

Abstract
Axotomized neurons have the innate ability to undergo regenerative sprouting but this is often impeded by the inhibitory central nervous system environment. To gain mechanistic insights into the key molecular determinates that specifically underlie neuronal regeneration at a transcriptomic level, we have undertaken a DNA microarray study on mature cortical neuronal clusters maintained in vitro at 8, 15, 24 and 48 hrs following complete axonal severance. A total of 305 genes, each with a minimum fold change of ±1.5 for at least one out of the four time points and which achieved statistical significance (one-way ANOVA, P < 0.05), were identified by DAVID and classified into 14 different functional clusters according to Gene Ontology. From our data, we conclude that post-injury regenerative sprouting is an intricate process that requires two distinct pathways. Firstly, it involves restructuring of the neurite cytoskeleton, determined by compound actin and microtubule dynamics, protein trafficking and concomitant modulation of both guidance cues and neurotrophic factors. Secondly, it elicits a cell survival response whereby genes are regulated to protect against oxidative stress, inflammation and cellular ion imbalance. Our data reveal that neurons have the capability to fight insults by elevating biological antioxidants, regulating secondary messengers, suppressing apoptotic genes, controlling ion-associated processes and by expressing cell cycle proteins that, in the context of neuronal injury, could potentially have functions outside their normal role in cell division. Overall, vigilant control of cell survival responses against pernicious secondary processes is vital to avoid cell death and ensure successful neurite regeneration.
Keywords: neurons, axotomy, microarray, regeneration, neurite cytoskeleton, secondary processes
Introduction
One of the striking features of the injured central nervous system (CNS) is the failure of severed axons to adequately regenerate to restore loss of function. This was initially believed to be caused by an intrinsic inability of injured axons to sprout regenerative processes. However, the seminal studies of Albert Aguayo and others using peripheral or cellular tissue grafts transplanted into the lesioned spinal cord have clearly demonstrated that the environment of the injured CNS is a critical determinant of whether injured axons can regenerate [1]. The molecular determinates of the inhibitory CNS environment are now well-understood, with major players being myelin-associated molecules (such as nogo, myelin-associated glycoprotein) and chondroitin sulphate proteoglycans [2].
Equally important has been the discovery that injured neurons have an intrinsic capacity to regenerate following complete axonal transection. Although bearing the limitation of being one-dimensional, in vitro experimental models have proven particularly insightful in this field of research. A particular advantage of these approaches is the ability to specifically evaluate intrinsic regeneration of injured axons in the absence of glial cells or inhibitory substrates and molecules. Elegant studies have demonstrated that severance of individual axons of cultured CNS neurons results in a rapid regenerative response [3, 4]. Furthermore, axotomy of thick fasciculated axonal bundles of mature (21 days in vitro) clusters of cortical neurons results in regenerative sprouting within 8 hrs after injury [5, 6].
Several studies have directly investigated the precise mechanisms that underlie the intrinsic regenerative sprouting response of injured CNS neurons. Microtubule stabilizing drugs significantly alter the regenerative sprouting response following axonal transection of cortical neurons in vitro, indicating that cytoskeletal re-organization is a key process underlying axonal regeneration [6]. This has recently been confirmed by in vivo studies reporting that the microtubule stabilizing drug taxol facilitates axonal regeneration of the injured optic nerve [7]. Highlighting that the regenerative sprouting response is an active process, Verma et al. [8] have reported that protein synthesis is essential for efficient generation of regenerative growth cones following axotomy in vitro. They demonstrated that axotomy leads to a four- to six-fold increase in 3H-leucine incorporation, and that the protein synthesis inhibitors cycloheximide and anisomycin both impair the ability of severed axons to form regenerative sprouts [8].
To identify key molecular determinants of successful axonal regeneration, a number of studies have utilized DNA microarray techniques to reveal groups of genes that are up- or down-regulated in response to axonal injury and during axonal regeneration. These studies have implicated a wide variety of genes in the regenerative response, including cytoskeletal, cell cycle and ion homeostasis genes. A notable feature of these studies is that they have generally been undertaken within in vivo injury situations, such as the injured optic nerve [9, 10], sciatic nerve [11, 12] or spinal cord [13, 14]. Hence, because of the presence of glia and other cells and the inability of microarray approaches to discriminate cell-specific gene expression, these studies do not give a clear indication of the key genes directly responsible for intrinsic regenerative sprouting of injured neurons. To address this, we have undertaken a microarray study following complete axonal severance of mature (21 days in vitro) cultured cortical neurons.
Materials and methods
Cell culture preparation for rat primary cortical neuronal clusters
All animal experimentation was performed under the guidelines stipulated by the University of Tasmania Animal Ethics Committee, which is in accordance with the Australian code of practice for the care and use of animals for scientific purposes. Cortical neuron cultures were prepared according to previously published protocols [6, 15, 16]. Briefly, cortical tissue was isolated from E17 hooded Wistar rat embryos and incubated in 0.1% trypsin (in HEPES buffer) for 15 min. Following trituration and filtration through a 20 μm filter, neurons were plated into 12-well tissue culture plates pre-coated overnight with 1mg/ml of L-lysine in borate buffer, pH 7.4, at a cell density of 4.5 × 105 cells/well. Neuronal cultures were maintained at 37°C in humidified air containing 5% CO2 for 21 days before experimental axotomy. Neurons were initially plated into a culture medium consisting of Neurobasal™ medium (Gibco; Life Technologies Corp., California, USA), supplemented with 10% foetal bovine serum, 0.1% B-27 supplement (Gibco), 0.1 mM L-glutamine (Gibco) and 200 U/ml gentamicin (Gibco). After 24 hrs, the media was replaced with medium lacking foetal bovine serum, and replaced every 3 or 4 days.
Axotomy of rat primary cortical neuronal clusters
Neurons were maintained in culture for 21 days, at which time they had formed large spherical clusters of neurons and an interconnected network of thick fasciculated bundles of axons. Using a fine blade microscalpel, axonal bundles were completely severed at approximately half way along the axonal bundles. All axonal bundles in each culture well were severed. At the appropriate time points, RNA from neuronal cells were collected, representing neurons both proximal and distal to the axotomy.
Total RNA extraction and isolation
RNA from neuronal clusters was extracted at indicated time points (8, 15, 24 and 48 hrs) after axotomy and control (no axotomy) using RNeasy Mini Kit (Qiagen Cat. No. 74104) as per the manufacturer’s instructions. The whole procedure was performed with RNase-free filtered pipette tips. 1.5 μl of the RNA sample was taken for spectrophotometric quantification using Nanodrop ND-1000 Version 3.2.1. Another 1 μl was used for RNA quality analysis using E-gene HDA-GT12 genetic analyser.
Real-time (RT)-PCR
Reverse transcription was carried out according to steps specified by manufacturer (Applied Biosystems Taqman reverse transcription reagents; Life Technologies Corp.). Each cDNA sample was duplicated with two No Template Control (NTC) for each probe used. All genes tested were normalized against either of the internal loading controls, 18S rRNA or GAPDH. Twenty microlitres of the Taqman master mix was pipetted to the bottom of each well of the optical 96-well fast reaction plate. Five microlitres of cDNA or water (NTC) was added to the designated reaction well. The plate was then read by the 7000 Fast Real-Time PCR System with conditions according to the manufacturer’s protocol.
DNA microarray
Transcriptomic profiling was performed on Illumina Rat Ref12 Ver.1 arrays for rat neuronal clusters 8, 15, 24 and 48 hrs after axonal severance. Six biological replicates were obtained for the control and three for each of the four time-points after injury. Five hundred nanograms of total RNA for each sample was brought up to an initial volume of 11 μl. RNA reverse transcription was performed with Illumina® TotalPrep RNA Amplification Kit and the concentration of cRNA was quantitated using the NanoDrop ND-1000. Seven hundred and fifty nanograms of cRNA topped up to 5 μl RNase-free water was mixed with 10 μl hybridization buffer. Hybridization using streptavidin-Cy3 labelling was carried out according to the manufacturer’s instruction (Illumina Inc., San Diego, USA). Subsequently, the beadchip was read on the Illumina scanner using Bead Studio software at Scan Factor = 0.65.
Microarray data collection and analysis
Analysis of the scanned images was performed with BeadScan (Illumina Inc.). Signal data generated from the Illumina® Bead Studio software was analysed using GeneSpring© v7.3 software. All differentially expressed genes in this study were selected based on the following parameters: (1) a minimum of ±1.5 fold change in at least one of post-injury time-points and (2) passed the statistical screening test of one-way ANOVA (P < 0.05) and Benjamini-Hochberg False Discovery Rate Correction. Genes which were differentially expressed were annotated according to Gene Ontology Biological process with the use of an online bioinformatics resource namely Database for Annotation, Visualization and Integrated Discovery (DAVID) 6.7 (http://david.abcc.ncifcrf.gov/) [17, 18]. All microarray data reported here are described in accordance with MIAME guidelines, and have been deposited in NCBIs Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO Super-Series accession number GSE 23653.
Statistical analysis
All experiments were repeated at least three times. Data were analysed using post-hoc Tukey test with one-way ANOVA to assess significant differences in multiple comparisons. Values of P < 0.05 were considered as statistically significant and presented as mean ± S.E.
Results
DNA microarray analysis
Gene expression profiles were obtained for 8, 15, 24 and 48 hrs post severance of axon bundles connecting rat neuronal clusters in culture. Genes with a minimum fold change of ±1.5 for at least one of the four time points were classified as differentially regulated and subjected to one-way ANOVA analysis. Those that reached significance with a P < 0.05 were subjected to grouping using the online bioinformatics resource DAVID. A total of 305 genes were identified and each of them was then classified into one out of the 14 functional clusters identified to avoid overestimation (Table 1). Figure 1 summarizes the number of identified genes in each biological process and the percentage these genes within the group occupied out of the total. Differences between up and down regulated genes across the four time points after axotomy were presented in Figure 2A and B, respectively. Generally, even though the genomic profiles across time were quite similar, it was evident from Figure 2A that there was a slight transient elevation of genes involved in certain biological processes such as axogenesis, morphogenesis, actin and cytoskeleton organization, cell cycle, response to oxidative stress, inflammation and chemotaxis at 8 hrs in comparison to the later time points and this was conversely true for the down-regulated genes as seen in Figure 2B.
