

Abstract
5′AMP-activated protein kinase (AMPK) activation occurs under a variety of stress conditions but the role of this enzyme in the promotion or inhibition of stress-induced cell death is unclear. To address this issue, we transformed two different cell lines with shRNA-expressing plasmids, targeting the alpha subunit of AMPK, and verified AMPKα downregulation. The cell lines were then stressed by exposure to medium without glucose (PC12 cells) or with the viral thymidine kinase-specific DNA replication inhibitors: acyclovir, penciclovir and ganciclovir (herpes simplex virus thymidine kinase-expressing Baby Hamster Kidney cells). In non-AMPK-downregulated cells, these stress treatments induced AMPK upregulation and phosphorylation, leaving open the question whether the association of AMPK activation with stress-induced cell death reflects a successful death-promoting or an ineffective death-inhibiting activity. In AMPKα-deficient cells (expressing AMPKα-specific shRNAs or treated with Compound C) exposure to low glucose medium or DNA replication inhibitors led to an enhancement of cell death, indicating that, under the conditions examined, the role of activated AMPK is not to promote, but to protect from or delay stress-induced cell death.
Keywords: AMPK, AICAR, apoptosis, glucose, ACV, PCV, GCV
Introduction
Two schools of thought have emerged concerning the involvement of the 5′AMP-activated protein kinase (AMPK) in cell death. One supports the accepted standpoint that AMPK, as a metabolic switch, permits cell survival under conditions of stress. Indeed, Russell et al.[1] report a protective role of AMPK against apoptosis during ischemia and Shaw et al.[2] demonstrate the protective effect of LKB-1 against apoptosis induced by agents that elevate intracellular AMP levels. AMPK has also been reported to promote neuronal and astrocyte survival under conditions which would normally lead to apoptosis [3] and 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR), an activator of AMPK, has also been reported to inhibit apoptosis induced by a variety of stimuli [4, 5].
The second school of thought associates AMPK activation with apoptosis, suggestive of a direct role for this enzyme in cell death. Contrasting the reported protective effects of AICAR, Saitoh et al.[6] report that AICAR treatment effects a reduction in gastric cancer cell viability. Further evidence supporting the argument that AMPK activation leads to apoptosis is provided in a study showing that both the AMPK activators, metformin, and AICAR induce β cell apoptosis and that ‘compound C’, an inhibitor of AMPK, reduced metformin-induced apoptosis. It was further shown that apoptosis associated with activated AMPK involves JNK and c-Jun phosphorylation and coincides with Caspase-3 activation [7]. Finally, in a study on AMPK β1 subunit induction in response to treatment with DNA-damaging agents, Li et al.[8] report that AMPKβ1 is upregulated during DNA damage-induced apoptosis in a variety of cell lines, that overexpression of AMPKβ1 causes growth arrest, and hypothesize that AMPKβ1 induction may facilitate cell killing.
Mammalian AMPK is a heterotrimeric complex comprising a catalytic α subunit and regulatory β and γ subunits. Multiple isoforms exist for each subunit (α1, α2, β1, β2, γ1, γ2, γ3) theoretically giving rise to 12 AMPK holoenzymes, differing in subunit isoform combination. Functional properties of the enzyme may also depend on isoform usage, for example, complexes containing the α1 isoform have an exclusively cytoplasmic location and are stimulated to a much lesser extent by AMP than complexes containing the α2 isoform which are predominantly nuclear, suggesting a role for α2 complexes in regulating gene expression [9]. Phosphorylation (activation) of AMPK occurs at the T172 residue on the α subunit and is promoted by AMP binding. Dephosphorylation (inactivation) of AMPK occurs when the enzyme is bound to ATP instead of AMP. The activation status of AMPK is therefore controlled by the intracellular AMP:ATP ratio, so that if intracellular ATP levels fall, and the ratio shifts in favour of AMP (during stress conditions such as glucose and/or oxygen depletion), the enzyme is predominantly bound to AMP and becomes activated.
Once activated, AMPK is known to phosphorylate several downstream targets including acetyl-CoA carboxylase, glycogen synthase, phosphofructokinase [10, 11] and the transcriptional coactivator p300 [12], with the overall effect of switching off ATP-consuming pathways (such as fatty acid, glycogen and protein synthesis) and switching on ATP-generating pathways (such as glycolysis and fatty acid oxidation). This in turn has the effect of restoring cellular energy levels, which may be important in maintaining cell survival under conditions of stress. However, when cell death is inevitable, AMPK may further be important in influencing whether death occurs via the necrotic or apoptotic route. It is suggested that the downstream controller capable of directing cells towards either necrosis or apoptosis is ATP, based on the accepted hypothesis that apoptosis is an active, energy-requiring process [13–15]. Evidence suggests that, when the cell's ATP levels fall below a critical threshold, apoptosis ensues as long as enough ATP is still available for energy-requiring apoptotic processes. Further falls in ATP levels below the critical threshold result in failure to maintain apoptotic processes and necrosis ensues [16]. Therefore, as a cell's ATP levels fall below the critical threshold for maintenance of survival, the effect of AMPK activation may be to promote apoptotic, rather than necrotic, death.
To address the paradox concerning the role of AMPK activation in cell death or survival, we investigated the phosphorylation status of AMPK in two in vitro death systems: in herpes simplex virus thymidine kinase-expressing Baby Hamster Kidney (HSVTK+ BHK) cells, which undergo cell death on treatment with the antiviral guanosine nucleoside analogues acyclovir (ACV), penciclovir (PCV) and ganciclovir (GCV) [17], and in PC12 cells exposed to glucose-free medium. In both systems cell death was associated with AMPK activation, and the magnitude of AMPK activation positively correlated with the extent of apoptotic death. Furthermore, transformation of the cells with shRNA-expressing vectors, which downregulated AMPK, resulted in accelerated and enhanced death, indicating that AMPK plays an important role in cell death inhibition under conditions of stress.
