

Abstract
The extracellular matrix plays a number of important roles, among them providing structural support and information to cellular structures such as blood vessels imbedded within it. As more complex organisms have evolved, the matrix ability to direct signalling towards the vasculature and remodel in response to signalling from the vasculature has assumed progressively greater importance. This review will focus on the molecules of the extracellular matrix, specifically relating to vessel formation and their ability to signal to the surrounding cells to initiate or terminate processes involved in blood vessel formation.
Keywords: extracellular matrix, blood vessel formation, angiogenesis
Introduction
Although originally articulated for the purposes of function’ also applies in a biological context. architecture theory [1], the adage of ‘form follows Examples of this concept are replete throughout biology (perhaps more accurately stated as ‘function follows form’) as natural selection hones the ‘forms’ (molecules, tissues, etc.) to better target the ‘functions’ being selected.The extracellular matrix (ECM) exemplifies this process from its initially discovered properties of providing a structural environment or scaffold for particular groups of cells to assemble into tissues. Later, research has uncovered that not only does the ECM provide a platform for these tissues, the composition of the ECM manifests as specific forms, which serve to further guide the production and assembly of specific tissues. Via receptors on the cells interacting with the ECM, a feedback-signalling process between the cells and the ECM takes place, which guides the developmental process of the tissue and, even after its maturity, allows tissues to respond to various environmental stimuli.
Phylogenetic data generated from relatively recent completion of genome sequencing projects have shown that the molecules of the ECM, especially ones related to cell–matrix adhesion, are ‘ancient and exquisitely conserved in multicellular animals’[2]. Even Coelenterates have basement membranes as well as more complex animals. Recent work involving the sequencing of other metazoans has revealed that the majority of ECM genes have evolved through duplication of pre-existing genes present in our vertebrate ancestors [3]. Thus, ECM genes are over-represented in the vertebrate genome giving ample grist for the evolutionary mill.
Control of blood vessel formation is illustrative of the signalling process between the ECM and cells. Recent work, which will be discussed in the review, demonstrating how the ECM is involved in regulating developmental and well as adult blood vessel vascularization programs will be the focus. The first part of the review will familiarize the reader with the major players in the ECM. In the second part, the focus will be on the signalling aspects of the ECM and what has been discovered concerning how those molecules signal to cells to mediate vascularization.
ECM: composition and structure
Like the cosmos, the tissues of the human body are composed of mostly (extracellular) space …and cells. The molecules secreted by the cells occupying these tissues interact to create a complex network, which constitutes the ECM. As there are many different tissues in the body, so are there different organizations of cells and matrices. Examples exist from where the proportions vary from mostly matrix to a few cells (connective tissue) and others where the cells are tightly packed together and the matrix is merely a thin sheet on which they rest (epithelia). Also, the composition of macromolecules gives rise to various forms of tissue. Calcification of the matrix occurs in bones and teeth for production of very dense materials while in tendons these structures are organized into elastic chords for tensile strength. What is it about the molecules of the ECM and their properties which allow for such varied properties?
Broadly speaking, two classes of molecules are produced by the ECM: fibrous proteins and glycosaminoglycans (GAGs). The fibrous proteins are mainly collagen, laminin and elastin. These fibrous proteins, which participate both as a scaffold and in adhesion in matrix structures, are embedded in GAGs. The chemical properties of GAGs enable them to be highly hydrated giving them gelatinous properties or making them what has been termed a ‘ground substance’. This substance imparts a physiological function by facilitating resistance of compressive forces while also allowing diffusion of vital nutrients between the blood and tissue. Further discussion of the structure and properties of the fibrous proteins and GAGs in the ECM is continued by molecule below. A thorough discussion of all the molecules would fill a book; therefore, brief descriptions will be given, which are pertinent to the role ECM in neovascularization, and reviews will be cited for those interested in more detailed information.
Collagen
The collagens are the most abundant proteins in mammals and in the ECM [4]. Primarily in skin and bone, collagens make up 25% of total protein mass. Their defining structural feature is a triple-stranded helical structure of three polypeptide chains, which are referred to as the chains. These chains are composed of a series of three amino acid (Gly-X-Y) sequences where X is typically proline and Y is, commonly, a post-translationally modified form of proline called 4-hydroxyproline. Because of the stereochemical constraints of triple-helix formation, glycine, the amino acid with the smallest side chain (a hydrogen), is the only amino acid small enough to occupy the interior of the triple-stranded, -chain structure termed procollagen. This three amino acid structure allows for tight packing of the triple-stranded chains into rod-like structures with the amino acid side chains of the X and Y molecules on the outside of the rod. These side chains are presumed to help direct the self-assembly of the procollagen molecule. Once secreted, proteolytic enzymes remove the propolypeptides from procollagen allowing it to form much longer collagen fibrils in the extracellular space. The driving force for this assembly is presumed to be the insolubility of the collagen fibrils versus that of procollagen. Aggregations of these fibrils (10–30 nm diameter) form collagen fibres (500–3000 nm in diameter), which are then organized to rein-force the tensile strength of the respective tissue in which they are present [4].
With 25 different collagen molecules and their ability to assemble with varied combination of chains, these molecules have been organized into classes based on their macromolecular properties. Fibril forming or fibrillar collagens (Types I, II, III, V and XI) are the most common and account for 90% of body collagen. These types are involved in forming the classical examples of collagen structure for functions in tissues such as bone, skin, tendons and ligaments. Fibril-associated or FACIT collagens (Types IX, XII, XIV, XVI and XIX) are collagens that do not form fibrils. They are found attached to the fibril-forming collagens forming lateral associations. Thus, it is thought that these collagens are involved with the ordering of collagen fibrils among each other and with other matrix molecules. Network-forming collagens (types IV, VII, VIII and X) form net-like or mesh-work structures used in creating sheet-like structures such as in the basement membrane (type IV) or in anchoring fibrils (type VII), which link basement membranes to underlying collagen and laminin in the ECM [5, 6]. Interruptions in the Gly-X-Y repeat amino acid sequence of collagen with non-collagenous sequences are what allow variability in forming different three-dimensional structural shapes (i.e. fibrillar versus network forming versus fibril associated). Type IV collagen is the most abundant member of the basement membrane and is one of the key molecules in its formation due to the ability of this collagen to self-assemble [7, 8]. Another class of collagens whose members seem to play a critical role in neo-vascularization have been named multiplexins (types XV and XVIII) [9, 10] as their structures contain collagen α chains but have different structural features from the classical collagens such as GAGs attached [11, 12]. Although it contains the classic triple-helical collagen domains and behaves as classical collagen [12], type XVIII collagen displays heparan sulfate GAGs [13], making it not only a collagen but also a proteoglycan [9, 14]. Type XVIII collagen also contains multiple interruptions in the triple-helical domain as well as a unique NC1 (non-collagenous) domain at the C-terminus. Two human variants are expressed resulting from transcription from two different promoters [15] creating characteristic tissue expression patterns with high levels in liver, lung and kidney. Furthermore, immunohistochemistry demonstrated that the short protein localizes to vascular and basement membranes while the majority of the long form is expressed in the liver [16, 17]. Type XV collagen has similar sequence similarity to type XVIII collagen suggesting a common ancestor (reviewed in ref. [11]). Type XV has a larger N-terminal and a smaller C-terminal domain with gene expression in all analysed tissues but high levels especially in heart, placenta and skeletal muscle [18, 19]. Further details concerning other classes and assembly of collagen have been described [4, 20].
Laminin
The first being discovered and isolated over 20 years ago, laminins are currently a family of 12 heteror-trimeric glycoproteins, which are usually found in basement membranes. They have been shown to have migratory, adhesive and signalling functions [21, 22]. From a combination of 5α, 4β and 6γ subunits, 1 each of these individually coded protein chains assemble to form a large coiled-coil, quarternary structure with 3 short arms and 1 long arm [23] of which 15 structures are currently known or predicted with certainly more to follow. Though all basement membranes contain laminin, each contains its own combination of selected isoforms, suggesting that this heterogeneity allows different basement membranes to have individual functional roles in tissues while having the same common structure [24]. Laminins have one or more small globular domains at the end of their short and long arms, which mediate interaction with other molecules or laminins. The laminin chains have large globular domains at their C-termini, which is the G-domain. This domain consists of five modules (LG1-LG5) being grouped in three (LG1-LG3) or two (LG4-LG5) and connected by a linker domain [24]. Most of the cell surface binding occurs through these domains. Blood vessels have been reported to express laminin 4α, 5α, 1β and γ1 chains, which suggest that laminin-8 and laminin-10 are involved in their formation. Also, vascular endothelial cells have been shown to produce these isoforms [21, 25, 26].
Fibronectin
Besides proteins that link together to form various scaffolds and structural features, the ECM is composed of other proteins with multiple protein binding domains, which adhere to the scaffolding molecules and to cell surface receptors, therefore contributing not only to the organization of the ECM but to the cells within it. One of the first of these proteins to be characterized was fibronectin (reviewed in refs. [27, 28]). Fibronectin is a large, secreted glycoprotein dimer (each chain ∼ 270 kD) with the chains joined by disulfide bonds at one end. It exists in multiple isoforms, one of which is a soluble form found in blood and other body fluids where it is thought to be involved with blood clotting and wound healing. The other forms of fibronectin are assembled on the surface of cells and then become part of the ECM as insoluble fibronectin fibrils [28]. Distinct from the collagen that undergoes self-assembly, it is thought that fibronectin must be activated to assemble into fibrils [29, 30]. This activation process is associated with integrin binding sites as well as other proteins, which prevent the fibril forming process.The process of fibril assembly also seems to be tied to the regulation of actin filaments inside the cell. Fibronectin fibrils that form near the surface are usually aligned with intracellular actin stress fibres. If these cells are treated with cytochalasin, a drug which disrupts actin filament formation, fibronectin fibrils dissociate from the cell surface by what seems to be an integrin mediated process [31].
The primary structure of the fibronectin protein domains consists of mostly three types of repeating modules [32]: type I, II and III are numbered according to their positions from N-terminus to C-terminus in the fibronectin protein (i.e. III2 is the second type III repeat in the molecule) [27, 28]. The other types of domains found in fibronectin are those which are utilized for binding to heparin, collagen and other fibronectin molecules as well as to other cell surface receptors (i.e. integrins, discussed below). Each of these domains of fibronectin is encoded by an exon. Fibronectin isoforms are composed typically of about 50 exons, each encoding either the repeating modules or specific structural binding domains, all of which can be rearranged by alternative splicing of the exons [33]. In fact, fibronectin, like collagen, is thought to have evolved through multiple exon duplication [34, 35]. Of all the modules, the most abundant is the type III fibronectin repeat, which is among the most common of all the domains in vertebrates and contains the sequence that binds integrins. This tripeptide motif [36, 37], which is composed of amino acids RGD (arginine, glycine, aspartic acid), is the central feature of the integrin binding site [38], and even very short peptide sequences containing this motif can effectively compete with fibronectin binding to cells.
Elastin
Elastin is a hydrophobic protein of the ECM, which allows tissues such as skin and blood vessels to have the required ability to transiently stretch. The 72 kD pre-cursor form of the protein, tropoelastin, is secreted into the extracellular space where these molecules become highly cross-linked to each other via a copper-requiring, lysyl oxidase enzyme, which form networks of elastin fibres and sheets [39]. The composition of elastin is mainly two types of small segments that alternate along a polypeptide chain: one segment, helical rich, is composed of alanine and lysine chains, which are where the cross-links form between the molecules, and the second segment is composed of hydrophobic segments responsible for the molecule's elastic properties. The cross-linking reaction leads to the formation of tetravalent bonding of elastin, lysinonorleucine, desmosines and isodesmosines (the last three are lysine derived products from cross-linking), which leads to the polymerization of tropoelastin into insoluble elastin [40, 41]. How the structural conformation of elastin fibres contributes to the functional elasticity of the fibres is still unknown, although it is thought that the random coil structure of the molecules cross-linked into a network gives it the ability to stretch like a rubber band [39]. Elastic fibres contain other molecules besides elastin. A sheath of microfibrils covers an elastin core. These microfibrils contain a number of distinct and different glycoproteins. One such family of microfibril resident proteins are the fibrillins whose members precede elastin in developing tissues and form a scaffolding structure on which elastin fibres are attached [39, 42]. Comprising 50% of the dry weight of major arteries, elastin is the most prominent ECM protein in arteries [43]. In vessels, elastic lamina alternate with rings of smooth muscle forming a strong, flexible part of the arterial wall [43, 44].
Nidogen
Nidogens (or entactins) are one of the four key components of basement membranes (the others being laminin, collagen IV and a proteoglycan) [45]. In mammals, there are two nidogens encoded by distinct genes [46]. Structurally, nidogens are sulfated glycoproteins that have three globular domains (G1–G3) separated by rod-like domain. G1 and G2 are at the amino terminus connected by a small link-age region while G3 is at the C-terminal domain connected by the rod-like region, which contains EGF repeats (reviewed in ref. [24]). Binding studies with nidogen-1 demonstrate a wide variety of basement membrane targets, and thus, it is suggested that these molecules perform a connection function between the collagen IV and laminin networks while integrating other basement membrane members into the ECM [47–49]. Nidogen-1's highest affinity binding site has been localized to one of the EGF repeats on the laminin 1 chain [50]. Nidogen-2 has been shown to be involved in cell adhesion, which also involves 3 1 and 61 integrins [51].
Glycosaminoglycans
As previously mentioned, GAGs are the molecules attributed to providing the ‘ground substance’ of the ECM. These molecules are unbranched carbohy-drate polymers composed of repeating disaccharide units consisting of N-acetylglucosamine (GlcNAc) or N-acetylgalactosamine (GalNAc) amino sugars AND glucuronic (GlcA) or iduronic (IdoA) acid. The combinations and linkages combine to produce different functional chains and nomenclatures. Hyaluronan, composed of GlcNAc β1-3GlcA β1-4 repeating link-age, is the simplest known GAG and is abundant in skin, synovial fluid and skeletal tissues and was originally isolated from the vitreous humour of the eye [52]. The remaining GAG structures are attached to protein cores, which result in a proteoglycan. Heparan sulfate (GlcNAc α1-4GlcA1α-4) and chondroitin sulfate (GalNAc α1-4GlcAα1-3) both are GAGs, which are attached to protein core molecules to produce proteoglycans such as perlecan, syndecans or glypicans. These heparan and chondroitin chains are further modified after their synthesis by epimerization (glucuronic acid to iduronic acid), N-deacetylation-N-sulfation, 2-O-sulfation, 3-O-suflation and 6-O-sulfation (reviewed in refs. [53, 54]). Modification of these core proteins (O-linked modification versus the N-linked modification of glycoproteins) occurs at serine or threonine sites in the protein. The hydroxyl side chains of these amino acids are modified first with a xylose residue followed by a linkage tetrasaccharide of which the final attachment of αβGalNAc or αGlcNAc determines whether the chain will be polymerized as a chondroitin sulfate or a heparan sulfate backbone, respectively (reviewed in ref. [53]). Matrix proteoglycans tend to display chondroitin sulfate or the further epimerized dermatan sulfate while the membrane proteoglycans tend to have heparan sulfate (as with most rules, these are only generally true as some syndecan family members have both heparan and chondroitin sulfate). Other ECM molecules can attach to the GAG part of the chain, which may be displayed on different core proteins. This fact can make sorting, which proteoglycan is responsible for certain molecular and phenotypic effects, especially in signalling, challenging.
Numerous proteoglycans are involved in the ECM. Agrin is an abundant proteoglycan of most basement membranes but mainly functions at the neuromuscular junction (reviewed in ref. [55]). As mentioned above, both collagen XV and collagen XVIII are proteoglycans with XV containing chondroitin sulfate [56] while XVIII has heparan sulfate [13]. Perlecan and the syndecans have been well characterized in terms of basement membrane and focal adhesion assembly with apparent roles to play in vascular formation and neoangiogenesis, and those will be discussed.
Perlecan
Perlecan is a large (∼470 kD protein core, up to ∼800 kD when glycanated) proteoglycan with many O-linked glycosylation and three to four heparan sulfate sequences attached. There is only one gene encoding perlecan (Hspg2) in mammals and, similar to other matrix proteins, due to its varied modules, probably arose by gene duplication and exon shuffling [57]. The protein core is composed of five modules, which allow for a variety of binding partners from classical ECM components such as laminin-1, collagen IV, nidogen-1 and fibronectin to growth factors, which include fibroblast growth factor-2 (FGF2), vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) (reviewed in ref. [58]) [59]. Although embedded in the basement membrane, perlecan also associates with the cell surface via interaction with α2β1 integrin, which also binds to fibrillar collagen [60].
Syndecan
Syndecans are a family of type I transmembrane pro-teoglycans, which have four family members in vertebrates (Syndecan-1, -2, -3, -4). These proteoglycans consist of a short, variable extracellular domain with attachment sites for three to five heparan sulfate or chondroitin sulfate chains, a single span transmem-brane domain and a conserved cytoplasmic tail, which contains two highly conserved (C1 and C2) regions and one variable (V) region. In the extracellular domain, the GAG chains are what mediate interaction between growth factors and their receptors such as FGF2/FGF receptor for which syndecan has been shown to function as a co-receptor (reviewed in ref. [61]). Also, these chains mediate interaction with ECM proteins such as fibronectin, and it has also been suggested that the chondroitin and heparan chains of syndecan-1 and syndecan-4 mediate ker-atinocyte binding to laminin-5 [62]. Through extracellular and intracellular domain swap experiments, the transmembrane domain has been shown to be sufficient for oligomerization of syndecan-2 and syndecan-4 [63]. Clustering of syndecan-4 (by antibodies and by Fc-receptor-Syndecan-4 chimeras) has been demonstrated to induce FGF2 endocytosis in a clathrin and dynamin independent process, which requires syndecan-4 independent activation of Rac1 [64]. In the cytoplasmic domain, a point mutation in the PDZ binding domain (a domain named for the motif associated with intracellular signalling proteins in which it was first described: Post synaptic density, Discs large, Zona occludens-1) located at the tail of the syndecan-4 molecules has been shown to reduce migration in endothelial cells suggesting a role for this domain in endothelial cell migration [65]. Syndecan-4 also participates with integrins and fibronectin in focal adhesion formation (sites of tight adhesion to the underlying extracellular matrix) by modulating focal adhesion kinase formation [66] and being recruited into focal adhesion complexes with fibronectin after activation by protein kinase C [67].