Table 1.
Gene profiles of neuronal clusters in vitro, 8, 15, 24 and 48 hrs after axon severance (P < 0.05)
| Time | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| GenBank | Symbols | Gene name | 8 hrs | S.E. (±) | 15 hrs | S.E. (±) | 24 hrs | S.E. (±) | 48 hrs | S.E. (±) |
| Regulation of axonogenesis | ||||||||||
| NM_013191 | S100b | S100 protein, polypeptide | 6.57 | 2.62 | 7.94 | 2.10 | 5.81 | 2.44 | 8.52 | 2.37 |
| NM_013166 | Cntf | Ciliary neurotrophic factor | 4.06 | 1.73 | 5.76 | 1.75 | 4.33 | 2.04 | 5.30 | 3.02 |
| NM_012809 | Cnp1 | Cyclic nucleotide phosphodiesterase 1 | 2.52 | 1.83 | 3.69 | 1.28 | 2.87 | 1.39 | 3.44 | 1.01 |
| NM_138828 | Apoe | Apolipoprotein E | 2.00 | 0.54 | 2.30 | 0.69 | 2.27 | 0.68 | 2.28 | 0.81 |
| XM_236640 | Plxnb1 | Plexin B1 (predicted) | 1.96 | 0.63 | 2.53 | 0.71 | 2.23 | 0.68 | 2.15 | 0.57 |
| NM_017026 | Mbp | Myelin basic protein | 1.85 | 1.42 | 2.40 | 1.34 | 1.48 | 0.44 | 2.11 | 0.68 |
| NM_013001 | Pax6 | Paired box gene 6 | 1.81 | 0.52 | 1.80 | 0.50 | 1.70 | 0.50 | 1.88 | 0.62 |
| NM_199407 | Unc5c | Unc-5 homologue C | 1.79 | 0.56 | 1.75 | 0.49 | 1.46 | 0.43 | 1.35 | 0.41 |
| NM_012829 | Cck | Cholecystokinin | 1.78 | 1.59 | −2.05 | 0.98 | 1.37 | 1.99 | −1.32 | 0.91 |
| NM_031066 | zygin I,Fez1 | Fasciculation and elongation protein | 1.51 | 0.41 | 1.58 | 0.47 | 1.73 | 0.46 | 1.59 | 0.63 |
| NM_181086 | Tnfrsf12a | Tumour necrosis factor receptor superfamily, member 12a | 1.46 | 0.43 | −1.56 | 0.21 | −1.18 | 0.50 | −1.67 | 0.29 |
| XM_217250 | Ephb1 | Eph receptor B1 | 1.40 | 0.46 | 2.31 | 0.67 | 1.85 | 0.63 | 1.40 | 0.46 |
| XM_236203 | Dscaml1 | Down syndrome cell adhesion molecule-like 1 (predicted) | 1.29 | 0.40 | 1.60 | 0.54 | 1.37 | 0.44 | 1.21 | 0.35 |
| XM_222794 | Tnn | Tenascin N (predicted) | 1.19 | 1.18 | −1.30 | 0.22 | −1.22 | 0.34 | −1.54 | 0.19 |
| NM_012934 | Dpysl3 | Dihydropyrimidinase-like 3 | −1.38 | 0.22 | −1.67 | 0.20 | −2.10 | 0.17 | −2.14 | 0.12 |
| NM_012610 | Ngfr | Nerve growth factor receptor (member 16) | −1.38 | 0.18 | −1.57 | 0.18 | −1.62 | 0.16 | −1.31 | 0.24 |
| XM_341201.2 | Epha5 | Eph receptor A5 (predicted) | −1.39 | 0.40 | −3.12 | 0.19 | −2.06 | 0.29 | −2.74 | 0.10 |
| NM_023023 | Dpysl5 | Dihydropyrimidinase-like 5 | −1.44 | 0.22 | −1.72 | 0.18 | −1.55 | 0.27 | −1.92 | 0.17 |
| NM_019272 | Sema4f | Semaphorin 4f | −1.46 | 0.32 | −2.56 | 0.34 | −1.62 | 0.46 | −2.74 | 0.32 |
| NM_017310 | Sema3a | Short basic domain, secreted, semaphorin 3A | −1.68 | 0.20 | −1.84 | 0.17 | −1.65 | 0.17 | −1.73 | 0.17 |
| NM_031073 | Ntf3 | Neurotrophin 3 | −1.69 | 0.16 | −1.73 | 0.18 | −1.66 | 0.17 | −1.80 | 0.16 |
| XM_236623 | Sema3f | Short basic domain, secreted, semaphorin 3 F (predicted) | −1.81 | 0.16 | −1.99 | 0.18 | −2.22 | 0.12 | −1.89 | 0.16 |
| XM_234514 | Bcl11b | B-cell leukaemia/lymphoma 11B (predicted) | −1.85 | 0.41 | −3.78 | 0.24 | −2.53 | 0.37 | −4.01 | 0.26 |
| NM_019288 | App | Amyloid (A4) precursor protein | −2.38 | 0.11 | −2.59 | 0.12 | −2.85 | 0.11 | −2.89 | 0.14 |
| XM_341567.2 | Plxdc2 | Plexin domain containing 2 (predicted) | −2.40 | 0.15 | −1.62 | 0.19 | −2.54 | 0.24 | −2.04 | 0.23 |
| NM_145098 | Nrp1 | Neuropilin 1 | −2.55 | 0.13 | −4.53 | 0.06 | −3.78 | 0.08 | −3.82 | 0.09 |
| NM_053485 | S100a6 | S100 calcium binding protein A6 (calcyclin) | −5.71 | 0.05 | −5.76 | 0.05 | −6.93 | 0.04 | −6.74 | 0.04 |
| Neurite development and morphogenesis | ||||||||||
| XM_215963 | Lama5 | Laminin, ·5 | 2.39 | 0.73 | 2.21 | 0.75 | 2.05 | 0.56 | 1.88 | 0.54 |
| XM_342392 | Notch1 | Notch gene homologue 1, | 2.35 | 0.65 | 2.43 | 0.64 | 2.00 | 0.55 | 1.81 | 0.48 |
| NM_030997.1 | Vgf | VGF nerve growth factor inducible | 2.25 | 0.58 | 1.42 | 0.40 | 1.71 | 0.64 | 1.15 | 0.35 |
| NM_053021 | Clu | Clusterin | 1.96 | 0.61 | 2.17 | 0.68 | 1.89 | 0.68 | 2.43 | 0.73 |
| NM_053911 | Pscd2 | Pleckstrin homology, Sec7 and coiled/coil domains 2 | 1.77 | 0.53 | 1.64 | 0.49 | 1.70 | 0.53 | 1.45 | 0.67 |
| XM_221672 | Tiam1 | T cell lymphoma invasion and metastasis 1 (predicted). | 1.66 | 0.60 | −1.07 | 0.46 | 1.20 | 0.71 | −1.14 | 0.52 |
| NM_031131 | Tgfb2 | Transforming growth factor, 2 | 1.58 | 0.60 | 1.13 | 0.67 | 1.08 | 0.39 | 1.04 | 0.72 |
| NM_012924 | Cd44 | CD44 antigen | 1.58 | 0.47 | −1.08 | 0.28 | −1.02 | 0.53 | −1.11 | 0.40 |
| NM_030991 | Snap25 | Synaptosomal-associated protein 25 | 1.18 | 0.93 | −2.69 | 0.65 | −1.00 | 1.52 | −2.46 | 0.80 |
| NM_019248 | Ntrk3 | Neurotrophic tyrosine kinase, receptor, type 3 | 1.06 | 0.46 | −1.33 | 0.23 | −1.18 | 0.27 | −1.56 | 0.21 |
| NM_012731 | Ntrk2 | Neurotrophic tyrosine kinase, receptor, type 2 | 1.03 | 0.32 | −2.14 | 0.17 | −1.27 | 0.44 | −2.15 | 0.37 |
| NM_012513 | Bdnf | Brain-derived neurotrophic factor | 1.01 | 0.68 | −1.84 | 0.17 | −1.24 | 0.51 | −1.79 | 0.15 |
| NM_012700.1 | Stx1b | Syntaxin 1B2 (2) | −1.00 | 0.27 | −1.41 | 0.31 | −1.03 | 0.42 | −1.51 | 0.35 |
| XM_232283 | Plxnb1 | Plexin D1 (predicted) | −1.05 | 0.25 | −1.30 | 0.22 | −1.19 | 0.24 | −1.65 | 0.18 |
| NM_153470 | Lzts1 | Leucine zipper, putative tumour suppressor 1 | −1.12 | 0.73 | −2.09 | 0.51 | −1.17 | 0.81 | −2.02 | 0.47 |
| NM_013038 | Stxbp1 | Syntaxin binding protein 1 | −1.20 | 0.54 | −2.54 | 0.37 | −1.47 | 0.66 | −2.62 | 0.35 |
| NM_133652 | Cspg5 | Chondroitin sulphate proteoglycan 5 | −1.28 | 0.22 | −1.59 | 0.19 | −1.55 | 0.21 | −1.30 | 0.37 |
| XM_579472 | Slit1 | Slit homologue 1 | −1.53 | 0.30 | −2.35 | 0.30 | −1.56 | 0.45 | −2.91 | 0.17 |
| NM_019334 | Pitx2 | Paired-like homeodomain transcription factor 2 | −1.53 | 0.18 | −1.54 | 0.20 | −1.54 | 0.19 | −1.44 | 0.19 |
| NM_017237 | Uchl1 | Ubiquitin carboxy-terminal hydrolase L1 | −1.60 | 0.35 | −2.32 | 0.27 | −1.75 | 0.32 | −2.56 | 0.31 |
| NM_012548 | Edn1 | Endothelin 1 | −1.61 | 0.19 | −1.78 | 0.21 | −1.80 | 0.16 | −1.71 | 0.17 |
| NM_134331 | Epha7 | Eph receptor A7 | −1.65 | 0.18 | −1.53 | 0.22 | −1.52 | 0.23 | −1.52 | 0.20 |
| XM_346464 | Slit2 | Slit homologue 2 | −1.69 | 0.17 | −1.56 | 0.18 | −1.83 | 0.17 | −1.88 | 0.22 |