Materials and methods
Cell culture
HSVTK-transformed BHK cells were grown in Dulbecco's Modified Minimal Essential Medium (DMEM) supplemented with 5% fetal bovine serum (FBS). PC12 cells were grown in RPMI1640 medium supplemented with 10% horse serum and 5% fetal calf serum. For glucose-deprivation experiments, PC12 cells were washed three times in PBS and maintained in RPMI1640 medium, with or without glucose, supplemented with 2% horse serum and 1% fetal calf serum.
Compounds
GCV, PCV, and ACV were obtained from GlaxoSmithkline Research and Development, Stevenage, UK. Guanosine nucleoside analogues were solubilized in water and, for experiments, were used at working concentrations of 1 μM and 10 μM.
The AMPK activator, AICAR, was obtained from Toronto Research Chemicals, solubilized in water and used at working concentrations of 100 μM and 1 mM. The AMPK inhibitor, ‘compound C’ (6-[4-(2-Piperidin-1-yl-ethoxy)-phenyl)]-3- pyridin-4-yl-pyrrazolo[1,5,-a]-pyrimidine), was obtained from Calbiochem, dissolved in 100 mM HCl and used at working concentrations of 0.1 μM, 1 μM, 5 μM, and 50 μM.
Western blotting
Adherent cells were harvested with a cell scraper. Nonadherent cells were harvested by centrifugation. Cell pellets were homogenized in a buffer consisting per 10 ml volume of urea (3 g), thiourea (1.52 g), Tris (42 mg), CHAPS (400 mg) and DTT (100 mg). Homogenized samples were quantitated by Bradford assay (BioRad). For SDS-PAGE, samples were diluted 1:1 in Laemmli buffer and run on 15% (cleaved Caspase-3) or 10% (AMPKα, PARP, cleaved Caspase-3) polyacrylamide minigels.
Semiquantitative Western blotting was carried out according to standard protocols. Rabbit polyclonal antibodies to cleaved Caspase-3 were obtained from the Hôpitaux Universitaires de Genève. Rabbit polyclonal anti-PARP antibody (reactive with both the 112 kD and 85 kD fragments) was obtained from Santa Cruz and used at a dilution of 1:1000. Rabbit polyclonal anti total AMPKα and anti phosphorylated AMPKα antibodies were obtained from Cell Signaling Technology and rabbit polyclonal antibodies specific to AMPKα1 or α2 were obtained from Upstate. All AMPKα antibodies were used at a dilution of 1:2000.
Flow cytometry analysis
To assess viability, cells were harvested by trypsinization, washed once in PBS and resuspended in 800 μl PBS containing 4μl of propidium iodide solution (5 mg/ml). After 20 min incubation at 4°C the extent of staining was compared among the treatment groups by flow cytometry.
For cell cycle analysis, cells were harvested by trypsinization, washed once in PBS and fixed in ice cold 70% ethanol for 1 hr. Cells were then washed once in PBS and resuspended in 800 μl PBS with 10 μg of DNase-free RNase and incubated for 30 min at room temperature. 4 μl of propidium iodide solution (5 mg/ml) were then added and after further 30 min incubation at 4°C, cells were analyzed by flow cytometry.
Production of stable shRNA-expressing cell lines
DNA templates encoding short hairpin (sh)RNAs, targeted against a region within the protein kinase domain of the α subunit of AMPK, were cloned into the Ambion pSilencer2.1-U6 neo vector, according to the kit manufacturer's instructions. The upper strand sequence of the AMPKα1 shRNA template (excluding the restriction sites for cloning into pSilencer) was: 5′CATGATGTCAGATGGTGAATTCTCAAGAGAAATTCACCATCTGACATCATGTTTTTTGGAA 3′ and the corresponding AMPKα2 shRNA template was: 5′GTATGATGTCAGATGGTGAATTCTCAAGAGAAATTCACCATCTGACATCATACTT TTTTGGAA 3′ where the sequences in italics correspond to the sense and antisense siRNA templates respectively, the underlined ‘G’ at the beginning of the α2 sequence is an additional nucleotide inserted to improve RNA polymerase III transcription (since the enzyme preferentially initiates with a purine residue), the underlined sequence between the sense and antisense templates corresponds to the loop joining the siRNA template strands and the terminal, nonunderlined sequence corresponds to the RNA polymerase III terminator sequence and ‘gene silencing’ element.
The vectors generated are referred to here as ‘pSilencer/AMPKα1shRNA’ or ‘pSilencer/AMPKα2sh RNA’. Vectors were linearized prior to transformation. Cells were electroporated with pSilencer/AMPK α1shRNA, pSilencer/AMPKα2shRNA or pSilencer/ negative control shRNA (the latter plasmid was provided by Ambion and contains a scrambled sequence of no known similarity to the human, rat or mouse genomes, for which the shRNA cassette is shown below). 5′ACTACCGTTGTTATAGGTG TTCAAGAGA CACCTATAACAACGGTAGT TTTTTGGA 3′.
Stable clones and cell populations were selected in G418 (1 mg/ml for BHK and 0.5 mg/ml for PC12 cells) and assayed by semi-quantitative Western blotting for reduced levels of AMPKα.
Assessment of cell growth and viability
For Trypan blue exclusion assays cells were resuspended in PBS and the same volume of Trypan blue (0.4%) was added to the cell suspension. Viable cells were counted in a Neubauer chamber.
Results
Confirmation of apoptosis in glucose-deprived PC12 and drug-treated HSVTK+ BHK cells
Detection of poly ADP ribose polymerase (PARP) cleavage by Western blotting confirmed that glucose deprivation induced apoptosis in PC12 cells (data not shown).