Receptors for ECM molecules
Assembly of the vast network of molecules into an ECM is only part of the picture, as cells must have some mechanism of attaching to the matricellular environment. The syndecans and other transmem-brane proteoglycans do function as coreceptors and provide some of the anchoring, but the majority of receptors on animal cells for binding ECM components are the integrins (reviewed in ref. [68]). Consisting of two transmembrane heterodimers (αandβ), which are non-covalently associated glyco-proteins, integrins have only been found in meta-zoans [69]. Indeed, worms only contain two and one subunits forming the organism's two integrins, while the number in mammals is estimated to be 18α and 8β subunits with 24 combinations of protein having been identified [70]. A considerable amount of redundancy exists in that many matrix proteins bind multiple integrins. Also, integrins have a lower binding affinity for their targets than other receptor molecules but compensate by being up to 100-fold higher in cell surface concentration. This weaker, multivalent binding has been suggested to allow for efficient but easily reversible interactions. Integrin binding also requires divalent cations (Mg2+ or Ca2+) as well as an aspartate or glutamate (the RGD sequence mentioned above in Fibronectins) residue, which are involved in coordination of the divalent cation [71–73]. To possess the ability to ‘ground’ cells in the membrane, on the cytoplasmic side, most integrins’ connection to the cytoskeleton is mediated by intra-cellular anchoring proteins (i.e. talin, β-actin, filamin). Attached to the integrin αsubunit, these anchoring proteins attach to actin or to other anchor proteins such as vinculin. Thus, integrin clustering leads to production of focal adhesions between the ECM and the cell [74]. Integrins also are involved in signalling processes between matrix proteins and the cell. The clustering of integrins by matrix proteins has been shown to activate focal adhesions, which are mediated by a tyrosine kinase, focal adhesion kinase (FAK). FAK is recruited to focal adhesions where these molecules cross-phosphorylate tyrosine molecules on each other creating docking sites for the Src family of tyrosine kinases, which produces even greater binding sites for other signalling proteins (reviewed in ref. [75]). This process could be referred to as outside-in signalling, which is the method by which most signalling occurs. Integrins, however, also have the ability to participate in a process called inside-out signalling [76, 77]. By allowing activation of inactive integrins already on the cell surface, inside-out signalling allows for an immediate adhesion response without having to permit time for secretion or synthesis of the integrins. This process allows platelets, for example, when activated by injury, to activate integrin binding to fibrinogen to form a platelet clot.
Although integrins are the major type of ECM receptor, other receptor molecules, especially in endothelial cells, have been shown to exist [78]. Dystroglycan is a membrane-spanning glycoprotein heterodimer composed of αandβ dystroglycan non-covalently associated subunits translated as a propeptide and proteolytically cleaved into two proteins [79]. It was originally isolated from the dystrophin-glycoprotein complex, which is thought to provide structural stability to the sarcolemma, and mutations in this complex have been associated with several muscular dystrophies (reviewed in ref. [80]). ECM attachment of dystroglycan is mediated through the subunit O-linked, mucin type carbohydrates to laminin-2, and this interaction has been shown to be dependent on the glycosylation of dystroglycan. Cytoplasmic attachment of the transmem-brane subunit is accomplished through dystrophin, which binds to the actin cytoskeleton [81, 82]. Dystroglycan has been shown by cDNA cloning and western blotting to be expressed in bovine aortic endothelial cells and this interaction was mediated by laminin-1 [83].
Evaluation of ECM molecules and their modulators in vessel formation
Up to this point, the molecules involved in the ECM, with particular attention to those in vascularization, have been described in terms of their synthesis and their relationship to the other ECM members in forming the matrix. Now, with the continued use of vessel formation as a framework, experiments involving these molecules to elucidate their role in the vascularization process will be examined.
Angiogenesis as a term is frequently referred to as the process of new blood vessel growth. In the field, however, as previously described [84], this term is associated with a specific biological process, which only partially covers the events involved in new blood vessel formation. In this review, neovascularization refers to the general process of blood vessel growth and broadly involves vasculogenesis, arteriogenesis and angiogenesis. Vasculogenesis describes the process of de novo formation or remodelling of pre-existing channels by angioblasts (embryonic development) or circulating vascular progenitor cells (adult neovascularization). Although it is the main player in embryonic development, the functional significance of vasculogenesis in ischemia or peripheral circulation has not been established (reviewed in ref. [84]). Arteriogenesis is defined as the process of maturation or remodelling of existing or newly formed arteries [85, 86]. The matter of whether arteriogenesis occurs via remodelling of pre-existing vessels or by formation of new vessels from progenitor cells is controversial. Lastly, angiogenesis refers to the process of forming new capillaries from post-capillary venules and is mostly driven by tissue hypoxia [84, 87].
As has been previously described and will be futher illustrated below, the ECM plays critical roles in most of the processes of blood vessel formation. The detailed processes involved in de novo and adult blood vessel formation have been extensively reviewed [88–94]. In blood vessel formation of embryonic development, vasculogenesis and angiogenesis participate in the formation of the vasculature. Briefly, in vasculogenesis, hemangioblasts are derived from groups of FGF2-stimulated splanchnic mesoderm cells as precursor cells of blood vessels and blood cells [95]. VEGF–VEGF receptor 2 (Flk1/KDR) signalling mediates the differentiation of hemangioblasts into cell aggregates or blood islands [96]. Outer cells of blood islands differentiate into angioblasts (blood vessel precursors) while the inner cells become hematopoietic stem cells (blood cell precursors). Next, angioblasts become endothelial cells, which form the internal lining of blood vessels and, later, organize into tube structures creating a network of capillaries or the primary capillary plexus. VEGF–VEGF receptor 1 (Flt1) controls the formation of tubes from blood islands [97, 98]. Lastly, angiopoietin-1-Tie2 receptor mediates recruitment of smooth muscle-like pericytes to cover capillaries [99–101]. During angiogenesis, this primary network is remodelled, pruned and specialized into arterial and vein capillary beds [88]. Organ capillary networks, which are formed from inside the organs and are not grown from larger vessels, are then joined to the other forming network to build the circulatory system (reviewed in ref. [93]). It is in the angiogenic process that research has primarily focused on how ECM components provide direction for regulating vessel cell migration, proliferation, differentiation and survival. Thus, these processes are the experimental measures used to gauge angiogenic potential, especially in vitro. Genetic manipulation of defined angiogeneic models and construction of mutations in cells and animals are also utilized to provide evidence as to whether confirmation or denial is appropriate in these in vitro hypotheses. An example is provided (Fig. 1) where arteriogenesis of synectin (an adapter protein found in the syndecan-4 pathway) null mice is examined. The femoral artery ligation model was employed to demonstrate a profound reduction in recovery of synectin in hindlimb reperfusion of synectin null versus wildtype mice. This hindlimb in vivo data correlated with previous in vitro data, which had revealed that primary endothelial cells isolated from synectin knockout mice showed reduced migration in comparison to cell isolated from wildtype mice in a cell wounding/migration assay.
1.
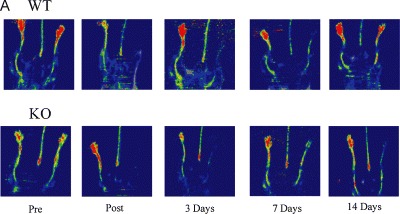
Effects on arteriogenesis by synectin disruption in mice. (A) Laser Doppler images of the time course of perfusion in hindlimbs of synectin wildtype (WT) and null (KO) mice (perfusion is indicated by colour where red > yellow > green > blue) (B) Quantitative analysis of laser Doppler images indicates significant alterations in hindlimb reperfusion immediate post and 14 days after femoral artery ligation in synectin null mice (black bars). Means +/- SEM, P < 0.05 from Chittenden et al. Dev. Cell. (2006), 10: 783–795.
Determination of essential ECM components of vessel formation
Since ECM components are the scaffolding that girds the vascular system, one collection of mutations that would be expected to effect vessel formation would be disruption of the basement membrane structure or attachment of components to the structure resulting in no or malformed configurations. While these phenotypes involving tube formation and vessel stability certainly do occur, other experiments show that certain ECM molecules are not necessary for structure, but are critically involved in cell guidance (migration or chemotaxis), proliferation or viability. In this section, recent experiments are high-lighted concerning ECM component(s) involved in vessel formation. Since certain components of the ECM can play necessary roles in certain initial embryonic developmental programs, it can be difficult to determine if said molecule is not also critical in later programs without clever genetic techniques (i.e. tissue specific and/or drug inducible, promoters and recombinases). Thus, examination will focus on experiments that have been determined to be related to vessel formation (Table 1). Experiments involving tumour angiogenesis will not be discussed except where there is overlap with the focus of the current paper (many reviews of this subject are available [102–106]). Also, matrix metalloproteases (MMPs) have been covered in a review in this series [107] and by others [108–111].
1.
Extracellular matrix molecule null mutations involved in blood vessel formation*
| Knockout | Phenotype(s) | Comments | References |
|---|---|---|---|
| α1(I) collagen | Mutants survive until E12-E14, death due to rupture of blood vessels | Fibrillar collagen type I may be important for mechanical stability of blood vessels | [113] |
| α1(III) collagen | Model of Ehlers–Danlos syndrome type IV, 90% die perinatally, 10% survive until adulthood with death due to rupture of blood vessels | Like type I, fibrillar collagen type III may be important for mechanical stability of blood vessels | [135] |
| α1(IV)2 a2(IV) collagen | Structural deficiencies in basement membrane, failure of Reichert's membrane, lethality at E10.5-E11.5 with vascular bleeding in the heart and arteries | Mechanical demands on basement membrane as size of embryo increased and circulation started, instability in basement membrane structure | [136] |
| α1(XVIII) collagen | No obvious defects in vascular patterning or capillary density in most organs two-fold increase in microvessels of knockouts in modified aortic explant assay, endostatin admin. negates increase to wildtype levels | Increased endothelial cell adhesion in null mice may allow for greater vessel stability, also large increase of adherence in knockout on fibronectin matrix | [171–173] |
| Heat shock protein 47 | Abnormally oriented epithelial tissues, embryonic lethality at E11.5 or earlier due to ruptured blood vessels | Hsp47 is a molecular chaperone of triple helix formation of type I collagen a chain, seems to disrupt proper formation of type IV collagen | [137] |
| α4 chain of Laminin | Lethal at day E11.5 and newborns, ruptures in microvascular cell wall result in subcutaneous hemorrhaging, which were exposed to greater stress during birth. In vivo angiogenesis assays result in 50% of mice sacrificed due to chronic hemorrhages | Weakened capillary basement membrane results in subcutaneous hemorrhaging. Small group survive may be due to expression of a5 laminin which begins after birth | [142] |
| Fibronectin | Range of phenotypes including deformed heart and embryonic vessels, E8.0 until death consisted of low numbers of primitive blood cells and embryonic vessels of larger size in the embryonic vasculature | Mesenchymal defects due to deficits in proliferation, adhesion and migration of cells | [145] |
| Perlecan | Between E10 and E12, 50% of mice die due to hemorrhage of pericardial cavity, cephalic and cartilaginous abnormalities result in death of respiratory failure at birth | Failure of basement membranes due to mechanical stress | [156] |
| Perlecan (reduced heparan sulfate) | Delayed wound healing, retarded FGF2-induced tumour growth and impaired angiogenesis in cornea micropocket assay, enhanced smooth muscle cell proliferation resulting in intimal hyperplasia | Reduced heparan chains disrupt modulation of FGF2, VEGF and other growth factors. | [158,159] |
| Syndecan-1 | Higher degree of inflammation, delayed wound healing | Delay in wound healing possibly due to defects in granulation tissue and angiogenesis | [162] |
| Syndecan-2 | Essential for sprouting angiogenesis in zebrafish based on morpholino knockdown | Modulation of VEGF signalling by syndecan-2 disrupted | [164] |
| Syndecan-4 | Delayed wound healing in heterozygous and homozygous mice, diffuse and degenerated vessels in placental labyrinth | Modulation of angiogenesis via heparan chains or disruption of fibronectin binding, reduces anti-thrombin binding sites by fewer heparan chains and may result in a more procoagulant state | [166,167] |
All mutations are in mice except for syndecan-2.
Collagen-I and laminins are important for vessel structural integrity and provide contrasting signals in angiogenesis
As previously described in Collagens, Collagen-I is a member of the fibril forming family of collagens, which make up a large percentage of the ECM in the body and is a constituent of many tissues that under-go embryonic angiogenesis during development [112]. In vivo, a mouse knockout model of type I collagen generated by insertional mutagenesis produced a lethal phenotype between day 12 and day 14 of gestation [113]. While distribution of other collagens, laminin and fibronectin were not effected, no type I collagen was detected in the mutant embryos. The mutants displayed a phenotype of mechanical instability of vessels due to rupture as well as defects in hemopoietic cells of the liver [113]. In vitro, bovine aortic endothelial cells synthesize type I collagen and its production is limited to the cells forming capillary tubes in the endothelial cell monolayer [114, 115]. Also, human umbilical vein endothelial cell monolayers have demonstrated a robust angiogenesis with type I collagen and sulfated GAGs [116, 117]. In the embryo, capillary development has been shown to begin with endothelial precursor cells transitioning to a spindle-shaped morphology, followed by alignment into cord-like structures that form a network [118–121] (reviewed in ref. [122]). These cord-like structures undergo maturation, forming lumens for blood transport and the endothelial cells attach to basal lamina [119, 123]. Inhibitors of collagen triple helix formation or fibrillogenesis prevent angiogenesis in the chick embryo [116]. Type I collagen, therefore, would seem to be involved in activation of endothelial cells for angiogenesis. Blood vessels, however, do not contain type I collagen but contain mainly type IV collagen and laminin. Thus, a model [94, 122] has been proposed, which suggests that ECM signalling regulates endothelial cell morphogenesis and sprouting. In the model, blood vessel endothelial cells are normally not exposed to type I collagen because of the continuous basement membrane. Induction of angiogenesis would then be coincident with degradation of the basement membrane and exposure of endothelial cells to type I collagen. This exposure would activate the endothelial cells and drive sprout formation until the regeneration of a continouus basement membrane. In support of the theory, molecular data show that, in vivo, in endothelial cells isolated from the microvasculature, type I collagen induced morphogenesis is mediated by selective VEGF induction [124] of α1β1 and α2β1 integrins [125]. Thus, molecularly, it is proposed that type I collagen via integrins activates Src and Rho pathways and suppresses Rac and protein kinase A pathways resulting in induction of actin stress fibres, disruption of VE-cadherin and precapillary cord formation while cells plated on laminin-1 induce Rac activation (which has been shown to reduce Rho) but do not induce endothelial cell morphogenesis [122]. Further support is demonstrated by Src kinases modulating vascular morphogenesis [126]. These kinases have been suggested to be involved in angiogenesis by regulating vascular permeability [127]. Also, Rho family members have been shown to be involved in actin cytoskeletal changes (reviewed in ref. [128]). Finally, through the use of Src and Rho inhibitors and mutants, activation of Src and Rho were shown to be directly involved in type I collagen activated human dermal endothelial cell capillary morphogenesis, while laminin-1 produced a consistent activation of Rac but did not induce capillary formation [122]. In vitro, induction of capillary morphogenesis has also been shown via the use of peptide mimetics to require the engagement of α2β1 integrin by a single type I collagen integrin binding site, which appears to signal via p38 mitogen activated protein kinase and FAK inactivation [112]. One problem with the model, however, is the use of laminin-1 as the substrate for the in vitro cell studies. Laminin-1 (α1β1γ1) is a prototype laminin, frequently used with in vitro cell adhesion and migration assays, and does not occur in capillary endothelia basement membranes, but is purified from Engelbreth–Holm–Swarm (Matrigel) tumour ([129, 130] (reviewed in ref. [131]). The endothelial basement membrane contains 4 and 5 chains of laminin, which would correspond to laminin-8 (α4β1γ1) and laminin-10 (α5β1γ1) for endothelial cell binding [131]. The 1α chain does occur in the meningeal epithelium and the parenchymal basement membrane (which is the collective name for the meningeal and astroglial basement membranes). Further experimental proof of this model of sprouting angiogenesis will have to wait for future experiments.
The function of collagens in the blood vessel basement membrane
In the same group as type I collagen, type III collagen is a fibrillar collagen composed of three α1(III) chains and is also a major component of the ECM. Mutations in the Col3A1 gene encoding type III collagen appears to result in Ehlers–Danlos syndrome type IV, which manifests in human beings as fragile skin and blood vessels with a frequent cause of death being aortic rupture in adult life (reviewed in ref. [132]). Previous work has shown that type III collagen is colocalized with type I collagen in the collagen fibril and that this may be essential for specific kinds of fibrillogenesis. Different ratios of type III to type I collagen are thought to modulate the size of the type I fibre, and a difference of size has been observed in various tissues or at different developmental stages of the same tissue [133, 134]. To assist in further study, a knockout mouse of the Col3A1 gene was constructed. Most of the homozygous Col3A1-/-died perinatally; however, 10% of the mutant mice survived until adulthood but manifested similar clincal symptoms as Ehlers–Danlos syndrome type IV patients including sudden death due to rupture of large vessels, which resulted in much earlier deaths than wildtype mice [135].
