

Abstract
The (pro)renin–renin receptor [(P)RR] was discovered as an important novel component of the renin–angiotensin system (RAS). The functional significance of (P)RR is widely studied in renal and vascular pathologies and has sparked interest for a potential role in cardiovascular disease. To investigate the role of (P)RR in cardiac pathophysiology, we aimed to assess (P)RR regulation in adverse cardiac remodelling of the failing heart. In particular, we evaluated the expression of (P)RR in different models of heart failure and across different species. Significantly increased levels of (P)RR mRNA were found in post-myocardial infarcted (MI) hearts of rats (1.6-fold, P < 0.05) and mice (5-fold, P < 0.01), as well as in transgenic rats with overexpression of the mouse renin gene (Ren2) (2.2-fold, P < 0.01). Moreover, we observed a strong increase of (P)RR expression in hearts of dilated cardiomyopathy (DCM) patients (5.3-fold, P < 0.001). Because none of the tested commercially available antibodies appeared to detect endogenous (P)RR, a (P)RR-specific polyclonal antibody was generated to study (P)RR protein levels. (P)RR protein levels were significantly increased in the post-MI rat heart (1.4-fold, P < 0.05) as compared to controls. Most interestingly in DCM patients, a significant 8.7-fold (P < 0.05) increase was observed. Thus, protein expression paralleled gene expression. These results demonstrate that (P)RR expression is strongly up-regulated both in rodent models of heart failure and in the failing human heart, hinting to a potential role for (P)RR in cardiac pathophysiology.
Keywords: (P)RR, renin–angiotensin system, heart failure, hypertrophy, myocardial infarction, remodelling, renin
Introduction
The renin angiotensin system (RAS) is a pivotal system in volume and blood pressure regulation. For a long time, it was assumed that its deleterious effects are exclusively mediated via its downstream effectors, angiotensin II and aldosterone [1, 2]. The cloning of the (pro)renin–renin receptor [(P)RR] has, however, challenged this paradigm [3]. Binding of both renin and its inactive precursor, prorenin, to the (P)RR results in enhanced (pro)renin activation in a non-proteolytic manner, and has an approximately five-fold increase in renin catalytic activity as a result. Thus, the netto tissue RAS activity by the classical signalling pathway involving angiotensin II formation and subsequent activation of angiotensin II receptors may be augmented by expression of (P)RR. Furthermore, evidence exists for angiotensin II–independent effects of (P)RR as it is capable of intracellular induction of the mitogen-activated protein kinase (MAPK) pathways, resulting in increased cell proliferation and up-regulation of profibrotic genes [3].
Considerable interest therefore exists for the functional consequences of (P)RR. (P)RR is expressed in kidney, specifically in renal mesangial cells and in heart, brain, blood vessels, macrophages, T cells and granulocytes [3]. Although evidence exists that (P)RR may be involved in renal pathophysiology [4-6], its potential role in heart disease remains unclear. Ichihara et al. showed increased mRNA expression of (P)RR in stroke-prone spontaneously hypertensive rats (SHRsp) [7] and Hirose et al. showed increased mRNA expression in rat hearts post-MI [8]. Whether other causes of cardiac remodelling are associated with increased (P)RR expression, whether (P)RR protein expression parallels mRNA expression and whether (P)RR is regulated in species other than rat, most importantly human beings, is currently unknown.
We herein present data on the transcript and protein expression of (P)RR in multiple models of murine and rat heart failure and cardiac remodelling, as well as data from human heart failure.
Materials and methods
Cloning of mouse (P)RR
A full length mouse (P)RR cDNA (1060 base pairs encoding 350 amino acids) was PCR amplified using a mouse kidney cDNA library (prepared from pooled RNA of three C57Bl6/J males, 10 weeks old). For amplification, the following primers were used: forward, CATGGCTGTGCTGGTCGTTCT and reverse, TCAATCTATTCGAATCTTCT. This PCR fragment was cloned into the XhoI and HindIII sites of a pcDNA3.1 expression vector (Invitrogen, Darmstadt, Germany). For antibody generation, a Sau3A DNA fragment (encoding amino acids 41–239) from the (P)RR binding domain, denoted as (P)RR-BD, was cloned into the BamHI sites of the Escherichia coli expression vector pQE31 (Qiagen, Venlo, Netherlands) generating a 6xHis-tag fusion.