Guanosine nucleoside analogue treatment of HSVTK-expressing cells is reported to induce both apoptotic and non-apoptotic death, depending on the analogue used: previously we showed that PCV and GCV-induced apoptosis of HSVTK+ BHK cells was associated with DNA laddering and phosphatidyl serine exposure, whereas death induced by ACV was not associated with these events [17]. To further clarify the GCV- and PCV-induced apoptotic process we show here that cleavage of proCaspase-3, and its substrate PARP, is also involved. Contrarily, PARP cleavage was not detected in ACV-treated cells and, although some residual Caspase-3 cleavage also occurred, this did not exceed the background levels observed in the non-treated control (Fig. 1).
1.
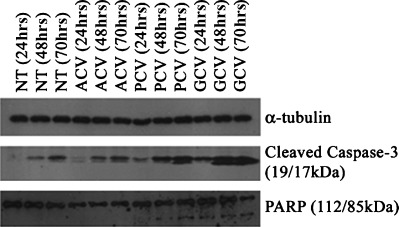
Apoptosis induction on treatment of HSVTK+ BHK cells with PCV and GCV. Western blot analysis shows high levels of cleaved Caspase-3 at 48 hrs and 70 hrs of treatment with PCV and GCV. PCV and GCV treatment also induces cleavage of the Caspase-3 substrate, PARP, which is not observed on treatment with ACV. (NT = non-treated control.)
AMPKα1/2 isoform expression
AMPKα isoforms are reported to exhibit differing cell type and subcellular expression. Western blotting using AMPKα1 and α2-specific antibodies revealed that the α1 isoform is predominantly expressed in PC12 and in HSVTK+ BHK cells, whereas significant levels of both isoforms are detectable in HeLa cells (Fig. 2). However, no changes in individual isoform ratios were detectable upon cell death induction in PC12 or HSVTK+ BHK cells by glucose deprivation or by guanosine nucleoside analogue treatment, respectively α data not shown).
2.
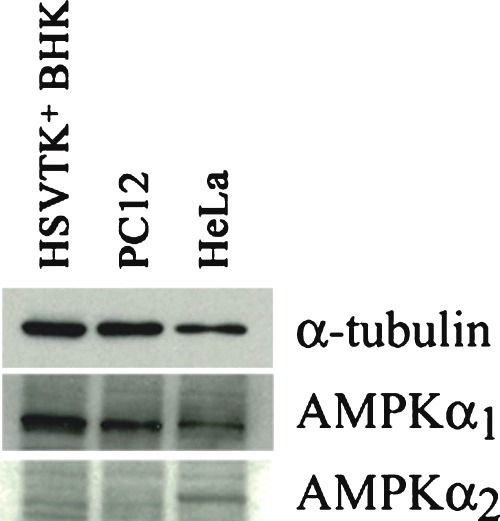
Western blot showing AMPKα1/2 isoform expression in different cell types. Whereas both isoforms are detectable in HeLa cells, only AMPKα1 is found in HSVTK+ BHK and PC12 cells. Samples were equalized to α-tubulin (upper panel).
AMPKα phosphorylation is associated with cell death in glucose-deprived PC12 cells, and in guanosine nucleoside analogue-treated HSVTK+ BHK cells
The extent of AMPKα phosphorylation occurring during glucose deprivation in PC12 cells and guanosine nucleoside analogue treatment of HSVTK+ BHK cells was investigated by Western blotting. Glucosedeprived PC12 cells displayed increased levels of phosphorylated AMPKα (Fig. 3A shows the effects of 48 hrs glucose deprivation on AMPKα phosphorylation). In GCV and PCV-treated HSVTK+ BHK cells, compared to the non-treated and ACV-treated samples, AMPKα is markedly phosphorylated by 24 hrs. Phosphorylation of the AMPKα subunit is further increased for all drug-treated samples, including ACV, on days 2 and 3 (Fig. 3B).
3.
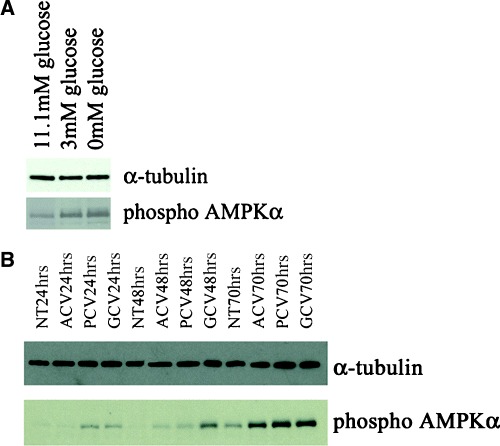
Western blot confirming enhanced phosphorylation of AMPKα in (A) PC12 cells exposed to low (3 mM) and glucose-free medium (11.1 mM glucose = normal culture conditions) and (B) HSVTK+ BHK cells treated with ACV, PCV or GCV.
