

Abstract
The pathways that control mesenchymal stem cells (MSCs) differentiation are not well understood, and although some of the involved transcription factors (TFs) have been characterized, the role of others remains unclear. We used human MSCs from tibial plateau (TP) trabecular bone, iliac crest (IC) bone marrow and Wharton’s jelly (WJ) umbilical cord demonstrating a variability in their mineral matrix deposition, and in the expression levels of TFs including Runx2, Sox9, Sox5, Sox6, STAT1 and Slug, all involved in the control of osteochondroprogenitors differentiation program. Because we reasoned that the basal expression level of some TFs with crucial role in the control of MSC fate may be correlated with osteogenic potential, we considered the possibility to affect the hMSCs behaviour by using gene silencing approach without exposing cells to induction media. In this study we found that Slug-silenced cells changed in morphology, decreased in their migration ability, increased Sox9 and Sox5 and decreased Sox6 and STAT1 expression. On the contrary, the effect of Slug depletion on Runx2 was influenced by cell type. Interestingly, we demonstrated a direct in vivo regulatory action of Slug by chromatin immunoprecipitation, showing a specific recruitment of this TF in the promoter of Runx2 and Sox9 genes. As a whole, our findings have important potential implication on bone tissue engineering applications, reinforcing the concept that manipulation of specific TF expression levels may elucidate MSC biology and the molecular mechanisms, which promote osteogenic differentiation.
Keywords: human mesenchymal stem cells, Slug, osteogenesis, transcription factors, gene silencing
Introduction
Adult human mesenchymal stem cells (hMSCs) are considered to be a promising candidate for use in cell-based therapy or drug development applications for bone and cartilage [1-4], as demonstrated by many pre-clinical and clinical studies [5]. However, the sources from which these cells can be obtained and the exact culture conditions for in vitro differentiation induction are still under discussion. This is partly due to the difficulty in obtaining homogeneous hMSC populations, and partly due to the lack of knowledge of the complex molecular mechanisms controlling self-renewal, proliferation, differentiation and senescence of hMSCs, although several key components have been characterized. The pathways and the factors implicated in the regulation of these processes are of crucial importance for advances in bone and cartilage tissue engineering. At present, a great number of experimental strategies are addressed to investigate the role of epigenetic mechanisms and key transcription factors implicated in the control of stem cell properties, as well as in the chondrogenic or osteoblast committed status [6-11].
In this context, the importance of fine-tuning–specific gene expression and appropriate culture system conditions, including the presence of specific growth factors, has emerged. It is, in fact, well-established that the response to specific biological response modifiers is affected by the cell microenvironment. For this reason, a specific modulation or treatment/drug administration may differently affect MSC behaviour considering the cell source and differentiation potential which is strictly correlated with maturation stage. In this regard, an important question to be addressed is whether hMSCs obtained from different sources have the same potential to differentiate, or if there is already a pre-commitment depending on the niche from which they were obtained. Indeed, many studies have drawn attention to the study of unequal proliferative and differentiation capacities of MSC cell population isolated from different compartments, which may contain a various number of primitive stem cells, progenitors and/or committed cells [12-17]. Therefore, to prevent the discrepancies between data collected by different laboratories, and to encourage a rapid translation of MSC-based therapy into clinical practice as an alternative to conventional orthopaedic procedures, a great effort is being made to compare the molecular and biological features of different MSC populations, as well as improving and standardizing the isolation and cell culture techniques.
As a contribution to these issues, we aimed to demonstrate the unique nature of MSCs from different sources (i) by examining the different osteoblastic differentiation potential of hMSCs obtained from bone marrow iliac crest (IC), bone marrow tibial plateau (TP) and Wharton’s jelly (WJ) umbilical cord and (ii) by comparing their response to Slug gene silencing.
Previously, our research group provided evidence implicating Slug transcription factor as a positive regulator of the maturation process of human osteoblasts, and as possible effector of Wnt/β-catenin signalling [18]. Human Slug belongs to the Snail family of genes encoding zinc-finger transcription factors; it is involved in a broad spectrum of biological functions, such as epithelial– mesenchymal transition, cell differentiation, cell motility, cell-cycle regulation and apoptosis [19-22]. Our previous findings also suggest that Slug depletion may have a potential pro-chondrogenic effect [18]. Therefore, because many observations have demonstrated that the correct levels of transcription factors (TFs) are crucial to achieving differentiation towards a desired lineage from stem cells [23-25], we found it intriguing to investigate the effect of Slug depletion in different hMSC populations.
Materials and methods
Isolation and culture of hMSCs
hTP-MSCs were isolated from bone marrow TP aspirates (nine samples) using Ficoll-Hypaque density gradient (Sigma-Aldrich, St. Louis, MO, USA) as previously reported [26]. Briefly, nucleated cells were collected at the interface, washed twice, suspended in α-MEM supplemented with 15% FBS and penicillin G (Sigma-Aldrich), counted and plated at a concentration of 2 × 106 cells/T150 flask. After 48 hrs non-adherent cells were removed and the adherent MSCs expanded in vitro.
hBM-MSCs were isolated from bone-marrow aspirates of the IC (six samples) and cultured as described for hTP-MSCs.
hWJ-MSCs were isolated from WJ of human umbilical cords (six samples) collected from full-term births. The cords were processed as previously described [27] within 4 hrs. Briefly, small pieces of cord (2–4 mm in length) were placed directly into 25 cm2 flask for culture expansion in DMEM low glucose media (Euroclone S.p.a., Milan, Italy), supplemented with 10% FCS (Euroclone). After 5–7 days, the culture medium was removed and changed twice a week.
Informed consent was obtained by the Ethical committee for TP and IC from patients undergoing total hip replacement or ankle surgery, and for WJ from the mother after either caesarean section or natural delivery.
Osteogenic induction and mineralization assay
Osteogenesis of hMSCs was induced 24 hrs after seeding (in 12-well plates) by incubating cells in α-MEM medium supplemented with 100 μM ascorbic acid, 10 mM β-glycerophosphate and 100 nM dexamethasone (Sigma-Aldrich) for three to four weeks and analysed after the 24 hrs (Day 0) and at days 7, 14 and 21. The extent of mineralized matrix in the plates was determined by Alizarin Red S staining (Sigma-Aldrich) as previously reported [18].
Mineralized matrix positive to Alizarin Red S was analysed by image analysis. In particular, 10 different RGB images were acquired under white transmitted light from each well by a linear (γ = 1) charge-coupled device camera (DS Camera Control Unit DS-L2; Nikon, Tokyo, Japan) mounted on an inverted microscope (Nikon Eclipse TS-100; Nikon Instruments Europe BV, Amstelveen, The Netherlands), using an objective at 10× magnification and maintaining the sampling area fixed. Photometric analyses were conducted on the monochrome red channel of the RGB stack. Images were calibrated by adjusting the brightest grey level to 255 and then analysed using the public domain ImageJ software (rsbweb.nih.gov/ij). The data were expressed as integrated optical density (O.D.).
Flow cytometry analysis
hMSC (at passage 1) were fixed in 4% paraformaldehyde and incubated at 4°C for 30 min. with 5 μg/ml of the following monoclonal antibodies: anti-human–CD3, –CD14, –CD34, –CD45 (DAKO Cytomation, Glostrup, Denmark), –CD31 (Chemicon International, Temecula, CA), –CD73, –CD90, –CD146 (Becton Dickinson, Mountaine View, CA, USA), –CD105 (produced from the hybridoma cell line, clone SH2; ATCC, Rochville, MD, USA). The cells were washed twice and incubated with 2.4 μg/ml of a polyclonal rabbit anti-mouse immunoglobulins/FITC conjugate (DAKO Cytomation) at 4°C for 30 min. After two final washes, the cells were analysed using a FACStar plus Cytometer (Becton Dickinson). For isotype control, FITC-coupled non-specific mouse IgG was substituted for the primary antibody. Data were expressed both as mean percentage of positive cells and as mean fluorescence intensity (MFI) ratio determined using the mean fluorescence of the specific marker analysed divided by the mean fluorescence of the isotype control.
