The work was supported by University of Foggia (Local Research Funds 2006–2008); Fondazione Banca del Monte Domenico Siniscalco Ceci-Foggia-Italy; Telethon-Italy (GGP07132 to L.C.). The authors have no financial or other conflict of interest related to this study.
Supporting Information
Characterization of the oxidation state of isolatedLDL. (A) Electrophoretic shift assay. A comparativeelectrophoretic mobility of native LDL, Cu2+-oxidizedLDL and following exposure to HK-2 cells is shown. Paired gels werestained to detect either proteins (Brilliant Blu G) or lipids(Oil-O-Red) as described in the supplementary material.(B) Differential UV spectra of HK-2-exposed LDL. IsolatedLDLs were suspended in DMEM at the concentration of 100 μg/mland added either to an empty culturing dish or to a dish platedwith HK-2 cell. After 24 hrs of incubation equal volumes from theculturing media were withdrawn from the two dishes and spectrallyscanned. The spectra shown was obtained subtracting the absorbanceof the LDL suspended in the sole medium from that of the LDL whichwere exposed to the HK-2 cells. Identical results were obtained atintermediate interval of incubation (3, 6, 12 h; not shown). Forcomparison the spectra of Cu2+-oxidized LDL is alsoshown with its second derivative spectra indicating the spectralfeatures of the peroxidation-linked dienes formed.
Effect of nLDL and oxLDL treatment on HK-2 cell lineviability. (A) Phase contrast micrographs of HK-2treated for 24 hrs with 100 μg/ml of either nLDL or oxLDL.(B) Cell-death assay. HK-2 cells treated with 100 μg/mlof either nLDL (24 hrs) or oxLDL (6 h) were tested by the AnnexinV/carboxy-fluorescein assay as indicated in the supplementarymaterial. The upper panel shows a representative confocalmicroscopy imaging with the red and green fluorescence indicatingmembrane binding of Annexin V and cell loading/maintenance ofcarboxy-fluorescein respectively. As compared to control HK-2 cells(CTRL) nLDL-treated cells resulted to be all CF positive but withan enhanced number of Annexin V-positive cells (pinpointed byarrows) indicative of early apoptosis. OxLDL-treatment resulted inCF negative and Annexin V-positive cells indicative of lateapoptosis or necrosis. The histogram shows the quantitativeanalysis expressed as fluorescence ratio of the two probes of atleast 100 cells for each conditions selected from three separateexperiments. The bars indicate the mean values (±SEM);*P < 0.01 versus CTRL, **P < 0.001versus CTRL, #P < 0.01 versus nLDL-treated.All the bars inside the micrographs are 30 μm.
Quantitative RT-PCR analysis of antioxidantenzymes. The quantification of transcripts abundance in treatedHK-2 cells (100 μg/ml of LDL for 24 hrs) was determinedperforming with the °Ct method, the untreated HK-2 cells beingthe reference sample and β-actin the internal control. SOD1and SOD2, superoxide dismutase isoform 1 and 2, respectively; CAT,catalase; GPX1 and GPX2, gluthatione peroxidase isoforms 1 and 4,respectively. The bars are means ± S.E.M. of three independent measurements. See supplementary material for primers used, PCR conditions and further details.
NOX expression in HK-2. (A) RT-PCRanalysis of NOX iso-form expression. The agarose gel shows theamplicon level of the catalytic subunits of the NADPH oxidaseisoforms NOX 1, 2, 4, 5 together with the splicing variant NOX 2s.MK, markers. See supplementary material for primers used, PCRconditions and further details. (B) Differentialspectrophotometric analysis of NOX-related cytochrome b. Thespectrum of Na2S2O4-treated minusuntreated cell lisates was first recorded by a split beamspec-trophotometer, then 20 mM of t-BICN was added to the reducedsample (measure cuvette) and a second spectra recorded. Theresulting ΔAbs spectrum, diagnostic for the presence ofNOX-linked cytochrome b, and the calculated amount of NOX is shownon the right. See supplementary material for further details.