As one of the major constituents of basement membranes, type IV collagen was suspected to play a major role in basement membrane stability.
Different models of genetic knockouts now indicate that, although important for stability and functionality, type IV collagen is not essential for formation of basement membrane assembly [136, 137]. Type IV collagen null mice, generated by disruption of the Col4a-/-locus, which removes α1(IV)2α2(IV) chains, developed until embryonic day 9.5 (E9.5) with examination of basement membrane proteins showing apparently normal deposition and assembly. Structural deficiencies in basement membranes at E10.5-E11.5, which were associated with vascular bleeding in the heart and arteries at cell–cell contacts and failure of Reichert's membrane as the size of the embryo increased suggested that these defects were manifested as increasing mechanical demands were required of the basement membranes. This evidence suggests that type IV collagen is involved in mechanical stability of membrane structure [136]. Additional proof is provided in another model of collagen dysregulation. Heat shock protein 47 (Hsp47) is a molecular chaperone that was discovered to chaperone triple-helical procollagen molecules in the endoplasmic reticulum. Genetic mouse models ablating the Hsp47 gene resulted in basement membrane disruption in homozygous mice due to deficiencies in the production of mature chain of type I collagen, which effects fibril formation resulting in a type I collagen unable to form a rigid, triple-helical structure. The mutation also perturbs the proper molecular form of type IV collagen by a pathway still under investigation [137, 138]. Due to the improper formation of type IV collagen in basement membranes, Hsp47-/-mice displayed abnormally oriented epithelial tissues and ruptured blood vessels, which led to death ≤11.5 days after coitus, which is in time frame of the lethality with the type IV collagen mutant [137]. Seemingly, the Hsp47 -/-mice have similar mechanically related basement membrane phenotypes as the type IV collagen null mice. Finally, in the ex vivo rat aortic model of angiogenesis using a type I collagen matrix, type IV collagen promoted new vessel elongation and survival in a dose-dependent manner compared with controls (no type IV collagen), which grew until day 9 and then regressed during the second and third week of culture (treatment with 3, 30, 300 g/ml of type IV collagen in 43, 57, 119% increase in microvascular length) [139]. In all then, type IV collagen seems important for stability of blood vessel formation.
Laminin a4 is a key molecule in basement membrane assembly, microvessel stability and maturation
Laminins are the other molecule in basement membrane that can polymerize into a network (the other being type IV collagen). Recall that they are composed of three, α, β and γ chains encoded by individual genes, which assemble into a disulfide bonded, cross-shaped structure (reviewed in ref. [45]). Tissue distribution differences and interaction with cells are usually mediated by the chains, which exhibit functional importance [131]. Two laminin α chains (α4 and β5), one chain (β1) and one β chain (γ1) have been shown to be expressed in blood vessels, which are found in two laminin molecules: laminin-8 (α4β1γ1) and laminin-10 (α5β1γ1) [21]. Using cell adhesion assays with antibodies to different integrin subunits and recombinantly generated laminin-8, HT-1080 cells (human fibrosarcoma) and bovine capillary endothelial cells were shown to use α6β1 and α6β4 integrins to mediate binding to laminin-8 [140]. Later, using similar techniques, recombinant laminin-10 promoted migration in vascular endothelial cells better than recombinant laminin-8 with 31 integrin being the major adhesion molecule for laminin-10 [141]. To examine the role of laminin α4 chains in basement membranes, mice were generated harboring Lama4 null alleles. In late embryonic E11.5 and particularly in newborns, the absence of laminin 4 weakened the capillary basement membranes with ruptures in the microvascular walls resulting in subcutaneous hemorrhaging, especially in newborn, which were subjected to greater mechanical stress during birth [142]. Immunohistochemistry revealed a loss of 1 and 1 laminin chains in the capillary basement membranes as well as large reduction in type IV collagen and nidogen while perlecan levels remained unchanged, suggesting that laminin-8 may be required for capillary basement membrane assembly of nidogen and type IV collagen [142]. After birth, there is a reduction in the vascular phenotype, which has been suggested to correlate with the known increase in production of laminin 5 chain (laminin-10) from 3–4 weeks post-birth to adulthood, which may partially rescue the phenotype [26]. The Lama4 null mice were also tested for their ability to form new capillaries using the cornea angiogenesis assay. Results from this assay led to greater than 50% of the mice having to be sacrificed due to chronic hemorrhages; however, in the surviving animals, the new vessels assumed a normal structure with time perhaps as part of a laminin α5 rescue [142].
The laminin α5 chain (in laminin-10) was also investigated in terms of constructing a null mouse. The Lama5-/-mouse results in an embyronic lethal phenotype with mice progressing normally until E9 with multiple defects occuring (exencephaly, syndactyly, placentopathy) with no mice surviving after E 16.5 with death attributed to defects in the placenta [143]. If laminin α5 is involved in vessel formation, it is most likely not embryonic related as its expression does not occur early enough in vessels (3–4 weeks post-birth to adulthood [see laminin-10 above]) and only mainly occurs in capillaries of endothelial cell basement membranes [26, 131]. It may, however, be associated with endothelial maturation. Future answers should arrive soon as construction of null mice that are targeted to laminin α5 and laminin α4 and α5 in endothelial basement membranes is currently in progress (reviewed in ref. [131]).
Fibronectin is essential for vascular development
As a major molecule in the ECM, which has been demonstrated to be involved in cellular adhesion, spreading and migration, the broad range of functions associated with fibronectin including embryonic development, immunological responses, fibrosis and wound healing seem appropriate (reviewed in ref. [144]). Because of the extensive purview of fibronectin function in vitro and in vivo, null mice were constructed to further ascertain the specific role of this molecule. Constructed in three independent mouse strains, the fibronectin gene resulted in a homozygous embryonic lethal phenotype in all strains and intercrosses between these strains [145]. A range of phenotypes were exhibited including shortened anterior–posterior axes, deformed neuro tubes, absence of notochord and somites, deformed heart and embryonic vessels among others whose common theme seemed to be derived from defects in mesodermal development thought to be due to deficits in cell proliferation, adhesion and migration [145]. Developmental abnormalities from E8.0 until death consisted of low numbers of primitive blood cells in the embryonic vasculature and embryonic vessels of large size and fewer in frequency than controls not allowing further analysis of vascular formation, which normally takes place around E8.5.
Blood cells and vascular endothelial cells are derived but blood vessels are not apparent suggesting that fibronectin is involved in vasculogenesis [145, 146]. Further detailed examination of fibronectin null embryos involving lineage analysis concluded that fibronectin is essential for organization of heart and blood vessels [144]. As discussed earlier, integrins are specifically involved in activation of fibronectin matrix assembly. One would then expect that the integrin associated with fibronectin, in this case α5β1 integrin, would also be critical for function in similar tissues as elucidated in the fibronectin null mouse. Indeed, reduced blood vessel formation has been seen in α5 integrin-negative embryoid bodies [147] and similar mesodermal defects are present in 5-integrin-deficient mice [148]. If fibronectin and α5β1 integrin receptor are involved in vasculogenesis and angiogenesis, how might these processes be mediated? Using surface plasmon resonance, co-immunoprecipitation and solid-phase binding assays, two binding domains on fibronectin were found to modulate the activity of VEGF [149]. These novel VEGF binding domains on fibronectin promote the association between α5β1 integrin and VEGF receptor 2 (Flk-1), which was confirmed by coprecipitation studies requiring full length but not fragments of fibronectin [149]. Association of the integrin with the tyrosine kinase receptor allowed enhanced amplification of mitogen activated protein kinase signalling (MAPK) for endothelial cell migration [149]. Later work by the same group has narrowed the binding site for VEGF to the fibronectin C-terminal domain within type III repeats 13–14 [150], which has previously been revealed to be the heparin II binding domain [151–153]. This domain has been previously demonstrated as a major syndecan binding site with roles in focal adhesion and stress fibre formation [154]. In a related study, VEGF binding sites are suggested to become available when fibronectin is prompted to adopt an extended confirmation by interaction with a hydrophilic surface or heparin/heparan sulfate [155]. Long GAGs of greater than 22 saccharides with sulfation on the 6-O and N positions of glucosamine were required for full activity of the complex. While the heparin/heparan sulfate interaction serves to enhance protein–protein interactions of VEGF and fibronectin, it also demonstrates a mechanism of how GAGs can attenuate matrix structure to alter growth factor binding [155].
Perlecan and syndecans modulate growth factors in angiogenesis
Perlecan contains various modules, which allow it to participate in a variety of cellular interactions as well as being a major constituent of basement membranes (reviewed in ref. [12]). To gather information concerning the in vivo functions of perlecan, Hspg2−/mice were constructed. Suprisingly, however, homozygous perlecan-null mice form normal basement membranes [156]. Between E10 and E12, nearly half the mice die, however, due to hemorrhage of the pericardial cavity, which possibly results from deterioration and failure of the basement membrane; therefore, suggesting a role for perlecan in mechanical stability of these membranes [156]. Surviving Hspg2-/-mice have cephalic and cartilaginous abnormalities and die of respiratory failure after birth. Also, perlecan null mice cartilage had severe disorganization of columnar structures of chondrocytes, defective endochondral ossification, and disorganized collagen fibrils and GAGs suggesting perlecan is important for organizational structure of the matrix [157]. Due to the variety of phenotypes and the possible number of interactions perlecan can have with other molecules, a second perlecan mutant was made, which removed only exon 3, Hspg2Δ3/Δ3, which ablates two of the three possible heparan sulfate attachment sites [158]. These null mice survived and grew into apparently healthy adults. The Hspg2Δ3/Δ3 mice did display delayed wound healing, retarded FGF2 induced tumour growth and impaired angio-genesis in the cornea micropocket assay [158] pre-sumably due to the reduction of heparan sulfate, which has been shown to sequester, modulate the release and attenuate FGF, VEGF and other growth factors related to neovascularization. Another study using these same Hspg2Δ3/Δ3 mice discovered that, in vitro, isolated mutant smooth muscle cells showed increased proliferation as compared with wildtype cells and, in vivo, enhanced smooth muscle cell pro-liferation culminating in intimal hyperplasia after interruption of carotid artery flow [159] implying that the heparan sulfate side chains of perlecan contribute to FGF2 mediated growth control of smooth muscle cells.
Syndecans can also modulate growth factor signalling via their heparan or chondroitin sulfate chains. Of the four members of the syndecan family, all have been identified in vascular cells and virtually all nucleated cells express one family member [61, 160]. Currently, syndecans are considered not to act independently but participate as co-receptors working with FGF tyrosine kinases or with integrins as with fibronectin (see section Fibronectin is essential for vascular development), but clustering of syndecan-4 molecules either with antibodies or using chimeric systems has been shown to cause endocytosis and activate Rac1, hinting that syndecans may have their own cellular functions that work in concert with other coreceptor partners [61, 64, 161]. Further investigation of the in vivo aspect of syndecan function has resulted in mouse null mutants being produced in three of the four family members (syndecan-1, -3 and -4) and a zebrafish model (syndecan-2) using morpholinos for knockdown. All the mouse models resulted in mice which appeared to have normal embryonic development and matured into apparently normal adults. Syndecan-1 null mice showed increased leukocyte adhesion in a retina perfusion model versus wildtype mice, especially after tumour necrosis factor β treatment [162]. Sdc1-/-mice also had a higher degree of inflammation, which may mediate corneal angiogenesis and also a delay in wound healing presumably due to a defect in keratinocytes [162]. Further analysis by production of a transgenic syndecan-1 to produce overexpressed syndecan-1 also demonstrated a delay in wound healing with defects in granulation tissue formation and angiogenesis [163]. The wound healing delay and the angiogenesis defects in the overexpressed systems were likely due to the ability of shed syndecan-1 ectodomain to inhibit cell proliferation during wounding repair and angiogenesis by binding growth factors competitively, which allowed for fewer factors to stimulate cells in the tissue [163]. Although there is no mouse model for syndecan-2 mice as of yet, morpholino knockdown technology was used in the zebrafish model to explore the function of syndecan-2 in vascular development. Syndecan-2 was shown to be essential for sprouting angiogenesis in zebrafish and, in later experiments, human syndecan-2 and fragments of the cytosolic portion of syndecan-2 could rescue the zebrafish knockout phenotype [164]. Also, through gain and loss of function genetic studies in zebrafish, VEGF and syndecan-2 were shown to genetically interact: VEGF mediated ectopic signalling is compromised in syndecan-2 morphants and ectopic syndecan-2 potentiates ectopic VEGF signalling [164]. Thus, syndecan-2 is essential for angiogenic sprouting in zebrafish. A similar phenotype was accomplished by using siRNA to syndecan-2 in mouse glioma cells. Downregulation of syndecan-2 expression resulted in inhibited cell mobility and reduced formation of capillary tubes in vivo in brain microvascular endothelial cells [165]. Bacterial recombinant (i.e. no GAG chains) syndecan-2 ectodomain activated membrane protrusion, migration and capillary tube formation in brain microvascular endothelial cells [165]. Syndecan-2 seems necessary for angiogenesis and the shed syndecan-2 ectodomain may increase angiogenic processes. Similar to syndecan-2, syndecan-4 null mice have delayed wound repair but in both heterozygous and homozygous genotypes [166]. Significant differences in the level of vascularization of the granulation tissue on day 6 after wounding were apparent in homozygous and heterozygous syndecan-4 null mice indicating defects in angiogenesis and/or wounding [166]. In another generated null model, sdc4-/-mice displayed a greater area of diffuse and degenerated vessels in the placental labyrinth with more calcium and fibrin deposition indicating a reduced anti-coagulation [167]. Heparan sulfate chains are known to provide an activation mechanism for anti-thrombin, which results in a procoagulant state (reviewed in ref. [168]). The absence of these heparan chains in syndecan-4 null mice may be responsible for their anti-coagulant phenotype. The last syndecan family member for which there is a null mouse is syndecan-3, but abnormalities in that model currently appear to be related to skele-togenesis and to neural phenotypes (reviewed in ref.[61]).
Fragments of ECM proteins regulate angiogenic processes
Several of the ECM proteins that have been discussed have arisen through an evolutionary selective process known as exon shuffling. Proteins assembled in this way are composed of modules or domains, which impart specific functions to the parent protein. It is logical then that fragments of these parent ECM proteins, whether by natural or synthetic fragmentation, might have the potential to function independently. Fragments of ECM proteins, which have been suggested to modulate angiogenesis, are in Table 2. Almost all of the fragments are involved in inhibition instead of activation of angiogenesis. Most of these fragments have been isolated or created for the purpose of inhibiting tumour angiogenesis. Others are produced naturally, in vivo, which suggests that they may have endogenous functions in angio-geneic pathways and/or other physiological purposes.
2.
Fragments of extracellular matrix molecules involved in angiogenesis
| Fragment | ECM parent | Size (kD) | Properties [Reference] |
|---|---|---|---|
| Endostatin | Type XVIII collagen | 20 | -Identification and cloning of endostatin as an angiogenesis inhibitor [170] -Angiogenic effect mediated by heparin binding domains [175] -p53 activates a transcriptional program increasing endostatin production [180] -Disassembly of actin cytoskeleton via RhoA inhibition [242] |
| Restin | Type XV collagen | 22 | -Inhibits angiogenesis, tumour growth supression [243–245] |
| Arresten | Type IV (α1) collagen | 26 | -Inhibit endothelial cell proliferation and tumour growth [103, 177] -Anti-angiogenic activity mediated by alb1 integrin [178] |
| Canstatin | Type IV (α2) collagen | 24 | -Inhibits angiogenesis, tumour growth suppression [246] |
| Tumstatin | Type IV (α3) collagen | 30 | -Induces apoptosis of proliferating endothelial cells [247] -Inhibits angiogenesis, suppresses tumour growth [248–251] -p53 activates a transcriptional program increasing tumstatin production [180] |
| Unnamed | Type IV (α6) collagen | ∼25 | -Inhibits angiogenesis, tumour growth suppression [177] |
| Anastellin | Fibronectin | 9 | -Inhibits tumour growth, angiogenesis and metastasis [182, 183] -Inhibits ERK signalling, G1 arrest in endothelial cells |
| Endorepellin | Perlecan | 81 | -Inhibits angiogenesis, can bind endostatin [184] -Disassembly of actin cytoskeleton and focal adhesions via 21 inte-grin [185] |
| γ-elastin | Elastin | unknown | -Proangiogenic in chick chorioallantoic membrane model [188] -Stimulate tube formation in endothelial cells in Matrigel and collagen [188] -Promote endothelial cell migration in in vitro wound healing assay [188] -Upregulate pro-MT1-MMP and pro-MMP2 expression (gene) and activation (protein) [188] |
The first protein on the list, endostatin, is probably the most famous of these fragments. Endostatin is a proteolytic fragment of C-terminal, non-collagenous (NC1) domain of type XVIII collagen (reviewed in ref. [169]). It was isolated as a proteolytic fragment from a hemangioendothelioma, cloned and shown to regress primary tumours in mice [170]. Null mice generated by inactivation of the collagen XVIII gene, Col18a1, appeared to have no major defect in vascular patterning or capillary density in most organs, and the experiments involving tumour growth showed no significant difference with wildtype animals [171, 172]. Further examination, however, using a modified aortic explant assay, revealed a two-fold increase in microvessel growth in knockout versus wildtype animals, which could be eliminated by administration of low levels (0.1 g/mL) of endostatin [173]. Analysis of endothelial cells from type XVIII collagen wildtype and null mice found that Col18a1-/-cells are more adhesive to fibronectin [173]. This increased adhesion in the null mice, considered in light of the two-fold microvessel outgrowth of the knockout in the modified aortic explant assay, indicates a possible stabilization in newly formed vessels allowing for reduced regression and an increase in microvessel outgrowth [173]. Endostatin added exogenously was able to reduce the increased adhesion of type XVIII collagen null cells to wildtype levels [173]. Due to striking differences in the increased adherence of fibronectin in knockout cells, an RGD peptide was used to examine the role of α5β1 integrin, which is a known binding partner of fibronectin. Both wildtype and knockout endothelial cells were proportionally inhibited in the binding assay, suggesting α5β1 inte-grin is probably not a cause of the fibronectin binding difference in the mouse model [173]. Another possible binding partner is heparan sulfate proteoglycans (see Fibronectin is essential for vascular development). In another study, endostatin has been demonstrated to regulate endothelial cell adhesion and cytoskeletal organization similar to that of FGF2 [174] and, in a later report, showed the inhibition of FGF2 and VEGFA mediated angiogenesis in the chick chorioallantoic membrane assay with endo-statin where inactivation of heparin binding domains by mutagenesis negated the effects of growth factor inhibition, implying that endostatin attenuates its anti-angiogenic effects via heparin/heparan sulfate [175]. Heparin or related GAGs could also mediate the adherence of endothelial cells from type XVIII collagen null mice.