Small interfering RNA (siRNA) transfection
The anti-human Slug siRNA and appropriate control, scramble (scr) siRNAs, were synthesized by Invitrogen Life Technologies (Carlsbad, CA, USA). The nucleotide sequences of the siRNA pair are as follows: sense: 5′-CCCUGGUUGCUUCAAGGACACAUUA-3′, anti-sense: 5′-UAAUGUGUCCUUGAAGCAACCAGGG-3′. 24 hours before siRNA transfection, hMSCs were seeded in triplicate at a density of 12 × 103/cm2. Cells were transfected with 30 nM siRNA using Lipofectamine RNAiMAX (Invitrogen Life Technologies) according to the manufacturer’s instructions. Transfected cells were incubated for six days at 37°C before gene silencing analysis. Medium GC Stealth RNAi Negative Control Duplex (Invitrogen, Life Technologies) was used as negative control.
Real-time quantitative RT-PCR
Cells from three wells were harvested and total RNA was extracted using an RNeasy Mini Kit (Qiagen, Hilden, Germany) in accordance with the manufacturer’s instruction and as previously described [18]. Real-time PCR was carried out using the ABI PRISM 7700 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). TaqMan technology, the Assays-On-Demand kit for human Slug, Runx2, Sox9, Sox5, Sox6 and STAT1 were used. The mRNA levels of target genes were corrected for GAPDH mRNA levels (endogenous control). All PCR reactions were performed in triplicate for each sample.
Western blotting
For Western blot analysis, the cells were washed twice with ice-cold PBS and cell lysates were prepared as previously reported [18]. Briefly, 10 μg of each sample was electrophoresed on a 12% SDS-PAGE. The proteins were then transferred onto an Immobilon-P PVDF membrane (Millipore, Billerica, MA, USA). After blocking with PBS-0.05% Tween 20 and 5% dried milk, the membranes were probed with the following antibodies: Runx2 (SC-10758) and Sox9 (SC-20095) from Santa Cruz Biotechnology (Santa Cruz, CA, USA), Slug (L40C6) and STAT1 from Cells Signaling Technology (Denvers, CA, USA), Sox5 (ab26041) and Sox6 (ab68316) from Abcam (Cambridge, UK). After washing with PBS-Tween, the membranes were incubated with peroxidase-conjugated anti-mouse (1:2000) (Dako, Glostrup, Denmark) or anti-rabbit (1:50,000) in 5% non-fat milk. Immunocomplexes were detected using Supersignal West Femto Substrate (Pierce, Rockford, IL, USA). Anti-IP3K (06-195) from Upstate Biotechnology (Lake Placid, NY, USA) was used to confirm equal protein loading.
Viability analysis (calcein-AM uptake assay)
Viability assay was performed as described previously [27]. For propidium iodide and calcein analysis the cells were visualized with a Nikon camera (DS Camera Control Unit DS-L1) mounted on a fluorescence microscope (Nikon Eclipse 50i; Nikon Corporation, Tokyo, Japan) using the filter block for fluorescein. Dead cells were stained in red, whereas viable ones appeared in green.
Measurement of apoptosis
After the indicated days of treatment with Slug siRNA, the cells were rinsed twice with PBS solution and fixed for 25 min. in 4% paraformaldehyde at room temperature. Apoptotic cells were detected by the DeadEnd Colorimetric Apoptosis Detection System (Promega) according to the manufacturer’s instructions. A dark brown DAB signal indicates positive staining, whereas shades of blue-green to greenish tan indicate a non-reactive cell.
Scratch wound assay
hMSCs were seeded at density of 12 × 103/cm2, grown to 90% confluency, transfected with siSlug or scr RNAs, and 72 hrs later were wounded by scratching with a pipette tip. 24 and 48 hrs after wounding the cells were incubated with Calcein AM (final 1 μM) at 37°C in the incubator for 30 min. Images were taken at 0, 24 and 48 hrs.
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was carried out as previously described [22], using the standard protocol supplied by Upstate Biotechnology with their ChIP assay reagents. The cells were cross-linked with 1% formaldehyde for 10 min. at 37°C, washed in ice-cold PBS and suspended in SDS lysis buffer for 10 min. on ice. Samples were sonicated, diluted 10-fold in dilution buffer supplemented with protease inhibitors and pre-cleared with 80 μl of DNA-coated protein A-agarose; supernatant was used directly for immunoprecipitation with 5 μg of anti-Slug, (sc-10436; Santa Cruz Biotechnology), overnight at 4°C. Immunocomplexes were mixed with 80 μl of DNA-coated protein A-agarose followed by incubation for 1 hr at 4°C. Beads were collected and sequentially washed three times with 1 ml each of the following buffers: low salt wash buffer (0.1% SDS,1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8.1, 150 mM NaCl), high salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8.1, 500 mM NaCl), LiCl wash buffer (0.25 mM LiCl, 1% IGEPAL-CA630, 1% deoxycholic acid, 1 mM EDTA,10 mM Tris-pH 8.1), and TE buffer. Immunocomplexes were eluted two times by adding a 250-μl aliquot of a freshly prepared solution of 1% SDS, 0.1M NaHCO3 and the cross-linking reactions were reversed by incubation at 65°C for four hrs. Samples were then digested with proteinase K (10 mg/ml) at 42°C for 1 hr, DNA was recovered by phenol/chloroform extractions, ethanol precipitated using 1 μl of 20 mg/ml glycogen as the carrier, and suspended in sterile water. For PCR analysis, aliquots of chromatin before immunoprecipitation were saved (input). PCR was performed to analyse the presence of DNA precipitated by Slug-specific antibody, and by using specific primers (Table 1) to amplify fragments of Runx2 and Sox9 gene promoters. Each PCR reaction was performed with 5 μl of the bound DNA fraction or 2 μl of the input, as follows: pre-incubation at 95°C for 5 min., 30 cycles of 1 min. denaturation at 95°C, 1 min. annealing at 62°C, and 1 min. at 72°C, with one final incubation at 72°C for 5 min. No antibody control was included in each experiment.
Table 1.
PCR primers used for chromatin immunoprecipitation assay (ChlP)
| Gene | Primer sequences | Product size (bp) |
|---|---|---|
| Runx2 | Forward F1: 5′-ATATCCTTCTGGATGCCAGG-3′ | 167 |
| Reverse R1: B′-AAGCACTATTACTGGAGAGGC-S′ | ||
| Forward F2: 5′-GTTTCAGTGAATGCTAATGTAG-3′ | 290 | |
| Reverse R2:5′-AAGCGTTCATTTAACATGCAG-3′ | ||
| Forward F3: 5′-CAAGAGCTTTATTTGCATTGAC-3′ | 282 | |
| Reverse R3:5′-TTGTCCTCTGTGAGGCCTAT-3′ | ||
| Sox9 | Forward F1: 5′-GATAGTGTCCTCACTTCGCA-3′ | 467 |
| Reverse R1: B′-TCCACTCTGGCGGAGTCATG-S′ | ||
| Forward F2: 5′-CAGCCACCACCATCCAAGTT-3′ | 470 | |
| Reverse R2:5′-GAAGGGCATTGTGTGTACAG-3′ |
Statistical analysis
The Kruskal–Wallis ANOVA and Mann–Whitney non-parametric unpaired tests were used to compare MSCs obtained from the three different sources (TP, IC and WJ), whereas the Wilcoxon-paired test was used to compare basal versus siSlug silenced MSCs. Values of P < 0.05 were considered significant. The analysis was performed with CSS Statistic Statistical software (Statsoft, Tulsa, OK, USA).