| XM_342172 | Thbs4 | Thrombospondin 4 | −1.70 | 0.16 | −1.74 | 0.19 | −1.95 | 0.15 | −2.19 | 0.12 |
| XM_223820 | Zfp312 | Zinc finger protein 312 (predicted) | −1.79 | 0.47 | −3.76 | 0.13 | −1.55 | 0.64 | −3.10 | 0.10 |
| XM_215451 | Cspg2 | Chondroitin sulfate proteoglycan 2 | −1.80 | 0.22 | −1.38 | 0.22 | −1.94 | 0.17 | −2.11 | 0.16 |
| XM_342591 | Bmp7 | Bone morphogenetic protein 7 | −2.12 | 0.25 | −1.27 | 0.23 | −1.76 | 0.32 | 1.22 | 0.58 |
| NM_017089 | Efnb1 | Ephrin B1 | −2.56 | 0.10 | −2.94 | 0.14 | −2.86 | 0.10 | −3.15 | 0.08 |
| NM_031321 | Slit3 | Slit homologue 3 | −2.84 | 0.11 | −3.26 | 0.10 | −3.19 | 0.09 | −3.52 | 0.08 |
| NM_053595 | Pgf | Placental growth factor | −5.49 | 0.05 | −4.06 | 0.08 | −5.27 | 0.06 | −4.70 | 0.06 |
| NM_012774 | Gpc3 | Glypican 3 | −9.15 | 0.10 | −4.92 | 0.06 | −9.44 | 0.04 | −5.87 | 0.05 |
| Actin filament formation | ||||||||||
| XM_341553 | PRKCQ | Protein kinase C, theta | 3.84 | 1.86 | 5.17 | 1.56 | 4.04 | 2.02 | 4.55 | 1.39 |
| XM_342648.2 | N-WASP | Neural Wiskott--Aldrich syndrome protein | 3.75 | 1.28 | 4.83 | 1.64 | 3.53 | 2.07 | 4.24 | 2.38 |
| XM_218617.3 | Myh14 | Myosin, heavy polypeptide 14 (predicted) | 2.96 | 0.70 | 3.44 | 0.99 | 2.87 | 0.87 | 2.51 | 1.06 |
| NM_019357 | Vil2 | Villin 2 | 2.16 | 0.67 | 1.57 | 0.46 | 1.66 | 0.41 | 1.73 | 0.58 |
| NM_053484 | Gas7 | Growth arrest specific 7 | 2.15 | 0.96 | 1.14 | 0.75 | 1.65 | 1.52 | 1.10 | 0.54 |
| NM_019131 | Tpm1 | Tropomyosin 1, α | 1.94 | 0.58 | 2.03 | 0.64 | 1.76 | 0.47 | 1.84 | 0.94 |
| NM_017117.1 | Capn3 | Calpain 3 | 1.7 | 0.5 | 1.89 | 0.62 | 1.49 | 0.49 | 1.53 | 0.42 |
| NM_022401.1 | Plec1 | Plectin 1 | 1.68 | 0.42 | 1.72 | 0.51 | 1.43 | 0.37 | 1.18 | 0.40 |
| NM_001005889 | Rdx | Radixi | 1.61 | 0.46 | 1.61 | 0.54 | 1.40 | 0.46 | 1.29 | 0.66 |
| NM_019167 | Spnb3 | β-Spectrin 3 | 1.45 | 1.17 | −1.73 | 0.69 | 1.22 | 1.53 | −1.91 | 0.56 |
| XM_221196 | Fscn2 | Fascin homologue 2, actin-bundling protein (predicted) | 1.20 | 0.39 | 1.32 | 0.45 | 1.30 | 0.43 | 1.58 | 0.52 |
| XM_236444.3 | LOC315840 | Similar to Myosin VI | 1.17 | 0.33 | 1.59 | 0.43 | 1.36 | 0.43 | 1.29 | 0.40 |
| NM_030873 | Pfn2 | Profilin 2 | −1.06 | 0.24 | −1.31 | 0.22 | −1.14 | 0.24 | −1.58 | 0.34 |
| XM_237115 | Nck2 | Non-catalytic region of tyrosine kinase adaptor protein 2 | −1.08 | 0.39 | −1.72 | 0.30 | −1.36 | 0.35 | −1.74 | 0.32 |
| NM_031144 | Actb | Actin, β | −1.30 | 0.24 | −1.36 | 0.24 | −1.59 | 0.21 | −1.44 | 0.36 |
| XM_579522 | Actn4 | Actinin ·4 | −1.31 | 0.22 | −1.65 | 0.17 | −1.81 | 0.17 | −1.75 | 0.18 |
| NM_013194 | Myh9 | Myosin, heavy polypeptide 9 | −1.35 | 0.25 | −1.59 | 0.17 | −1.66 | 0.18 | −1.42 | 0.20 |
| XM_215862 | Dstn | Destrin (predicted) | −1.38 | 0.22 | −1.34 | 0.28 | −1.64 | 0.21 | −1.55 | 0.37 |
| NM_001009689 | Cdc42ep2 | CDC42 effector protein (Rho GTPase binding) 2 | −1.54 | 0.18 | −1.59 | 0.20 | −1.83 | 0.16 | −1.74 | 0.16 |
| NM_019289.2 | Arpc1b | Actin-related protein 2/3 complex, subunit 1B | −1.61 | 0.16 | −1.70 | 0.17 | −1.81 | 0.15 | −1.92 | 0.16 |
| NM_053814 | Mrip | Myosin phosphatase-Rho interacting protein | −1.64 | 0.17 | −2.16 | 0.13 | −2.43 | 0.13 | −2.78 | 0.14 |
| XM_579484 | Evl | Ena-vasodilator stimulated phosphoprotein | −1.79 | 0.14 | −2.45 | 0.14 | −1.96 | 0.19 | −2.88 | 0.17 |
| NM_019212 | Acta1 | Actin, α1, skeletal muscle | −1.91 | 0.16 | −2.27 | 0.40 | −2.22 | 0.21 | −2.49 | 0.42 |
| NM_023992 | Gpr54 | G protein-coupled receptor 54 | −2.05 | 0.16 | −2.10 | 0.13 | −2.01 | 0.15 | −2.07 | 0.14 |
| XM_579179 | Arhe | Ras homologue gene family, member E | −2.21 | 0.13 | −2.45 | 0.13 | −2.45 | 0.13 | −2.62 | 0.14 |
| NM_001007554 | Cal | CSX-associated LIM | −4.19 | 0.08 | −4.57 | 0.06 | −4.66 | 0.07 | −3.90 | 0.08 |
| NM_012722 | Eln | Elastin | −4.62 | 0.12 | −3.69 | 0.12 | −4.63 | 0.09 | −3.98 | 0.08 |
| NM_012893.1 | Actg2 | Actin, γ2 | −33.7 | 0.01 | −28.1 | 0.01 | −22.3 | 0.03 | −26.5 | 0.01 |
| Microtubule cytoskeleton organization and biogenesis | ||||||||||
| XM_341694.2 | Dncl2b | Dynein, cytoplasmic, light chain 2B (predicted) | 20.8 | 6.64 | 23.8 | 7.22 | 16.4 | 13.4 | 22.5 | 10.6 |
| NM_053508 | Tekt1 | Tektin 1 | 6.81 | 2.11 | 8.84 | 2.55 | 6.01 | 4.81 | 8.01 | 4.91 |
| NM_001007726 | Dnai2 | Dynein, axonemal, intermediate polypeptide 2 | 6.65 | 2.05 | 8.07 | 2.65 | 5.66 | 3.42 | 6.58 | 3.78 |
| XM_224615.3 | Dnah1 | Dynein, axonemal, heavy polypeptide 1 | 4.83 | 1.30 | 7.20 | 2.05 | 5.82 | 3.20 | 5.22 | 2.69 |
| XM_576475.1 | vOC501059 | Similar to kinesin-like protein 9 | 2.52 | 0.78 | 2.70 | 0.77 | 2.35 | 0.96 | 2.72 | 0.86 |
| NM_012935 | Cryab | Crystallin, ·B | 2.44 | 1.35 | 2.33 | 0.88 | 2.12 | 0.65 | 2.03 | 1.25 |
| XM_234720.3 | Dnah11 | Dynein, axonemal, heavy polypeptide 11 | 2.41 | 0.67 | 3.06 | 0.86 | 2.37 | 0.80 | 2.56 | 0.80 |
| XM_213354.3 | Dnah9 | Dynein, axonemal, heavy polypeptide 9 | 2.02 | 0.67 | 2.45 | 0.82 | 2.03 | 0.82 | 2.30 | 0.79 |
| NM_001007004 | Tuba4 | Tubulin, ·4 | 1.97 | 0.59 | 1.03 | 0.31 | 1.53 | 0.64 | −1.08 | 0.52 |
| NM_199094.1 | Tubb2 | Tubulin, 2 | 1.96 | 0.57 | 1.81 | 0.54 | 1.77 | 0.58 | 1.74 | 0.56 |
| NM_206950 | Mig12 | MID1 interacting G12-like protein | 1.96 | 0.52 | 2.20 | 0.66 | 2.00 | 0.53 | 1.94 | 0.69 |
| XM_575908 | Tekt2 | Tektin 2 (predicted) | 1.92 | 0.56 | 2.07 | 0.62 | 1.66 | 0.87 | 2.32 | 0.68 |
| NM_031763 | Pafah1b1 | Platelet-activating factor acetylhydrolase, isoform Ib | 1.74 | 0.46 | 1.76 | 0.55 | 1.58 | 0.43 | 1.56 | 0.39 |
| XM_218820 | Prc1 | Protein regulator of cytokinesis 1 (predicted) | 1.65 | 0.48 | 1.46 | 0.43 | 1.43 | 0.37 | 1.63 | 0.79 |
| NM_001009666 | Dnalc4 | Dynein, axonemal, light chain 4 | 1.56 | 0.46 | 1.43 | 0.36 | 1.28 | 0.42 | 1.24 | 0.51 |
| NM_001009645 | Kif22 | Kinesin family member 22 | 1.53 | 0.39 | 1.06 | 0.31 | 1.16 | 0.31 | 1.17 | 0.50 |
| XM_240978.3 | Kifc3 | Kinesin family member C3 (predicted) | 1.50 | 0.41 | 1.50 | 0.38 | 1.65 | 0.47 | 1.06 | 0.32 |
| XM_341686 | Ap1g1 | Adaptor-related protein complex 1, gamma 1 subunit | 1.49 | 0.42 | 1.41 | 0.51 | 1.51 | 0.43 | 1.21 | 0.44 |