Other NC domains of chains of type IV collagen can also generate anti-angiogenic fragments (Table 2) (reviewed in ref. [176]). Arresten, derived from (β1(IV)NC1), has been shown to inhibit endothelial cell proliferation and tumour growth [103, 177]. Mechanistically, arresten also mediates its effects viaα1β1 integrin, which inhibits migration, proliferation and tube formation in endothelial cells [178]. Similarly, tumstatin, canstatin and an unnamed type IV (6) chain have been shown to be inhibitors of angiogenesis [176]. Another recent report links the p53 tumour suppressor pathway to production of collagen XVIII and collagen IV anti-angiogenic fragments, endostatin and tumstatin (reviewed in ref. [179]) [180]. The enzyme, α(II) collagen prolyl-4-hydroxylase (α2PH), is responsible for the hydroxylation of proline, which is critical for triple-helical strand formation in collagen biosynthesis and is a rate-limiting step in the pathway [181]. p53 transcriptionally activates α2PH gene, which results in the extracellular release of collagen fragments, endostatin and tumstatin [180]. In the case of mutant p53, α2PH is not activated and there is a reduction in the level of secreted anti-angiogenic fragments. In vivo testing consisted of α2PH expressing H1299 cells grafted in to the flanks of nude mice where ectopic expression resulted in almost complete suppression of tumour growth as compared to two control lines, which developed larger tumours within one month [180]. Also, in xenografted tumours from HCT116 cells, tumour cells from p53-/-cells had reduced tumstatin staining and increased vascularization versus tumours derived from wildtype HCT116 cell [180], suggesting that the production of collagen derived anti-angiogenic fragments may be in concert with a p53-dependent mechanism of anti-angiogenesis.
Fragments derived from fibronectin, perlecan and elastin have also been shown to modulate tumour angiogenesis. Anastellin, which is a fragment of the III1c repeat of fibronectin, has been shown to suppress tumour growth and metastasis in mouse and human models of cancer [182, 183]. Endorepellin, from the C-terminal, domain V fragment of perlecan, has also shown to inhibit angiogenesis and block migration of endothelial cells [184], which ultimately leads to actin disassembly (reviewed in ref. [12]) [185]. γ-elastins are peptides generated by partial hydrolysis of ligamentum nuchae γelastin in 1M KOH in 80% aqueous ethanol [186]. The sequence, VGVAPG–found in γ-elastins, has been found to be one of the angiogeneic motifs that are repeated multiple times in tropoelastin [187]. These peptides were then employed in various angiogenesis assays where they enhanced angiogenesis in the chick chorioallantoic membrane assay, enhanced capillary tube formation of human microvascular endothelial cells and human umbilical vein endothelial cells on type I collagen gel, and increased pro-MMP2 and pro-MT-MMP in human microvascular endothelial cells, leading to the conclusion that elastin peptides can promote endothelial cell migration and tubulogenesis through upregulation of MT1-MMP [108, 188].
Proteins involved in signalling through ECM molecules
Endogenous ECM components and their fragments can regulate blood vessel formation. Other molecules, also have been found which can modulate, inhibit or coordinate the signalling functions of ECM residents. Among the many that have been reported, exploration of two prominent families is presented.
Thrombospondins
Composed of five modular, secreted glycoproteins with no known orthologs in flies or worms, the mechanism of the thrombospondin (TSP) family are not well understood [189]. The TSP family can be divided into two groups: TSP1 and TSP2 form one subgroup of the larger chain mass at ∼145 kD and TSP3, TSP4 and TSP5 form a second subgroup with chain mass of ∼100 kD [189]. TSP1 and TSP2 have demonstrated ability to inhibit angiogenesis, reduce endothelial cell migration and mediate endothelial cell survival (reviewed in ref. [190]) and will be the focus of discussion. Eight receptors have been identified for TSP1 and, of these, five (CD36 [191, 192], CD47 [193], heparan sulfate proteoglycans [194], α3β1 integrin [195], and α4β1 integrin [196]) are thought to be involved in angiogenesis. In addition, TSP1 has also been shown to participate in platelet aggregation [197]. TSP1 and TSP2 have been detected in endothelial cells, fibroblasts and platelets under varying conditions, which have made the tissue source of the participating TSP in angiogenesis difficult to identify (reviewed in ref. [198]). The generation of TSP1 null mice revealed viable mice that had increased inflammatory cell infiltrates and epithelial cell hyperplasia in the lungs resulting in pneumonia and none of the other abnormalities that might be expected from TSP1 in vitro data suggesting other TSPs or proteins compensate for the lack of TSP1 [199]. Further work with muscle tissue explant culture from TSP1 null mice demonstrated enhanced neo-vascularization associated with endotheolial cell out-growth but perivascular smooth muscle cells displayed decreased outgrowth [200]. In the same study, examination of the mRNA extracted from out-growths revealed that endogenous TSP1 increased levels of type IV collagen (αl) and decreased levels of type I collagens (α1, α2) and type III collagen (α1) suggesting that TSP1 regulates a set of molecules, which control the activation of angiogenesis [200].
To the untrained eye, TSP2 null mice look normal and reproduced in the appropriate frequency. With closer examination, however, numerous subtle defects were apparent (i.e. greater elasticity in skin, ability to fashion knots in the tail) in null mice, which were determined to be improper organization of collagen fibrils [201]. Also, an abnormal bleeding time led to discovery that TSP2 null mice had greater vascular density than wildtype, which correlated with previous in vitro data suggesting that TSP2 was an inhibitor of angiogenesis [201]. Recently, a TSP1-TSP2 double knockout (TSP-DKO) was generated, which has provided some insight into the role of TSP in angiogenesis [202]. Immunostaining with TSP1 in wildtype versus double knockout mice of bone marrow revealed that megakaryocytes rather than other cell types express TSPs [202]. In addition, megakaryocytes from wildtype animals stimulated angiogenesis in a Matrigel plug assay but cells from TSP-DKO showed 1.8-fold greater stimulation over a 21 day period, suggesting TSPs in wildtype animals are controlling the level of angiogenesis [202]. Bone marrow myelosuppression studies were also used to demonstrate that TSP-DKO mice, which had enhanced recovery in hind limb ischemia model, could donate bone marrow via irradiation and then transplantation to wildtype mice resulting in the transfer of the enhanced neoangiogenesis seen in the knockout mice had been conferred to the wildtype mice [202]. Experimental results suggested that TSP1 and TSP2 control platelet numbers in the blood by regulating the amount of megakaryocyte production in the marrow through regulating vascular density. Thus, thrombopoietic cells can modulate angiogenesis by releasing inhibitory TSPs [198, 202].
CCN proteins
The CCN family of secreted proteins were individually discovered and characterized but were later grouped into a protein family based on the high degree of similarity in their predicted secondary structure [203, 204]. These six proteins, the family name of which is derived from the first initials of the first three members (Cysteine-rich protein 61-cyr61, CCN1; Connective tissue growth factor—CTGF, CCN2; Nephroblastoma overexpressed protein— Nov, CCN3; the other members are, respectively, Wnt-inducible secreted protein-1, -2, -3—WISP-1, -2, -3, CCN4, CCN5, CCN6) contain four distinct modules, which have been demonstrated to react with a wide variety of ECM molecules. From N-terminus to C-terminus, the four modules are an insulin-like growth factor binding protein (IGFBP-module I), a Von Willebrand factor domain (VWC- module II), a thrombospondin-homology domain (TSP1-module III) and a cysteine knot, heparin binding carboxy terminal domain (CT-module IV). Being individually coded on single exons, these modules most likely evolved through exon shuffling [203] and binding studies have shown that most of the individual domains can function in the absence of the others (reviewed in ref. [205]). Experiments analyzing this protein family have yielded data relating to many cellular physiological processes including migration, proliferation, adhesion, differentiation, apoptosis (and/or anoikis) and chondrogenesis. There are several recent reviews [205–209] and a book [204] discussing the CCN family. Specific to this review, experimental data demonstrating the involvement of CCN family members in vasculogenic and angio-geneic processes will be discussed below.
Being associated with the cell through numerous binding partners allows CCN1 (Cyr61, Cysteine rich protein 61) to be involved in many different biological functions, although its role in neovascularization seems to be the most studied. CCN1 binding is currently known to be associated with five integrins (αvβ3, αvβ5, a6β1, αIIIbβ3, αMβ2) and heparan sulfate proteoglycans [210]. From a gene expression perspective, CCN1 can regulate expression of genes involved in angiogenesis and the ECM (i.e. type I collagen, MMP1, MMP3, TIMPs, VEGF-A and VEGF-C) [211]. Interest in CCN1 as a mediator of angiogenesis was demonstrated [212] in the rat corneal pocket angiogenesis assay where CCN1 promoted corneal neovascularization as did FGF2 but not vehicle or pre-incubated CCN1 with anti-CCN1 antibodies. Later, in the rabbit hind limb ischemia model, adenoviral mediated human CCN1 gene transfer was shown to be a potent stimulus of revascularization of the limb even exceeding that of the positive control of adenoviral mediated VEGF165 [213]. In studies of wound healing, CCN1 shows high induction in dermal fibroblasts in granulation tissues during cutaneous wound repair and, consequently, is presumed to activate a genetic program of wound repair in skin fibroblasts [211, 214]. CCN1 has also been associated in regulation of expression of other tissues response to injury such as bone fracture repair and liver regeneration [214, 215]. In the context of embryogenesis, CCN1 has been visualized at sites of mesenchymal condensation and neovascularization as well as being involved in promoting uterine vessel growth [216]. Construction of a CCN1 null mouse corroborated the results of the above experimental observations in that 30% of null mice suffered failure of chorioallantoic membrane fusion while the remainder died due to placental vascular defects and compromised vascular development [217]. Further investigation revealed that CCN1 deficiency resulted in a problem associated with vessel bifurcation (non-sprouting angiogenesis) at the chorioallantoic junction, which causes reduced vascularization of the placenta. Correlation of this phenotype with that of null mutants of VEGF receptor 3 [218], which is deficient in one of the VEGF-C receptors and results in a dysregulated Vegf-C expression phenotype in the allantoic mesoderm, suggests that CCN1 regulated expression of Vegf-C is involved in vessel bifurcation [217]. Also, in v integrin null mice, 80% of the mice die due to undervascularization of the placenta. In the placental labyrinth zone, the αv integrin null mice appear morphologically similar to CCN1-null mice with foetal vessels at the chorionic plate appearing collapsed or absent [219]. Since embryonic vessels and the primary vascular plexus of the yolk sac successfully form, CCN1 appears not to be required for vasculogenesis [217].
In vitro experiments at the cellular and molecular level involving CCN1 have revealed differing effects on many cell types, which are believed to be receptor specific. Most of these differences have been attributed to binding of different integrins associated with multiple cell types. In fibroblasts and smooth muscle cells, for example, CCN1 induces cell adhesion by binding to a61 integrin and heparan sulfate proteoglycans as coreceptors [220] while its promotion of angiogenesis in endothelial cells has been shown to be through binding the αvβ3 integrin [221]. Also interesting is the data that activated endothelial cell adhesion to matrix CCN1 via αvβ3 integrin protects cells by suppressing apoptosis while fibroblast adhesion to matrix CCN1 via α6β1 integrin and a specific heparan sulfate proteoglycan, syndecan-4 (antibodies to syndecan-4 inhibited the effect and syndecan-4 was found in focal adhesions with CCN1), induces apoptosis by a non-transcription dependent p53 mechanism leading to caspase-9 and caspase-3 activation [222]. Thus, certain matrix proteins in combination with syndecan-4 and/or heparan sulfate proteoglycans can suppress or induce apoptosis by adhesion in a cell-type dependent manner and, therefore, at the tissue level, may play key roles in de novo or neovascular vessel formation [205, 206, 222].
Not surprisingly, due to their related structure, CCN2 (CTGF, connective tissue growth factor) has similar properties of CCN1. Both proteins play key regulatory roles in survival and function of endothelial cells and in angiogenesis [208] by promoting endothelial cell growth, adhesion and migration [212, 223–225]. As for their roles in embryonic angiogenesis, CCN2 null mice are also embryonic lethal but not due to placental vascular defects or vessel integrity but because of defects in endochondral ossification resulting in skeletal defects from dysregulation of bone growth [226] of which one result is the inability to breathe at birth due to abnormal rib formation. Closer examination of these mice, however, also reveals that CCN2 null mice have expansion of the hypertrophic zones in growth plates, which has been previously demonstrated to result from an impairment of angiogenesis [226–228]. Immunostaining for PECAM confirmed that in neonatal CCN2 wildtypes, a fine network of capillaries was present in well-developed ossification zones while in CCN2 null mice capillary formation was much less extensive [226]. These defects were ultimately suggested to be due to decreased expression of VEGF in the hypertrophic zones of CCN2-null mice. Reduced CCN2 results in inefficient infiltration of osteoclasts/chondroclasts, which express MMP-9. By these cells not being present, the level of MMP-9 was reduced and MMP-9 is required for growth plate angiogenesis [229]. MMP mediated degradation of the matrix activates VEGF, which is chemotactic for osteoclasts [226, 227]. In other work with CCN2 related to VEGF regulation, a yeast two-hybrid binding study screening potential binding inhibitors to VEGF165 from a human chondrocyte cDNA library resulted in CCN2 as a candidate inhibitor molecule [230]. CCN2 was determined to bind to VEGF165 through the thrombospondin-1 domain of CCN2 and the exon-7 coding region of VEGF165. Further studies demonstrated that (1) CCN2 could inhibit the binding of VEGF165 to human umbilical vein endothelial cells and to recombinant VEGF Receptor 2 but not VEGF Receptor 1; (2) in vitro, CCN2 can inhibit VEGF165 induced tube formation of bovine aortic endothelial cells by <45% versus VEGF165 alone and the effect was reversed by addition of anti-CCN2 antibody; (3) in vivo, CCN2 inhibited VEGF165 stimulation of vessel formation in the Matrigel injection model in mice [230]. Although these results of CCN2 being pro- and anti-angiogeneic may seem contradictory, the context and quantity of the molecule may be important for which effect CCN2 mediates [205]. Finally, in an investigation examining the physiological relevance of CCN2 mediated cell adhesion, CCN2 null fibroblasts were discovered to have reduced extracellular signal-regulated kinase (ERK) and focal adhesion kinase (FAK) phosphorylation, reduced -smooth muscle actin stress fibre formation and impaired spreading on fibronectin [231]. Given that CCN2 binds fibronectin and 4 integrins, 1 integrins, 5 integrins and syndecan-4 and is involved in tissue development and wound healing (reviewed in ref. [206]), these results suggest that the physiological role of CCN2 may be in regulation of fibroblast adhesion [231].
CCN3 (Nov, nephroblastoma overexpressed), the third member of the CNN family, was originally discovered as an upregulated gene in avian nephrob-lastomas induced by myeloblastosis associated virus (MAV) (reviewed in ref. [232]) [233]. Thus, early analyses of CCN3 were focused in cancer [232]. Later, with the identification of the CCN protein family, CCN3 was characterized as CCN1 and CCN2 and was shown to be involved in cell migration, adhesion and survival in endothelial cells [234]. CCN3 is also involved in the angiogenic process as it induced neo-vascularization in the rat cornea model [234] as well as being highly expressed in granulation tissue of cutaneous wounds five to seven days after injury [235], and rat aortic vascular smooth muscle cells express CCN3, in vivo and in vitro with high expression in the aorta, and start with low expression at day 7 but increase to high expression in the intima at day 14 in the rat carotid artery balloon injury model implicating CCN3 in vascular injury [236]. There are some distinct differences between CCN3 and the other CCNs. CCN3 from yeast has been shown to interact with Notch1 and influence Notch signalling through the Notch C-terminus [237] and Notch as been implicated in vessel development. Unlike CCN1& CCN2, CCN3 does not seem to mediate its wound healing effects via VEGF-A, but via another mechanism [235]. Further delineation of the role of CCN3 will most likely be provided by production of a CCN3 null mouse.
The remaining family members CCN4, CCN5 and CCN6 (Wnt inducible secreted proteins-1,-2,-3, Wisp -1,-2,-3) have had less examination due to a lack of readily available reagents (i.e. recombinant material and antibodies) [206]. CCN5 (also known as COP-1), which is the only CCN family member that lacks the CT module, was found to inhibit proliferation of vascular smooth muscle cells [238]. Human mutations in CCN6 have been reported to be associated with progressive pseudorheumatoid dysplasia [239], an autosomal recessive skeletal disorder where patients experience cartilage loss and destructive bone changes as they age. By modulation of the various molecules of the ECM, some members of the CCN family of proteins appear to play significant roles in neovascularization. Further work involving a CCN1-CCN2 double null mouse is already underway [205].
ECM signalling in vessel formation: what next?