Results
Characterization of hMSCs
Adult hMSCs isolated from TP trabecular bone, IC bone marrow and WJ umbilical cord, were examined. When MSC suspension was first plated, the cells derived from the three sources displayed both a monocyte-like and a spindle-shaped morphology. After one passage (P1), the spindle-shaped cells were predominant and proliferated to confluence. hTP- and hIC-MSCs usually reached the confluence after 2–3 weeks from seeding, whereas hWJ-MSCs formed adherent colonies more rapidly, reaching confluence after 10–12 days and demonstrating a major expansion capability (Fig. 1A). To evaluate MSC characteristics from the different sources, the immunophenotypic profile of adherent cells from each culture was determined by testing a panel of surface markers using flow cytometry (Fig. 1B). MSC from all samples were positive for CD90, CD73, CD105, CD146 (mesenchymal cell markers), but negative for CD31 (endothelial cell marker), CD3, CD14, CD34, CD45 (haematopoietic cell markers). In particular, we observed that the expression of CD90, CD73 and CD105 was not significantly different in all the three types of MSC analysed (Fig. 1C; total mean ± S.D. 97 ± 3), whereas the percentage of positive cells for CD146 was significantly lower in hTP-MSC (mean ± S.D. = 25.77 ± 17.82) and in hWJ-MSC (mean ± S.D. = 23.4 ± 19.09) compared to hIC-MSC (mean ± S.D. = 53.12 ± 23.01, P = 0.000007 and 0.01, respectively). Moreover, as shown in Figure 1C, the MFI ratio of the positive markers was not significantly different in all the three types of MSC analysed.
Fig 1.
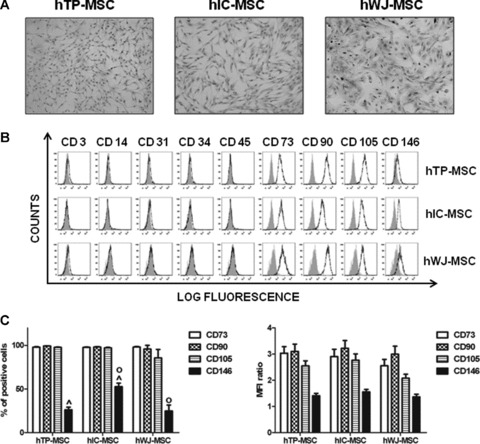
Isolation and phenotypical characterization of hMSCs. (A) Optical micrographs of hMSCs isolated from tibial plateau (TP) trabecular bone, iliac crest (IC) bone marrow and Wharton’s jelly (WJ) umbilical cord. After one passage (P1), cells show a fibroblast-like morphology (original magnification: 10×). (B) Flow cytometric analysis of a representative case of hTP-MSC, hIC-MSC and hWJ-MSC. Open histograms represent the isotype control antibody, dotted histograms represent anti-CD3, -CD14, -CD31, -CD34, -CD45, -CD73, -CD90, -CD105 and -CD146 antibodies. X-axis: mean fluorescent channel; Y-axis: number of events. (C) Flow cytometric data of anti-CD73, -CD90, -CD105 and -CD146 antibodies expressed both as mean percentage of positive cells ± S.E. and as mean of MFI ratio ± S.E. Statistical analysis was performed hTP-MSC versus hIC-MSC^ and hIC-MSC versus hWJ-MSCo, as described in Results.
Effect of Slug knockdown
In the complexity of the MSC genetic program, we investigated the effect of silencing of a specific transcription factor, Slug, that our recent studies have determined as a novel positive regulator of osteogenesis in human primary osteoblasts [18].
Specifically, hMSCs were transfected with siRNA sequence targeting Slug gene transcript [18]. Non-targeting siRNA was used as a negative control. We first checked that siRNA/Slug was efficient in decreasing Slug expression in all hMSCs. Quantitative real-time RT-PCR showed that basal Slug expression levels are higher in hTP- and hIC-MSCs than in hWJ-MSCs (Fig. 2A). Nevertheless, in all cases siRNA/Slug reduced Slug mRNA expression by about 80% (P < 0.05). The efficiency of Slug silencing was also validated at protein level in all hMSCs by Western blot analysis (Fig. 2B).
Fig 2.
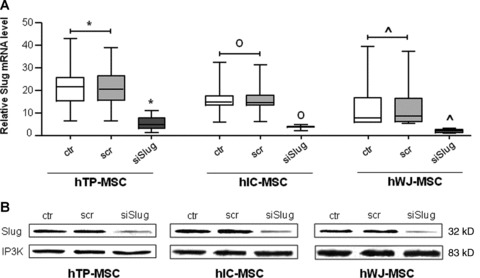
Silencing of Slug gene expression by siSlug in hMSCs. hMSCs were transfected with siSlug or a non-relevant siRNA (scr). (A) Slug expression was determined at mRNA level, and revealed by quantitative RT-PCR analysis. RT-PCR results were calculated using the DDCt method, using GAPDH as the housekeeping gene, and WJ-MSCs siSlug transfected sample as the calibrator. Statistical analysis was performed control and a non-relevant siRNA versus siSlug-silenced cells (*, o and ^ for hTP-MSC, hIC-MSC and hWJ-MSC, respectively), as described in Results. (B) Slug expression was determined at protein level, and revealed by Western blot. Ten μg of whole cell lysates were assayed on a 12% SDS-PAGE, and the proteins were visualized using Supersignal Femto Substrate (Pierce). Size markers are reported (kD). IP3K was used as loading control.
To rule out any adverse effects of treatment with Slug siRNA, viability assays and apoptosis assays at different time points (1, 2, 3 and 6 days) post-transfection with Slug siRNA were carried out. The double staining with a Calcein-AM cell viability assay kit and TUNEL test demonstrated that transfected hMSCs were highly viable (Fig. 3A) and did not show apoptotic nuclei (Fig. 3B) up to 6 days. The same photomicrographs suggest that Slug-silenced cells slightly reduced their proliferation capacity, and changed their morphology by becoming rounded in response to siRNA/Slug treatment.
Fig 3.
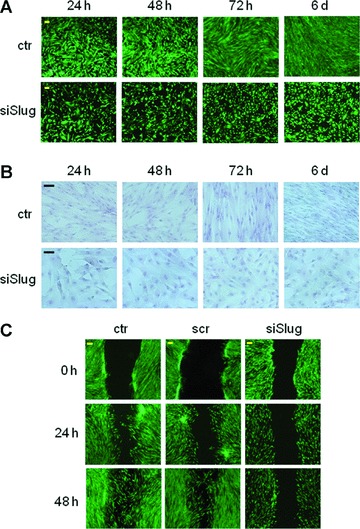
Effect of Slug siRNA treatment on viability (A) and apoptosis (B) of hMSCs cultured for the indicated length of time with Slug siRNA (siSlug) or not (ctr). (A) Viability was determined by double staining assay with Calcein-AM and propidium iodide. Fluorescence photomicrographs (4× magnification) are representative merged images showing the presence of green fluorescence (calcein-AM)-labelled live cells and the absence of red fluorescence (PI)-labelled dead cells. (B) Apoptosis was determined by Tunel assay; the absence of brown colour reaction indicates that the cells did not undergo apoptosis. Representative photomicrographs (10× magnification) are reported. (C) Analysis of hMSCs migration by in vitro scratch assay. hMSCs were transfected with 30 nM siSlug or a non-relevant siRNA (scr). 72 hrs after silencing treatment, hMSCs monolayers were scratch wounded with a pipet tip (0 hr), and observed over the indicated time periods, 0, 24 and 48 hrs (4× magnification). Images show siSlug treated cells exhibited a reduced capacity to cover the scratch area compared to control cells.