| NM_053618 | Bbs2 | Bardet--Biedl syndrome 2 homologue | 1.45 | 0.37 | 1.58 | 0.48 | 1.44 | 0.43 | 1.44 | 0.45 |
| XM_224232 | Cenpj | Centromere protein J | 1.33 | 0.38 | 1.51 | 0.43 | 1.33 | 0.45 | 1.32 | 0.41 |
| NM_022507 | Prkcz | Protein kinase C, zeta | 1.00 | 0.28 | −1.36 | 0.26 | −1.11 | 0.40 | −1.60 | 0.35 |
| NM_198752.1 | KIFC2 | Kinesin family member C2 | −1.01 | 0.36 | −2.00 | 0.38 | −1.26 | 0.74 | −2.40 | 0.27 |
| NM_053947 | Mark1 | MAP/microtubule affinity-regulating kinase 1 | −1.11 | 0.25 | −1.55 | 0.22 | −1.34 | 0.25 | −1.80 | 0.17 |
| XM_343018.2 | Spg4 | Spastic paraplegia 4 (spastin) (predicted) | −1.25 | 0.23 | −1.38 | 0.21 | −1.43 | 0.19 | −1.68 | 0.25 |
| NM_024346 | Stmn3 | Stathmin-like 3 | −1.43 | 0.37 | −2.39 | 0.30 | −1.56 | 0.48 | −2.66 | 0.28 |
| NM_133320 | Ndel1 | nudE nuclear distribution gene E homologue like 1 | −1.60 | 0.18 | −2.32 | 0.12 | −1.79 | 0.20 | −2.55 | 0.20 |
| XM_215469 | Map1b | Microtubule-associated protein 1b | −1.63 | 0.35 | −2.22 | 0.18 | −2.12 | 0.23 | −2.74 | 0.17 |
| NM_017166 | Stmn1 | Stathmin 1 | −2.11 | 0.25 | −2.84 | 0.21 | −2.29 | 0.28 | −3.10 | 0.17 |
| Apical protein localization | ||||||||||
| XM_223229 | Shrm | PDZ domain actin binding protein Shroom | 1.67 | 0.52 | 1.74 | 0.57 | 1.45 | 0.53 | 1.55 | 0.48 |
| XM_222896 | Ltap | Loop tail associated protein (predicted) | 1.41 | 0.52 | 1.48 | 0.35 | 1.23 | 0.54 | 1.33 | 0.55 |
| Cell cycle | ||||||||||
| NM_053749 | Aurkb | Aurora kinase B | 4.99 | 1.95 | 5.90 | 1.71 | 4.01 | 1.58 | 5.80 | 1.65 |
| XM_574943 | Ccna1 | Cyclin A1 (predicted) | 3.94 | 1.18 | 4.67 | 1.39 | 3.01 | 1.56 | 3.69 | 1.88 |
| XM_573172 | Pnutl2 | Peanut-like 2 (predicted) | 3.33 | 1.34 | 3.48 | 1.10 | 2.47 | 1.18 | 2.79 | 2.11 |
| XM_579390 | Hrasls3 | HRAS-like suppressor | 3.29 | 1.20 | 4.37 | 1.23 | 3.32 | 1.57 | 4.59 | 1.43 |
| XM_220423 | Sept8 | Septin 8 (predicted) | 2.40 | 1.16 | 1.88 | 1.06 | 2.26 | 1.05 | 1.71 | 1.41 |
| NM_053677 | Chek2 | Protein kinase Chk2 | 2.33 | 0.84 | 2.58 | 0.75 | 1.91 | 0.70 | 2.45 | 1.03 |
| NM_021863 | Hspa2 | Heat shock protein 2 | 2.17 | 0.64 | 2.09 | 0.59 | 1.62 | 0.48 | 2.12 | 0.74 |
| XM_215222 | Cetn2 | Centrin 2 | 2.15 | 0.60 | 2.43 | 0.71 | 1.97 | 0.81 | 2.15 | 1.16 |
| XM_574892 | E2f5 | E2F transcription factor 5 | 1.86 | 0.54 | 1.54 | 0.45 | 1.69 | 0.51 | 1.44 | 0.45 |
| NM_001005765 | Rap1a | RAS-related protein 1a | 1.76 | 0.54 | 1.65 | 0.45 | 1.49 | 0.49 | 1.49 | 0.41 |
| XM_579383 | vOC501059 | Adenylate cyclase activating polypeptide 1 | 1.74 | 1.96 | −1.90 | 0.30 | 1.14 | 2.35 | −2.01 | 0.25 |
| NM_001004107 | Tacc1a | Transforming, acidic coiled-coil containing protein 1a | 1.58 | 0.47 | 1.51 | 0.42 | 1.48 | 0.40 | 1.20 | 0.47 |
| NM_133396 | Tesk2 | Testis-specific kinase 2 | 1.50 | 0.44 | 1.34 | 0.40 | 1.35 | 0.40 | 1.33 | 0.36 |
| NM_138873 | Nbn | Nibrin | 1.50 | 0.49 | 1.26 | 0.32 | 1.37 | 0.42 | 1.16 | 0.37 |
| NM_013058 | Id3 | inhibitor of DNA binding 3 | 1.21 | 0.31 | 1.53 | 0.40 | 1.44 | 0.41 | 1.30 | 0.35 |
| XM_579239 | RT1-CE7 | RT1 class I, CE7 | 1.20 | 0.37 | 1.62 | 0.70 | 1.09 | 0.29 | 1.39 | 0.42 |
| NM_021739 | Camk2b | Calcium/calmodulin-dependent protein kinase II subunit | 1.16 | 0.51 | −2.35 | 0.33 | −1.22 | 0.73 | −2.37 | 0.43 |
| XM_341907 | Eif4g2 | Eukaryotic translation initiation factor 4, gamma 2 | −1.16 | 0.28 | −1.35 | 0.26 | −1.56 | 0.21 | −1.32 | 0.38 |
| XM_342804 | Ccne2 | Cyclin E2 (predicted) | −1.40 | 0.20 | −1.50 | 0.19 | −1.51 | 0.21 | −1.39 | 0.21 |
| XM_222157 | Kntc1 | Kinetochore associated 1 (predicted) | −1.41 | 0.24 | −1.50 | 0.26 | −1.52 | 0.19 | −1.68 | 0.18 |
| XM_214152 | Cdkn3 | Cyclin-dependent kinase inhibitor 3 (predicted) | −1.47 | 0.21 | −1.70 | 0.20 | −1.95 | 0.15 | −1.76 | 0.25 |
| NM_053653 | Vegfc | Vascular endothelial growth factor C | −1.48 | 0.29 | −1.73 | 0.20 | −1.61 | 0.20 | −1.77 | 0.31 |
| XM_223270 | Ccng2 | Cyclin G2 (predicted) | −1.53 | 0.19 | −1.47 | 0.21 | −1.57 | 0.20 | −1.39 | 0.22 |
| NM_053703 | Map2k6 | Mitogen-activated protein kinase kinase 6 | −1.66 | 0.18 | −1.51 | 0.17 | −1.46 | 0.20 | −1.82 | 0.19 |
| NM_021693 | Snf1lk | SNF1-like kinase | −1.67 | 0.27 | −2.16 | 0.15 | −1.84 | 0.33 | −3.06 | 0.08 |
| NM_139087 | Cgref1 | Cell growth regulator with EF hand domain 1 | −1.69 | 0.23 | −2.70 | 0.12 | −1.86 | 0.27 | −2.54 | 0.11 |
| NM_052807 | Igf1r | Insulin-like growth factor 1 receptor | −1.81 | 0.17 | −2.06 | 0.16 | −2.08 | 0.14 | −2.52 | 0.11 |
| NM_033099 | Ptprv | Protein tyrosine phosphatase, receptor type, V | −2.37 | 0.11 | −1.77 | 0.21 | −2.19 | 0.14 | −2.29 | 0.12 |
| NM_053713.1 | Klf4 | Kruppel-like factor 4 | −3.30 | 0.15 | −3.81 | 0.10 | −3.45 | 0.09 | −4.09 | 0.10 |
| Negative regulation of programmed cell death | ||||||||||
| NM_053819 | Timp1 | Inhibitor of metalloproteinase 1 | 1.79 | 0.51 | 1.83 | 0.53 | 1.46 | 0.43 | 1.13 | 0.30 |
| XM_235497 | Mkl1 | Megakaryoblastic leukaemia (translocation) 1 (predicted) | 1.05 | 0.27 | −1.25 | 0.26 | −1.09 | 0.31 | −1.68 | 0.27 |
| NM_033230 | Akt1 | Thymoma viral proto-oncogene 1 | −1.20 | 0.26 | −1.50 | 0.24 | −1.43 | 0.23 | −1.49 | 0.30 |
| NM_207592 | Gpi | Glucose phosphate isomerase | −1.34 | 0.19 | −1.44 | 0.19 | −1.37 | 0.21 | −1.59 | 0.22 |
| NM_031345 | Dsipi | Delta sleep inducing peptide, immunoreactor | −1.37 | 0.21 | −1.40 | 0.21 | −1.69 | 0.18 | −1.68 | 0.33 |
| XM_216377 | Bag1 | Bcl2-associated athanogene 1 (predicted) | −1.48 | 0.20 | −1.44 | 0.19 | −1.53 | 0.20 | −1.84 | 0.21 |
| NM_057130.1 | Bid3 | BH3 interacting (with BCL2 family) domain | −1.58 | 0.18 | −2.35 | 0.31 | −1.74 | 0.27 | −2.44 | 0.22 |
| NM_013083 | Hspa5 | Heat shock 70 kD protein 5 | −1.64 | 0.16 | −1.51 | 0.17 | −1.50 | 0.19 | −1.74 | 0.24 |
| XM_225257 | Txndc5 | Thioredoxin domain containing 5 (predicted) | −1.69 | 0.15 | −1.52 | 0.21 | −1.67 | 0.16 | −1.52 | 0.21 |
| NM_012992 | Npm1 | Nucleophosmin 1 | −1.72 | 0.16 | −2.25 | 0.16 | −2.19 | 0.13 | −2.35 | 0.18 |