References
- 1.Kannel WB, Gordon T, Castelli WP. Role of lipid and lipoprotein fractions in atherogenesis: the Framingham Study. Prog Lipid Res. 1981;20:339–48. doi: 10.1016/0163-7827(81)90067-9. [DOI] [PubMed] [Google Scholar]
- 2.Steinberg D, Parthasarathy S, Carew TE, et al. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N Engl J Med. 1989;320:915–24. doi: 10.1056/NEJM198904063201407. [DOI] [PubMed] [Google Scholar]
- 3.Berliner JA, Heinecke JW. The role of oxidized lipoproteins in atherogenesis. Free Radic Biol Med. 1996;20:707–27. doi: 10.1016/0891-5849(95)02173-6. [DOI] [PubMed] [Google Scholar]
- 4.Lee HS, Song CH. Oxidized Low-Density Lipoprotein and Oxidative Stress in the Development of Glomerulosclerosis. Am J Nephrol. 2009;29:62–70. doi: 10.1159/000151277. [DOI] [PubMed] [Google Scholar]
- 5.Saland JM, Ginsberg HN. Lipoprotein metabolism in chronic renal insufficiency. Pediatr Nephrol. 2007;22:1095–112. doi: 10.1007/s00467-007-0467-5. [DOI] [PubMed] [Google Scholar]
- 6.Fogo AB. Mechanisms of progression of chronic kidney disease. Pediatr Nephrol. 2007;22:2011–22. doi: 10.1007/s00467-007-0524-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cases A, Coll E. Dyslipidemia and the progression of renal disease in chronic renal failure patients. Kidney Int Suppl. 2005;99:S87–S93. doi: 10.1111/j.1523-1755.2005.09916.x. [DOI] [PubMed] [Google Scholar]
- 8.Kairaitis LK, Harris DCH. Tubularinterstitial interactions in proteinuric renal diseases. Nephrology. 2001;6:198–207. [Google Scholar]
- 9.Wardle EN. Cellular oxidative processes in relation to renal disease. Am J Nephrol. 2005;25:13–22. doi: 10.1159/000083477. [DOI] [PubMed] [Google Scholar]
- 10.Nistala R, Whaley-Connell A, Sowers R. Redox control of renal function and hypertension. Antioxid Redox Signal. 2008;10:2047–89. doi: 10.1089/ars.2008.2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robinson KM, Janes MS, Beckman JS. The selective detection of mitochondrial superoxide by live cell imaging. Nat Protoc. 2008;3:941–7. doi: 10.1038/nprot.2008.56. [DOI] [PubMed] [Google Scholar]
- 12.Brown GC. Control of respiration and ATP synthesis in mammalian mitochondria and cells. Biochem J. 1992;284:1–13. doi: 10.1042/bj2840001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sumimoto H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J. 2008;275:3249–77. doi: 10.1111/j.1742-4658.2008.06488.x. [DOI] [PubMed] [Google Scholar]
- 14.Cave AC, Brewer AC, Narayanapanicker A, et al. NADPH oxidases in cardiovascular health and disease. Antioxid Redox Signal. 2006;8:691–728. doi: 10.1089/ars.2006.8.691. [DOI] [PubMed] [Google Scholar]
- 15.Li Y, Trush MA. Diphenyleneiodonium, an NAD(P)H oxidase inhibitor, also potently inhibits mitochondrial reactive oxygen species production. Biochem Biophys Res Commun. 1998;253:295–9. doi: 10.1006/bbrc.1998.9729. [DOI] [PubMed] [Google Scholar]
- 16.Johnson DK, Schillinger KJ, Kwait DM, et al. Inhibition of NADPH oxidase activation in endothelial cells by ortho-methoxy-substituted catechols. Endothelium. 2002;9:191–203. doi: 10.1080/10623320213638. [DOI] [PubMed] [Google Scholar]
- 17.Gill PS, Wilcox CS. NADPH oxidases in the kidney. Antioxid Redox Signal. 2006;8:1597–07. doi: 10.1089/ars.2006.8.1597. [DOI] [PubMed] [Google Scholar]
- 18.Adam-Vizi V, Chinopoulos C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol Sci. 2006;27:639–45. doi: 10.1016/j.tips.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 19.Hawkins BJ, Madesh M, Kirkpatrick CJ, et al. Superoxide flux in endothelial cells via the chloride channel-3 mediates intracellular signaling. Mol Biol Cell. 2007;18:2002–12. doi: 10.1091/mbc.E06-09-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Uchida S, Sasaki S. Function of chloride channels in the kidney. Annu Rev Physiol. 2005;67:759–78. doi: 10.1146/annurev.physiol.67.032003.153547. [DOI] [PubMed] [Google Scholar]