To further explore the effects of Slug knockdown on cell function, we performed the scratch-wound healing assay. Scratching of the hMSCs monolayer triggers a migratory event similar to the events that happen in fracture healing. We found that Slug-repressed cells had an impaired ability to close the wounded area compared with control and scrambled cells. After 48 hrs, gap closure in Slug-repressed hMSCs was significantly reduced because migration from the border of the wound was very slow. In Figure 3(C), a representative scratch assay performed on hWJ-MSCs is shown; the same results were obtained by using hMSCs from TP and IC.
The next step was to investigate whether Slug has a specific role in determining the molecular signature of hMSCs. This was tested by analysing the effect of Slug knockdown on the expression of transcription factors, which are required in the control of the differentiation program of osteochondroprogenitors. These genes include (i) Runx2, a member of runt family playing a pivotal role in osteoblast differentiation decision and hypertrophic chondrocyte maturation [28, 29]; (ii) Sox9, which is particularly necessary for chondrogenic differentiation commitment [30, 31]; (iii) Sox5 and Sox6, whose main function in chondrocytes is to boost the ability of Sox9 to activate major chondrocyte markers [32]; (iv) STAT1, which inhibits chondrocyte proliferation and regulates bone development [33, 34].
Quantitative RT-PCR analysis revealed that, after Slug silencing, all genes, except Runx2, showed the same modulation that was independent of cell type (Fig. 4). Slug-silenced cells always showed an increase in Sox9 and Sox5 when compared to the control condition. In particular, this up-regulation was significant for hTP-MSCs (for Sox9, P = 0.035; for Sox5, P = 0.027) and hWJ-MSCs (for Sox9, P = 0.027; for Sox5, P = 0.027). On the contrary, Sox6 and STAT1 expression declined in all hMSCs treated with siRNA/Slug compared to the control. In particular, down-regulation of Sox6 was significant for hWJ-MSCs (P = 0.027), whereas down-regulation of STAT1 was significant for hTP-MSCs (P = 0.027) and hIC-MSCs (P = 0.043). Finally, Runx2 expression did not significantly change after the Slug knockdown, even if slightly increased in hIC-MSCs, and decreased in hWJ-MSCs. The effect of Slug silencing on Runx2, Sox5, Sox6, Sox9 and Stat1 expression was confirmed also at protein level by Western blot analysis (see the inserts in Fig. 4). As a whole, the trend we observed suggests that Slug may act as a negative regulator of Sox9 and Sox5 expression, and as a positive regulator of Sox6 and STAT1 genes, in hMSCs. On the contrary, as regards Runx2, the role of Slug seems to be influenced by cell type. This is in agreement with our previous data [18] demonstrating that the same Slug knockdown increased Sox9 expression, but decreased Runx2 in human primary osteoblasts. This different role of Slug in mature committed osteoblasts and in their undifferentiated progenitors suggests a cell stage-specific mechanism of control of Runx2 and osteoblast differentiation by Slug.
Fig 4.
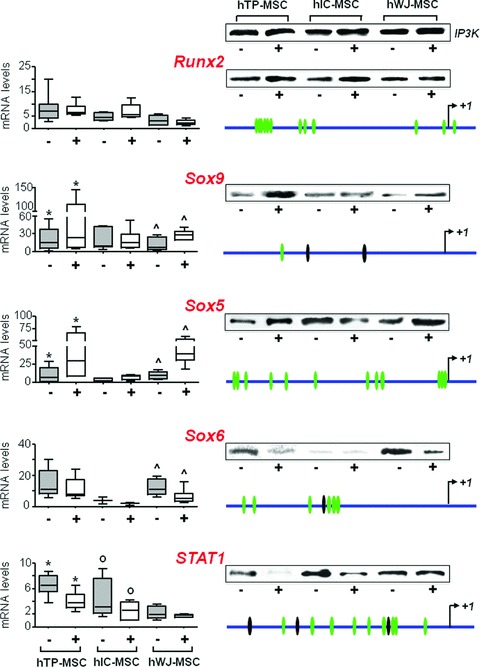
Effect of Slug knockdown on the expression of specific genes. Runx2, Sox9, Sox5, Sox6 and STAT1 gene expression was analysed after siSlug treatment in hMSCs isolated from tibial plateau (TP) trabecular bone iliac crest (IC) bone marrow and Wharton’s jelly (WJ) umbilical cord. On the left, quantitative RT-PCR analysis for the detection of mRNA levels are reported. RT-PCR results were calculated using the DDCt method, and the sample with the highest ΔCt as the calibrator for each gene analysis. Statistical analysis was performed control versus siSlug-silenced cells (*, o and ^ for hTP-MSC, hIC-MSC and hWJ-MSC, respectively) as described in Results. In the inserts on the right, Western blot analysis for the detection of protein expression are reported. Ten μg of whole cell lysates were assayed on a 12% SDS-PAGE, and the proteins were visualized using Supersignal Femto Substrate (Pierce). IP3K was used as loading control. On the right schematic representations of Runx2, Sox9, Sox5, Sox6 and STAT1 human gene promoters (3000 bp upstream +1 transcription site) are also reported. Using the TFSEARCH predicting transcription factor binding sites program, several potential Slug binding motifs have been identified in the promoter regions of all five genes (green ovals). Sites showing 100% homology with consensus-binding site (CAGGTG/CACCTG) are indicated with black ovals.
The human genomic DNA sequences belonging to 5′ regulatory regions of Runx2, Sox9, Sox5, Sox6 and STAT1 were analysed for the presence of putative Slug binding sites (E boxes) [19] by TFSEARCH predicting transcription factor binding sites program (www.cbrc.jp/research/db/TFSEARCH.html). This analysis revealed the presence of E boxes in the promoter regions of all five genes (Fig. 4).
Slug transcription factor is in vivo recruited at Runx2 and Sox9 gene promoters
On the basis of these findings, we tried to dissect the relationship between Slug and some of the analysed genes by investigating in vivo Slug recruitment at specific promoter sequences. We focused our attention on the promoter of Runx2 and Sox9 genes, and the functionality of their E boxes was analysed by ChIP (Fig. 5) on six samples of hMSCs from the three sources.
Fig 5.
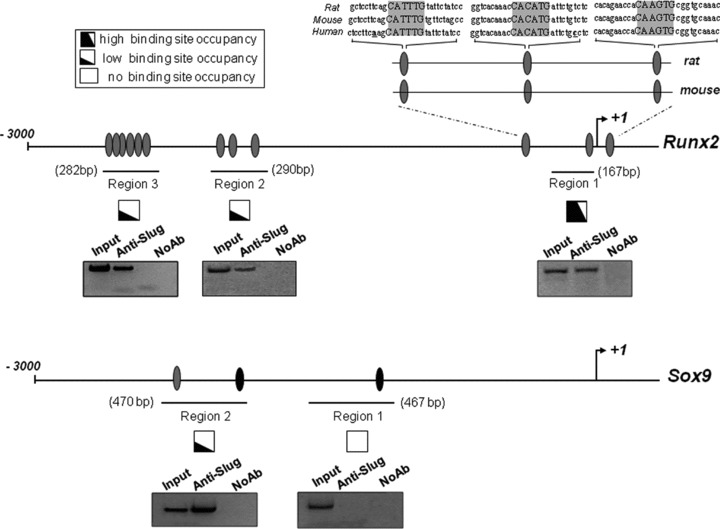
In vivo recruitment of Slug on Runx2 and Sox9 human gene promoters. Protein–DNA complexes were in vivo formaldehyde–cross-linked in six samples of hMSC isolated from tibial plateau (TP) trabecular bone, iliac crest (IC) bone marrow and Wharton’s jelly (WJ) umbilical cord. Chromatin fragments were subjected to immunoprecipitation with antibody against Slug. After cross-link reversal, the coimmunoprecipitated DNA was amplified by PCR using the primers pairs spanning the reported regions of Runx2 and Sox9 promoters. Aliquots of chromatin taken before immunoprecipitation were used as input positive controls whereas chromatin eluted from immunoprecipitations lacking antibody were used as no antibody controls (NoAb). PCR fragments of all samples analysed were resolved in 1.5% agarose gels and subjected to densitometric analysis for a semi-quantitative determination of occupancy of binding sites. High, low or no ability to recruit Slug is represented by the extent of the black portion of the square. A representative PCR analysis is reported below each investigated region. The relative positions of Slug putative binding sites (grey ovals) are indicated. Sites showing 100% homology with consensus-binding site (CAGGTG/CACCTG) are indicated with black ovals. The alignment of sequences from rat, mouse and human of Runx2 promoter region 1 is reported, showing the highly conserved E boxes.