Protein expression and antibody generation
His6-(P)RR-BD expression in E. coli cells was induced by isopropyl-β-D-thiogalactopyranoside (IPTG) for 4 hrs at 37°C. His6-(P)RR-BD inclusion bodies isolated in T10N50E1 buffer (10 mM Tris, pH 8.0; 50 mM NaCl; 1 mM EDTA, 0.05% NP40) were dissolved in 6M Guanidine buffer (100 mM NaH2PO4, 10 mM Tris-HCl and 6M Guanidine HCl, pH 8.0). His6-(P)RR-BD was allowed to bind to Ni-NTA agarose beads (Qiagen) for 1 hr at room temperature on a rotating shaker. Unbound protein was removed by washing the beads with 8M urea buffer [100 mM NaH2PO4, 10 mM Tris-HCl and 8M urea, pH 8.0). His6-(P)RR-BD was subsequently eluted with elution buffer (100 mM NaH2PO4, 10 mM Tris-HCl and 8M urea, pH 4.5]. Finally, purified His6-(P)RR-BD was separated on a SDS-PAGE gel, stained with 0.25M KCl and isolated from the gel by electrophoresis in a dialysis bag for 3 hrs at 4°C.
To generate a polyclonal antibody against (P)RR, purified recombinant His6-(P)RR-BD was used for immunization of two rabbits. Immunization was performed for every other week, and after the second, third and fourth boosts blood samples were drawn to generate serum.
Titre test and purification of the antibody
(P)RR polyclonal antibody was purified using Schleicher and Schuell nitrocellulose membranes (0.45 μm) containing immobilized purified recombinant (P)RR. This membrane was blocked with [PBST (PBS+0.5% Tween)+5% BSA] for 1 hr at room temperature and subsequently incubated with serum for 2 hrs at 4°C on a rotating wheel. Unbound antibody was removed by washing with PBST six times. Finally, antibody was eluted with 300 μl glycine (100 mM, pH 2.8), and this was repeated five times.
Cell culture
HeLa S3 cells were cultured in DMEM with 4.5 g/l glucose supplemented with 10% FCS, 100 U/ml penicillin and 100 μg/ml streptomycin. Cells were grown in a humidified incubator at 5% CO2 and 37°C. For plasmid transfection of HeLa S3 cells, lipofectamine LTX reagent (Invitrogen) was used. Cells in a 12-wells plate with 60–80% confluency were transfected with 0.5 μg DNA for 24 hrs.
mRNA expression and quantitative RT-PCR
Relative gene expression of (P)RR and atrial natriuretic peptide (ANP) were determined by real-time polymerase chain reaction (RT-PCR). Gene expression was normalized using reference genes 36B4 and GAPDH. Values were expressed relative to appropriate control groups. The primers used for the RT-PCR are summarized in Table S1.
Western blot
A total of 35 μg of whole cell protein lysates were separated on a SDS-PAGE gel and proteins were immunoblotted onto nitrocellulose membranes. Membranes were incubated with rabbit polyclonal (P)RR primary antibody (2 μg/ml), goat polyclonal (P)RR primary antibody (Cat. no. ab5959, 1 μg/ml; Abcam, Cambridge, UK), goat polyclonal (P)RR primary antibody (Cat. no. IMG-3561, 1 μg/ml; Imgenex, San Diego, CA, USA), mouse monoclonal p-ERK antibody (Cat. no. sc-7383, 0.2 μg/ml; Santa Cruz, CA, USA), mouse monoclonal ERK 1/2 antibody (Cat. no. sc-135900, 0.1 μg/ml; Santa Cruz) or with rabbit monoclonal GAPDH primary antibody (400 ng/ml; Fitzgerald Inc., North Acton, MA, USA). After incubation with secondary polyclonal goat anti-rabbit (1:2000) or polyclonal rabbit anti-goat (1:4000) or polyclonal rabbit antimouse (1:2000; Dako Inc. Glostrup, Denmark) antibodies, proteins were detected using Super Signal West Pico Chemiluminescent Substrate (Thermo Scientific Inc., Rockford, IL, USA). The band densities were scanned and quantified using Syngene Bio Imaging device and normalized to GAPDH.