| NM_021691 | Twist2 | twist homologue 2 | −2.47 | 0.12 | −2.30 | 0.16 | −2.63 | 0.10 | −2.39 | 0.12 |
| XM_225940 | Tnfaip8 | Tumour necrosis factor, α-induced protein 8 (predicted) | −3.13 | 0.09 | −2.37 | 0.14 | −3.10 | 0.09 | −2.70 | 0.11 |
| Regulation of apoptosis | ||||||||||
| NM_031970 | Hspb1 | Heat shock 27 kD protein 1 | 3.35 | 1.04 | 4.01 | 1.24 | 3.34 | 1.33 | 4.61 | 1.37 |
| NM_001007617 | SNF1/AMPK | SNF1/AMP-activated protein kinase | 2.97 | 1.47 | 3.44 | 1.10 | 2.96 | 1.35 | 3.28 | 2.28 |
| NM_031775 | Casp6 | Caspase 6 | 2.13 | 0.80 | 2.61 | 0.78 | 2.00 | 0.73 | 2.64 | 1.00 |
| NM_017180 | Phlda1 | Pleckstrin homology-like domain, family A, member 1 | 1.94 | 0.78 | 1.22 | 0.34 | 1.48 | 0.52 | 1.23 | 0.34 |
| XM_231692 | Braf | v-raf murine sarcoma viral oncogene homologue B1 | 1.69 | 0.52 | 1.98 | 0.70 | 1.63 | 0.52 | 1.57 | 0.70 |
| NM_139261.1 | Hspbp1 | hsp70-interacting protein | 1.68 | 0.51 | 1.39 | 0.45 | 1.54 | 0.45 | 1.26 | 0.72 |
| NM_001004199 | Tax1bp1 | Tax1 (human T cell leukaemia virus type I) binding protein 1 | 1.41 | 0.38 | 1.67 | 0.54 | 1.42 | 0.43 | 1.31 | 0.41 |
| XM_579385 | G6pdx | Glucose-6-phosphate dehydrogenase | 1.37 | 0.35 | 1.57 | 0.48 | 1.51 | 0.44 | 1.32 | 0.46 |
| NM_012660 | Eef1a2 | Eukaryotic translation elongation factor 1 α2 | 1.16 | 0.63 | −2.50 | 0.53 | −1.10 | 0.94 | −2.26 | 0.69 |
| XM_217191 | Bnip2 | BCL2/adenovirus E1B 19kDa-interacting protein 1, NIP2 | −1.24 | 0.25 | −1.30 | 0.26 | −1.48 | 0.23 | −1.53 | 0.29 |
| NM_012922 | Casp3 | Caspase 3, apoptosis related cysteine protease | −1.35 | 0.19 | −1.47 | 0.19 | −1.39 | 0.19 | −1.51 | 0.21 |
| NM_023979 | Apaf1 | Apoptotic peptidase activating factor 1 | −1.40 | 0.21 | −1.68 | 0.18 | −1.52 | 0.20 | −1.66 | 0.17 |
| NM_080897 | Bnip1 | BCL2/adenovirus E1B 19 kD-interacting protein 1 | −1.54 | 0.18 | −1.81 | 0.15 | −1.70 | 0.16 | −2.00 | 0.15 |
| NM_053420 | Bnip3 | BCL2/adenovirus E1B 19 kD-interacting protein 3 | −1.62 | 0.17 | −2.19 | 0.15 | −2.10 | 0.14 | −2.27 | 0.18 |
| NM_053736 | Casp11 | Caspase 11 | −2.31 | 0.14 | −2.00 | 0.16 | −2.46 | 0.13 | −2.50 | 0.12 |
| NM_130422 | Casp12 | Caspase 12 | −2.54 | 0.12 | −1.89 | 0.19 | −2.52 | 0.12 | −2.22 | 0.12 |
| NM_172336 | Atf5 | Activating transcription factor 5 | −2.62 | 0.15 | −2.17 | 0.15 | −2.14 | 0.20 | −3.14 | 0.08 |
| Response to oxidative stress | ||||||||||
| NM_017014 | Gstm1 | Glutathione S-transferase, Ì1 | 4.46 | 1.35 | 5.22 | 1.76 | 4.07 | 2.04 | 5.46 | 1.62 |
| NM_012580 | Hmox1 | Heme oxygenase (decycling) 1 | 4.10 | 2.56 | 1.71 | 0.88 | 2.12 | 1.09 | 1.16 | 0.52 |
| NM_134349 | Mgst1 | Microsomal glutathione S-transferase 1 | 2.90 | 0.87 | 3.39 | 1.00 | 3.08 | 1.07 | 2.72 | 0.95 |
| NM_012880 | Sod3 | Superoxide dismutase 3, extracellular | 2.38 | 0.73 | 2.51 | 0.58 | 2.36 | 1.05 | 2.64 | 1.00 |
| NM_053576 | Prdx6 | Peroxiredoxin 6 | 2.21 | 0.66 | 2.64 | 0.83 | 2.65 | 0.99 | 2.93 | 0.83 |
| NM_012837 | Cst3 | Cystatin C | 2.06 | 0.52 | 2.18 | 0.63 | 2.07 | 0.62 | 2.08 | 0.63 |
| XM_214236 | Nudt15 | Nudix (nucleoside diphosphate linked moiety X)-type 15 | 1.90 | 0.59 | 1.57 | 0.53 | 1.51 | 0.49 | 1.55 | 0.48 |
| NM_017051 | Sod2 | Superoxide dismutase 2, mitochondrial | 1.70 | 0.47 | 1.87 | 0.59 | 1.32 | 0.43 | 1.62 | 0.51 |
| NM_017305 | Gclm | Glutamate cysteine ligase, modifier subunit | 1.69 | 0.44 | 1.31 | 0.43 | 1.32 | 0.42 | 1.08 | 0.42 |
| NM_012815 | Gclc | Glutamate-cysteine ligase, catalytic subunit | 1.67 | 0.49 | 1.14 | 0.32 | 1.23 | 0.33 | −1.04 | 0.47 |
| NM_057143 | Park7 | Fertility protein SP22 | 1.56 | 0.43 | 1.68 | 0.45 | 1.52 | 0.45 | 1.53 | 0.56 |
| NM_012962 | Gss | Glutathione synthetase | 1.48 | 0.42 | 1.66 | 0.52 | 1.61 | 0.46 | 1.22 | 0.38 |
| NM_030826 | Gpx1 | Glutathione peroxidase 1 | 1.48 | 0.36 | 1.67 | 0.42 | 1.42 | 0.37 | 1.53 | 0.57 |
| NM_019354 | Ucp2 | Uncoupling protein 2 | 1.47 | 0.43 | 1.60 | 0.47 | 1.17 | 0.56 | 2.34 | 0.70 |
| NM_031614 | Txnrd1 | Thioredoxin reductase 1 | 1.46 | 0.47 | −1.15 | 0.23 | −1.13 | 0.28 | −1.60 | 0.18 |
| XM_216452 | Dhcr24 | 24-Dehydrocholesterol reductase (predicted) | 1.20 | 0.36 | −1.41 | 0.27 | −1.05 | 0.65 | −1.66 | 0.31 |
| XM_579339 | Serpine1 | Serine (or cysteine) Proteinase inhibitor, clade E, member 1 | −1.10 | 0.31 | −1.69 | 0.20 | −1.67 | 0.31 | −2.11 | 0.13 |
| NM_017365 | elfin,Pdlim1 | PDZ and LIM domain 1 | −1.18 | 0.26 | −1.31 | 0.21 | −1.62 | 0.21 | −1.46 | 0.20 |
| NM_017258 | Btg1 | B-cell translocation gene 1, anti-proliferative | −1.33 | 0.25 | −1.32 | 0.22 | −1.27 | 0.27 | −1.54 | 0.28 |
| NM_031056 | Mmp14 | Matrix metalloproteinase 14 (membrane-inserted) | −1.42 | 0.20 | −1.71 | 0.16 | −2.04 | 0.14 | −2.01 | 0.13 |
| NM_053769 | Dusp1 | Dual specificity phosphatase 1 | −1.54 | 0.32 | −2.61 | 0.18 | −1.73 | 0.36 | −2.61 | 0.24 |
| XM_216403 | Xpa | Xeroderma pigmentosum, complementation group A | −1.55 | 0.18 | −1.44 | 0.22 | −1.57 | 0.20 | −1.55 | 0.18 |
| NM_017025 | Ldha | Lactate dehydrogenase A | −1.55 | 0.18 | −2.35 | 0.12 | −2.20 | 0.12 | −2.45 | 0.12 |
| NM_024134 | Ddit3 | DNA-damage inducible transcript 3 | −2.00 | 0.15 | −2.41 | 0.12 | −2.38 | 0.12 | −2.74 | 0.11 |
| NM_001008767 | Txnip | Up-regulated by 1,25-dihydroxyvitamin D-3 | −3.59 | 0.22 | −2.32 | 0.11 | −4.98 | 0.22 | −1.97 | 0.15 |
| NM_019292 | Ca3 | Carbonic anhydrase 3 | −9.36 | 0.06 | −5.14 | 0.05 | −12.0 | 0.13 | −6.89 | 0.08 |
| XM_213440 | Col1a1 | Collagen, type 1, α1 | −16.8 | 0.02 | −8.92 | 0.03 | −19.1 | 0.04 | −19.8 | 0.02 |
| Response to unfolded protein | ||||||||||
| NM_053612 | Hspb8 | Heat shock 22 kD protein 8 | 3.64 | 0.89 | 2.98 | 0.67 | 2.91 | 0.72 | 2.81 | 0.83 |
| XM_215549 | OSP94 | Osmotic stress protein 94 kD (predicted) | 3.55 | 1.23 | 3.21 | 0.97 | 3.00 | 0.78 | 2.98 | 1.53 |
| NM_138887 | Hspb6 | Heat shock protein, α-crystallin-related, B6 | 1.98 | 0.58 | 2.25 | 0.69 | 1.89 | 0.69 | 2.03 | 0.92 |
| NM_175761 | Hspca | HEAT shock protein 1, α | 1.81 | 0.48 | 1.80 | 0.60 | 1.61 | 0.57 | 1.42 | 0.78 |