- 21.Levy R. The role of cytosolic phospholipase A2-alfa in regulation of phagocytic functions. Biochim Biophys Acta. 2006;1761:1323–34. doi: 10.1016/j.bbalip.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 22.Rimessi A, Giorgi C, Pinton P, et al. The versatility of mitochondrial calcium signals: from stimulation of cell metabolism to induction of cell death. Biochim Biophys Acta. 2008;1777:808–16. doi: 10.1016/j.bbabio.2008.05.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerencsér AA, Adam-Vizi V. Selective, high-resolution fluorescence imaging of mitochondrial Ca2+ concentration. Cell Calcium. 2001;30:311–21. doi: 10.1054/ceca.2001.0238. [DOI] [PubMed] [Google Scholar]
- 24.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 25.Porubsky S, Schmid H, Bonrouhi M, et al. Influence of native and hypochlorite-modified low-density lipoprotein on gene expression in human proximal tubular epithelium. Am J Pathol. 2004;164:2175–87. doi: 10.1016/S0002-9440(10)63775-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zager RA, Johnson ACM, Hanson SY, et al. Acute tubular injury causes dysreg-ulation of cellular cholesterol transport proteins. Am J Pathol. 2003;163:313–20. doi: 10.1016/S0002-9440(10)63655-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hayashi K, Wakino S, Sugano N, et al. Ca2+ channel subtypes and pharmacology in the kidney. Circ Res. 2007;100:342–53. doi: 10.1161/01.RES.0000256155.31133.49. [DOI] [PubMed] [Google Scholar]
- 28.Kathryn JM, Freeman MW. Scavenger receptors in atherosclerosis beyond lipid uptake. Arterioscler Thromb Vasc Biol. 2006;26:1702–11. doi: 10.1161/01.ATV.0000229218.97976.43. [DOI] [PubMed] [Google Scholar]
- 29.Tanaka T, Nangaku M, Miyata T, et al. Blockade of Calcium Influx through L-Type Calcium Channels Attenuates Mitochondrial Injury and Apoptosis in Hypoxic Renal Tubular Cells. J Am Soc Nephrol. 2004;15:2320–33. doi: 10.1097/01.ASN.0000138287.46849.82. [DOI] [PubMed] [Google Scholar]
- 30.Hirabayashi T, Murayama T, Shimizu T. Regulatory mechanism and physiological role of cytosolic phospholipase A2. Biol Pharm Bull. 2004;27:1168–73. doi: 10.1248/bpb.27.1168. [DOI] [PubMed] [Google Scholar]
- 31.Mankelow TJ, Pessach E, Levy R, et al. The requirement of cytosolic phospholipase A2 for the PMA activation of proton efflux through the N-terminal 230-amino acid fragment of gp91phox. Biochem J. 2003;374:315–9. doi: 10.1042/BJ20030495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol. 2003;15:247–54. doi: 10.1016/s0955-0674(03)00002-4. [DOI] [PubMed] [Google Scholar]
- 33.Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol. 2008;4:278–86. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 34.Naqui A, Chance B, Cadenas E. Reactive oxygen intermediates in biochemistry. Annu Rev Biochem. 1986;55:137–66. doi: 10.1146/annurev.bi.55.070186.001033. [DOI] [PubMed] [Google Scholar]
- 35.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Madamanchi NR, Runge MS. Mitochondrial dysfunction in atherosclerosis. Circ Res. 2007;100:460–73. doi: 10.1161/01.RES.0000258450.44413.96. [DOI] [PubMed] [Google Scholar]
- 37.Zager RA, Johnson AC, Hanson SY. Proximal tubular cholesterol loading after mitochondrial, but not glycolytic, blockade. Am J Physiol Renal Physiol. 2003;285:F1092–9. doi: 10.1152/ajprenal.00187.2003. [DOI] [PubMed] [Google Scholar]
- 38.Zorov DB, Filburn CR, Klotz LO, et al. Reactive oxygen species (ROS)- induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:1001–14. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brady NR, Hamacher-Brady A, Westerhoff HV, Gottlieb RA. A wave of reactive oxygen species (ROS)-induced ROS release in a sea of excitable mitochondria. Antioxid Redox Signal. 2006;8:1651–65. doi: 10.1089/ars.2006.8.1651. [DOI] [PubMed] [Google Scholar]