Myocardial tissue specimen from patients with heart failure
We have previously described the study population in detail [9]. Briefly, in our diagnostic work-up, patients with DCM underwent cardiac catheterization and endomyocardial biopsies (EMB). EMB from the right ventricular septum were taken and snap-frozen. All patients had dilated, poorly contractile left ventricles and were stable on medication. To study mRNA levels, we tested EMB from 20 patients. For protein analysis, we used heart tissue that was obtained during left ventriculectomy (n = 3). Control tissue was obtained from healthy human beings who donated their hearts as organ donors, but whose hearts were refused because of surgical reasons (n = 5).
Animal models and experimental protocols
We employed the established model of MI in both rats (n = 5; Munich Wistar Frömter, MWF) for 10 weeks and mice (n = 6) for 4 weeks (C57Bl6/J) by inducing coronary artery occlusion as previously described [10]. Furthermore, we employed 6-week-old homozygous male TGR (mREN2)27 rats (n = 5), a model of fast-forward heart failure and Sprague–Dawley (SD) control rats (n = 5) as previously described [11]. Rats were treated for six weeks with ACE inhibitor, lisinopril (10 μg/ml), in drinking water. Rats had ad libitum access to regular chow and drinking water. At the end of six weeks, phenotyping was performed. Non-infarcted part of hearts from post-MI animals and apex from the heart of Ren2 rats were collected under anaesthesia and stored at −80°C for further analysis.
All experiments were approved by the Animal Ethical Committee of the University of Groningen, The Netherlands, and conducted in accordance with existing guidelines for the care and use of laboratory animals.
Statistics
Data are expressed as mean ± S.E.M. Differences were tested using Student’s t-test or ANOVA, and results were considered statistically significant when P-values were <0.05.
Results
Detection of (P)RR protein expression using various antibodies
Full length (P)RR consists of four domains, an N-terminal signal peptide (a.a. 1–19), the extracellular ligand binding domain for renin/(pro)renin binding (a.a. 20–303), (P)RR-BD, a single transmembrane domain (a.a. 304–326) and a short cytoplasmic domain (a.a. 327–350; Fig. 1). For antibody generation, a fragment of (P)RR-BD (a.a. 41–239) was expressed and purified and used for generating a rabbit polyclonal antibody (Fig. 1). To test this antibody, a Western blot with mouse heart tissue was probed with the affinity purified antibody. As a positive control, a lysate from transiently (P)RR expressing HeLa S3 cells was used and as a control, non-overexpressing (P)RR HeLa S3 was used. Expression of (P)RR in the transiently transfected HeLaS3 cells was confirmed by RT-PCR (Fig. S1). As shown in Figure 2, this (P)RR antibody recognized a band of 37 kD [the expected size of (P)RR] in mouse heart lysates, which was also strongly present in the positive control and barely present in the control HeLa S3 cells. This confirms the expression of (P)RR in the heart and the specificity of our antibody. It is noteworthy that we also tested two commercial available antibodies that have been used in some other studies, but we could not detect a specific (P)RR signal with these antibodies.
Fig 1.

Schematic depiction of (pro)renin–renin receptor. SP: signal peptide (1–19 amino acids); (P)RR-BD: (P)RR binding domain (20–303 amino acids); TM: transmembrane domain (304–326 amino acids); CD: cytoplasmic domain 327–350 amino acids). The domain for antibody generation is indicated (41–239).
(P)RR expression in different organs
Having established (P)RR expression in the heart, we next investigated (P)RR expression in other organs. At the mRNA level, maximal (P)RR expression was observed in brain, followed by kidney, intestine, liver and heart (Fig. 3A). In muscle and lung, expression was nearly absent. At the protein level, maximal expression was observed in kidney followed by liver, brain, heart and intestine (Fig. 3B and C). Again, expression was low in muscle and lung. Except for brain, mRNA and protein expression levels paralleled each other.
Fig 3.