| XM_579648 | Ero1l | Rattus norvegicus ERO1-like | −1.40 | 0.20 | −1.53 | 0.20 | −1.51 | 0.20 | −1.59 | 0.19 |
| NM_031789 | Nfe2l2 | Nuclear factor, erythroid derived 2, like 2 | −1.53 | 0.21 | −1.79 | 0.16 | −1.88 | 0.15 | −1.72 | 0.20 |
| NM_053523 | Herpud1 | Homocysteine-inducible, endoplasmic reticulum stress-inducible, ubiquitin-like domain member 1 | −1.64 | 0.17 | −1.57 | 0.18 | −1.77 | 0.18 | −1.64 | 0.24 |
| XM_579186 | Txndc4 | Thioredoxin domain containing 4 (endoplasmic reticulum) | −1.65 | 0.18 | −1.56 | 0.18 | −1.79 | 0.17 | −1.78 | 0.22 |
| NM_022232 | Dnajc3 | Protein kinase inhibitor p58 | −1.75 | 0.18 | −1.78 | 0.18 | −1.94 | 0.17 | −1.99 | 0.21 |
| NM_001004210 | Xbp1 | X-box binding protein 1 | −2.09 | 0.15 | −2.32 | 0.12 | −2.49 | 0.11 | −2.61 | 0.11 |
| NM_001005562 | Creb3l1 | cAMP responsive element binding protein 3-like 1 | −7.08 | 0.04 | −6.23 | 0.05 | −7.43 | 0.04 | −7.65 | 0.04 |
| Calcium ion homeostasis | ||||||||||
| NM_017333 | Ednrb | Endothelin receptor type B | 4.64 | 1.30 | 4.72 | 1.49 | 4.41 | 1.46 | 4.94 | 1.35 |
| NM_013179 | Hcrt | Hypocretin | 3.07 | 2.31 | 3.31 | 1.04 | 2.64 | 1.93 | 4.51 | 1.27 |
| NM_017338 | Calca | Calcitonin/calcitonin-related polypeptide, · | 1.93 | 0.65 | 2.11 | 0.62 | 1.85 | 0.57 | 1.99 | 0.68 |
| XM_341418 | Edg4 | Endothelial differentiation, lysophosphatidic acid G-protein-coupled receptor 4 (predicted) | 1.90 | 0.58 | 2.50 | 0.77 | 2.04 | 0.66 | 1.96 | 1.07 |
| NM_031648 | Fxyd1 | FXYD domain-containing ion transport regulator 1 | 1.83 | 0.70 | 2.46 | 0.81 | 1.88 | 0.80 | 2.88 | 1.20 |
| XM_579178 | S100a1 | S100 calcium binding protein A1 | 1.74 | 0.79 | 2.04 | 0.62 | 1.86 | 0.85 | 2.38 | 0.85 |
| NM_012713 | Prkcb1 | Protein kinase C, β1 | 1.59 | 0.80 | −1.14 | 0.55 | 1.27 | 0.82 | −1.24 | 0.68 |
| NM_017010 | Grin1 | Glutamate receptor, ionotropic, N-methyl D-aspartate 1 | 1.56 | 0.84 | −1.47 | 0.70 | 1.16 | 1.31 | −1.71 | 0.63 |
| NM_016991 | Adra1b | Adrenergic receptor, α1b | −1.24 | 0.24 | −1.57 | 0.19 | −1.44 | 0.22 | −1.72 | 0.16 |
| NM_001007235 | Itpr1 | Inositol 1,4,5-triphosphate receptor 1 | −1.31 | 0.31 | −1.54 | 0.31 | −1.44 | 0.31 | −1.93 | 0.33 |
| NM_138535.1 | Grip2 | Glutamate receptor interacting protein 2 | −1.39 | 0.21 | −1.59 | 0.17 | −1.13 | 0.27 | −1.81 | 0.18 |
| NM_030987 | Gnb1 | Guanine nucleotide binding protein, β1 | −1.42 | 0.21 | −1.88 | 0.18 | −1.64 | 0.20 | −1.92 | 0.22 |
| NM_053867 | Tpt1 | Tumour protein, translationally-controlled 1 | −1.43 | 0.23 | −1.53 | 0.21 | −1.42 | 0.21 | −1.61 | 0.18 |
| NM_021666 | Trdn | Triadin | −1.57 | 0.17 | −1.58 | 0.19 | −1.52 | 0.18 | −1.55 | 0.18 |
| NM_031123 | Stc1 | Stanniocalcin 1 | −1.62 | 0.37 | −1.84 | 0.19 | −1.49 | 0.24 | −2.10 | 0.25 |
| NM_021663 | Nucb2 | Nucleobindin 2 | −1.69 | 0.18 | −1.67 | 0.21 | −1.75 | 0.18 | −1.82 | 0.33 |
| NM_023970 | Trpv4 | Transient receptor potential cation channel, subfamily V, member 4 | −1.71 | 0.16 | −1.44 | 0.25 | −1.72 | 0.17 | −1.62 | 0.14 |
| NM_017022 | Itgb1 | Integrin β1 (fibronectin receptor β) | −1.77 | 0.18 | −1.87 | 0.21 | −2.15 | 0.14 | −2.42 | 0.23 |
| NM_012714 | Gipr | Gastric inhibitory polypeptide receptor | −2.23 | 0.14 | −3.09 | 0.10 | −2.34 | 0.18 | −3.46 | 0.08 |
| NM_053936 | Edg2 | Endothelial differentiation, lysophosphatidic acid G-protein--coupled receptor, 2 | −3.13 | 0.28 | −2.26 | 0.23 | −3.85 | 0.17 | −2.17 | 0.14 |
| XM_579462 | Pln | Phospholamban | −7.16 | 0.04 | −6.34 | 0.05 | −6.56 | 0.05 | −6.18 | 0.04 |
| NM_053019 | Avpr1a | Arginine vasopressin receptor 1A | −10.1 | 0.03 | −9.82 | 0.03 | −9.53 | 0.03 | −10.9 | 0.03 |
| Vesicle transport | ||||||||||
| NM_053555.1 | Vamp5 | Vesicle-associated membrane protein 5 | 1.65 | 0.52 | 2.02 | 0.71 | 1.35 | 0.50 | 1.98 | 0.59 |
| NM_057097.1 | Vamp3 | Vesicle-associated membrane protein 3 | 1.55 | 0.38 | 1.69 | 0.40 | 1.47 | 0.45 | 1.48 | 0.60 |
| NM_138835.1 | Syt12 | Synaptotagmin 12 | 1.56 | 0.46 | 1.15 | 0.35 | 1.27 | 0.35 | 1.17 | 0.33 |
| XM_343205.2 | Syt1 | Synaptotagmin 1 | 1.33 | 1.08 | −2.13 | 0.90 | 1.09 | 1.59 | −1.96 | 0.85 |
| Inflammatory response | ||||||||||
| NM_133624.1 | Gbp2 | Guanylate nucleotide binding protein 2 | 17.5 | 7.13 | 20.0 | 6.93 | 14.0 | 3.77 | 12.9 | 4.69 |
| NM_212466 | Bf | B-factor, properdin | 6.47 | 3.81 | 9.28 | 9.00 | 7.12 | 1.98 | 10.9 | 4.90 |
| NM_172222 | C2 | Complement component 2 | 3.55 | 0.97 | 4.03 | 1.22 | 3.17 | 0.90 | 4.50 | 1.51 |
| XM_215095.3 | Gprc5b | G protein-coupled receptor, family C, group 5, member B | 3.07 | 1.03 | 2.75 | 0.78 | 2.29 | 0.69 | 3.32 | 1.43 |
| NM_199093 | Serping1 | Serine (or cysteine) peptidase inhibitor, clade G, member 1 | 2.80 | 2.06 | 2.48 | 1.23 | 2.40 | 1.15 | 3.67 | 2.05 |
| NM_012488 | A2m | ·2-Macroglobulin | 2.61 | 0.64 | 3.49 | 0.82 | 2.60 | 1.26 | 3.76 | 1.10 |
| NM_019363 | Axo1 | Aldehyde oxidase 1 | 2.53 | 0.70 | 5.69 | 3.33 | 4.03 | 2.37 | 2.83 | 0.84 |
| XM_573457.1 | Gpr37l1 | G protein-coupled receptor 37-like 1 (predicted) | 2.52 | 0.85 | 3.05 | 0.98 | 2.63 | 0.88 | 3.62 | 1.94 |
| NM_138502 | Mgll | Monoglyceride lipase | 2.51 | 1.08 | 2.16 | 0.81 | 2.21 | 0.79 | 2.31 | 0.72 |
| NM_152242.1 | Gpr56 | G protein-coupled receptor 56 | 2.42 | 0.73 | 2.16 | 0.53 | 2.23 | 0.64 | 2.10 | 0.68 |
| NM_212541 | Ng22 | Ng22 protein | 1.94 | 0.64 | 2.16 | 0.68 | 1.66 | 0.51 | 2.12 | 0.65 |
| NM_138900 | C1s | Complement component 1, s subcomponent | 1.91 | 0.54 | 2.18 | 0.82 | 1.75 | 0.51 | 1.92 | 0.70 |
| NM_031351 | Atrn | Attractin | 1.89 | 0.52 | 1.68 | 0.52 | 1.84 | 0.53 | 1.42 | 0.43 |
| NM_031007.1 | Adcy2 | Adenylate cyclase 2 | 1.88 | 0.51 | 1.78 | 0.55 | 1.80 | 0.52 | 1.78 | 0.56 |
| XM_579383.1 | Adcyap1 | Adenylate cyclase activating polypeptide 1 (predicted) | 1.74 | 1.96 | −1.90 | 0.30 | 1.14 | 2.35 | −2.01 | 0.25 |
| NM_013157 | Ass | Argininosuccinate synthetase | 1.71 | 2.33 | 1.05 | 0.36 | 1.52 | 1.49 | −1.05 | 0.30 |
| NM_031634 | Mefv | MEDITERRANEAN fever | 1.61 | 0.47 | 1.77 | 0.48 | 1.73 | 0.62 | 1.97 | 0.56 |