Confirmation of AMPKα downregulation, and reduction in stress-induced phospho AMPKα levels, in pSilencer/AMPKα shRNA-transformed cell populations
Stable transformants, expressing shRNAs targeting AMPKα, were generated in a number of ways: (a) in HSVTK+ BHK cells, cell populations were electroporated with the constructed pSilencer vectors and individual clones were grown up under G418 selection. Reduction in levels of AMPKα in individual clones was verified by Western blotting (Fig. 4A, panel (ii)). The clones were then treated with 10 μM GCV and, compared to GCV-treated clones transformed with the negative control vector, also displayed reduced levels of total and phospho AMPKα (Fig. 4A, panels (v) and (vi)). Individual clones were then tested for differences in susceptibility to cell death during guanosine nucleoside analogue treatment. (b) HSVTK+ BHK cells were first ‘cloned out’ to obtain a homogeneous, GCV-sensitive, population, prior to electroporation with the constructed pSilencer vectors. The electroporated populations were then selected with G418, tested for reduction in AMPKα expression (Fig. 4B), and assayed for differences in susceptibility to guanosine nucleoside analogueinduced death. (c) The homogeneous, GCV sensitive, HSVTK+ BHK cell population generated in ‘b’ above, was electroporated with pSilencer vectors, followed by cloning out under G418 selection. Individual clones, with reduced or undetectable levels of AMPKα, were then assayed for differences in susceptibility to guanosine nucleoside analogueinduced death. (d) PC12 cells were electroporated with the constructed pSilencer vectors, selected with G418, tested for reduction in levels of AMPKα, and of phospho AMPKα during glucose deprivation (Fig. 4C), and assayed for differences in susceptibility to glucose deprivation-induced death.
4.
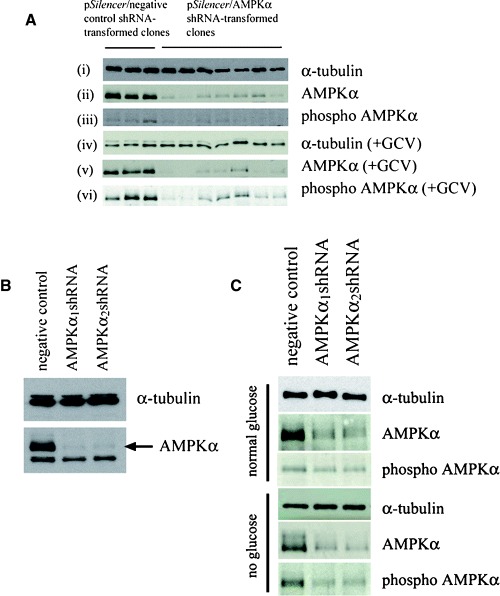
(A) Western blot confirming reduction in levels of AMPKα in pSilencer-transformed HSVTK+ BHK cell clones, expressing the α1 or α2-targeting shRNA sequences (panel (ii)). Treatment with 10 μM GCV did not significantly increase total or phospho AMPKα expression in the AMPKα-downregulated clones (panels (v) and (vi)). Sample loadings in panels (ii) and (iii) were equalized to α-tubulin (panel (i)). Sample loadings in panels (v) and (vi) were equalized to α-tubulin (panel (iv) is a reprobe of the blot in panel (vi)). (B) Western blot confirming AMPKα downregulation in pSilencer/AMPKαshRNA-transformed HSVTK+ BHK cell populations, compared to the pSilencer/negative control shRNA-transformed population. (C) Western blot confirming AMPKα downregulation in pSilencer/AMPKαshRNA-transformed PC12 cells. Both total and phospho AMPKα remained downregulated during glucose deprivation. Sample loadings from the respective treatment groups were equalized to α-tubulin.
AMPKα downregulation slows cell growth and enhances cell death under stress conditions
shRNA-expressing, AMPK-downregulated HSVTK+ BHK cell clones, generated by method ‘a’ described above (where the cell population was not rendered homogeneous by ‘cloning out’ prior to electroporation), displayed disparate differences in the rate and extent of GCV-induced apoptosis, observed morphologically and by flow cytometry analysis of propidium iodide-stained cells (data not shown). These differences were unrelated to AMPK expression. However, since one of the clones generated was no longer sensitive to GCV due to loss of the HSVTK gene, it is likely that the disparate results between individual clones reflected differences in HSVTK expression. Despite this, it is of interest that, regardless of the method used to generate the AMPK-targeted, shRNAexpressing cell population, and regardless of the cell type transformed (to date, we have tested HSVTK+ BHK, HeLa and PC12 cell lines), the populations transformed with the pSilencer/AMPKα2shRNA vector were the most difficult to grow and displayed a slower growth rate compared to the pSilencer/ AMPKα1shRNA–transformed populations which also displayed a slightly slower growth rate than the pSilencer/negative control shRNA-transformed cells (Fig. 5). In some cases, however, recovery from slowed growth occurred within 3-5 passages, but this regain of growth rate was generally accompanied by regain–or increase in–AMPKα expression, despite maintained G418 resistance.
5.
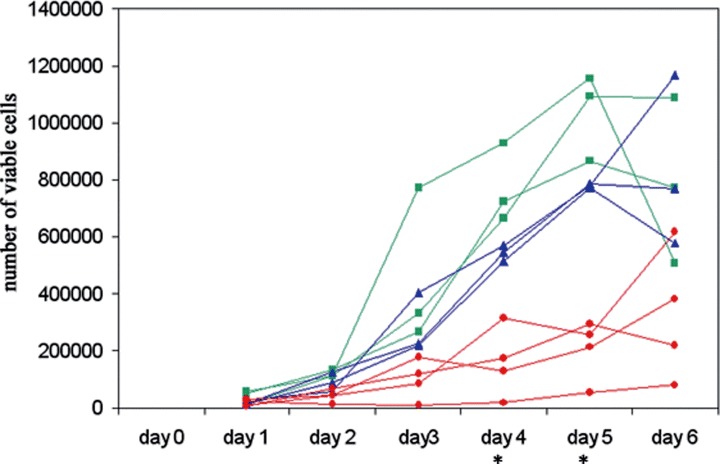
Growth curves of HSVTK+ BHK cell clones transformed with pSilencer/negative control shRNA (▪), pSilencer/AMPKα1shRNA (▴) or pSilencer/AMPKα2sh RNA (•), showing a slower growth rate in the pSilencer/ AMPKαshRNA-transformed clones, with most significant slowed growth in the α2 clones. (The differences between the individual groups (negative control, α1, α2) on days 4 and 5 are statistically significant, as measured by two-tail t-test, assuming equal variance (P < 0.05)). Growth was monitored by viable counts of Trypan blue-stained cells.