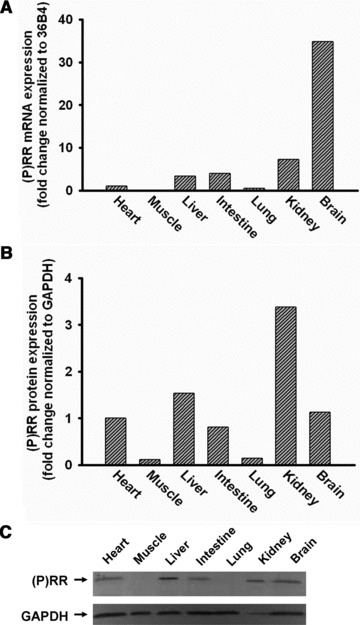
Expression of (P)RR mRNA and protein in different organs of C57Bl6/J mouse. (P)RR mRNA is highly expressed in brain, kidney, intestine and liver and moderate in heart and undetectable in skeletal muscle and lung (n = 4, A). (P)RR protein is highly expressed in kidney, brain, liver and intestine, moderate in heart and undetectable in skeletal muscle and lung (n = 4, B and C).
Characteristics and expression of (P)RR in rats and mice with post-MI heart failure
To investigate potential changes in (P)RR expression in post-MI cardiac tissue, a coronary artery occlusion was performed in rats and mice and cardiac remodelling ensued for 10 weeks in rats and 4 weeks in mice. Post-MI rats showed an infarct size of 46.9 ± 2.5%. Exact infarct sizes of the mouse hearts were not assessed, but we scanned the mice with MRI and left ventricular ejection fraction was 32% (normal mice: ∼80%; Table S2). Heart weights were significantly increased as compared to sham operated animals due to cardiac hypertrophy and remodelling. Ejection fraction and fraction shortening were significantly reduced for both rats and mice. In addition, in the non-infarcted parts of post-MI rat hearts, ANP mRNA expression (14.7-fold, P < 0.001) was strongly increased and maximal contraction (P < 0.05) and relaxation (P < 0.05) of the left ventricular were all significantly decreased in post-MI rats as compared to sham-operated animals (Table S2).
Post-MI rat hearts showed a significant increase in (P)RR mRNA of about 1.6-fold (P < 0.05) in the non-infarcted hypertrophied areas of the heart compared to sham-operated rats (Fig. 4A). In mice, we demonstrate a more robust up-regulation of approximately 5-fold (P < 0.01; Fig. 4B) in the non-infarcted area. Figure 4C displays (P)RR protein levels in non-infarcted area of post-MI rat hearts, which were also significantly elevated by approximately 1.4-fold (P < 0.05; Fig. 4D).
Fig 4.
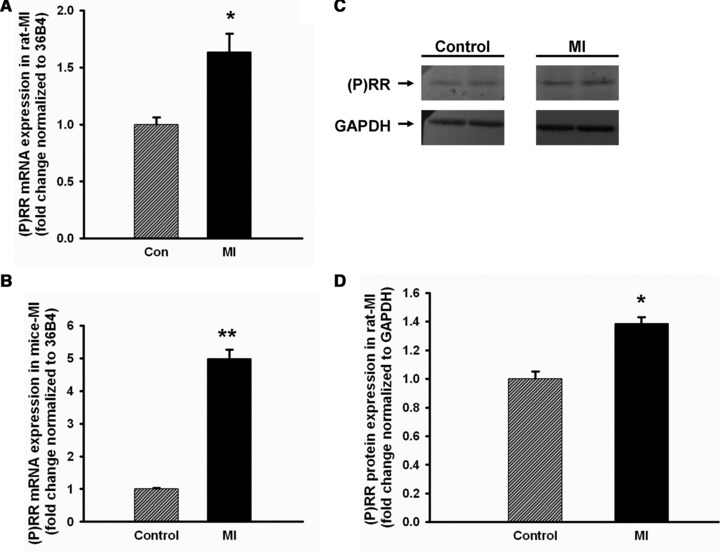
Elevation of (P)RR in post-MI heart. Both mRNA (in rat, n = 5, A; and in mouse, n = 6, B) and protein (in rat, n = 4, C and D) expression of (P)RR were significantly increased in non-infarcted areas of hearts of post-MI compared to sham-operated rats and mice. Data are expressed as mean ± S.E.M. *P < 0.05, **P < 0.01 versus control.