- 40.Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta. 2006;1757:509–17. doi: 10.1016/j.bbabio.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 41.Rathore R, Zheng YM, Niu CF, et al. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCepsilon signaling axis in pulmonary artery smooth muscle cells. Free Radic Biol Med. 2008;45:1223–31. doi: 10.1016/j.freeradbiomed.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wosniak JJ, Santos CX, Kowaltowski AJ, et al. Cross-talk between mitochondria and NADPH oxidase: Effects of mild mitochondrial dysfunction on angiotensin II mediated increase in Nox isoform expression and activity in vascular smooth muscle cells. Antioxid Redox Signal. 2009;11:1265–78. doi: 10.1089/ars.2009.2392. [DOI] [PubMed] [Google Scholar]
- 43.Waring P. Redox active calcium ion channels and cell death. Arch Biochem Biophys. 2005;434:33–42. doi: 10.1016/j.abb.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 44.Huser J, Blatter LA. Fluctuation in mitochondrial membrane potential caused by repetitive gating of the permeability transition pore. Biochem J. 1999;343:311–17. [PMC free article] [PubMed] [Google Scholar]
- 45.Rustenbeck I, Münster W, Lenzen S. Relation between accumulation of phos-pholipase A2 reaction products and Ca2+ release in isolated liver mitochondria. Biochim Biophys Acta. 1996;1304:129–38. doi: 10.1016/s0005-2760(96)00113-0. [DOI] [PubMed] [Google Scholar]
- 46.Penzo D, Petronilli V, Angelin A, et al. Arachidonic acid released by phospholi-pase A(2) activation triggers Ca(2+)-dependent apoptosis through the mitochondrial pathway. J Biol Chem. 2004;279:25219–25. doi: 10.1074/jbc.M310381200. [DOI] [PubMed] [Google Scholar]
- 47.Di Paola M, Zaccagnino P, Oliveros-Celis C, et al. Arachidonic acid induces specific membrane permeability increase in heart mitochondria. FEBS Lett. 2006;580:775–81. doi: 10.1016/j.febslet.2005.12.090. [DOI] [PubMed] [Google Scholar]
- 48.Cocco T, Di Paola M, Papa S, et al. Arachidonic acid interaction with the mito-chondrial electron transfer chain promotes reactive oxygen species generation. Free Radic Biol Med. 1999;27:51–9. doi: 10.1016/s0891-5849(99)00034-9. [DOI] [PubMed] [Google Scholar]
- 49.Fearon IM. OxLDL enhances l-type Ca2+ currents via lysophosphatidylcholineinduced mitochondrial reactive oxygen species (ROS) production. Cardiovasc Res. 2006;69:855–64. doi: 10.1016/j.cardiores.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 50.Cui XL, Ding Y, Alexander LD, et al. Oxidative signaling in renal epithelium: Critical role of cytosolic phospholipase A2 and p38(SAPK) Free Radic Biol Med. 2006;41:213–21. doi: 10.1016/j.freeradbiomed.2006.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kowaltowski AJ, Castilho RF, Vercesi AE. Mitochondrial permeability transition and oxidative stress. FEBS Lett. 2001;495:12–5. doi: 10.1016/s0014-5793(01)02316-x. [DOI] [PubMed] [Google Scholar]
- 52.Watanabe N, Zmijewski JW, Takabe W, et al. Activation of Mitogen-Activated Protein Kinases by LysophosphatidylcholineInduced Mitochondrial Reactive Oxygen Species Generation in Endothelial Cells. Am J Pathol. 2006;168:1737–48. doi: 10.2353/ajpath.2006.050648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang W, Fang H, Groom L, et al. Superoxide flashes in single mitochondria. Cell. 2008;134:279–90. doi: 10.1016/j.cell.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li XH, Wu YJ. Characteristics of lysophosphatidylcholine-induced Ca2+ response in human neuroblastoma SH-SY5Y cells. Life Sci. 2007;80:886–92. doi: 10.1016/j.lfs.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 55.Liu L, Barrett CF, Rittenhouse AR. Arachidonic acid both inhibits and enhances whole cell calcium currents in rat sympathetic neurons. Am J Physiol Cell Physiol. 2001;280:1293–305. doi: 10.1152/ajpcell.2001.280.5.C1293. [DOI] [PubMed] [Google Scholar]
- 56.Fried LF. Effects of HMG-CoA reductase inhibitors (statins) on progression of kidney disease. Kidney Int. 2008;74:571–6. doi: 10.1038/ki.2008.231. [DOI] [PubMed] [Google Scholar]