shRNA-expressing, AMPK-downregulated HSVTK+ BHK cells, generated by method ‘b’ described above (where the cell population was rendered homogeneous by ‘cloning out’ prior to electroporation) displayed obvious differences in susceptibility to 10 μM PCV and GCV on days 2 and 3 of treatment, with an increased proportion of dead cells detected in the AMPK-downregulated populations compared to the negative control. However, no increased susceptibility to cell death was observed on treatment with the less potent compound, ACV.
shRNA-expressing, AMPK-downregulated HSVTK+ BHK cell clones, generated by method ‘c’ described above, displayed differences in susceptibility to 10 μM PCV and GCV on day 2 and to 10 μM PCV on day 3 of treatment, with an increased proportion of dead cells detected in the AMPK-downregulated populations compared to the negative control (Fig. 6A). Again, no increased susceptibility to cell death was observed with ACV.
6.
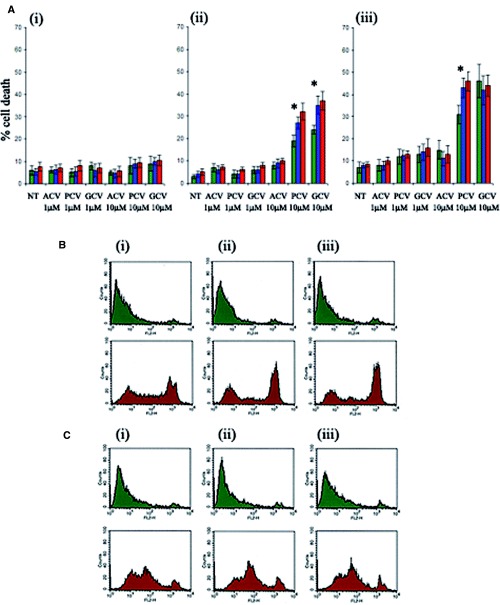
(A) Effect of AMPK downregulation on the extent of guanosine nucleoside analogue-induced death in shRNA-transformed HSVTK+ BHK cells at (i) 24 hrs, (ii) 48 hrs and (iii) 72 hrs of treatment. Cell death was measured by propidium iodide staining and flow cytometry analysis. ‘Dead’ cells were counted as propidium iodide-positive events scoring above the 102 value on the FL2 scale. Data are averaged from n = 3 clones from the respective shRNA-expressing groups (green = pSilencer/negative control shRNA, blue = pSilencer/AMPKα1shRNA, red = pSilencer/ AMPKα2shRNA-transformed cells). Asterisk indicates significant difference between levels of cell death in the negative control shRNA and AMPKα shRNAexpressing cells, treated with 10 μM PCV or GCV (p < 0.05, Student's t-test). (B) Flow cytometry analysis of propidium iodide-stained pSilencer/negative control (i), AMPKα1 (ii) or AMPKα2 (iii) shRNA-transformed PC12 cells, cultured for 4 days in medium with (green plots) or without (red plots) glucose. Enhanced cell death is evident in the pSilencer/AMPKαshRNAtransformed cells. (C) Flow cytometry analysis of propidium iodide-stained, non-transformed PC12 cells, cultured for 4 days without compound C (i), with 0.1 μM compound C (ii) or with 1μM compound C (iii), in medium with (green plots) or without (red plots) glucose. Glucose deprivation-induced death is enhanced in the compound C-treated cells.
shRNA-expressing, AMPK-downregulated PC12 cell populations displayed increased susceptibility to glucose deprivation-induced death, with a greater proportion of non-viable cells in the AMPKα-downregulated groups at days 4 and 5 of glucose deprivation. In all cases the percentages of propidium iodide-staining cells were compared between pSilencer/negative control and AMPKα1 or AMPKα2 shRNA-transformed PC12 cell populations at the following cut-off values on the FL2 scale: 101, 2 × 101, 102, 2 × 102, 3 × 102, 4 × 102, 5 × 102, 6 × 102, 7 × 102, 8 × 102, 9 × 102, 103. Statistically significant increases in the number of propidium iodide-stained AMPKα-depleted cells were observed at the FL2 cut-off values examined ranging from 102 to 103 (P < 0.05). On day 4 of culture in glucose- deprived medium the greatest increase was observed at the 6 × 102 cut-off value with 32.91% (±1.52%) of pSilencer/negative control shRNA-transformed cells scoring above the cut-off compared to 46.92% (±0.92%) and 53.74% (±1.18%) of pSilencer/ AMPKα1 and AMPKα2shRNA-transformed cells respectively. The smallest statistically significant increase at this timepoint was observed at the 102 cutoff value with 46.85% (±1.23%) of pSilencer/ negative control shRNA-transformed cells scoring above the cut-off compared to 56.62% (±1.21%) and 61.3% (±1.23%) of pSilencer/AMPKα1 and AMPK α2shRNAtransformed cells (Fig. 6B).
Compound C treatment of PC12 cells was found to be toxic at concentrations of 50 μM (100% cell death within 24 hrs) and 5 μM (50% cell death within 2–3 days of treatment). Marginal toxicity was observed with 1 μM Compound C from day 3 of treatment. For this reason experiments were conducted with 1 μM Compound C with the additional inclusion of a nontoxic concentration of 0.1 μM. Treatment with 0.1 μM Compound C resulted in an enhancement of cell death during glucose deprivation (Fig. 6C).
These results strongly suggest that, while activated AMPK is increased under stress conditions, its role is to protect from cell death.