ERK kinase activation during post-MI remodelling
Phosphorylated ERK kinase (expressed as p-ERK/ERK ratio) was significantly increased by about 2.2-fold (P < 0.05) in the rat post-MI model. A significant positive correlation (r = 0.730, Pearson correlation coefficient; P = 0.04) between (P)RR expression and ERK activation was observed, when compared to sham-operated rats (Fig. S2).
Characteristics and (P)RR regulation in Ren2 rats and effects of ACE inhibition
The Ren2 rat is a model that overexpresses mouse renin, resulting in hypertension and heart failure development. The HW/BW ratio was significantly increased in Ren2 rats as compared to the control SD group (P < 0.01). ACE inhibition attenuated hypertension and prevented heart failure development in Ren2 rats. dp/dt max (P <0.01) and dp/dt min (P <0.01) were significantly decreased in Ren2 group when compared to control group. To confirm the beneficial effects of ACEi treatment, we also measured the expression of ANP mRNA levels which we report to be approximately 12-fold (P < 0.001) higher in untreated Ren2 rats, whereas ACEi blunted the increase (2.7-fold, P < 0.05) in expression (Table S2).
In the Ren2 rats, no significant differences in cardiac (P)RR mRNA expression were detected compared to controls. However, treatment with the ACEi, lisinopril, up-regulated (P)RR mRNA by 2.2-fold (P < 0.01; Fig. 5A). To determine if the up-regulation of (P)RR mRNA of lisinopril-treated Ren2 rats parallels (P)RR protein expression, we measured the expression in these groups and found no significant difference (Fig. 5B). Thus, mRNA expression does not always parallel protein expression.
Fig 5.
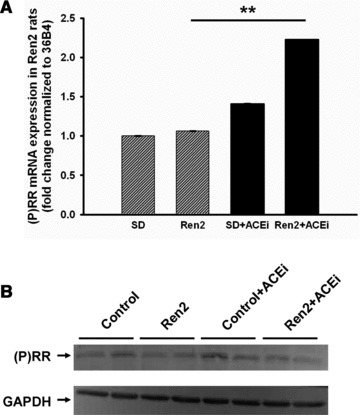
Regulation of (P)RR due to hypertensive therapy in rat heart Comparison of the expression of (P)RR mRNA (A) and protein (B) in control (SD), Ren2, control (SD)+ACEi, Ren2+ACEi rats (n = 5). Data are expressed as mean ± S.E.M. **P < 0.01 versus Ren2.
Dilated cardiomyopathy up-regulates (P)RR expression in human heart
To elucidate the regulation of (P)RR in human heart failure, we first evaluated (P)RR expression of heart tissue from patients with DCM. (P)RR mRNA levels were up-regulated significantly about 5.3-fold (P < 0.001) in the hearts of DCM patients compared to healthy controls (Fig. 6A). In addition, protein levels of (P)RR showed a similar up-regulation and was about 8.8-fold (P < 0.05) higher in DCM patients compared to healthy controls (Fig. 6B and C).
Fig 6.
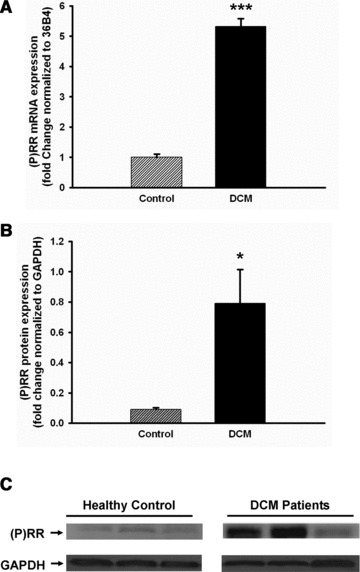
Up-regulation of (P)RR in the heart of human DCM. Heart tissues were obtained from healthy control and dilated cardiomyopathy patients. Both mRNA (n = 20, A) and protein (n = 3, B and C) expression were significantly increased in DCM patients compared to healthy controls. Data are expressed as mean ± S.E.M. *P < 0.05, ***P < 0.001 versus control.