- 57.Wang CY, Liu PY, Liao JK. Pleiotropic effects of statin therapy: molecular mechanisms and clinical results. Trends Mol Med. 2008;14:37–44. doi: 10.1016/j.molmed.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Characterization of the oxidation state of isolatedLDL. (A) Electrophoretic shift assay. A comparativeelectrophoretic mobility of native LDL, Cu2+-oxidizedLDL and following exposure to HK-2 cells is shown. Paired gels werestained to detect either proteins (Brilliant Blu G) or lipids(Oil-O-Red) as described in the supplementary material.(B) Differential UV spectra of HK-2-exposed LDL. IsolatedLDLs were suspended in DMEM at the concentration of 100 μg/mland added either to an empty culturing dish or to a dish platedwith HK-2 cell. After 24 hrs of incubation equal volumes from theculturing media were withdrawn from the two dishes and spectrallyscanned. The spectra shown was obtained subtracting the absorbanceof the LDL suspended in the sole medium from that of the LDL whichwere exposed to the HK-2 cells. Identical results were obtained atintermediate interval of incubation (3, 6, 12 h; not shown). Forcomparison the spectra of Cu2+-oxidized LDL is alsoshown with its second derivative spectra indicating the spectralfeatures of the peroxidation-linked dienes formed.
Effect of nLDL and oxLDL treatment on HK-2 cell lineviability. (A) Phase contrast micrographs of HK-2treated for 24 hrs with 100 μg/ml of either nLDL or oxLDL.(B) Cell-death assay. HK-2 cells treated with 100 μg/mlof either nLDL (24 hrs) or oxLDL (6 h) were tested by the AnnexinV/carboxy-fluorescein assay as indicated in the supplementarymaterial. The upper panel shows a representative confocalmicroscopy imaging with the red and green fluorescence indicatingmembrane binding of Annexin V and cell loading/maintenance ofcarboxy-fluorescein respectively. As compared to control HK-2 cells(CTRL) nLDL-treated cells resulted to be all CF positive but withan enhanced number of Annexin V-positive cells (pinpointed byarrows) indicative of early apoptosis. OxLDL-treatment resulted inCF negative and Annexin V-positive cells indicative of lateapoptosis or necrosis. The histogram shows the quantitativeanalysis expressed as fluorescence ratio of the two probes of atleast 100 cells for each conditions selected from three separateexperiments. The bars indicate the mean values (±SEM);*P < 0.01 versus CTRL, **P < 0.001versus CTRL, #P < 0.01 versus nLDL-treated.All the bars inside the micrographs are 30 μm.
Quantitative RT-PCR analysis of antioxidantenzymes. The quantification of transcripts abundance in treatedHK-2 cells (100 μg/ml of LDL for 24 hrs) was determinedperforming with the °Ct method, the untreated HK-2 cells beingthe reference sample and β-actin the internal control. SOD1and SOD2, superoxide dismutase isoform 1 and 2, respectively; CAT,catalase; GPX1 and GPX2, gluthatione peroxidase isoforms 1 and 4,respectively. The bars are means ± S.E.M. of three independent measurements. See supplementary material for primers used, PCR conditions and further details.
NOX expression in HK-2. (A) RT-PCRanalysis of NOX iso-form expression. The agarose gel shows theamplicon level of the catalytic subunits of the NADPH oxidaseisoforms NOX 1, 2, 4, 5 together with the splicing variant NOX 2s.MK, markers. See supplementary material for primers used, PCRconditions and further details. (B) Differentialspectrophotometric analysis of NOX-related cytochrome b. Thespectrum of Na2S2O4-treated minusuntreated cell lisates was first recorded by a split beamspec-trophotometer, then 20 mM of t-BICN was added to the reducedsample (measure cuvette) and a second spectra recorded. Theresulting ΔAbs spectrum, diagnostic for the presence ofNOX-linked cytochrome b, and the calculated amount of NOX is shownon the right. See supplementary material for further details.