Occupancy of these E boxes by Slug was compared among hMSCs. ChIP data were collectively considered for the presence or absence of PCR signals and the different promoter regions containing the E boxes were characterized for their high, low or no ability to recruit Slug. The results revealed that Slug can, in vivo, associate with multiple sites across Runx2 and Sox9 promoters to a different extent regardless of the hMSCs source.
Considering Runx2 gene, Slug associated with regions 2 and 3 to a similar extent, but more localized to a discrete region proximal to the transcription start site (region 1). Interestingly, the E boxes within region 1 are highly conserved, as revealed by the alignment of sequences from rat, mouse and human.
Furthermore, with regard to Slug occupancy of Sox9 promoter, region 2 was the only one involved in the interaction, whereas no chromatin was immunoprecipitated from the region 1. The alignment of Sox9 sequence promoters from rat, mouse and human revealed no significant homology.
As a whole, these ChIP experiments revealed that both Runx2 and Sox9 are Slug target genes in hMSCs, and that the E boxes present in these promoters are differently involved in the Slug recruitment.
Gene expression profiling and relationship to osteogenic potential
We then investigated the osteogenic potential of hMSCs from the three sources. At 21 days of differentiation, the cultures showed the presence of mineralized nodules following alizarin red staining analysis (Fig. 6A). These data support the hypothesis that MSCs can be successfully differentiated towards osteogenic lineage when appropriately stimulated in vitro. Interestingly, mineralization occurred at day 7 only for hWJ-MSC whereas for hTP- and hIC-MSC started from day 14. In addition, we noticed that, both at day 14 and 21, mineralization was significantly increased in hTP-MSC (P = 0.003 and 0.006, respectively) and hIC-MSC (P = 0.00001 and 0.006, respectively) compared to hWJ-MSC. Comparing hTP- and hIC-MSC we observed that, whereas at day 14 mineralization was significantly higher in hIC-MSC than in hTP-MSC (P = 0.008), at day 21 it reached approximately the same values in both cell sources.
Fig 6.
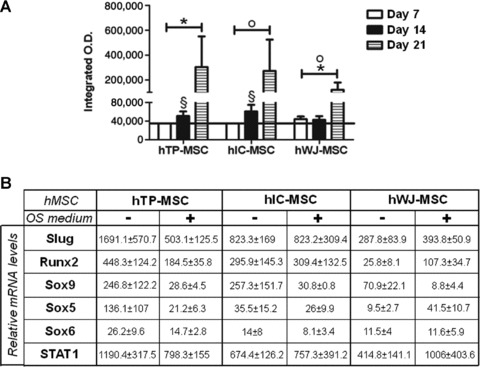
Osteogenic differentiation of hMSCs. (A) Differentiation potential of hMSCs from the three sources was assessed by evaluating the mineralization of the cell cultures in the presence of osteogenic induction medium, up to 21 days. Data were expressed as integrated optical density (O.D.) ± S.E. Statistical analysis was performed hTP-MSC versus hWJ-MSC*, hTP-MSC versus hIC-MSC^ and hIC-MSC versus hWJ-MSCo, as described in Results. (B) Slug, Runx2, Sox9, Sox5, Sox6 and STAT1 gene expression was valuated in hMSCs isolated from tibial plateau (TP) trabecular bone, iliac crest (IC) bone marrow and Wharton’s jelly (WJ) umbilical cord and cultured up to 21 days in osteogenic medium. mRNA level was revealed by quantitative RT-PCR analysis. RT-PCR results were calculated using the DDCt method. Data were expressed as fold difference value between the calibrator (Sox9 expression level at day 21 in hWJ-MSCs) and the other samples. Data were expressed as median ± S.E.M. It was not possible to apply statistical tests because the N was equal to four samples for each group analysed.
The expression levels of transcription factors were then assessed (Fig. 6B). As expected, Sox9 expression was down-regulated during osteoblastic differentiation of all hMSCs, but the presence of osteogenic medium had different effects on the other transcription factors, depending on the MSC source. After 21 days in osteogenic medium Slug, Runx2, Sox5, Sox6 and STAT1 expression showed a general decrease in hTP-MSC, whereas remained approximately constant in hIC-MSC, although these cells are identical in mineralization level. On the contrary, an appreciable increase in the expression levels of Slug, Runx2, Sox5 and STAT1 genes was observed in hWJ-MSC after osteogenic induction.
Interestingly, when hWJ-MSCs cultured in osteogenic medium were forced to overexpress Slug the deposition of mineralized matrix was significantly increased (Fig. S1).
Discussion
Bone tissue regeneration and repair through therapeutic use of cells requires special attention to two important issues: (i) the search for new crucial molecules for the commitment of MSCs toward osteogenic lineage, and (ii) the definition of optimal osteoprogenitor source and cell culture conditions. This is strictly related to the development of innovative experimental targeted strategies for human cultured MSCs. In this context, the concept of manipulating specific TFs is creating great interest. Overexpression or depletion of TFs, including Runx2, Osterix, Sox9, Twist1, NFATc1, Foxo1, Sp1, HIF-1 c/EBP and Rex-1, was recently performed in MSCs [35-42]. This approach is largely based on the knowledge of the molecular networks implicated in osteogenic differentiation, and, at the same time, it allows the identification of the role of a specific TF in mediating the fate and maturation of hMSCs. In this respect, many studies have demonstrated that the level of modulation of several osteoblast-associated genes may differ depending on culture conditions and source from which the cells are taken [12, 13, 16, 17, 43]. These observations may have significant implications for cell-based bone tissue regeneration, particularly when much evidence in literature suggests that the basal expression levels of specific genes in uninduced MSCs may have a crucial role in this scenario [7, 44, 45].
In this study, we used hMSCs isolated from TP trabecular bone, IC bone marrow and WJ umbilical cord, and demonstrated that the cell response to a same ‘in vitro’ microenvironment may be different.