Discussion
The main findings of this study are that (P)RR mRNA and protein expression are increased in cardiac remodelling and heart failure. We tested heart tissue derived from a broad spectrum of experimental models of cardiac remodelling and heart failure, from various aetiologies (pressure overload, ischaemia) and in different species (mouse, rat). We corroborated our findings in human tissue specimens in which we also show an increased expression of (P)RR. Finally, we report to have generated an anti-(P)RR antibody with higher specificity than currently available commercial antibodies, which we believe will be very helpful in future studies into (P)RR.
The (P)RR was cloned in 2002 and has changed the paradigm in thinking about the RAS considerably. Until then, it was believed that circulating renin and (pro)renin themselves would be unharmful, as their effects were thought to be conferred via downstream elements of the RAS, specifically angiotensin II and aldosterone. However, binding of renin/prorenin to the (P)RR has specific downstream effects in cell culture systems that mimic AngII [12, 13], for example DNA synthesis, activation of stress-related kinases (MAPK, ERK), activation of PAI-1 and phosphorylation of heat shock proteins [3, 14, 15]. Furthermore, when (pro)renin or renin binds the (P)RR, renin activity is higher so that ‘classical’ signalling of the RAS is also increased. Thus, high levels of local (P)RR may theoretically have profound impact on local RAS activity.
Despite this theorem, little knowledge exists on the role of (P)RR in the heart, one of the main target organs of the RAS. Transcript (mRNA) expression of (P)RR is up-regulated in the hearts of stroke-prone spontaneous hypertensive rats (SHRsp) [7] and in the hearts of rats with congestive heart failure [8]. So far, no data are available on protein expression of (P)RR, especially in hearts of human beings and rodents. We herein confirm previous reports on transcriptional regulation of (P)RR in the rat heart with adverse cardiac remodelling, and extend these findings to the protein level. Moreover, we showed that this phenomenon is conserved in mice and man. In fact, we observed that mRNA expression in post-MI mice hearts and cardiomyopathic human hearts were even more strongly up-regulated than in rat hearts. Again, using our new antibody, we could show for the first time that protein levels of (P)RR are increased as well. Finally, we confirm the potential role of (P)RR in adverse signalling pathways such as ERK kinase.
Although (P)RR was up-regulated in all models we tested, we observed no increase in the TGR(mRen2) 27 rat (Ren-2 rat). To assess further insight of (P)RR regulation in a high renin model, we used this monogenic rat model with overexpression of the murine renin-2 gene, resulting in hypertension and concomitant heart failure development. No significant up-regulation of (P)RR in Ren2 compared to control rats was observed. It is possible that the extreme high renin status does not represent a normal RAS system, thereby not allowing typical RAS regulation, including (P)RR. However, ACEi treatment of Ren2 rats increased (P)RR mRNA expression significantly. This is in line with previous observations that blood pressure lowering drugs may increase (P)RR expression in target organs [16, 17].
The study of the (P)RR has been hampered by the lack of a specific antibody. We could not detect reliable results with currently commercial available antibodies. We therefore took the initiative to generate a specific antibody. Ongoing studies have revealed that (P)RR is encoded by the ATP6AP2 gene and exists in a truncated form that is 95% homologous to the ATP6AP2 gene product, vacuolar-type H+-ATPase (V-ATPase), the function of which is to maintain acidic pH of intracellular vesicles [18]. This function resides in C-terminal domain (amino acid 283–350). We chose to raise an antibody with specific activity against the domain of (P)RR (41–239 amino acids) that binds prorenin. We report that the antibody detects a specific band at expected size (37 kD). We compared our antibody with two other commercially available antibodies and confirmed that only our self-generated antibody detects (P)RR at the correct size. This new antibody may therefore strengthen future studies into (P)RR.