By providing a signalling as well as a structural network, the ECM is a major player in blood vessel formation. Evolution, through gene (or genome) duplication and exon shuffling, has a rich environment of protein modules from which to select new ECM proteins. Aside from the new proteins waiting to be discovered, the current ones are still waiting to relin-quish more secrets. Several themes have emerged. Integrins, certainly the king of ECM receptor function, do not mediate all the communication from the ECM to the cell. GAGs seem to be involved in transmitting some of the ECM signalling (i.e. VEGF and endostatin signalling via fibronectin) and with their diversity of structure may be more involved in ECM signalling as co-receptors or bona fide receptors in the future. Also, as the proteome is smaller than expected, more molecular functions may be related to either post-translational modification (i.e. glycosylation, glycanation, hydroxylation) or in fragmentation of larger molecules to be used to employ new functions.
From a therapeutic prospective, one of the goals of angiogenic study should be to harness these ECM molecules to either target the growth of new vessels or use nature's paradigms to create artifical ones. Progress is being made in these areas. Tissue engineered blood vessels (TEBVs) have been constructed from a patient's own cells which serve as arterial bypass grafts in long term animal models [240]. Also, biodegradable polymers such as the biodegradable elastomer, which mimic the natural network of collagen and elastin have potential for uses in tissue engineering and drug delivery [241]. As more knowledge is accumulated concerning how the ECM functions in neovascularization, more amazing technical and medical breakthroughs should be possible.
Acknowledgments
The authors would like to thank the members of the Angiogenesis Research Center, Dartmouth Medical School for insightful comments and discussions. Supported in part by NIH grant HL62289 (MS).
References
- 1.Sullivan L. In: Athey I, Kindergarten chats (revised 1918) and other writings. New York: Peter Smith; >The tall office building artistically considered. pp. 202–13. [Google Scholar]
- 2.Hynes RO, Zhao Q. The evolution of cell adhesion. J Cell Biol. 2000;150:F89–96. doi: 10.1083/jcb.150.2.f89. [DOI] [PubMed] [Google Scholar]
- 3.Huxley-Jones J, Robertson DL, Boot-Handford RP. On the origins of the extracellular matrix in vertebrates. Matrix Biol. 2007;26:2–11. doi: 10.1016/j.matbio.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 4.Prockop DJ, Kivirikko KI. Collagens: molecular biology, diseases, and potentials for therapy. Annu Rev Biochem. 1995;64:403–34. doi: 10.1146/annurev.bi.64.070195.002155. [DOI] [PubMed] [Google Scholar]
- 5.Christiano AM, Greenspan DS, Lee S, Uitto J. Cloning of human type VII collagen. Complete primary sequence of the alpha 1(VII) chain and identification of intragenic polymorphisms. J Biol Chem. 1994;269:20256–62. [PubMed] [Google Scholar]
- 6.Uitto J, Chung-Honet LC, Christiano AM. Molecular biology and pathology of type VII collagen. Exp Dermatol. 1992;1:2–11. doi: 10.1111/j.1600-0625.1992.tb00065.x. [DOI] [PubMed] [Google Scholar]
- 7.Kuhn K, Wiedemann H, Timpl R, Risteli J, Dieringer H, Voss T, Glanville RW. Macromolecular structure of basement membrane collagens. FEBS Lett. 1981;125:123–8. doi: 10.1016/0014-5793(81)81012-5. [DOI] [PubMed] [Google Scholar]
- 8.Timpl R, Wiedemann H, Van Delden V, Furthmayr H, Kuhn K. A network model for the organization of type IV collagen molecules in basement membranes. Eur J Biochem. 1981;120:203–11. doi: 10.1111/j.1432-1033.1981.tb05690.x. [DOI] [PubMed] [Google Scholar]
- 9.Oh SP, Kamagata Y, Muragaki Y, Timmons S, Ooshima A, Olsen BR. Isolation and sequencing of cDNAs for proteins with multiple domains of Gly-Xaa- Yaa repeats identify a distinct family of collagenous proteins. Proc Natl Acad Sci USA. 1994;91:4229–33. doi: 10.1073/pnas.91.10.4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oh SP, Warman ML, Seldin MF, Cheng SD, Knoll JH, Timmons S, Olsen BR. Cloning of cDNA and genomic DNA encoding human type XVIII collagen and localization of the alpha 1(XVIII) collagen gene to mouse chromosome 10 and human chromosome 21. Genomics. 1994;19:494–9. doi: 10.1006/geno.1994.1098. [DOI] [PubMed] [Google Scholar]
- 11.Marneros AG, Olsen BR. The role of collagenderived proteolytic fragments in angiogenesis. Matrix Biol. 2001;20:337–45. doi: 10.1016/s0945-053x(01)00151-2. [DOI] [PubMed] [Google Scholar]
- 12.Iozzo RV. Basement membrane proteoglycans: from cellar to ceiling. Nat Rev Mol Cell Biol. 2005;6:646–56. doi: 10.1038/nrm1702. [DOI] [PubMed] [Google Scholar]
- 13.Halfter W, Dong S, Schurer B, Cole GJ. Collagen XVIII is a basement membrane heparan sulfate proteoglycan. J Biol Chem. 1998;273:25404–12. doi: 10.1074/jbc.273.39.25404. [DOI] [PubMed] [Google Scholar]
- 14.Rehn M, Pihlajaniemi T. Alpha 1(XVIII), a collagen chain with frequent interruptions in the collagenous sequence, a distinct tissue distribution, and homology with type XV collagen. Proc Natl Acad Sci USA. 1994;91:4234–8. doi: 10.1073/pnas.91.10.4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rehn M, Pihlajaniemi T. Identification of three N-terminal ends of type XVIII collagen chains and tissuespecific differences in the expression of the corresponding transcripts. The longest form contains a novel motif homologous to rat and Drosophilafrizzled proteins. J Biol Chem. 1995;270:4705–11. doi: 10.1074/jbc.270.9.4705. [DOI] [PubMed] [Google Scholar]
- 16.Muragaki Y, Timmons S, Griffith CM, Oh SP, Fadel B, Quertermous T, Olsen BR. Mouse Col18a1 is expressed in a tissue-specific manner as three alternative variants and is localized in basement membrane zones. Proc Natl Acad Sci USA. 1995;92:8763–7. doi: 10.1073/pnas.92.19.8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saarela J, Rehn M, Oikarinen A, Autio-Harmainen H, Pihlajaniemi T. The short and long forms of type XVIII collagen show clear tissue specificities in their expression and location in basement membrane zones in humans. Am J Pathol. 1998;153:611–26. doi: 10.1016/S0002-9440(10)65603-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Myers JC, Dion AS, Abraham V, Amenta PS. Type XV collagen exhibits a widespread distribution in human tissues but a distinct localization in basement membrane zones. Cell Tissue Res. 1996;286:493–505. doi: 10.1007/s004410050719. [DOI] [PubMed] [Google Scholar]
- 19.Kivirikko S, Saarela J, Myers JC, Autio-Harmainen H, Pihlajaniemi T. Distribution of type XV collagen transcripts in human tissue and their production by muscle cells and fibroblasts. Am J Pathol. 1995;147:1500–9. [PMC free article] [PubMed] [Google Scholar]
- 20.Ricard-Blum S, Ruggiero F. The collagen superfamily: from the extracellular matrix to the cell membrane. Pathol Biol. 2005;53:430–42. doi: 10.1016/j.patbio.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 21.Patarroyo M, Tryggvason K, Virtanen I. Laminin isoforms in tumor invasion, angiogenesis and metastasis. Semin Cancer Biol. 2002;12:197–207. doi: 10.1016/S1044-579X(02)00023-8. [DOI] [PubMed] [Google Scholar]
- 22.Colognato H, Yurchenco PD. Form and function: the laminin family of heterotrimers. Dev Dyn. 2000;218:213–34. doi: 10.1002/(SICI)1097-0177(200006)218:2<213::AID-DVDY1>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 23.Beck K, Dixon TW, Engel J, Parry DA. Ionic interactions in the coiled-coil domain of laminin determine the specificity of chain assembly. J Mol Biol. 1993;231:311–23. doi: 10.1006/jmbi.1993.1284. [DOI] [PubMed] [Google Scholar]
- 24.Yurchenco PD, Amenta PS, Patton BL. Basement membrane assembly, stability and activities observed through a developmental lens. Matrix Biol. 2004;22:521–38. doi: 10.1016/j.matbio.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 25.Frieser M, Nockel H, Pausch F, Roder C, Hahn A, Deutzmann R, Sorokin LM. Cloning of the mouse laminin alpha 4 cDNA. Expression in a subset of endothelium. Eur J Biochem. 1997;246:727–35. doi: 10.1111/j.1432-1033.1997.t01-1-00727.x. [DOI] [PubMed] [Google Scholar]
- 26.Sorokin LM, Pausch F, Frieser M, Kroger S, Ohage E, Deutzmann R. Developmental regulation of the laminin alpha5 chain suggests a role in epithelial and endothelial cell maturation. Dev Biol. 1997;189:285–300. doi: 10.1006/dbio.1997.8668. [DOI] [PubMed] [Google Scholar]
- 27.Ruoslahti E. Fibronectin and its receptors. Annu Rev Biochem. 1988;57:375–413. doi: 10.1146/annurev.bi.57.070188.002111. [DOI] [PubMed] [Google Scholar]
- 28.Wierzbicka-Patynowski I, Schwarzbauer JE. The ins and outs of fibronectin matrix assembly. J Cell Sci. 2003;116:3269–76. doi: 10.1242/jcs.00670. [DOI] [PubMed] [Google Scholar]
- 29.Johnson KJ, Sage H, Briscoe G, Erickson HP. The compact conformation of fibronectin is determined by intramolecular ionic interactions. J Biol Chem. 1999;274:15473–9. doi: 10.1074/jbc.274.22.15473. [DOI] [PubMed] [Google Scholar]
- 30.Schwarzbauer JE, Sechler JL. Fibronectin fibrillogenesis: a paradigm for extracellular matrix assembly. Curr Opin Cell Biol. 1999;11:622–7. doi: 10.1016/s0955-0674(99)00017-4. [DOI] [PubMed] [Google Scholar]
- 31.Hynes RO, Destree AT. Relationships between fibronectin (LETS protein) and actin. Cell. 1978;15:875–86. doi: 10.1016/0092-8674(78)90272-6. [DOI] [PubMed] [Google Scholar]
- 32.Pankov R, Yamada KM. Fibronectin at a glance. J Cell Sci. 2002;115:3861–3. doi: 10.1242/jcs.00059. [DOI] [PubMed] [Google Scholar]
- 33.Engel J, Odermatt E, Engel A, Madri JA, Furthmayr H, Rohde H, Timpl R. Shapes, domain organizations and flexibility of laminin and fibronectin, two multifunctional proteins of the extracellular matrix. J Mol Biol. 1981;150:97–120. doi: 10.1016/0022-2836(81)90326-0. [DOI] [PubMed] [Google Scholar]
- 34.Tamkun JW, Schwarzbauer JE, Hynes RO. A single rat fibronectin gene generates three different mRNAs by alternative splicing of a complex exon. Proc Natl Acad Sci USA. 1984;81:5140–4. doi: 10.1073/pnas.81.16.5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwarzbauer JE, Patel RS, Fonda D, Hynes RO. Multiple sites of alternative splicing of the rat fibronectin gene transcript. EMBO J. 1987;6:2573–80. doi: 10.1002/j.1460-2075.1987.tb02547.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 37.Ruoslahti E, Pierschbacher MD. New perspectives in cell adhesion: RGD and integrins. Science. 1987;238:491–7. doi: 10.1126/science.2821619. [DOI] [PubMed] [Google Scholar]
- 38.Leahy DJ. Implications of atomic-resolution structures for cell adhesion. Annu Rev Cell Dev Biol. 1997;13:363–93. doi: 10.1146/annurev.cellbio.13.1.363. [DOI] [PubMed] [Google Scholar]
- 39.Rosenbloom J, Abrams WR, Mecham R. Extracellular matrix 4: the elastic fiber. FASEB J. 1993;7:1208–18. [PubMed] [Google Scholar]
- 40.Partridge SM, Elsden DF, Thomas J, Dorfman A, Telser A, Ho PL. Biosynthesis of the desmosine and isodesmosine cross-bridges in elastin. Biochem J. 1964;93:30C–3C. [PubMed] [Google Scholar]
- 41.Francis G, John R, Thomas J. Biosynthetic pathway of desmosines in elastin. Biochem J. 1973;136:45–55. doi: 10.1042/bj1360045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sakai LY, Keene DR, Engvall E. Fibrillin, a new 350- kD glycoprotein, is a component of extracellular microfibrils. J Cell Biol. 1986;103:2499–509. doi: 10.1083/jcb.103.6.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brooke BS, Bayes-Genis A, Li DY. New insights into elastin and vascular disease. Trends Cardiovasc Med. 2003;13:176–81. doi: 10.1016/s1050-1738(03)00065-3. [DOI] [PubMed] [Google Scholar]
- 44.Wolinski H, Glagov S. Lamellar unit of aortic medial structure and function in mammals. Circ Res. 1967;20:99–111. doi: 10.1161/01.res.20.1.99. [DOI] [PubMed] [Google Scholar]
- 45.Miner JH, Yurchenco PD. Laminin functions in tissue morphogenesis. Annu Rev Cell Dev Biol. 2004;20:255–84. doi: 10.1146/annurev.cellbio.20.010403.094555. [DOI] [PubMed] [Google Scholar]
- 46.Durkin ME, Wewer UM, Chung AE. Exon organization of the mouse entactin gene corresponds to the structural domains of the polypeptide and has regional homology to the low-density lipoprotein receptor gene. Genomics. 1995;26:219–28. doi: 10.1016/0888-7543(95)80204-y. [DOI] [PubMed] [Google Scholar]
- 47.Timpl R. Macromolecular organization of basement membranes. Curr Opin Cell Biol. 1996;8:618–24. doi: 10.1016/s0955-0674(96)80102-5. [DOI] [PubMed] [Google Scholar]
- 48.Aumailley M, Battaglia C, Mayer U, Reinhardt D, Nischt R, Timpl R, Fox JW. Nidogen mediates the formation of ternary complexes of basement membrane components. Kidney Int. 1993;43:7–12. doi: 10.1038/ki.1993.3. [DOI] [PubMed] [Google Scholar]
- 49.Hopf M, Gohring W, Kohfeldt E, Yamada Y, Timpl R. Recombinant domain IV of perlecan binds to nidogens, laminin-nidogen complex, fibronectin, fibulin-2 and heparin. Eur J Biochem. 1999;259:917–25. doi: 10.1046/j.1432-1327.1999.00127.x. [DOI] [PubMed] [Google Scholar]
- 50.Takagi J, Yang Y, Liu JH, Wang JH, Springer TA. Complex between nidogen and laminin fragments reveals a paradigmatic beta-propeller interface. Nature. 2003;424:969–74. doi: 10.1038/nature01873. [DOI] [PubMed] [Google Scholar]
- 51.Salmivirta K, Talts JF, Olsson M, Sasaki T, Timpl R, Ekblom P. Binding of mouse nidogen-2 to basement membrane components and cells and its expression in embryonic and adult tissues suggest complementary functions of the two nidogens. Exp Cell Res. 2002;279:188–201. doi: 10.1006/excr.2002.5611. [DOI] [PubMed] [Google Scholar]
- 52.Varki A, Cummings R, Esko J, Freeze H, Hart G, Marth M, editors. Essentials of glycobiology. Cold Spring Harbor, NY: Cold Spring Harbor Press; 1999. [PubMed] [Google Scholar]
- 53.Esko JD, Selleck SB. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem. 2002;71:435–71. doi: 10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- 54.Hacker U, Nybakken K, Perrimon N. Heparan sulphate proteoglycans: the sweet side of development. Nat Rev Mol Cell Biol. 2005;6:530–41. doi: 10.1038/nrm1681. [DOI] [PubMed] [Google Scholar]
- 55.Bezakova G, Ruegg MA. New insights into the roles of agrin. Nat Rev Mol Cell Biol. 2003;4:295–308. doi: 10.1038/nrm1074. [DOI] [PubMed] [Google Scholar]
- 56.Li D, Clark CC, Myers JC. Basement membrane zone type XV collagen is a disulfide-bonded chondroitin sulfate proteoglycan in human tissues and cultured cells. J Biol Chem. 2000;275:22339–47. doi: 10.1074/jbc.M000519200. [DOI] [PubMed] [Google Scholar]
- 57.Cohen IR, Grassel S, Murdoch AD, Iozzo RV. Structural characterization of the complete human perlecan gene and its promoter. Proc Natl Acad Sci USA. 1993;90:10404–8. doi: 10.1073/pnas.90.21.10404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iozzo RV, San Antonio JD. Heparan sulfate proteoglycans: heavy hitters in the angiogenesis arena. J Clin Invest. 2001;108:349–55. doi: 10.1172/JCI13738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Knox S, Merry C, Stringer S, Melrose J, Whitelock J. Not all perlecans are created equal: interactions with fibroblast growth factor (FGF) 2 and FGF receptors. J Biol Chem. 2002;277:14657–65. doi: 10.1074/jbc.M111826200. [DOI] [PubMed] [Google Scholar]