First, we found that hMSCs isolated from the three sources possess similar surface marker profiles, but the basal expression levels of TFs, which control differentiation program of osteochondroprogenitors, such as Runx2, Sox9, Sox5, Sox6, STAT1 and Slug, are different. In particular, hWJ-MSCs showed the lowest expression levels of Runx2, Sox9, STAT1 and Slug. Further investigations, aimed at the analysis of differentiation potential, should resolve whether the transcripts we have analysed, together with those found by others, are merely expressed as part of a differentiation program, or whether their presence, as well as their expression levels, have functional relevance for hMSC behaviour. In this regard, when the osteogenic potential of hMSCs isolated from the three sources was investigated, we found that osteogenically induced hWJ-MSCs showed the characteristic staining of bone-like nodules beginning from day 7, whereas the same signs of mineralization were evident in hTP- and hIC-MSCs only from day 14. Nevertheless, hWJ-MSCs were not able to reach at day 21 the level of mineralization that we found in the other hMSCs, but, when Slug was overexpressed, their mineralization level was significantly increased (Fig. S1) strengthening our hypothesis on the role of Slug in hMSCs. Therefore, our findings indicate that there is a variability in the extent of osteogenic differentiation among the analysed hMSCs and that it may be specifically modulated. Interestingly, Runx2 expression showed a trend to decrease or remain unchanged in hTP- and hIC-MSCs respectively, but not in hWJ-MSCs after 21 days in osteogenic medium. It is, in fact, well established that, during bone development, Runx2 induces osteoblast differentiation and increases the number of immature osteoblasts, which form immature bone, whereas Runx2 expression has to be down-regulated for differentiation into mature osteoblasts, which form mature bone [30, 46]. Therefore, different osteogenic potential of hWJ-MSCs, which quickly respond to osteogenic medium, but do not reach a complete ‘end-stage’ differentiation, may be due to Runx2 expression, which increased during differentiation. This is in accordance with many other previously reported features of umbilical cord-derived stem cells [13, 47, 48]. Thanks to their properties, the hWJ-MSCs may be a particularly promising cell population, supporting new concepts in cellular therapy. Overall, these cells appear to possess the proper stage of development that makes them preferable candidates when a bone regeneration under endogenous factors control is required. In many cases, to promote tissue integration, it is, in fact, critical that the pre-differentiated osteogenic progenitors to be implanted are able not only to differentiate, but also to interact with the endogenous microenvironment and respond to local differentiation signals ‘in vivo’. This capability can be carried on by cells that have not completely reached the terminal differentiation such as the hWJ-MSCs here described. Conversely, it is conceivable that TP or IC, from which we obtained the other hMSCs, are already committed compartments containing stem cells with a higher maturation stage and particularly prone to form mature bone. Probably, these kind of cells are to be preferred when large bone defects have to be repaired.
Another aspect to take into account is that the achievement of pre-differentiated osteogenic progenitors requires the employment of induction media containing exogenous recombinant growth factors, foetal bovine serum, hormones and other reagents whose effects on long-term ‘in vivo’ differentiation are not known, and which may be potentially negative by transmitting infectious agents and triggering an immune response. Therefore, in the light of the considerations and issues presented at the beginning of this discussion, it is interesting to investigate the possibility that the change in the levels of specific gene transcription can replace the standard method of induction to differentiation. In this regard and to establish assumptions for next investigations, we considered the possibility to affect the behaviour of hMSCs by using gene silencing approach without exposing cells to induction media. We have chosen to silence Slug protein because our previous findings demonstrated that depletion of this transcription factor negatively affects the maturation process of osteoblasts, but has a positive effect on chondrocyte differentiation [18]. In addition, some indirect evidence from other laboratories suggests that Slug protein negatively regulates the proliferation of chondroprogenitors [34], whereas Slug forced overexpression in chick limb mesenchymal cells can induce apoptosis [49].
Our results obtained from RT-PCR analysis depicted a complex scenario where all Slug-silenced hMSCs from the three sources showed generally a higher expression of Sox9 and Sox5, and a lower expression of Sox6 and STAT1 in comparison with control cells. This suggests that Slug acts as a negative regulator of Sox9 and Sox5 expression, and as a positive regulator of Sox6 and STAT1 genes. Sox5, Sox6 and Sox9 constitute the so-called SOX trio and are essential factors for the development of embryonic cartilage, and are mainly associated with the commitment of undifferentiated MSCs into chondrocytes [50, 51]. In particular, Sox9 is the first essential transcription factor for chondrocyte differentiation [30, 31]. Sox 5 and Sox6 are indicated as downstream genes of Sox9 in chondrocytes, but are not absolutely necessary for chondrocyte differentiation even if they strongly potentiate Sox9 activity [32, 50]. Recent studies in MSCs have demonstrated that SOX trio family members may be differently regulated. For example, BMP-2, which was reported to control chondrogenic differentiation, increased Sox6 and Sox9, but not Sox5 mRNA expression [52]. Therefore, our results strengthen the evidence that a specific factor may differently affect SOX trio regulation, suggesting also a novel role for Slug transcription factor. In future studies, we will determine whether Slug acts directly on Sox5 and Sox6 promoters or the up-regulation of Sox5 by Slug gene silencing was mediated by Sox9.
Another interesting finding after Slug silencing is STAT1 down-regulation. This result demonstrates the specificity of knockdown treatment and suggests that STAT1 expression is positively regulated by Slug. Accordingly, in chondrogenesis, STAT1 acts downstream in relation to Slug and, as Slug, negatively regulates the proliferation of chondroprogenitors [34]; in mature chondrocytes, STAT1 is implicated as a key signalling molecule that mediates the anti-proliferative and apoptotic activity of FGFR-3 [53]. In MSCs, STAT1 is essential in interferon-γ (IFN-γ) induced signalling process and cooperates with many cytokines for regulation of gene expression [54]. Conversely, a recent study demonstrates that STAT1 is a negative regulator for osteoblast differentiation, and suggests that inhibition of STAT1 activity may be beneficial for skeletal fracture treatment [55]. However, in contrast to these observations, we demonstrated here that STAT1 expression is differently modulated in hMSCs induced towards osteogenic lineage. As a whole, these findings support the hypothesis that (i) modulating the expression of one or more specific transcription factors a preferential selection of osteo- or chondro-precursors may be obtained; and (ii) although the mechanisms of this process are currently unknown, nevertheless it seems that they are dependent on experimental models and developmental stages of the cells. Therefore, to clarify the role of STAT1 in osteochondroprogenitors and bone mature cells, and to understand the possible discrepancies between the data collected so far, further investigations are required.
These observations are consistent with the results obtained regarding Runx2 in Slug-silenced hMSCs. In fact, concerning Runx2, the role of Slug seems to be influenced by cell type. We found that, after Slug knockdown, Runx2 expression did not significantly change. This can be explained by the evidence that Runx2 has a broader spectrum of phenotype control of a cell in comparison with SOX trio or STAT1. In fact, in addition to its role in osteoblast differentiation, Runx2 also promotes chondrocyte maturation [29, 34]. This confirms that, for certain genes, including Runx2, their susceptibility to be modulated in expression levels depends on gene function, and is strictly correlated with heterogeneity and properties of MSC population. This corroborates the evidence supporting the unique nature of MSCs from different sources through their divergent responses to a specific biological modifier.
To elucidate Slug function in hMSCs and analyse the biological phenomena affected by Slug knockdown it is mandatory to investigate the ability of Slug-silenced hMSCs to undergo chondrogenic terminal differentiation or true osteogenesis. This can be properly performed only in Slug stably knocked down cells, which we are now creating with lentiviral systems, and in three-dimensional culture conditions. Alternatively, the consequences of loss of appropriate Slug expression on skeletal apparatus can be in vivo explored in transgenic Slug knockout mice [56, 57].
In conclusion, considering that an important issue for tissue regeneration is to identify new crucial molecules in hMSCs whose targeting may promote differentiation in vitro, our study encourages the development of alternative strategies to induce efficient differentiation for clinical use of hMSCs in bone and cartilage tissue engineering and repair.
Acknowledgments
This work was supported by grants from Telethon GGP10214, Regione Emilia Romagna, Programma di Ricerca Regione Università 2007–2009, the Fondazione Cassa di Risparmio di Padova e Rovigo. E.L. is a recipient of a fellowship from the Fondazione Cassa di Risparmio di Ferrara. We are very grateful to Prof. Fortunato Vesce and the staff of the Section of Obstetric and Gynaecological Clinic, Department of Biomedical Sciences and Advanced Therapies, University of Ferrara for sample recruitment of umbilical cord samples.