A significant role for (P)RR in renal and vascular pathologies has been proposed. (P)RR expression is increased in patients with diabetic nephropathy and end-stage kidney disease [4], and animal studies demonstrate up-regulation of renal (P)RR in rat models of hypertension [6], diabetes [19] and renal injury [5]. Moreover, transgenic rats overexpressing (P)RR in smooth muscle tissue with strong transgene overexpression in the vasculature exhibit a cardiovascular phenotype of elevated blood pressure and increased heart rate [20]. However, definite proof of a role of (P)RR in cardiac disease and heart failure is lacking. Mice in which the gene encoding (P)RR is disrupted are not viable. The vacuolar-type H+-ATPase (V-ATPase) function of (P)RR seems an important conserved function that cannot be manipulated. Mice with cardiomyocyte-specific disruption of (P)RR inevitably die of heart failure [21]. This study design does not allow for linking the measured (P)RR transcript and protein levels to functional changes, which represents a limitation. Future studies should unravel the role of the (up-regulated) (P)RR in cardiac remodelling and heart failure.
In conclusion, we report both increased mRNA and protein expression of (P)RR, in the failing hearts of various species and with various aetiologies. We generated an antibody with specificity against (P)RR that we believe will facilitate further elucidation of (P)RR regulation. Our findings therefore implicate a role for (P)RR in cardiac remodelling and heart failure.
Acknowledgments
This work was supported by the Netherlands Heart Foundation (grant 2007T046) and the Innovational Research Incentives Scheme program of the Netherlands Organization for Scientific Research (NWO VENI, grant 916.10.117), both to Dr. de Boer.
Conflict of interest
The authors declare no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article: Fig S1 RT-PCR analysis of (P)RR mRNA expression in control HeLa S3 cells and (P)RR–overexpressed HeLa S3 cells.
(A) Representative Western blots showing activation of ERK (phosphorylation and total ERK) in the non-infarcted myocardium during post MI remodelling. (B) Changes in p-ERK/ERK ratio of sham (gray fine bar) and MI (black bar) animals. Data are expressed as means ± SEM. N = 4, *P < 0.05 versus sham. (C) Correlation analysis between (P)RR and ERK activation during post MI.
Primer sequences used in qRT-PCR.
Animal Characteristics.
Please note: Wiley-Blackwell is not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Schroten NF, Gaillard CA, van Veldhuisen DJ, et al. New roles for renin and prorenin in heart failure and cardiorenal crosstalk. Heart Fail Rev. 2011 doi: 10.1007/s10741-011-9262-2. ; doi: 10.1007/s10741-011-9262-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Unger T, Li J. The role of the renin-angiotensin-aldosterone system in heart failure. J Renin Angiotensin Aldosterone Syst. 2004;5:S7–10. doi: 10.3317/jraas.2004.024. [DOI] [PubMed] [Google Scholar]
- 3.Nguyen G, Delarue F, Burckle C, et al. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest. 2002;109:1417–27. doi: 10.1172/JCI14276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takahashi K, Yamamoto H, Hirose T, et al. Expression of (pro)renin receptor in human kidneys with end-stage kidney disease due to diabetic nephropathy. Peptides. 2010;31:1405–8. doi: 10.1016/j.peptides.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 5.Hirose T, Mori N, Totsune K, et al. Increased expression of (pro)renin receptor in the remnant kidneys of 5/6 nephrectomized rats. Regul Pept. 2010;159:93–9. doi: 10.1016/j.regpep.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 6.Prieto MC, Williams DE, Liu L, et al. Enhancement of renin and prorenin receptor in the collecting ducts of Cyp1a1-Ren2 Rats contribute to development and progression of malignant hypertension. Am J Physiol Renal Physiol. 2010;300:581–8. doi: 10.1152/ajprenal.00433.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ichihara A, Kaneshiro Y, Takemitsu T, et al. Nonproteolytic activation of prorenin contributes to development of cardiac fibrosis in genetic hypertension. Hypertension. 2006;47:894–900. doi: 10.1161/01.HYP.0000215838.48170.0b. [DOI] [PubMed] [Google Scholar]