- 60.Bix G, Iozzo RV. Matrix revolutions: “tails” of basement- membrane components with angiostatic functions. Trends Cell Biol. 2005;15:52–60. doi: 10.1016/j.tcb.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 61.Tkachenko E, Rhodes JM, Simons M. Syndecans: new kids on the signaling block. Circ Res. 2005;96:488–500. doi: 10.1161/01.RES.0000159708.71142.c8. [DOI] [PubMed] [Google Scholar]
- 62.Okamoto O, Bachy S, Odenthal U, Bernaud J, Rigal D, Lortat-Jacob H, Smyth N, Rousselle P. Normal human keratinocytes bind to the alpha3LG4/5 domain of unprocessed laminin-5 through the receptor syndecan-1. J Biol Chem. 2003;278:44168–77. doi: 10.1074/jbc.M300726200. [DOI] [PubMed] [Google Scholar]
- 63.Choi S, Lee E, Kwon S, Park H, Yi JY, Kim S, Han IO, Yun Y, Oh ES. Transmembrane domain-induced oligomerization is crucial for the functions of syndecan- 2 and syndecan-4. J Biol Chem. 2005;280:42573–9. doi: 10.1074/jbc.M509238200. [DOI] [PubMed] [Google Scholar]
- 64.Tkachenko E, Lutgens E, Stan RV, Simons M. Fibroblast growth factor 2 endocytosis in endothelial cells proceed via syndecan-4-dependent activation of Rac1 and a Cdc42-dependent macropinocytic pathway. J Cell Sci. 2004;117:3189–99. doi: 10.1242/jcs.01190. [DOI] [PubMed] [Google Scholar]
- 65.Tkachenko E, Elfenbein A, Tirziu D, Simons M. Syndecan-4 clustering induces cell migration in a PDZ-dependent manner. Circ Res. 2006;98:1398–404. doi: 10.1161/01.RES.0000225283.71490.5a. [DOI] [PubMed] [Google Scholar]
- 66.Wilcox-Adelman SA, Denhez F, Goetinck PF. Syndecan-4 modulates focal adhesion kinase phosphorylation. J Biol Chem. 2002;277:32970–7. doi: 10.1074/jbc.M201283200. [DOI] [PubMed] [Google Scholar]
- 67.Baciu PC, Goetinck PF. Protein kinase C regulates the recruitment of syndecan-4 into focal contacts. Mol Biol Cell. 1995;6:1503–13. doi: 10.1091/mbc.6.11.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–87. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 69.Whittaker CA, Hynes RO. Distribution and evolution of von Willebrand/integrin A domains: widely dispersed domains with roles in cell adhesion and elsewhere. Mol Biol Cell. 2002;13:3369–87. doi: 10.1091/mbc.E02-05-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Humphries JD, Byron A, Humphries MJ. Integrin ligands at a glance. J Cell Sci. 2006;119:3901–3. doi: 10.1242/jcs.03098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee JO, Bankston LA, Arnaout MA, Liddington RC. Two conformations of the integrin A-domain (Idomain): a pathway for activation? Structure. 1995;3:1333–40. doi: 10.1016/s0969-2126(01)00271-4. [DOI] [PubMed] [Google Scholar]
- 72.Lee JO, Rieu P, Arnaout MA, Liddington R. Crystal structure of the A domain from the alpha subunit of integrin CR3 (CD11b/CD18) Cell. 1995;80:631–8. doi: 10.1016/0092-8674(95)90517-0. [DOI] [PubMed] [Google Scholar]
- 73.Emsley J, Knight CG, Farndale RW, Barnes MJ, Liddington RC. Structural basis of collagen recognition by integrin alpha2beta1. Cell. 2000;101:47–56. doi: 10.1016/S0092-8674(00)80622-4. [DOI] [PubMed] [Google Scholar]
- 74.Zamir E, Geiger B. Components of cell-matrix adhesions. J Cell Sci. 2001;114:3577–9. doi: 10.1242/jcs.114.20.3577. [DOI] [PubMed] [Google Scholar]
- 75.Midwood KS, Mao Y, Hsia HC, Valenick LV, Schwarzbauer JE. Modulation of cell-fibronectin matrix interactions during tissue repair. J Invest Dermatol. 2006;126:73–8. doi: 10.1038/sj.jidsymp.5650005. [DOI] [PubMed] [Google Scholar]
- 76.Lu C, Takagi J, Springer TA. Association of the membrane proximal regions of the alpha and beta subunit cytoplasmic domains constrains an integrin in the inactive state. J Biol Chem. 2001;276:14642–8. doi: 10.1074/jbc.M100600200. [DOI] [PubMed] [Google Scholar]
- 77.Takagi J, Erickson HP, Springer TA. C-terminal opening mimics ‘inside-out’ activation of integrin alpha5beta1. Nat Struct Biol. 2001;8:412–6. doi: 10.1038/87569. [DOI] [PubMed] [Google Scholar]
- 78.Basson CT, Knowles WJ, Bell L, Albelda SM, Castronovo V, Liotta LA, Madri JA. Spatiotemporal segregation of endothelial cell integrin and nonintegrin extracellular matrix-binding proteins during adhesion events. J Cell Biol. 1990;110:789–801. doi: 10.1083/jcb.110.3.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- 80.Barresi R, Campbell KP. Dystroglycan: from biosynthesis to pathogenesis of human disease. J Cell Sci. 2006;119:199–207. doi: 10.1242/jcs.02814. [DOI] [PubMed] [Google Scholar]
- 81.Michele DE, Barresi R, Kanagawa M, Saito F, Cohn RD, Satz JS, Dollar J, Nishino I, Kelley RI, Somer H, Straub V, Mathews KD, Moore SA, Campbell KP. Post-translational disruption of dystroglycan- ligand interactions in congenital muscular dystrophies. Nature. 2002;418:417–22. doi: 10.1038/nature00837. [DOI] [PubMed] [Google Scholar]
- 82.Michele DE, Campbell KP. Dystrophin-glycoprotein complex: post-translational processing and dystroglycan function. J Biol Chem. 2003;278:15457–60. doi: 10.1074/jbc.R200031200. [DOI] [PubMed] [Google Scholar]
- 83.Shimizu H, Hosokawa H, Ninomiya H, Miner JH, Masaki T. Adhesion of cultured bovine aortic endothelial cells to laminin-1 mediated by dystroglycan. J Biol Chem. 1999;274:11995–2000. doi: 10.1074/jbc.274.17.11995. [DOI] [PubMed] [Google Scholar]
- 84.Simons M. Angiogenesis: where do we stand now? Circulation. 2005;111:1556–66. doi: 10.1161/01.CIR.0000159345.00591.8F. [DOI] [PubMed] [Google Scholar]
- 85.Helisch A, Schaper W. Arteriogenesis: the development and growth of collateral arteries. Microcirculation. 2003;10:83–97. doi: 10.1038/sj.mn.7800173. [DOI] [PubMed] [Google Scholar]
- 86.Schaper W, Scholz D. Factors regulating arteriogenesis. Arterioscler Thromb Vasc Biol. 2003;23:1143–51. doi: 10.1161/01.ATV.0000069625.11230.96. [DOI] [PubMed] [Google Scholar]
- 87.Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–60. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 88.Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9:685–93. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- 89.Jain RK, Carmeliet PF. Vessels of death or life. Sci Am. 2001;285:38–45. doi: 10.1038/scientificamerican1201-38. [DOI] [PubMed] [Google Scholar]
- 90.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–6. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 91.Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6:389–95. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- 92.Folkman J. Fundamental concepts of the angiogenic process. Curr Mol Med. 2003;3:643–51. doi: 10.2174/1566524033479465. [DOI] [PubMed] [Google Scholar]
- 93.Gilbert SF. Developmental Biology. Sunderland, MA: Sinauer Associates, Inc; 2000. [Google Scholar]
- 94.Davis GE, Senger DR. Endothelial extracellular matrix: biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ Res. 2005;97:1093–107. doi: 10.1161/01.RES.0000191547.64391.e3. [DOI] [PubMed] [Google Scholar]
- 95.Shalaby F, Ho J, Stanford WL, Fischer KD, Schuh AC, Schwartz L, Bernstein A, Rossant J. A requirement for Flk1 in primitive and definitive hematopoiesis and vasculogenesis. Cell. 1997;89:981–90. doi: 10.1016/s0092-8674(00)80283-4. [DOI] [PubMed] [Google Scholar]
- 96.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O'Shea KS, Powell-Braxton L, Hillan KJ, Moore MW. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–42. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 97.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in Flk-1- deficient mice. Nature. 1995;376:62–6. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 98.Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;376:66–70. doi: 10.1038/376066a0. [DOI] [PubMed] [Google Scholar]
- 99.Davis S, Aldrich TH, Jones PF, Acheson A, Compton DL, Jain V, Ryan TE, Bruno J, Radziejewski C, Maisonpierre PC, Yancopoulos GD. Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell. 1996;87:1161–9. doi: 10.1016/s0092-8674(00)81812-7. [DOI] [PubMed] [Google Scholar]
- 100.Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell. 1996;87:1171–80. doi: 10.1016/s0092-8674(00)81813-9. [DOI] [PubMed] [Google Scholar]
- 101.Vikkula M, Boon LM, Carraway KL, 3rd, Calvert JT, Diamonti AJ, Goumnerov B, Pasyk KA, Marchuk DA, Warman ML, Cantley LC, Mulliken JB, Olsen BR. Vascular dysmorphogenesis caused by an activating mutation in the receptor tyrosine kinase TIE2. Cell. 1996;87:1181–90. doi: 10.1016/s0092-8674(00)81814-0. [DOI] [PubMed] [Google Scholar]
- 102.Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology. 2005;69:4–10. doi: 10.1159/000088478. [DOI] [PubMed] [Google Scholar]
- 103.Colorado PC, Torre A, Kamphaus G, Maeshima Y, Hopfer H, Takahashi K, Volk R, Zamborsky ED, Herman S, Sarkar PK, Ericksen MB, Dhanabal M, Simons M, Post M, Kufe DW, Weichselbaum RR, Sukhatme VP, Kalluri R. Anti-angiogenic cues from vascular basement membrane collagen. Cancer Res. 2000;60:2520–6. [PubMed] [Google Scholar]
- 104.Sund M, Xie L, Kalluri R. The contribution of vascular basement membranes and extracellular matrix to the mechanics of tumor angiogenesis. APMIS. 2004;112:450–62. doi: 10.1111/j.1600-0463.2004.t01-1-apm11207-0806.x. [DOI] [PubMed] [Google Scholar]
- 105.Nyberg P, Xie L, Kalluri R. Endogenous inhibitors of angiogenesis. Cancer Res. 2005;65:3967–79. doi: 10.1158/0008-5472.CAN-04-2427. [DOI] [PubMed] [Google Scholar]
- 106.Folkman J. Angiogenesis. Annu Rev Med. 2006;57:1–18. doi: 10.1146/annurev.med.57.121304.131306. [DOI] [PubMed] [Google Scholar]
- 107.Rundhaug JE. Matrix metalloproteinases and angiogenesis. J Cell Mol Med. 2005;9:267–85. doi: 10.1111/j.1582-4934.2005.tb00355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bellon G, Martiny L, Robinet A. Matrix metalloproteinases and matrikines in angiogenesis. Crit Rev Oncol Hematol. 2004;49:203–20. doi: 10.1016/j.critrevonc.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 109.Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006;25:9–34. doi: 10.1007/s10555-006-7886-9. [DOI] [PubMed] [Google Scholar]
- 110.Van Hinsbergh VW, Engelse MA, Quax PH. Pericellular proteases in angiogenesis and vasculogenesis. Arterioscler Thromb Vasc Biol. 2006;26:716–28. doi: 10.1161/01.ATV.0000209518.58252.17. [DOI] [PubMed] [Google Scholar]
- 111.Roy R, Zhang B, Moses MA. Making the cut: protease- mediated regulation of angiogenesis. Exp Cell Res. 2006;312:608–22. doi: 10.1016/j.yexcr.2005.11.022. [DOI] [PubMed] [Google Scholar]
- 112.Sweeney SM, DiLullo G, Slater SJ, Martinez J, Iozzo RV, Lauer-Fields JL, Fields GB, San Antonio JD. Angiogenesis in collagen I requires alpha2beta1 ligation of a GFP*GER sequence and possibly p38 MAPK activation and focal adhesion disassembly. J Biol Chem. 2003;278:30516–24. doi: 10.1074/jbc.M304237200. [DOI] [PubMed] [Google Scholar]
- 113.Lohler J, Timpl R, Jaenisch R. Embryonic lethal mutation in mouse collagen I gene causes rupture of blood vessels and is associated with erythropoietic and mesenchymal cell death. Cell. 1984;38:597–607. doi: 10.1016/0092-8674(84)90514-2. [DOI] [PubMed] [Google Scholar]
- 114.Fouser L, Iruela-Arispe L, Bornstein P, Sage EH. Transcriptional activity of the alpha 1(I)-collagen promoter is correlated with the formation of capillary-like structures by endothelial cells in vitro. J Biol Chem. 1991;266:18345–51. [PubMed] [Google Scholar]
- 115.Iruela-Arispe ML, Diglio CA, Sage EH. Modulation of extracellular matrix proteins by endothelial cells undergoing angiogenesis in vitro. Arterioscler Thromb. 1991;11:805–15. doi: 10.1161/01.atv.11.4.805. [DOI] [PubMed] [Google Scholar]
- 116.Ingber D, Folkman J. Inhibition of angiogenesis through modulation of collagen metabolism. Lab Invest. 1988;59:44–51. [PubMed] [Google Scholar]
- 117.Sweeney SM, Guy CA, Fields GB, San Antonio JD. Defining the domains of type I collagen involved in heparin- binding and endothelial tube formation. Proc Natl Acad Sci USA. 1998;95:7275–80. doi: 10.1073/pnas.95.13.7275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Drake CJ, Brandt SJ, Trusk TC, Little CD. TAL1/SCL is expressed in endothelial progenitor cells/angioblasts and defines a dorsal-to-ventral gradient of vasculogenesis. Dev Biol. 1997;192:17–30. doi: 10.1006/dbio.1997.8751. [DOI] [PubMed] [Google Scholar]
- 119.Drake CJ, Little CD. VEGF and vascular fusion: implications for normal and pathological vessels. J Histochem Cytochem. 1999;47:1351–6. doi: 10.1177/002215549904701101. [DOI] [PubMed] [Google Scholar]
- 120.Drake CJ, Fleming PA. Vasculogenesis in the day 6.5 to 9.5 mouse embryo. Blood. 2000;95:1671–9. [PubMed] [Google Scholar]
- 121.Vernon RB, Sage EH. Between molecules and morphology .Extracellular matrix and creation of vascular form. Am J Pathol. 1995;147:873–83. [PMC free article] [PubMed] [Google Scholar]
- 122.Liu Y, Senger DR. Matrix-specific activation of Src and Rho initiates capillary morphogenesis of endothelial cells. FASEB J. 2004;18:457–68. doi: 10.1096/fj.03-0948com. [DOI] [PubMed] [Google Scholar]
- 123.Aloisi M, Schiaffino S. Growth of elementary blood vessels in diffusion chambers II. Electron microscopy of capillary morphogenesis. Virchows Arch B Cell Pathol. 1971;8:328–41. doi: 10.1007/BF02893542. [DOI] [PubMed] [Google Scholar]
- 124.Senger DR, Claffey KP, Benes JE, Perruzzi CA, Sergiou AP, Detmar M. Angiogenesis promoted by vascular endothelial growth factor: regulation through alpha1beta1 and alpha2beta1 integrins. Proc Natl Acad Sci USA. 1997;94:13612–7. doi: 10.1073/pnas.94.25.13612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Whelan MC, Senger DR. Collagen I initiates endothelial cell morphogenesis by inducing actin polymerization through suppression of cyclic AMP and protein kinase A. J Biol Chem. 2003;278:327–34. doi: 10.1074/jbc.M207554200. [DOI] [PubMed] [Google Scholar]
- 126.Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- 127.Eliceiri BP, Paul R, Schwartzberg PL, Hood JD, Leng J, Cheresh DA. Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol Cell. 1999;4:915–24. doi: 10.1016/s1097-2765(00)80221-x. [DOI] [PubMed] [Google Scholar]
- 128.Ridley AJ. Rho family proteins: coordinating cell responses. Trends Cell Biol. 2001;11:471–7. doi: 10.1016/s0962-8924(01)02153-5. [DOI] [PubMed] [Google Scholar]
- 129.Paulsson M, Aumailley M, Deutzmann R, Timpl R, Beck K, Engel J. Laminin-nidogen complex. Extraction with chelating agents and structural characterization. Eur J Biochem. 1987;166:11–9. doi: 10.1111/j.1432-1033.1987.tb13476.x. [DOI] [PubMed] [Google Scholar]
- 130.Timpl R, Rohde H, Robey PG, Rennard SI, Foidart JM, Martin GR. Laminin–a glycoprotein from basement membranes. J Biol Chem. 1979;254:9933–7. [PubMed] [Google Scholar]
- 131.Hallmann R, Horn N, Selg M, Wendler O, Pausch F, Sorokin LM. Expression and function of laminins in the embryonic and mature vasculature. Physiol Rev. 2005;85:979–1000. doi: 10.1152/physrev.00014.2004. [DOI] [PubMed] [Google Scholar]
- 132.Mao JR, Bristow J. The Ehlers-Danlos syndrome: on beyond collagens. J Clin Invest. 2001;107:1063–9. doi: 10.1172/JCI12881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Fleischmajer R, Perlish JS, Timpl R, Olsen BR. Procollagen intermediates during tendon fibrillogenesis. J Histochem Cytochem. 1988;36:1425–32. doi: 10.1177/36.11.3049791. [DOI] [PubMed] [Google Scholar]