Conflict of interest
The authors confirm that there are no conflicts of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Effect of Slug overexpression on osteogenic differentiation of hWJ-MSCs
Please note: Wiley-Blackwell is not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Caplan AI. Mesenchymal stem cell: cell-based reconstructive therapy in orthopaedics. Tiss Eng. 2005;11:1198–211. doi: 10.1089/ten.2005.11.1198. [DOI] [PubMed] [Google Scholar]
- 2.Arthur A, Zannettino A, Gronthos S. The therapeutic applications of multipotential mesenchymal/stromal stem cells in skeletal tissue repair. J Cell Physiol. 2009;218:237–45. doi: 10.1002/jcp.21592. [DOI] [PubMed] [Google Scholar]
- 3.Arvidson K, Abdallah BM, Applegate LA, et al. Bone regeneration and stem cells. J Cell Mol Med. 2011;15:718–46. doi: 10.1111/j.1582-4934.2010.01224.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robey PG, Kuznetsov SA, Riminucci M, et al. Skeletal (“mesenchymal”) stem cells for tissue engineering. Methods Mol Med. 2007;140:83–99. doi: 10.1007/978-1-59745-443-8_5. [DOI] [PubMed] [Google Scholar]
- 5.Satija NK, Singh VK, Verma YK, et al. Mesenchymal stem cell-based therapy: a new paradigm in regenerative medicine. J Cell Mol Med. 2009;13:4385–402. doi: 10.1111/j.1582-4934.2009.00857.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Augello A, De Bari C. The regulation of differentiation in mesenchymal stem cells. Hum Gene Ther. 2010;21:1226–38. doi: 10.1089/hum.2010.173. [DOI] [PubMed] [Google Scholar]
- 7.Satija NK, Gurudutta GU, Sharma S, et al. Mesenchymal stem cells: molecular targets for tissue engineering. Stem Cells Dev. 2007;16:7–23. doi: 10.1089/scd.2006.9998. [DOI] [PubMed] [Google Scholar]
- 8.Squillaro T, Alessio N, Cipollaro M, et al. Partial silencing of methyl cytosine protein binding 2 (MECP2) in mesenchymal stem cells induces senescence with an increase in damaged DNA. FASEB J. 2010;24:1593–603. doi: 10.1096/fj.09-143057. [DOI] [PubMed] [Google Scholar]
- 9.Qi H, Aguiar DJ, Williams SM, et al. Identification of genes responsible for osteoblast differentiation from human mesodermal progenitor cells. Proc Natl Acad Sci USA. 2003;100:3305–10. doi: 10.1073/pnas.0532693100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karsenty G. The complexities of skeletal biology. Nature. 2003;423:316–8. doi: 10.1038/nature01654. [DOI] [PubMed] [Google Scholar]
- 11.Shibata KR, Aoyama T, Shima Y, et al. Expression of the p16INK4A gene is associated closely with senescence of human mesenchymal stem cells and is potentially silenced by DNA methylation during in vitro expansion. Stem Cells. 2007;25:2371–82. doi: 10.1634/stemcells.2007-0225. [DOI] [PubMed] [Google Scholar]
- 12.Mosna F, Senseb L, Krampera M. Human bone marrow and adipose tissue mesenchymal stem cells: a user’s guide. Stem Cells Dev. 2010;19:1449–70. doi: 10.1089/scd.2010.0140. [DOI] [PubMed] [Google Scholar]
- 13.Hsieh JY, Fu YS, Chang SJ, et al. Functional module analysis reveals differential osteogenic and stemness potentials in human mesenchymal stem cells from bone marrow and Wharton’s jelly of umbilical cord. Stem Cells Dev. 2010;19:1895–910. doi: 10.1089/scd.2009.0485. [DOI] [PubMed] [Google Scholar]
- 14.Bianco P, Riminucci M, Gronthos S, et al. Bone marrow stromal stem cells: nature, biology, and potential applications. Stem Cells. 2001;19:180–92. doi: 10.1634/stemcells.19-3-180. [DOI] [PubMed] [Google Scholar]
- 15.Lavrentieva A, Majore I, Kasper C, et al. Effects of hypoxic culture conditions on umbilical cord-derived human mesenchymal stem cells. Cell Commun Signal. 2010;8:18–26. doi: 10.1186/1478-811X-8-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Noël D, Caton D, Roche S, et al. Cell specific differences between human adipose-derived and mesenchymal-stromal cells despite similar differentiation potentials. Exp Cell Res. 2008;314:1575–84. doi: 10.1016/j.yexcr.2007.12.022. [DOI] [PubMed] [Google Scholar]
- 17.Mannello F, Tonti GA. Concise review: no breakthroughs for human mesenchymal and embryonic stem cell culture: conditioned medium, feeder layer, or feeder-free; medium with foetal calf serum, human serum, or enriched plasma; serum-free, serum replacement nonconditioned medium, or ad hoc formula? All that glitters is not gold! Stem Cells. 2007;25:1603–9. doi: 10.1634/stemcells.2007-0127. [DOI] [PubMed] [Google Scholar]
- 18.Lambertini E, Lisignoli G, Torreggiani E, et al. Slug gene expression supports human osteoblast maturation. Cell Mol Life Sci. 2009;66:3641–53. doi: 10.1007/s00018-009-0149-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nieto MA. The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol. 2002;3:155–66. doi: 10.1038/nrm757. [DOI] [PubMed] [Google Scholar]
- 20.Conacci-Sorrell M, Simcha I, Ben-Yedidia T, et al. Autoregulation of E-cadherin expression by cadherin-cadherin interactions: the roles of beta catenin signaling, Slug, and MAPK. J Cell Biol. 2003;163:847–57. doi: 10.1083/jcb.200308162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barrallo-Gimeno A, Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 2005;132:3151–61. doi: 10.1242/dev.01907. [DOI] [PubMed] [Google Scholar]
- 22.Lambertini E, Franceschetti T, Torreggiani E, et al. SLUG: a new target of lymphoid enhancer factor-1 in human osteoblasts. BMC Mol Biol. 2010;11:13–24. doi: 10.1186/1471-2199-11-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lian JB, Stein GS. The temporal and spatial subnuclear organization of skeletal gene regulatory machinery: integrating multiple levels of transcriptional control. Calcif Tissue Int. 2003;72:631–7. doi: 10.1007/s00223-002-1058-x. [DOI] [PubMed] [Google Scholar]
- 24.Barzilay R, Melamed E, Offen D. Introducing transcription factors to multipotent mesenchymal stem cells: making transdifferentiation possible. Stem Cells. 2009;27:2509–15. doi: 10.1002/stem.172. [DOI] [PubMed] [Google Scholar]
- 25.Chang HH, Hemberg M, Barahona M, et al. Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature. 2008;453:544–7. doi: 10.1038/nature06965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lisignoli G, Codeluppi K, Todoerti K, et al. Gene array profile identifies collagen type XV as a novel human osteoblast-secreted matrix protein. J Cell Physiol. 2009;220:401–9. doi: 10.1002/jcp.21779. [DOI] [PubMed] [Google Scholar]
- 27.Penolazzi L, Tavanti E, Vecchiatini R, et al. Encapsulation of mesenchymal stem cells from Wharton’s jelly in alginate microbeads. Tissue Eng Part C Methods. 2010;16:141–55. doi: 10.1089/ten.TEC.2008.0582. [DOI] [PubMed] [Google Scholar]
- 28.Komori T. Regulation of bone development and extracellular matrix protein genes by RUNX2. Cell Tissue Res. 2010;339:189–95. doi: 10.1007/s00441-009-0832-8. [DOI] [PubMed] [Google Scholar]