- 8.Hirose T, Mori N, Totsune K, et al. Gene expression of (pro)renin receptor is upregulated in hearts and kidneys of rats with congestive heart failure. Peptides. 2009;30:2316–22. doi: 10.1016/j.peptides.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 9.de Boer RA, Henning RH, Suurmeijer AJ, et al. Early expression of natriuretic peptides and SERCA in mild heart failure: association with severity of the disease. Int J Cardiol. 2001;78:5–12. doi: 10.1016/s0167-5273(00)00440-x. [DOI] [PubMed] [Google Scholar]
- 10.de Boer RA, Pokharel S, Flesch M, et al. Extracellular signal regulated kinase and SMAD signaling both mediate the angiotensin II driven progression towards overt heart failure in homozygous TGR(mRen2)27. J Mol Med. 2004;82:678–87. doi: 10.1007/s00109-004-0579-3. [DOI] [PubMed] [Google Scholar]
- 11.de Boer RA, Pinto YM, Suurmeijer AJ, et al. Increased expression of cardiac angiotensin II type 1 (AT(1)) receptors decreases myocardial microvessel density after experimental myocardial infarction. Cardiovasc Res. 2003;57:434–42. doi: 10.1016/s0008-6363(02)00704-6. [DOI] [PubMed] [Google Scholar]
- 12.Batenburg WW, Krop M, Garrelds IM, et al. Prorenin is the endogenous agonist of the (pro)renin receptor. Binding kinetics of renin and prorenin in rat vascular smooth muscle cells overexpressing the human (pro)renin receptor. J Hypertens. 2007;25:2441–53. doi: 10.1097/HJH.0b013e3282f05bae. [DOI] [PubMed] [Google Scholar]
- 13.Nabi AH, Kageshima A, Uddin MN, et al. Binding properties of rat prorenin and renin to the recombinant rat renin/prorenin receptor prepared by a baculovirus expression system. Int J Mol Med. 2006;18:483–8. [PubMed] [Google Scholar]
- 14.Nguyen G, Delarue F, Berrou J, et al. Specific receptor binding of renin on human mesangial cells in culture increases plasminogen activator inhibitor-1 antigen. Kidney Int. 1996;50:1897–903. doi: 10.1038/ki.1996.511. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi K, Hirose T, Mori N, et al. The renin-angiotensin system, adrenomedullins and urotensin II in the kidney: possible renoprotection via the kidney peptide systems. Peptides. 2009;30:1575–85. doi: 10.1016/j.peptides.2009.05.018. [DOI] [PubMed] [Google Scholar]
- 16.Feldt S, Maschke U, Dechend R, et al. The putative (pro)renin receptor blocker HRP fails to prevent (pro)renin signaling. J Am Soc Nephrol. 2008;19:743–8. doi: 10.1681/ASN.2007091030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krebs C, Hamming I, Sadaghiani S, et al. Antihypertensive therapy upregulates renin and (pro)renin receptor in the clipped kidney of Goldblatt hypertensive rats. Kidney Int. 2007;292:876–87. doi: 10.1038/sj.ki.5002408. [DOI] [PubMed] [Google Scholar]
- 18.Batenburg WW, Danser AH. The (pro)renin receptor: a new addition to the renin-angiotensin system? Eur J Pharmacol. 2008;585:320–4. doi: 10.1016/j.ejphar.2008.02.092. [DOI] [PubMed] [Google Scholar]
- 19.Siragy HM, Huang J. Renal (pro)renin receptor upregulation in diabetic rats through enhanced angiotensin AT1 receptor and NADPH oxidase activity. Exp Physiol. 2008;93:709–14. doi: 10.1113/expphysiol.2007.040550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burckle CA, Danser AH, Muller DN, et al. Elevated blood pressure and heart rate in human renin receptor transgenic rats. Hypertension. 2006;47:552–6. doi: 10.1161/01.HYP.0000199912.47657.04. [DOI] [PubMed] [Google Scholar]
- 21.Kinouchi K, Ichihara A, Sano M, et al. The (pro)renin receptor/ATP6AP2 is essential for vacuolar H+-ATPase assembly in murine cardiomyocytes. Circ Res. 2010;107:30–4. doi: 10.1161/CIRCRESAHA.110.224667. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Representative Western blots showing activation of ERK (phosphorylation and total ERK) in the non-infarcted myocardium during post MI remodelling. (B) Changes in p-ERK/ERK ratio of sham (gray fine bar) and MI (black bar) animals. Data are expressed as means ± SEM. N = 4, *P < 0.05 versus sham. (C) Correlation analysis between (P)RR and ERK activation during post MI.
Primer sequences used in qRT-PCR.
Animal Characteristics.