| NM_021690.1 | Rapgef3 | cAMP-regulated guanine nucleotide exchange factor I | 1.58 | 0.40 | 1.78 | 0.48 | 1.71 | 0.52 | 1.77 | 0.48 |
| XM_579359 | Cd59 | CD59 antigen | 1.40 | 0.45 | 1.47 | 0.39 | 1.23 | 0.79 | 2.00 | 0.56 |
| NM_053734 | Ncf1 | Neutrophil cytosolic factor 1 | 1.32 | 0.42 | 1.18 | 0.44 | 1.36 | 0.74 | 2.06 | 1.32 |
| NM_212490 | Atp6v1g2 | ATPase, H+ transporting, V1 subunit G isoform 2 | 1.31 | 0.75 | −1.50 | 0.62 | 1.18 | 0.88 | −1.50 | 0.78 |
| NM_022216.1 | Gpr20 | G protein-coupled receptor 20 | 1.25 | 0.37 | 1.70 | 0.56 | 1.34 | 0.44 | 1.64 | 0.46 |
| NM_153318 | Cyp4f6 | Cytochrome P450 4F6 | 1.21 | 0.30 | 1.51 | 0.39 | 1.25 | 0.37 | 1.42 | 0.41 |
| NM_001009353 | Pla2g7 | Phospholipase A2, group VII (platelet-activating factor acetylhydrolase, plasma) | 1.20 | 0.43 | 1.24 | 0.41 | 1.27 | 0.78 | 1.74 | 1.25 |
| NM_024157 | Cfi | Complement factor I | 1.19 | 0.33 | 1.20 | 0.33 | 1.20 | 0.34 | 1.59 | 0.50 |
| NM_022236.1 | Pde10a | Phosphodiesterase 10A | 1.08 | 0.30 | −1.52 | 0.38 | −1.30 | 0.29 | −1.70 | 0.36 |
| XM_579389 | Tf | Transferrin | −1.05 | 1.91 | −1.50 | 0.82 | −1.04 | 0.76 | 2.02 | 6.19 |
| NM_017196 | Aif1 | Allograft inflammatory factor 1 | −1.15 | 0.56 | −1.29 | 0.92 | −1.11 | 1.11 | 1.79 | 1.40 |
| NM_024125 | Cebpb | CCAAT/enhancer binding protein (C/EBP), β | −1.25 | 0.21 | −1.55 | 0.21 | −1.27 | 0.22 | −1.53 | 0.21 |
| XM_342092 | Vars2 | Valyl-tRNA synthetase 2 | −1.31 | 0.21 | −1.56 | 0.19 | −1.38 | 0.19 | −2.08 | 0.14 |
| NM_052809 | Cdo1 | Cysteine dioxygenase 1, cytosolic | −1.34 | 0.23 | −1.99 | 0.15 | −1.81 | 0.16 | −1.62 | 0.17 |
| NM_024486 | Acvr1 | Activin A receptor, type 1 | −1.38 | 0.23 | −1.46 | 0.21 | −1.65 | 0.19 | −1.61 | 0.17 |
| NM_012666 | Tac1 | Tachykinin 1 | −1.50 | 0.23 | −2.32 | 0.14 | −1.60 | 0.34 | −2.47 | 0.13 |
| XM_239239 | Map2k3 | Mitogen-activated protein kinase kinase 3 (predicted) | −1.67 | 0.18 | −2.12 | 0.14 | −2.08 | 0.15 | −2.46 | 0.15 |
| NM_130409 | Cfh | Complement component factor H | −1.68 | 0.18 | −1.25 | 0.25 | −1.57 | 0.19 | 1.02 | 0.32 |
| XM_579400 | Lbp | Lipopolysaccharide binding protein | −1.81 | 0.19 | −1.39 | 0.23 | −1.63 | 0.21 | −1.09 | 0.44 |
| XM_343169 | And | Adipsin | −2.07 | 0.14 | −2.14 | 0.15 | −1.83 | 0.15 | −1.88 | 0.14 |
| NM_019143 | Fn1 | Fibronectin 1 | −2.22 | 0.17 | −2.15 | 0.20 | −2.43 | 0.11 | −3.73 | 0.22 |
| NM_019262 | C1qb | Complement component 1, q subcomponent, β polypeptide | −2.42 | 0.22 | −2.81 | 0.29 | −2.08 | 0.71 | −1.38 | 0.54 |
| NM_001008524 | C1qg | Complement component 1, q subcomponent, gamma polypeptide | −2.49 | 0.15 | −2.97 | 0.18 | −2.17 | 0.39 | −1.44 | 0.47 |
| NM_017232 | Ptgs2 | Prostaglandin-endoperoxide synthase 2 | −3.59 | 0.16 | −6.51 | 0.06 | −3.84 | 0.13 | −6.18 | 0.08 |
| XM_240184 | Nfatc4 | Nuclear factor of activated T cells, cytoplasmic, calcineurin-dependent 4 (predicted) | −15.4 | 0.06 | −8.20 | 0.06 | −14.8 | 0.04 | −10.4 | 0.03 |
| Chemotaxis | ||||||||||
| NM_031530 | Ccl2 | Chemokine (C-C motif) ligand 2 | 6.01 | 8.84 | 2.87 | 2.48 | 3.68 | 9.35 | 2.66 | 2.08 |
| NM_182952.2 | Cxcl11 | Chemokine (C-X-C motif) ligand 11 | 5.24 | 1.72 | 6.02 | 8.94 | 2.80 | 0.81 | 2.34 | 1.31 |
| NM_022205 | Cxcr4 | Chemokine (C-X-C motif) receptor 4 | 3.14 | 1.20 | 4.19 | 1.11 | 2.81 | 0.82 | 3.56 | 2.11 |
| NM_012953 | Fosl1 | Fos-like antigen 1 | 2.97 | 1.17 | −1.04 | 0.27 | 1.47 | 1.19 | 1.02 | 0.25 |
| NM_001007612 | Ccl7 | Chemokine (C-C motif) ligand 7 | 2.80 | 3.76 | 2.09 | 1.76 | 2.01 | 3.77 | 1.56 | 0.75 |
| NM_139089 | Cxcl10 | Chemokine (C-X-C motif) ligand 10 | 2.67 | 1.43 | 10.3 | 23.6 | 3.87 | 2.87 | 3.86 | 3.14 |
| NM_053953.1 | Il1r2 | Interleukin 1 receptor, type II | 2.42 | 1.05 | 2.23 | 0.71 | 2.28 | 0.68 | 1.87 | 0.85 |
| NM_145672 | Cxcl9 | Chemokine (C-X-C motif) ligand 9 | 2.13 | 1.58 | 3.09 | 2.97 | 2.42 | 0.75 | 1.94 | 1.24 |
| NM_012747.2 | Stat3 | Signal transducer and activator of transcription 3 | 2.0 | 0.5 | 1.57 | 0.38 | 1.64 | 0.46 | 1.45 | 0.38 |
| NM_030845 | Cxcl1 | Chemokine (C-X-C motif) ligand 1 | 1.96 | 4.51 | −1.30 | 0.32 | 1.64 | 4.95 | 1.18 | 1.07 |
| XM_234422.3 | c-fos | c-Fos oncogene | 1.8 | 0.5 | 1.61 | 0.45 | 1.52 | 0.52 | 1.54 | 0.51 |
| NM_134455 | Cx3cl1 | Chemokine (C-X3-C motif) ligand 1 | 1.58 | 0.62 | 1.01 | 0.46 | 1.32 | 0.88 | −1.22 | 0.74 |
| NM_031512.1 | Il1b | Interleukin 1β | 1.57 | 0.77 | 1.30 | 0.48 | 1.30 | 0.42 | 1.46 | 0.62 |
| NM_017020.1 | Il6r | Interleukin 6 receptor | 1.54 | 0.44 | 1.21 | 0.36 | 1.28 | 0.37 | 1.28 | 0.33 |
| NM_031116 | Ccl5 | Chemokine (C-C motif) ligand 5 | 1.43 | 0.40 | 2.96 | 2.76 | 1.80 | 0.48 | 3.03 | 2.40 |
| NM_139111 | Cklf1 | Chemokine-like factor 1 | 1.24 | 0.38 | 1.39 | 0.44 | 1.13 | 0.31 | 1.89 | 0.56 |
| NM_031643 | Map2k1 | Mitogen-activated protein kinase kinase 1 | 1.08 | 0.30 | −1.27 | 0.29 | −1.14 | 0.22 | −1.55 | 0.49 |
| XM_342823 | Ccl27 | Chemokine (C-C motif) ligand 27 (predicted) | −1.12 | 0.28 | −1.55 | 0.25 | −1.16 | 0.38 | −1.62 | 0.19 |
| XM_579590 | Enpp2 | Ectonucleotide pyrophosphatase/phosphodiesterase 2 | −1.61 | 0.29 | −1.22 | 0.26 | −1.80 | 0.21 | −1.59 | 0.18 |
| NM_031327 | Cyr61 | Cysteine rich protein 61 | −1.65 | 0.24 | −2.00 | 0.19 | −1.90 | 0.26 | −2.22 | 0.25 |
| NM_031836 | Vegfa | Vascular endothelial growth factor A | −1.69 | 0.16 | −1.79 | 0.14 | −1.78 | 0.16 | −1.87 | 0.15 |
| NM_022177 | Cxcl12 | Chemokine (C-X-C motif) ligand 12 | −1.81 | 0.18 | −1.71 | 0.17 | −1.70 | 0.17 | −1.16 | 0.29 |
| NM_001007729 | Pf4 | Platelet factor 4 | −1.92 | 0.33 | −2.98 | 0.10 | −1.95 | 0.23 | −2.36 | 0.12 |
| NM_012881 | Spp1 | Secreted phosphoprotein 1 | −3.05 | 0.19 | −1.86 | 0.29 | −3.08 | 0.10 | −1.40 | 0.64 |
| NM_019233 | Ccl20 | Chemokine (C-C motif) ligand 20 | −3.09 | 0.44 | −2.17 | 0.41 | −1.94 | 0.94 | −2.62 | 0.30 |
Genes with a minimum fold change of ±1.5 for at least one out of the four time points passed one-way ANOVA analysis and are significant with P < 0.05. Data are expressed as fold-change ± S.E. Blue = genes that were up-regulated. Red = genes that were down-regulated.