The AMPK activator, AICAR, induces apoptosis in HSVTK+ BHK cells, but via mechanisms not involving AMPK
To investigate whether AMPK activation, independent of guanosine nucleoside analogue treatment, could induce apoptosis in HSVTK+ BHK cells, nontransformed HSVTK+ clones were treated with the AMPK activator, AICAR, at concentrations of 100 μM and 1 mM. AICAR induced significant AMPK phosphorylation at the 1 mM, but not at the 100 μM dose (Fig. 7A). While cells were unaffected by 100 μM AICAR, the 1 mM concentration induced cell death associated with Caspase-3 cleavage (Fig. 7B). In order to determine whether cell death was a direct result of AICAR-induced AMPK phosphorylation, the effects of AICAR treatment were investigated using an AMPKα-targeted shRNA-expressing clone, in which phosphorylated AMPK was undetectable, both in the absence or presence of 1 mM AICAR (Fig. 7C). This AMPKα-deficient clone nevertheless underwent cell death on treatment with 1 mM AICAR, associated with Caspase-3 cleavage (Fig. 7D).
7.
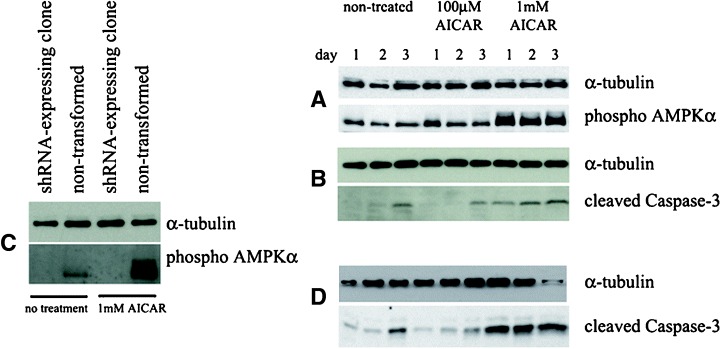
Western blots indicating (A) induction of AMPKα phosphorylation in HSVTK+ BHK cells treated with 1 mM (but not 100 μM) AICAR, (B) Caspase-3 cleavage in lysates from the same AICAR-treated cells, (C) absence of phospho AMPKα in an AICAR-treated pSilencer/AMPKαshRNA-transformed HSVTK+ BHK cell clone (compared to non-transformed cells) in which AMPKα expression was not detectable, (D) Caspase-3 cleavage in the same AMPKα-deficient pSilencer/AMPKα shRNA-transformed clone, induced by treatment with 1 mM AICAR.
To further investigate this, we additionally compared the effects of 100 μM and 1 mM AICAR on the extent of cell death in nine HSVTK+ BHK cell clones (n = 3 pSilencer/negative control shRNA, n = 3 pSilencer/AMPKα1shRNA and n = 3 pSilencer/ AMPKα2shRNA-expressing clones) at 18 hrs, 42 hrs, and 64 hrs). As before, 100 μM AICAR was found to have no effect on cell death or survival. However, 1 mM AICAR increased levels of cell death in all clones regardless of expression of AMPKα. While, at later timepoints (42 hrs and 64 hrs of treatment), 1 mM AICAR caused no significant differences in extent of death between AMPKexpressing and AMPKα-downregulated clones, significant differences were observed between clones at 18 hrs of treatment with increased levels of death in the AMPK-downregulated clones (Fig. 8A, B). The observed increased levels of death in AICARtreated AMPK-downregulated cells not only provides an additional cell stress model supporting the increased sensitivity of AMPK-deficient cells, but also suggests that AICAR functions to kill cells via additional mechanisms other than activation of AMPK.
8.
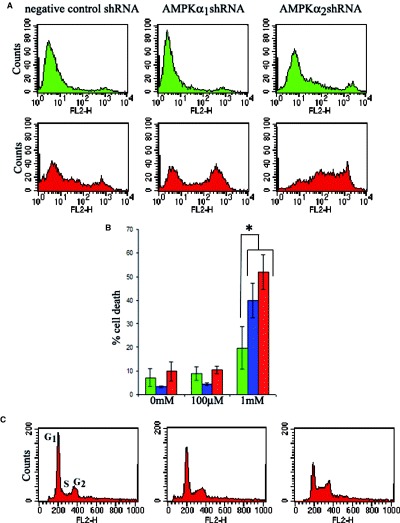
Effect of AICAR on AMPK-expressing and AMPK-downregulated HSVTK+ BHK cells. (A) Representative flow cytometry plots from propidium iodide-stained clones showing increased levels of death in AMPK-downregulated clones treated for 18 hrs with 1 mM AICAR. Top panel (green), non-treated cells. Bottom panel (red), 1 mM AICAR-treated cells. (B) Bar chart comparing extents of cell death in shRNA-expressing HSVTK+ BHK cells (green = negative, blue = AMPKα1, red = AMPKα2 shRNA-expressing cells) treated for 18 hrs with 0, 100 μM or 1 mM AICAR. Dead cells were counted as those scoring above the 102 value on the FL2 scale. Data are averaged from n = 3 clones per shRNA-expressing group. Asterisk indicates significant difference between levels of cell death in the negative control shRNA and AMPKα shRNA-expressing cells, treated with 1 mM AICAR (P < 0.05, Student's t-test). (C) Cell cycle analysis of propidium iodide-stained HSVTK+ BHK cells at 42 hrs of treatment with 100 μM or 1 mM AICAR. AICAR induces G1 phase exit and S-phase accumulation. AMPK expressiondependent differences in AICAR-mediated cell cycle distribution were not observed.
AICAR treatment induces S-phase accumulation in HSVTK+ BHK cells
Cell cycle analysis, carried out at 18 hrs, 42 hrs, and 64 hrs on HSVTK+ BHK cells exposed to 100 μM or 1 mM AICAR, revealed a dose-dependent overall reduction in the G1 peak and an increase in the proportion of cells in S-phase at the 42 hrs and 64 hrs timepoints (Fig. 8C). However, cell cycle stage accumulation appeared to be independent of AMPK expression level.