- 134.Keene DR, Sakai LY, Bachinger HP, Burgeson RE. Type III collagen can be present on banded collagen fibrils regardless of fibril diameter. J Cell Biol. 1987;105:2393–402. doi: 10.1083/jcb.105.5.2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Liu X, Wu H, Byrne M, Krane S, Jaenisch R. Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proc Natl Acad Sci USA. 1997;94:1852–6. doi: 10.1073/pnas.94.5.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Poschl E, Schlotzer-Schrehardt U, Brachvogel B, Saito K, Ninomiya Y, Mayer U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development. 2004;131:1619–28. doi: 10.1242/dev.01037. [DOI] [PubMed] [Google Scholar]
- 137.Nagai N, Hosokawa M, Itohara S, Adachi E, Matsushita T, Hosokawa N, Nagata K. Embryonic lethality of molecular chaperone hsp47 knockout mice is associated with defects in collagen biosynthesis. J Cell Biol. 2000;150:1499–506. doi: 10.1083/jcb.150.6.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Matsuoka Y, Kubota H, Adachi E, Nagai N, Marutani T, Hosokawa N, Nagata K. Insufficient folding of type IV collagen and formation of abnormal basement membrane-like structure in embryoid bodies derived from Hsp47-null embryonic stem cells. Mol Biol Cell. 2004;15:4467–75. doi: 10.1091/mbc.E04-01-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bonanno E, Iurlaro M, Madri JA, Nicosia RF. Type IV collagen modulates angiogenesis and neovessel survival in the rat aorta model. In vitro Cell Dev Biol Anim. 2000;36:336–40. doi: 10.1290/1071-2690(2000)036<0336:TICMAA>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 140.Kortesmaa J, Yurchenco P, Tryggvason K. Recombinant laminin-8 (alpha(4)beta(1)gamma(1)). Production, purification,and interactions with integrins. J Biol Chem. 2000;275:14853–9. doi: 10.1074/jbc.275.20.14853. [DOI] [PubMed] [Google Scholar]
- 141.Doi M, Thyboll J, Kortesmaa J, Jansson K, Iivanainen A, Parvardeh M, Timpl R, Hedin U, Swedenborg J, Tryggvason K. Recombinant human laminin-10 (alpha5beta1gamma1). Production, purification, and migration-promoting activity on vascular endothelial cells. J Biol Chem. 2002;277:12741–8. doi: 10.1074/jbc.M111228200. [DOI] [PubMed] [Google Scholar]
- 142.Thyboll J, Kortesmaa J, Cao R, Soininen R, Wang L, Iivanainen A, Sorokin L, Risling M, Cao Y, Tryggvason K. Deletion of the laminin alpha4 chain leads to impaired microvessel maturation. Mol Cell Biol. 2002;22:1194–202. doi: 10.1128/MCB.22.4.1194-1202.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Miner JH, Cunningham J, Sanes JR. Roles for laminin in embryogenesis: exencephaly, syndactyly, and placentopathy in mice lacking the laminin alpha5 chain. J Cell Biol. 1998;143:1713–23. doi: 10.1083/jcb.143.6.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.George EL, Baldwin HS, Hynes RO. Fibronectins are essential for heart and blood vessel morphogenesis but are dispensable for initial specification of precursor cells. Blood. 1997;90:3073–81. [PubMed] [Google Scholar]
- 145.George EL, Georges-Labouesse EN, Patel-King RS, Rayburn H, Hynes RO. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development. 1993;119:1079–91. doi: 10.1242/dev.119.4.1079. [DOI] [PubMed] [Google Scholar]
- 146.Risau W. Vasculogenesis, angiogenesis and endothelial cell differentiation during embyronic development. In: Feinberg RN, Sherer GK, Auerbach R, editors. The development of the vascular system. 1991. pp. 58–68. [Google Scholar]
- 147.Taverna D, Hynes RO. Reduced blood vessel formation and tumor growth in alpha5-integrin-negative teratocarcinomas and embryoid bodies. Cancer Res. 2001;61:5255–61. [PubMed] [Google Scholar]
- 148.Yang JT, Rayburn H, Hynes RO. Embryonic mesodermal defects in alpha 5 integrin-deficient mice. Development. 1993;119:1093–105. doi: 10.1242/dev.119.4.1093. [DOI] [PubMed] [Google Scholar]
- 149.Wijelath ES, Murray J, Rahman S, Patel Y, Ishida A, Strand K, Aziz S, Cardona C, Hammond WP, Savidge GF, Rafii S, Sobel M. Novel vascular endothelial growth factor binding domains of fibronectin enhance vascular endothelial growth factor biological activity. Circ Res. 2002;91:25–31. doi: 10.1161/01.res.0000026420.22406.79. [DOI] [PubMed] [Google Scholar]
- 150.Wijelath ES, Rahman S, Namekata M, Murray J, Nishimura T, Mostafavi-Pour Z, Patel Y, Suda Y, Humphries MJ, Sobel M. Heparin-II domain of fibronectin is a vascular endothelial growth factorbinding domain: enhancement of VEGF biological activity by a singular growth factor/matrix protein synergism. Circ Res. 2006;99:853–60. doi: 10.1161/01.RES.0000246849.17887.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Barkalow FJ, Schwarzbauer JE. Localization of the major heparin-binding site in fibronectin. J Biol Chem. 1991;266:7812–8. [PubMed] [Google Scholar]
- 152.Kimizuka F, Taguchi Y, Ohdate Y, Kawase Y, Shimojo T, Hashino K, Kato I, Sekiguchi K, Titani K. Production and characterization of functional domains of human fibronectin expressed in Escherichia coli. J Biochem. 1991;110:284–91. doi: 10.1093/oxfordjournals.jbchem.a123572. [DOI] [PubMed] [Google Scholar]
- 153.Lyon M, Rushton G, Askari JA, Humphries MJ, Gallagher JT. Elucidation of the structural features of heparan sulfate important for interaction with the Hep-2 domain of fibronectin. J Biol Chem. 2000;275:4599–606. doi: 10.1074/jbc.275.7.4599. [DOI] [PubMed] [Google Scholar]
- 154.Woods A, McCarthy JB, Furcht LT, Couchman JR. A synthetic peptide from the COOH-terminal heparinbinding domain of fibronectin promotes focal adhesion formation. Mol Biol Cell. 1993;4:605–13. doi: 10.1091/mbc.4.6.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mitsi M, Hong Z, Costello CE, Nugent MA. Heparin-mediated conformational changes in fibronectin expose vascular endothelial growth factor binding sites. Biochemistry. 2006;45:10319–28. doi: 10.1021/bi060974p. [DOI] [PubMed] [Google Scholar]
- 156.Costell M, Gustafsson E, Aszodi A, Morgelin M, Bloch W, Hunziker E, Addicks K, Timpl R, Fassler R. Perlecan maintains the integrity of cartilage and some basement membranes. J Cell Biol. 1999;147:1109–22. doi: 10.1083/jcb.147.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Arikawa-Hirasawa E, Watanabe H, Takami H, Hassell JR, Yamada Y. Perlecan is essential for cartilage and cephalic development. Nat Genet. 1999;23:354–8. doi: 10.1038/15537. [DOI] [PubMed] [Google Scholar]
- 158.Zhou Z, Wang J, Cao R, Morita H, Soininen R, Chan KM, Liu B, Cao Y, Tryggvason K. Impaired angiogenesis, delayed wound healing and retarded tumor growth in perlecan heparan sulfate-deficient mice. Cancer Res. 2004;64:4699–702. doi: 10.1158/0008-5472.CAN-04-0810. [DOI] [PubMed] [Google Scholar]
- 159.Tran PK, Tran-Lundmark K, Soininen R, Tryggvason K, Thyberg J, Hedin U. Increased intimal hyperplasia and smooth muscle cell proliferation in transgenic mice with heparan sulfate-deficient perlecan. Circ Res. 2004;94:550–8. doi: 10.1161/01.RES.0000117772.86853.34. [DOI] [PubMed] [Google Scholar]
- 160.Alexopoulou AN, Multhaupt HA, Couchman JR. Syndecans in wound healing, inflammation and vascular biology. Int J Biochem Cell Biol. 2006 doi: 10.1016/j.biocel.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 161.Tkachenko E, Simons M. Clustering induces redistribution of syndecan-4 core protein into raft membrane domains. J Biol Chem. 2002;277:19946–51. doi: 10.1074/jbc.M200841200. [DOI] [PubMed] [Google Scholar]
- 162.Gotte M, Joussen AM, Klein C, Andre P, Wagner DD, Hinkes MT, Kirchhof B, Adamis AP, Bernfield M. Role of syndecan-1 in leukocyte-endothelial interactions in the ocular vasculature. Invest Ophthalmol Vis Sci. 2002;43:1135–41. [PubMed] [Google Scholar]
- 163.Elenius V, Gotte M, Reizes O, Elenius K, Bernfield M. Inhibition by the soluble syndecan-1 ectodomains delays wound repair in mice overexpressing syndecan- 1. J Biol Chem. 2004;279:41928–35. doi: 10.1074/jbc.M404506200. [DOI] [PubMed] [Google Scholar]
- 164.Chen E, Hermanson S, Ekker SC. Syndecan-2 is essential for angiogenic sprouting during zebrafish development. Blood. 2004;103:1710–9. doi: 10.1182/blood-2003-06-1783. [DOI] [PubMed] [Google Scholar]
- 165.Fears CY, Gladson CL, Woods A. Syndecan-2 is expressed in the microvasculature of gliomas and regulates angiogenic processes in microvascular endothelial cells. J Biol Chem. 2006;281:14533–6. doi: 10.1074/jbc.C600075200. [DOI] [PubMed] [Google Scholar]
- 166.Echtermeyer F, Streit M, Wilcox-Adelman S, Saoncella S, Denhez F, Detmar M, Goetinck P. Delayed wound repair and impaired angiogenesis in mice lacking syndecan-4. J Clin Invest. 2001;107:9–14. doi: 10.1172/JCI10559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ishiguro K, Kadomatsu K, Kojima T, Muramatsu H, Nakamura E, Ito M, Nagasaka T, Kobayashi H, Kusugami K, Saito H, Muramatsu T. Syndecan-4 deficiency impairs the fetal vessels in the placental labyrinth. Dev Dyn. 2000;219:539–44. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1081>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 168.De Agostini A. An unexpected role for anticoagulant heparan sulfate proteoglycans in reproduction. Swiss Med Wkly. 2006;136:583–90. doi: 10.4414/smw.2006.11368. [DOI] [PubMed] [Google Scholar]
- 169.Marneros AG, Olsen BR. Physiological role of collagen XVIII and endostatin. FASEB J. 2005;19:716–28. doi: 10.1096/fj.04-2134rev. [DOI] [PubMed] [Google Scholar]
- 170.O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, Folkman J. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–85. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 171.Fukai N, Eklund L, Marneros AG, Oh SP, Keene DR, Tamarkin L, Niemela M, Ilves M, Li E, Pihlajaniemi T, Olsen BR. Lack of collagen XVIII/endostatin results in eye abnormalities. EMBO J. 2002;21:1535–44. doi: 10.1093/emboj/21.7.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sertie AL, Sossi V, Camargo AA, Zatz M, Brahe C, Passos-Bueno MR. Collagen XVIII, containing an endogenous inhibitor of angiogenesis and tumor growth, plays a critical role in the maintenance of retinal structure and in neural tube closure (Knobloch syndrome) Hum Mol Genet. 2000;9:2051–8. doi: 10.1093/hmg/9.13.2051. [DOI] [PubMed] [Google Scholar]
- 173.Li Q, Olsen BR. Increased angiogenic response in aortic explants of collagen XVIII/endostatin-null mice. Am J Pathol. 2004;165:415–24. doi: 10.1016/S0002-9440(10)63307-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Dixelius J, Cross M, Matsumoto T, Sasaki T, Timpl R, Claesson-Welsh L. Endostatin regulates endothelial cell adhesion and cytoskeletal organization. Cancer Res. 2002;62:1944–7. [PubMed] [Google Scholar]
- 175.Olsson AK, Johansson I, Akerud H, Einarsson B, Christofferson R, Sasaki T, Timpl R, Claesson-Welsh L. The minimal active domain of endostatin is a heparin-binding motif that mediates inhibition of tumor vascularization. Cancer Res. 2004;64:9012–7. doi: 10.1158/0008-5472.CAN-04-2172. [DOI] [PubMed] [Google Scholar]
- 176.Kalluri R. Basement membranes: structure, assembly and role in tumour angiogenesis. Nat Rev Cancer. 2003;3:422–33. doi: 10.1038/nrc1094. [DOI] [PubMed] [Google Scholar]
- 177.Petitclerc E, Boutaud A, Prestayko A, Xu J, Sado Y, Ninomiya Y, Sarras MP, Jr, Hudson BG, Brooks PC. New functions for non-collagenous domains of human collagen type IV. Novel integrin ligands inhibiting angiogenesis and tumor growth in vivo. J Biol Chem. 2000;275:8051–61. doi: 10.1074/jbc.275.11.8051. [DOI] [PubMed] [Google Scholar]
- 178.Sudhakar A, Nyberg P, Keshamouni VG, Mannam AP, Li J, Sugimoto H, Cosgrove D, Kalluri R. Human alpha1 type IV collagen NC1 domain exhibits distinct antiangiogenic activity mediated by alpha1beta1 integrin. J Clin Invest. 2005;115:2801–10. doi: 10.1172/JCI24813. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 179.Folkman J. Tumor suppression by p53 is mediated in part by the antiangiogenic activity of endostatin and tumstatin. Sci STKE. 2006;2006:pe35. doi: 10.1126/stke.3542006pe35. [DOI] [PubMed] [Google Scholar]
- 180.Teodoro JG, Parker AE, Zhu X, Green MR. p53- mediated inhibition of angiogenesis through up-regulation of a collagen prolyl hydroxylase. Science. 2006;313:968–71. doi: 10.1126/science.1126391. [DOI] [PubMed] [Google Scholar]
- 181.Kivirikko KI, Myllyla R. Posttranslational enzymes in the biosynthesis of collagen: intracellular enzymes. Methods Enzymol. 1982;82:245–304. doi: 10.1016/0076-6879(82)82067-3. [DOI] [PubMed] [Google Scholar]
- 182.Pasqualini R, Bourdoulous S, Koivunen E, Woods VL, Jr, Ruoslahti E. A polymeric form of fibronectin has antimetastatic effects against multiple tumor types. Nat Med. 1996;2:1197–203. doi: 10.1038/nm1196-1197. [DOI] [PubMed] [Google Scholar]
- 183.Yi M, Ruoslahti E. A fibronectin fragment inhibits tumor growth, angiogenesis, and metastasis. Proc Natl Acad Sci USA. 2001;98:620–4. doi: 10.1073/pnas.98.2.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Mongiat M, Sweeney SM, San Antonio JD, Fu J, Iozzo RV. Endorepellin, a novel inhibitor of angiogenesis derived from the C terminus of perlecan. J Biol Chem. 2003;278:4238–49. doi: 10.1074/jbc.M210445200. [DOI] [PubMed] [Google Scholar]
- 185.Bix G, Fu J, Gonzalez EM, Macro L, Barker A, Campbell S, Zutter MM, Santoro SA, Kim JK, Hook M, Reed CC, Iozzo RV. Endorepellin causes endothelial cell disassembly of actin cytoskeleton and focal adhesions through alpha2beta1 integrin. J Cell Biol. 2004;166:97–109. doi: 10.1083/jcb.200401150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Menasche M, Jacob MP, Godeau G, Robert AM, Robert L. Pharmacological studies on elastin peptides (kappa-elastin). Blood clearance, percutaneous penetration and tissue distribution. Pathol Biol. 1981;29:548–54. [PubMed] [Google Scholar]
- 187.Nackman GB, Karkowski FJ, Halpern VJ, Gaetz HP, Tilson MD. Elastin degradation products induce adventitial angiogenesis in the Anidjar/Dobrin rat aneurysm model. Surgery. 1997;122:39–44. doi: 10.1016/s0039-6060(97)90262-2. [DOI] [PubMed] [Google Scholar]
- 188.Robinet A, Fahem A, Cauchard JH, Huet E, Vincent L, Lorimier S, Antonicelli F, Soria C, Crepin M, Hornebeck W, Bellon G. Elastin-derived peptides enhance angiogenesis by promoting endothelial cell migration and tubulogenesis through upregulation of MT1-MMP. J Cell Sci. 2005;118:343–56. doi: 10.1242/jcs.01613. [DOI] [PubMed] [Google Scholar]
- 189.Bornstein P. Thrombospondins as matricellular modulators of cell function. J Clin Invest. 2001;107:929–34. doi: 10.1172/JCI12749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Armstrong LC, Bornstein P. Thrombospondins 1 and 2 function as inhibitors of angiogenesis. Matrix Biol. 2003;22:63–71. doi: 10.1016/s0945-053x(03)00005-2. [DOI] [PubMed] [Google Scholar]
- 191.Jimenez B, Volpert OV, Crawford SE, Febbraio M, Silverstein RL, Bouck N. Signals leading to apoptosis- dependent inhibition of neovascularization by thrombospondin-1. Nat Med. 2000;6:41–8. doi: 10.1038/71517. [DOI] [PubMed] [Google Scholar]
- 192.Jimenez B, Volpert OV, Reiher F, Chang L, Munoz A, Karin M, Bouck N. c-Jun N-terminal kinase activation is required for the inhibition of neovascularization by thrombospondin-1. Oncogene. 2001;20:3443–8. doi: 10.1038/sj.onc.1204464. [DOI] [PubMed] [Google Scholar]
- 193.Kanda S, Shono T, Tomasini-Johansson B, Klint P, Saito Y. Role of thrombospondin-1-derived peptide, 4N1K, in FGF-2-induced angiogenesis. Exp Cell Res. 1999;252:262–72. doi: 10.1006/excr.1999.4622. [DOI] [PubMed] [Google Scholar]