- 29.Kim IS, Otto F, Zabel B, et al. Regulation of chondrocyte differentiation by Cbfa1. Mech Dev. 1999;80:159–70. doi: 10.1016/s0925-4773(98)00210-x. [DOI] [PubMed] [Google Scholar]
- 30.Lefebvre V. Toward understanding SOX9 function in chondrocyte differentiation. Matrix Biol. 1998;16:529–40. doi: 10.1016/s0945-053x(98)90065-8. [DOI] [PubMed] [Google Scholar]
- 31.Bi W, Deng JM, Zhang Z, et al. Sox9 is required for cartilage formation. Nat Genet. 1999;22:85–9. doi: 10.1038/8792. [DOI] [PubMed] [Google Scholar]
- 32.Han Y, Lefebvre V. L-Sox5 and Sox6 drive expression of the aggrecan gene in cartilage by securing binding of Sox9 to a far-upstream enhancer. Mol Cell Biol. 2008;28:4999–5013. doi: 10.1128/MCB.00695-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiao L, Naganawa T, Obugunde E, et al. Stat1 controls postnatal bone formation by regulating fibroblast growth factor signaling in osteoblasts. J Biol Chem. 2004;279:27743–52. doi: 10.1074/jbc.M314323200. [DOI] [PubMed] [Google Scholar]
- 34.Goldring MB, Tsuchimochi K, Ijiri K. The control of chondrogenesis. J Cell Biochem. 2006;97:33–44. doi: 10.1002/jcb.20652. [DOI] [PubMed] [Google Scholar]
- 35.Lin L, Chen L, Wang H, et al. Adenovirus-mediated transfer of siRNA against Runx2/Cbfa1 inhibits the formation of heterotopic ossification in animal model. Biochem Biophys Res Commun. 2006;349:564–72. doi: 10.1016/j.bbrc.2006.08.089. [DOI] [PubMed] [Google Scholar]
- 36.Gordeladze JO, Noël D, Bony C, et al. Transient down-regulation of cbfa1/Runx2 by RNA interference in murine C3H10T1/2 mesenchymal stromal cells delays in vitro and in vivo osteogenesis, but does not overtly affect chondrogenesis. Exp Cell Res. 2008;314:1495–506. doi: 10.1016/j.yexcr.2007.12.023. [DOI] [PubMed] [Google Scholar]
- 37.Miraoui H, Severe N, Vaudin P, et al. Molecular silencing of Twist1 enhances osteogenic differentiation of murine mesenchymal stem cells: implication of FGFR2 signaling. J Cell Biochem. 2010;110:1147–54. doi: 10.1002/jcb.22628. [DOI] [PubMed] [Google Scholar]
- 38.Tominaga H, Maeda S, Miyoshi H, et al. Expression of osterix inhibits bone morphogenetic protein-induced chondrogenic differentiation of mesenchymal progenitor cells. J Bone Miner Metab. 2009;27:36–45. doi: 10.1007/s00774-008-0003-0. [DOI] [PubMed] [Google Scholar]
- 39.Deng ZL, Sharff KA, Tang N, et al. Regulation of osteogenic differentiation during skeletal development. Front Biosci. 2008;13:2001–21. doi: 10.2741/2819. [DOI] [PubMed] [Google Scholar]
- 40.Yano F, Kugimiya F, Ohba S, et al. The canonical Wnt signaling pathway promotes chondrocyte differentiation in a Sox9-dependent manner. Biochem Biophys Res Commun. 2005;333:1300–8. doi: 10.1016/j.bbrc.2005.06.041. [DOI] [PubMed] [Google Scholar]
- 41.Wan C, Shao J, Gilbert SR, et al. Role of HIF-1alpha in skeletal development. Ann N Y Acad Sci. 2010;1192:322–6. doi: 10.1111/j.1749-6632.2009.05238.x. . 42. Bhandari DR, Seo KW, Roh KH, et al. REX-1 expression and p38 MAPK activation status can determine proliferation/differentiation fates in human mesenchymal stem cells. PLoS One 2010; 5: e10493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.da Silva Meirelles L, Caplan AI, Beyer Nardi N. In search of the in vivo identity of mesenchymal stem cells. Stem Cells. 2008;26:2287–99. doi: 10.1634/stemcells.2007-1122. [DOI] [PubMed] [Google Scholar]
- 44.Karsenty G. Transcriptional control of skeletogenesis. Annu Rev Genomics Hum Genet. 2008;9:183–96. doi: 10.1146/annurev.genom.9.081307.164437. [DOI] [PubMed] [Google Scholar]
- 45.Marie PJ. Transcription factors controlling osteoblastogenesis. Arch Biochem Biophys. 2008;473:98–105. doi: 10.1016/j.abb.2008.02.030. [DOI] [PubMed] [Google Scholar]
- 46.Maruyama Z, Yoshida CA, Furuichi T, et al. Runx2 determines bone maturity and turnover rate in postnatal bone development and is involved in bone loss in estrogen deficiency. Dev Dyn. 2007;236:1876–90. doi: 10.1002/dvdy.21187. [DOI] [PubMed] [Google Scholar]
- 47.Moretti P, Hatlapatka T, Marten D, et al. Mesenchymal stromal cells derived from human umbilical cord tissues: primitive cells with potential for clinical and tissue engineering applications. Adv Biochem Eng Biotechnol. 2010;123:29–54. doi: 10.1007/10_2009_15. [DOI] [PubMed] [Google Scholar]
- 48.Troyer DL, Weiss ML. Wharton’s jelly-derived cells are a primitive stromal cell population. Stem Cells. 2008;26:591–9. doi: 10.1634/stemcells.2007-0439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim D, Song J, Jin EJ. MicroRNA-221 regulates chondrogenic differentiation through promoting proteosomal degradation of slug by targeting Mdm2. J Biol Chem. 2010;285:26900–7. doi: 10.1074/jbc.M110.115105. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 50.Ikeda T, Kamekura S, Mabuchi A, et al. The combination of SOX5, SOX6, and SOX9 (the SOX trio) provides signals sufficient for induction of permanent cartilage. Arthritis Rheum. 2004;50:3561–73. doi: 10.1002/art.20611. [DOI] [PubMed] [Google Scholar]
- 51.Park JS, Yang HN, Woo DG, et al. Chondrogenesis of human mesenchymal stem cells mediated by the combination of SOX trio SOX5, 6, and 9 genes complexed with PEI-modified PLGA nanoparticles. Biomaterials. 2011;32:3679–88. doi: 10.1016/j.biomaterials.2011.01.063. [DOI] [PubMed] [Google Scholar]
- 52.Fernandez-Lloris R, Vinals F, Lopez-Rovira T, et al. Induction of the Sry-related factor SOX6 contributes to bone morphogenetic protein-2-induced chondroblastic differentiation of C3H10T1/2 cells. Mol Endocrinol. 2003;17:1332–43. doi: 10.1210/me.2002-0254. [DOI] [PubMed] [Google Scholar]
- 53.Spagnoli A, Torello M, Nagalla SR, et al. Identification of STAT-1 as a molecular target of IGFBP-3 in the process of chondrogenesis. J Biol Chem. 2002;277:18860–7. doi: 10.1074/jbc.M200218200. [DOI] [PubMed] [Google Scholar]
- 54.Durbin JE, Hackenmiller R, Simon MC, et al. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell. 1996;84:443–50. doi: 10.1016/s0092-8674(00)81289-1. [DOI] [PubMed] [Google Scholar]
- 55.Tajima K, Takaishi H, Takito J, et al. Inhibition of STAT1 accelerates bone fracture healing. J Orthop Res. 2010;28:937–41. doi: 10.1002/jor.21086. [DOI] [PubMed] [Google Scholar]
- 56.Newkirk KM, Duncan FJ, Brannick EM, et al. The acute cutaneous inflammatory response is attenuated in Slug-knockout mice. Lab Invest. 2008;88:831–41. doi: 10.1038/labinvest.2008.37. [DOI] [PubMed] [Google Scholar]
- 57.Zhao P, Iezzi S, Carver E, et al. Slug is a novel downstream target of MyoD. Temporal profiling in muscle regeneration. J Biol Chem. 2002;277:30091–101. doi: 10.1074/jbc.M202668200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effect of Slug overexpression on osteogenic differentiation of hWJ-MSCs