Discussion
The results of this study confirm a role for activated AMPK in delaying and reducing the extent of apoptotic cell death occurring as a result of glucose deprivation or of DNA damage. Activated AMPK may diminish apoptotic processes by influencing gene expression, via phosphorylation of transcriptional (co-)activators, or via inhibitory phosphorylation of proteins directly involved in apoptosis. This may have implications in cancer therapy, where specific inhibitors of AMPK might improve the outcome of standard chemotherapy. Here we used two different cell lines and two different cell death models and showed in both cases that AMPK downregulation resulted in enhanced cell death under the stress conditions imposed.
The first model, in which HSVTK-expressing BHK cells were treated with antiherpesvirus guanosine nucleoside analogues, was originally described by Moolten in 1986 [18] and is employed as a suicide gene therapy approach for treating cancer. The majority of studies confirm that GCV induces apoptotic death in HSVTK-transformed cancer cell lines. Few studies have additionally employed ACV and PCV, probably because GCV is the most effective of these analogues. The pathway of GCV-induced apoptosis in HSVTK-transformed cells is not entirely elucidated. In the BHK cell model described, we confirm Caspase-3 and PARP cleavage, accompanied by cell shrinkage, DNA laddering and phosphatidyl serine exposure [17] during both GCV and PCV treatment. However, in this model the cell-killing effect of ACV treatment has always been less potent, appeared to induce sustained cell cycle arrest leading eventually to cell death but did not involve any significant Caspase-3 and PARP cleavage, phosphatidyl serine exposure or DNA laddering. In addition we previously reported ACV treatment of HSVTK+ BHK cells to induce cell swelling, typical of necrosis, rather than the cell shrinkage characteristic of apoptosis. The slowed growth rate associated with AMPK depletion in untreated HSVTK+ BHK cells might be predicted to render these cells more resistant to guanosine nucleoside analogue-induced death (due to the accompanying reduced rate of DNA replication). However, in the case of GCV and PCV, the AMPK-depleted cells display greater sensitivity to these analogues.
It is of interest that, while AMPK downregulation enhanced apoptotic death by GCV and PCV treatment, it failed to influence ACV-mediated necrosis. This might be explained by the fact that GCV and PCV-treated cells die by an ATP-dependent mechanism: apoptosis, a pre-requisite for which would be a reduction in ATP levels in order to reach the ‘apoptotic threshold’, coupled with sufficient ATP levels to permit the apoptotic process [16]. In AMPK-deficient cells, ATP levels are likely to fall faster to the apoptotic threshold resulting in earlier or enhanced induction of apoptosis. ACV-mediated death appears to occur via an ATP-independent mechanism and is therefore less likely to be influenced by reduced AMPK activity.
In the second model, in which glucose deprivation has been shown to induce apoptosis of PC12 cells [19, 20], we show again that the apoptotic process is enhanced by AMPK depletion. Therefore we tentatively conclude that AMPK depletion may enhance certain apoptotic death processes but is unlikely to influence necrotic processes. Further studies are, however, necessary to establish whether this phenomenon is extendable to the majority of cell types.
It should be considered, that these studies were carried out on cell types in which, using the Upstate isoform-specific antibodies, only AMPKα1 could be detected. The fact that the AMPKα2-targeted shRNA was able to achieve downregulation of total AMPKα in these cell lines, suggests that the α2 sequence may exhibit some cross-specificity, resulting in additional α1 knock-down. (This is conceivable, since the target α1 and α2 sequences differ in only one nucleotide.) Since the work was carried out only on α1-expressing cell lines, the apoptosis-enhancing effect of AMPK depletion may be specific only to α1 complexes. We have additionally not investigated β or γ isoform usage of the AMPK complexes within these cell types, which may again affect the outcome of the stress-induced cell death process.
The alternative school of thought supporting a proapoptotic role for activated AMPK is generally supported by observations that the AMPK activator, AICAR, induces apoptosis at concentrations which also induce AMPK phosphorylation. Our study, however, which showed that an shRNA-expressing clone, with no detectable AMPK expression, not only underwent apoptosis on AICAR treatment, but also appeared to be more susceptible, than AMPK-expressing cells, to AICAR-induced death, provides evidence that AICARinduced apoptosis occurs via mechanisms independent of AMPK activation and possibly also that AICARinduced AMPK activation may delay AICAR-induced death. A candidate mechanism by which AICAR may induce cell death, independently of AMPK activation, could be inhibition of DNA synthesis. This is evidenced by the AICAR-induced S-phase accumulation, observed in our cell cycle study, which supports the hypothesis that ZMP, the intracellular monophosphorylated form of AICAR, which mediates AMPK activation, is further converted to ZTP, the triphosphorylated form. dZTP could then interfere in DNA synthesis, resulting in cell death as a result of triggering of DNA damage pathways. The additional reduced levels of AMPK in the AMPK-downregulated clones, might subsequently render them more susceptible to the DNA synthesis-inhibition mechanism of cell death, which would provide an explanation for the AMPK expression- dependent differences in cell viability in the absence of AMPK expression-dependent differences in cell cycle stage accumulations.
While we provide evidence from these studies, that AMPK depletion enhances stress-induced apoptosis, suggesting that AMPK inhibitors might be beneficial in combination with certain cancer therapies, the additional likelihood that AMPK upregulation/activation may delay stress-induced apoptosis, may be of clinical benefit in reducing damage induced by conditions such as hypoglycaemic shock or stroke.
Acknowledgments
This work was funded in part by a grant from the NIH (DK 20495) and by a mentor fellowship grant from the American Diabetes Association.