- 194.Iruela-Arispe ML, Lombardo M, Krutzsch HC, Lawler J, Roberts DD. Inhibition of angiogenesis by thrombospondin-1 is mediated by 2 independent regions within the type 1 repeats. Circulation. 1999;100:1423–31. doi: 10.1161/01.cir.100.13.1423. [DOI] [PubMed] [Google Scholar]
- 195.Chandrasekaran L, He CZ, Al-Barazi H, Krutzsch HC, Iruela-Arispe ML, Roberts DD. Cell contactdependent activation of alpha3beta1 integrin modulates endothelial cell responses to thrombospondin- 1. Mol Biol Cell. 2000;11:2885–900. doi: 10.1091/mbc.11.9.2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Calzada MJ, Zhou L, Sipes JM, Zhang J, Krutzsch HC, Iruela-Arispe ML, Annis DS, Mosher DF, Roberts DD. Alpha4beta1 integrin mediates selective endothelial cell responses to thrombospondins 1 and 2 in vitro and modulates angiogenesis in vivo. Circ Res. 2004;94:462–70. doi: 10.1161/01.RES.0000115555.05668.93. [DOI] [PubMed] [Google Scholar]
- 197.Dixit VM, Haverstick DM, O’Rourke KM, Hennessy SW, Grant GA, Santoro SA, Frazier WA. A monoclonal antibody against human thrombospondin inhibits platelet aggregation. Proc Natl Acad Sci USA. 1985;82:3472–6. doi: 10.1073/pnas.82.10.3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Varner JA. The sticky truth about angiogenesis and thrombospondins. J Clin Invest. 2006;116:3111–3. doi: 10.1172/JCI30685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Lawler J, Sunday M, Thibert V, Duquette M, George EL, Rayburn H, Hynes RO. Thrombospondin-1 is required for normal murine pulmonary homeostasis and its absence causes pneumonia. J Clin Invest. 1998;101:982–92. doi: 10.1172/JCI1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Zhou L, Isenberg JS, Cao Z, Roberts DD. Type I collagen is a molecular target for inhibition of angiogenesis by endogenous thrombospondin-1. Oncogene. 2006;25:536–45. doi: 10.1038/sj.onc.1209069. [DOI] [PubMed] [Google Scholar]
- 201.Kyriakides TR, Zhu YH, Smith LT, Bain SD, Yang Z, Lin MT, Danielson KG, Iozzo RV, LaMarca M, McKinney CE, Ginns EI, Bornstein P. Mice that lack thrombospondin 2 display connective tissue abnormalities that are associated with disordered collagen fibrillogenesis, an increased vascular density, and a bleeding diathesis. J Cell Biol. 1998;140:419–30. doi: 10.1083/jcb.140.2.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kopp HG, Hooper AT, Broekman MJ, Avecilla ST, Petit I, Luo M, Milde T, Ramos CA, Zhang F, Kopp T, Bornstein P, Jin DK, Marcus AJ, Rafii S. Thrombospondins deployed by thrombopoietic cells determine angiogenic switch and extent of revascularization. J Clin Invest. 2006;116:3277–91. doi: 10.1172/JCI29314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Bork P. The modular architecture of a new family of growth regulators related to connective tissue growth factor. FEBS Lett. 1993;327:125–30. doi: 10.1016/0014-5793(93)80155-n. [DOI] [PubMed] [Google Scholar]
- 204.Perbal B, Takigawa M. CCN proteins: a new family of cell growth and differentiation regulators. London: World Scientific Publishers; 2005. [Google Scholar]
- 205.Kubota S, Takigawa M. CCN family proteins and angiogenesis: from embryo to adulthood. Angiogenesis. 2006 doi: 10.1007/s10456-006-9058-5. [DOI] [PubMed] [Google Scholar]
- 206.Leask A, Abraham DJ. All in the CCN family: essential matricellular signaling modulators emerge from the bunker. J Cell Sci. 2006;119:4803–10. doi: 10.1242/jcs.03270. [DOI] [PubMed] [Google Scholar]
- 207.Perbal B. CCN proteins: multifunctional signalling regulators. Lancet. 2004;363:62–4. doi: 10.1016/S0140-6736(03)15172-0. [DOI] [PubMed] [Google Scholar]
- 208.Brigstock DR. Regulation of angiogenesis and endothelial cell function by connective tissue growth factor (CTGF) and cysteine-rich 61 (CYR61) Angiogenesis. 2002;5:153–65. doi: 10.1023/a:1023823803510. [DOI] [PubMed] [Google Scholar]
- 209.Chen Y, Du XY. Functional properties and intracellular signaling of CCN1/Cyr61. J Cell Biochem. 2006 doi: 10.1002/jcb.21194. [DOI] [PubMed] [Google Scholar]
- 210.Leu SJ, Liu Y, Chen N, Chen CC, Lam SC, Lau LF. Identification of a novel integrin alpha 6 beta 1 binding site in the angiogenic inducer CCN1 (CYR61) J Biol Chem. 2003;278:33801–8. doi: 10.1074/jbc.M305862200. [DOI] [PubMed] [Google Scholar]
- 211.Chen CC, Mo FE, Lau LF. The angiogenic factor Cyr61 activates a genetic program for wound healing in human skin fibroblasts. J Biol Chem. 2001;276:47329–37. doi: 10.1074/jbc.M107666200. [DOI] [PubMed] [Google Scholar]
- 212.Babic AM, Kireeva ML, Kolesnikova TV, Lau LF. CYR61, a product of a growth factor-inducible immediate early gene, promotes angiogenesis and tumor growth. Proc Natl Acad Sci USA. 1998;95:6355–60. doi: 10.1073/pnas.95.11.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Fataccioli V, Abergel V, Wingertsmann L, Neuville P, Spitz E, Adnot S, Calenda V, Teiger E. Stimulation of angiogenesis by Cyr61 gene: a new therapeutic candidate. Hum Gene Ther. 2002;13:1461–70. doi: 10.1089/10430340260185094. [DOI] [PubMed] [Google Scholar]
- 214.Latinkic BV, Mo FE, Greenspan JA, Copeland NG, Gilbert DJ, Jenkins NA, Ross SR, Lau LF. Promoter function of the angiogenic inducer Cyr61gene in transgenic mice: tissue specificity, inducibility during wound healing, and role of the serum response element. Endocrinology. 2001;142:2549–57. doi: 10.1210/endo.142.6.8208. [DOI] [PubMed] [Google Scholar]
- 215.Hadjiargyrou M, Ahrens W, Rubin CT. Temporal expression of the chondrogenic and angiogenic growth factor CYR61 during fracture repair. J Bone Miner Res. 2000;15:1014–23. doi: 10.1359/jbmr.2000.15.6.1014. [DOI] [PubMed] [Google Scholar]
- 216.O’Brien TP, Lau LF. Expression of the growth factorinducible immediate early gene cyr61 correlates with chondrogenesis during mouse embryonic development. Cell Growth Differ. 1992;3:645–54. [PubMed] [Google Scholar]
- 217.Mo FE, Muntean AG, Chen CC, Stolz DB, Watkins SC, Lau LF. CYR61 (CCN1) is essential for placental development and vascular integrity. Mol Cell Biol. 2002;22:8709–20. doi: 10.1128/MCB.22.24.8709-8720.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Dumont DJ, Jussila L, Taipale J, Lymboussaki A, Mustonen T, Pajusola K, Breitman M, Alitalo K. Cardiovascular failure in mouse embryos deficient in VEGF receptor-3. Science. 1998;282:946–9. doi: 10.1126/science.282.5390.946. [DOI] [PubMed] [Google Scholar]
- 219.Bader BL, Rayburn H, Crowley D, Hynes RO. Extensive vasculogenesis, angiogenesis, and organogenesis precede lethality in mice lacking all alpha v integrins. Cell. 1998;95:507–19. doi: 10.1016/s0092-8674(00)81618-9. [DOI] [PubMed] [Google Scholar]
- 220.Chen N, Chen CC, Lau LF. Adhesion of human skin fibroblasts to Cyr61 is mediated through integrin alpha 6beta 1 and cell surface heparan sulfate proteoglycans. J Biol Chem. 2000;275:24953–61. doi: 10.1074/jbc.M003040200. [DOI] [PubMed] [Google Scholar]
- 221.Leu SJ, Lam SC, Lau LF. Pro-angiogenic activities of CYR61 (CCN1) mediated through integrins alphavbeta3 and alpha6beta1 in human umbilical vein endothelial cells. J Biol Chem. 2002;277:46248–55. doi: 10.1074/jbc.M209288200. [DOI] [PubMed] [Google Scholar]
- 222.Todorovicc V, Chen CC, Hay N, Lau LF. The matrix protein CCN1 (CYR61) induces apoptosis in fibroblasts. J Cell Biol. 2005;171:559–68. doi: 10.1083/jcb.200504015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Kireeva ML, Mo FE, Yang GP, Lau LF. Cyr61, a product of a growth factor-inducible immediate-early gene, promotes cell proliferation, migration, and adhesion. Mol Cell Biol. 1996;16:1326–34. doi: 10.1128/mcb.16.4.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Babic AM, Chen CC, Lau LF. Fisp12/mouse connective tissue growth factor mediates endothelial cell adhesion and migration through integrin alphavbeta3, promotes endothelial cell survival, and induces angiogenesis in vivo. Mol Cell Biol. 1999;19:2958–66. doi: 10.1128/mcb.19.4.2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Shimo T, Nakanishi T, Kimura Y, Nishida T, Ishizeki K, Matsumura T, Takigawa M. Inhibition of endogenous expression of connective tissue growth factor by its antisense oligonucleotide and antisense RNA suppresses proliferation and migration of vascular endothelial cells. J Biochem. 1998;124:130–40. doi: 10.1093/oxfordjournals.jbchem.a022071. [DOI] [PubMed] [Google Scholar]
- 226.Ivkovic S, Yoon BS, Popoff SN, Safadi FF, Libuda DE, Stephenson RC, Daluiski A, Lyons KM. Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development. Development. 2003;130:2779–91. doi: 10.1242/dev.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med. 1999;5:623–8. doi: 10.1038/9467. [DOI] [PubMed] [Google Scholar]
- 228.Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, Shapiro SD, Senior RM, Werb Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–22. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Reponen P, Sahlberg C, Munaut C, Thesleff I, Tryggvason K. High expression of 92-kDa type IV collagenase (gelatinase) in the osteoclast lineage during mouse development. Ann N Y Acad Sci. 1994;732:472–5. doi: 10.1111/j.1749-6632.1994.tb24789.x. [DOI] [PubMed] [Google Scholar]
- 230.Inoki I, Shiomi T, Hashimoto G, Enomoto H, Nakamura H, Makino K, Ikeda E, Takata S, Kobayashi K, Okada Y. Connective tissue growth factor binds vascular endothelial growth factor (VEGF) and inhibits VEGF-induced angiogenesis. FASEB J. 2002;16:219–21. doi: 10.1096/fj.01-0332fje. [DOI] [PubMed] [Google Scholar]
- 231.Chen Y, Abraham DJ, Shi-Wen X, Pearson JD, Black CM, Lyons KM, Leask A. CCN2 (connective tissue growth factor) promotes fibroblast adhesion to fibronectin. Mol Biol Cell. 2004;15:5635–46. doi: 10.1091/mbc.E04-06-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Perbal B. NOV story: the way to CCN3. Cell Commun Signal. 2006;4:3. doi: 10.1186/1478-811X-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Joliot V, Martinerie C, Dambrine G, Plassiart G, Brisac M, Crochet J, Perbal B. Proviral rearrangements and overexpression of a new cellular gene (nov) in myeloblastosis-associated virus type 1-induced nephroblastomas. Mol Cell Biol. 1992;12:10–21. doi: 10.1128/mcb.12.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Lin CG, Leu SJ, Chen N, Tebeau CM, Lin SX, Yeung CY, Lau LF. CCN3 (NOV) is a novel angiogenic regulator of the CCN protein family. J Biol Chem. 2003;278:24200–8. doi: 10.1074/jbc.M302028200. [DOI] [PubMed] [Google Scholar]
- 235.Lin CG, Chen CC, Leu SJ, Grzeszkiewicz TM, Lau LF. Integrin-dependent functions of the angiogenic inducer NOV(CCN3): implication in wound healing. J Biol Chem. 2005;280:8229–37. doi: 10.1074/jbc.M404903200. [DOI] [PubMed] [Google Scholar]
- 236.Ellis PD, Chen Q, Barker PJ, Metcalfe JC, Kemp PR. Nov gene encodes adhesion factor for vascular smooth muscle cells and is dynamically regulated in response to vascular injury. Arterioscler Thromb Vasc Biol. 2000;20:1912–9. doi: 10.1161/01.atv.20.8.1912. [DOI] [PubMed] [Google Scholar]
- 237.Sakamoto K, Yamaguchi S, Ando R, Miyawaki A, Kabasawa Y, Takagi M, Li CL, Perbal B, Katsube K. The nephroblastoma overexpressed gene (NOV/ccn3) protein associates with Notch1 extracellular domain and inhibits myoblast differentiation via Notch signaling pathway. J Biol Chem. 2002;277:29399–405. doi: 10.1074/jbc.M203727200. [DOI] [PubMed] [Google Scholar]
- 238.Delmolino LM, Stearns NA, Castellot JJ., Jr COP- 1, a member of the CCN family, is a heparin-induced growth arrest specific gene in vascular smooth muscle cells. J Cell Physiol. 2001;188:45–55. doi: 10.1002/jcp.1100. [DOI] [PubMed] [Google Scholar]
- 239.Hurvitz JR, Suwairi WM, Van Hul W, El-Shanti H, Superti-Furga A, Roudier J, Holderbaum D, Pauli RM, Herd JK, Van Hul EV, Rezai-Delui H, Legius E, Le Merrer M, Al-Alami J, Bahabri SA, Warman ML. Mutations in the CCN gene family member WISP3 cause progressive pseudorheumatoid dysplasia. Nat Genet. 1999;23:94–8. doi: 10.1038/12699. [DOI] [PubMed] [Google Scholar]
- 240.L’Heureux N, Dusserre N, Konig G, Victor B, Keire P, Wight TN, Chronos NA, Kyles AE, Gregory CR, Hoyt G, Robbins RC, McAllister TN. Human tissueengineered blood vessels for adult arterial revascularization. Nat Med. 2006;12:361–5. doi: 10.1038/nm1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Wang Y, Ameer GA, Sheppard BJ, Langer R. A tough biodegradable elastomer. Nat Biotechnol. 2002;20:602–6. doi: 10.1038/nbt0602-602. [DOI] [PubMed] [Google Scholar]
- 242.Wickstrom SA, Alitalo K, Keski-Oja J. Endostatin associates with lipid rafts and induces reorganization of the actin cytoskeleton viadown-regulation of RhoA activity. J Biol Chem. 2003;278:37895–901. doi: 10.1074/jbc.M303569200. [DOI] [PubMed] [Google Scholar]
- 243.Ramchandran R, Dhanabal M, Volk R, Waterman MJ, Segal M, Lu H, Knebelmann B, Sukhatme VP. Antiangiogenic activity of restin, NC10 domain of human collagen XV: comparison to endostatin. Biochem Biophys Res Commun. 1999;255:735–9. doi: 10.1006/bbrc.1999.0248. [DOI] [PubMed] [Google Scholar]
- 244.Sasaki T, Hohenester E, Timpl R. Structure and function of collagen-derived endostatin inhibitors of angiogenesis. IUBMB Life. 2002;53:77–84. doi: 10.1080/15216540211466. [DOI] [PubMed] [Google Scholar]
- 245.Sasaki T, Larsson H, Tisi D, Claesson-Welsh L, Hohenester E, Timpl R. Endostatins derived from collagens XV and XVIII differ in structural and binding properties, tissue distribution and anti-angiogenic activity. J Mol Biol. 2000;301:1179–90. doi: 10.1006/jmbi.2000.3996. [DOI] [PubMed] [Google Scholar]
- 246.Kamphaus GD, Colorado PC, Panka DJ, Hopfer H, Ramchandran R, Torre A, Maeshima Y, Mier JW, Sukhatme VP, Kalluri R. Canstatin, a novel matrixderived inhibitor of angiogenesis and tumor growth. J Biol Chem. 2000;275:1209–15. doi: 10.1074/jbc.275.2.1209. [DOI] [PubMed] [Google Scholar]
- 247.Maeshima Y, Colorado PC, Torre A, Holthaus KA, Grunkemeyer JA, Ericksen MB, Hopfer H, Xiao Y, Stillman IE, Kalluri R. Distinct antitumor properties of a type IV collagen domain derived from basement membrane. J Biol Chem. 2000;275:21340–8. doi: 10.1074/jbc.M001956200. [DOI] [PubMed] [Google Scholar]
- 248.Maeshima Y, Colorado PC, Kalluri R. Two RGDindependent alpha vbeta 3 integrin binding sites on tumstatin regulate distinct anti-tumor properties. J Biol Chem. 2000;275:23745–50. doi: 10.1074/jbc.C000186200. [DOI] [PubMed] [Google Scholar]
- 249.Maeshima Y, Manfredi M, Reimer C, Holthaus KA, Hopfer H, Chandamuri BR, Kharbanda S, Kalluri R. Identification of the anti-angiogenic site within vascular basement membrane-derived tumstatin. J Biol Chem. 2001;276:15240–8. doi: 10.1074/jbc.M007764200. [DOI] [PubMed] [Google Scholar]
- 250.Maeshima Y, Sudhakar A, Lively JC, Ueki K, Kharbanda S, Kahn CR, Sonenberg N, Hynes RO, Kalluri R. Tumstatin, an endothelial cell-specific inhibitor of protein synthesis. Science. 2002;295:140–3. doi: 10.1126/science.1065298. [DOI] [PubMed] [Google Scholar]
- 251.Maeshima Y, Yerramalla UL, Dhanabal M, Holthaus KA, Barbashov S, Kharbanda S, Reimer C, Manfredi M, Dickerson WM, Kalluri R. Extracellular matrixderived peptide binds to alpha(v)beta(3) integrin and inhibits angiogenesis. J Biol Chem. 2001;276:31959–68. doi: 10.1074/jbc.M103024200. [DOI] [PubMed] [Google Scholar]