References
- 1.Russell RR, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, Giordano FJ, Mu J, Birnbaum MJ, Young LH. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis and injury. J Clin Invest. 2004;114:495–503. doi: 10.1172/JCI19297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA. 2004;101:3329–35. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Culmsee C, Monnig J, Kemp BE, Mattson MP. AMP-activated protein kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. J Mol Neurosci. 2001;17:45–58. doi: 10.1385/JMN:17:1:45. [DOI] [PubMed] [Google Scholar]
- 4.Stefanelli C, Stanic I, Bonavita F, Flamigni F, Pignatti C, Guarnieri C, Caldarera CM. Inhibition of glucocorticoid-induced apoptosis with 5-aminoimidazole- 4-carboxamide ribonucleoside, a cell-permeable activator of AMP-activated protein kinase. Biochem Biophys Res Commun. 1998;243:821–6. doi: 10.1006/bbrc.1998.8154. [DOI] [PubMed] [Google Scholar]
- 5.Ido Y, Carling D, Ruderman N. Hyperglycemiainduced apoptosis in human umbilical vein endothelial cells: inhibition by the AMP-activated protein kinase activation. Diabetes. 2002;51:159–67. doi: 10.2337/diabetes.51.1.159. [DOI] [PubMed] [Google Scholar]
- 6.Saitoh M, Nagai K, Nakagawa K, Yamamura T, Yamamoto S, Nishizaki T. Adenosine induces apoptosis in the human gastric cancer cells via an intrinsic pathway relevant to activation of AMP-activated protein kinase. Biochem Pharmacol. 2004;67:2005–11. doi: 10.1016/j.bcp.2004.01.020. [DOI] [PubMed] [Google Scholar]
- 7.Kefas BA, Cai Y, Kerckhofs K, Ling Z, Martens G, Heimberg H, Pipeleers D, Van de Casteele M. Metformin-induced stimulation of AMP-activated protein kinase in beta cells impairs their glucose responsiveness and can lead to apoptosis. Biochem Pharmacol. 2004;68:409–16. doi: 10.1016/j.bcp.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 8.Li J, Jiang P, Robinson M, Lawrence TS, Sun Y. AMPKβ1 subunit is a p53-independent stress-responsive protein that inhibits tumour cell growth upon forced expression. Carcinogenesis. 2003;24:827–34. doi: 10.1093/carcin/bgg032. [DOI] [PubMed] [Google Scholar]
- 9.Salt I, Celler JW, Hawley SA, Prescott A, Woods A, Carling D, Hardie G. AMP-activated protein kinase: greater AMP dependence, and preferential nuclear localization, of complexes containing the α2 isoform. Biochem J. 1998;334:177–87. doi: 10.1042/bj3340177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saha AK, Ruderman NB. Malonyl-CoA and AMP-activated protein kinase: an expanding partnership. Mol Cell Biochem. 2003;253:65–70. doi: 10.1023/a:1026053302036. [DOI] [PubMed] [Google Scholar]
- 11.Hue L, Beauloye C, Marsin AS, Bertrand L, Horman S, Rider MH. Insulin and ischemia stimulate glycolysis by acting on the same targets through different and opposing signaling pathways. J Mol Cell Cardiol. 2002;34:1091–7. doi: 10.1006/jmcc.2002.2063. [DOI] [PubMed] [Google Scholar]
- 12.Yang W, Hong YH, Shen X-Q, Frankowski C, Camp HS, Leff T. Regulation of transcription by AMP-activated protein kinase. J Biol Chem. 2001;276:38341–4. doi: 10.1074/jbc.C100316200. [DOI] [PubMed] [Google Scholar]
- 13.Eguchi Y, Shimizu S, Tsujimoto Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997;57:1835–40. [PubMed] [Google Scholar]
- 14.Lelli JL, Becks LL, Dabrowska MI, Hinshaw DB. ATP converts necrosis to apoptosis in oxidant-injured endothelial cells. Free Radic Biol Med. 1998;25:694–702. doi: 10.1016/s0891-5849(98)00107-5. [DOI] [PubMed] [Google Scholar]
- 15.Lieberthal W, Menza SA, Levine JS. Graded ATP depletion can cause necrosis or apoptosis of cultured mouse proximal tubular cells. Am J Physiol. 1998;274:F315–27. doi: 10.1152/ajprenal.1998.274.2.F315. [DOI] [PubMed] [Google Scholar]
- 16.Richter C, Schweizer M, Cossarizza A, Franceschi C. Control of apoptosis by the cellular ATP level. FEBS Lett. 1996;378:107–10. doi: 10.1016/0014-5793(95)01431-4. [DOI] [PubMed] [Google Scholar]
- 17.Shaw MM, Gurr WK, Watts PA, Littler E, Field HJ. Ganciclovir and penciclovir, but not acyclovir, induce apoptosis in herpes simplex virus thymidine kinasetransformed baby hamster kidney cells. Antiviral Chem & Chemother. 2001;12:175–86. doi: 10.1177/095632020101200305. [DOI] [PubMed] [Google Scholar]
- 18.Moolten FL. Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: paradigm for a prospective cancer control strategy. Cancer Res. 1986;46:5276–81. [PubMed] [Google Scholar]
- 19.Tong L, Perez-Polo JR. Transcription factor DNA binding activity in PC12 cells undergoing apoptosis after glucose deprivation. Neurosci Lett. 1995;191:137–40. doi: 10.1016/0304-3940(95)11557-d. [DOI] [PubMed] [Google Scholar]
- 20.Liu Y, Song XD, Lui W, Zhang TY, Zuo J. Glucose deprivation induces mitochondrial dysfunction and oxidative stress in PC12 cell line. J Cell Mol Med. 2003;7:49–56. doi: 10.1111/j.1582-4934.2003.tb00202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]