Fig 1.

Pie chart annotating number and percentage of genes identified in each of the 14 biological processes. A total of 305 statistically significant regulated genes were identified by DAVID. Each gene was classified into one of its most relevant biological processes categorized according to Gene Ontology to avoid overestimation.
Fig 2.
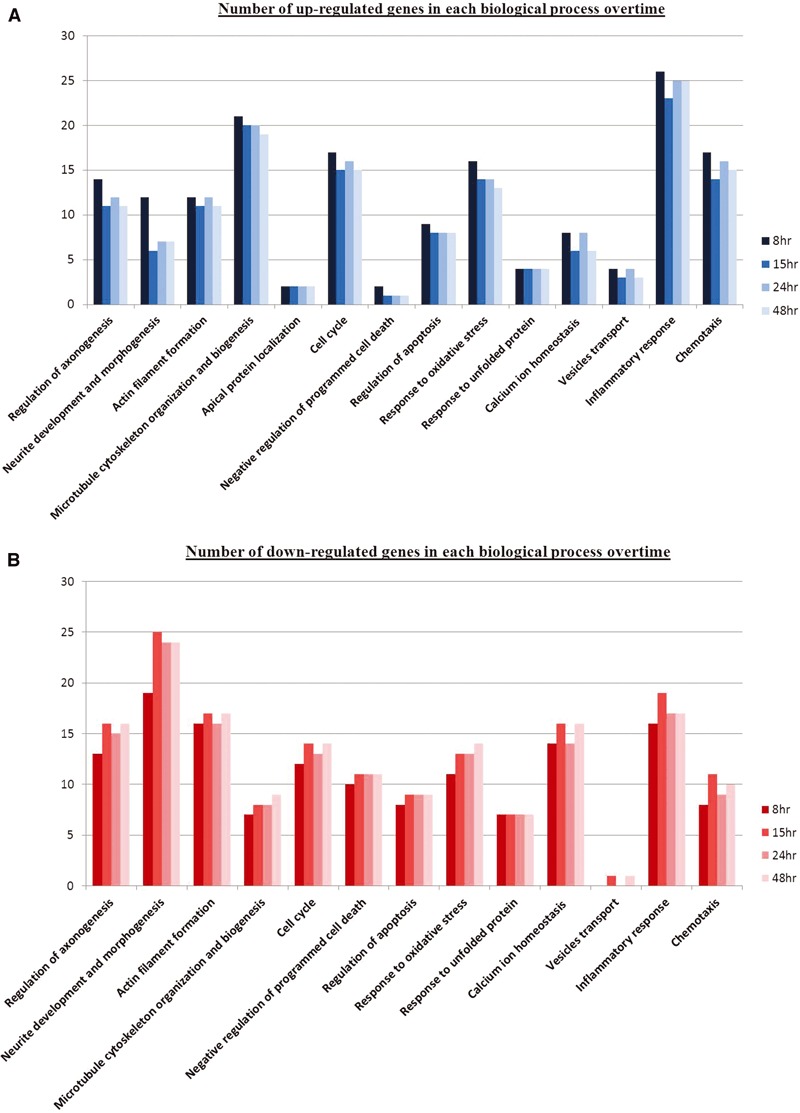
Comparison of biological processes associated with axotomized neurons across 8, 15, 24 and 48 hrs post-injury. Bar chart shows the distribution of up-regulated (A) and down-regulated (B) genes. Differentially transcribed genes involved in the number count were statistically significant. Overall, the genomic profiles for particular biological processes at 8 hrs were slightly distinct as compared to the other time points.
Association of genes with biological processes
Neurite network associated genes
There was a down-regulation of genes responsible for repulsive axon guidance cues and their receptors that are known to limit axonal regeneration and these include Efn, Sema, Slit, Bmp, Nrp, Plxd and Eph. Conversely, there was an increased expression of neurite outgrowth promoting genes such as Apoe, Cntf, Vgf and Lama5. In addition, there was a transient elevation in gene expression at 8 hrs that faltered with time for Snap, Tnn and neurotrophic-associated factors such as Nrtk and Bdnf. A varied expression of genes involved in actin filament formation and microtubule organization was observed and they include Tuba, Tubb, Acta, Actb, Spnb, Pfn, Stmn, Arp2/3, N-WASP and Map1b. Our data also reflected on the importance of protein trafficking during axonal repair as genes known to facilitate this process, mainly motor (Dnc, Dna, Kif, Myh, Tpm) and vesicle proteins (Vamp, Syt, Snap) were also differentially expressed.
Oxidative insult-related genes
Transcriptomic data revealed an elevated expression of genes related to GSH biosynthesis namely Gstm1, Mgst1, Gss and Gpx1. This was accompanied by a brief expression of Txnrd1 at 8 hrs and a prolonged expression of antioxidant enzyme genes which include superoxide dismutase (Sod2 and Sod3) and peroxiredoxin (Prdx6). Moreover, heat shock proteins (Hspa2, Hspb1, Hspb8, Hspb6, Hspca), that serve as molecular chaperones were also significantly up-regulated (except Hspa5).
Cell cycle and death genes
There was a diminished expression of various pro-apoptosis genes such as Apaf1 and Casp 3, 11 and 12 with the exception of Csap6.
In addition, Bnips, a group of pro-apoptosis Bcl-2 family proteins, were also significantly down-regulated. Accordingly, increased gene expression of Rap1a and Braf might represent activation of MEK/ERK pro-survival signalling pathway. Intriguingly, a series of cell cycle proteins were differentially regulated and some examples of which include Aurkb, a range of cyclins and few transcriptional and translational factors. The significance of changes in expression of these cell cycle genes in the absence of visible cell death upon axotomy is intriguing as there is increasing evidence showing that cell cycle proteins do play novel, alternate neuronal functions [19].
Ion homeostasis response genes
Disruption to ion balance after axotomy was reflected in the varied expression of genes that control ion entry channels such as Fxyd1, Adra1b, Edg2, Itpr1, Trpv4 and Avpr1a. In particular, we noticed an elevated gene expression of calcium-activated proteins such as Capn3 and S100A1 that play pivotal roles in cell death, axonal resealing and cytoskeleton remodelling. Temporal expression of calcium-associated members such as Camk2b, PKC and Grin1 further demonstrated a likely link between calcium levels and neurorepair. Furthermore, elevated gene expression of calcium-activated AMPK, a key regulator of energy balance, and its upstream activators namely Cntf and interleukins might signify a mechanism in place to compensate energy deficits during ion and/or metabolites imbalance states.
Inflammatory response genes
Inflammation is part of a complex ‘double-edged sword’ response to injury that facilitates healing and/or elicits death. Quite interestingly, the presence of inflammation-causing genes such as Bf, C2, Atrn and Ncf1 was accompanied by an increased expression of cAMP regulatory genes namely Adcy, Adcyap, Gpr and Rapgef. Interplay of various chemokines and cytokines was evident from the gene regulation profiles of numerous chemokine ligands (Ccl and Cxcl families), cytokine (II1b), and their receptors (Cxcr, II1r2 and II6r). Cytokine-activated transcription factors such as Stat3 and c-fos that drive cell proliferation, signal transduction and cytoskeleton restructuring were also increased.
Validation of microarray analysis
Microarray data for all time points after axonal injury were validated by RT-PCR against selected genes as presented in Table 2. The trend of gene expression changes for each selected target obtained from RT-PCR was similar to its microarray data. In general, this confirmed the reliability of the gene expression profiles attained.
Table 2.
Validation of microarray data using real-time PCR technique on rat neuronal samples, 8, 15, 24 and 48 hrs after axotomy
| 8 hrs | 15 hrs | 24 hrs | 48 hrs | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Gene title | Symbol | Microarray | Real-time PCR | Microarray | Real-time PCR | Microarray | Real-time PCR | Microarray | Real-time PCR |
| Neural Wiskott–Aldrich syndrome protein | N-WASP | 3.7 ± 1.3 | 7.4 ± 2.4 | 4.8 ± 1.6 | 2.7 ± 0.5 | 3.5 ± 2.1 | 1.7 ± 0.4 | 4.2 ± 2.4 | 2.9 ± 0.3 |
| Chemokine (C-X-C motif) ligand 10 | Cxcl10 | 2.7 ± 1.4 | 2.5 ± 0.8 | 10.3 ± 23.6 | 11.1 ± 3.3 | 3.9 ± 2.9 | 3.9 ± 2.5 | 3.9 ± 3.1 | 2.6 ± 0.8 |
| Dynein, cytoplasmic, light chain 2B (predicted) | Dncl2b | 20.8 ± 6.6 | 9.3 ± 2.3 | 23.9 ± 7.2 | 10.4 ± 3.5 | 16.4 ± 13.5 | 10.4 ± 3.9 | 22.5 ± 10.6 | 10.0 ± 1.7 |
| Guanylate nucleotide binding protein 2 | Gbp2 | 17.5 ± 7.1 | 7.8 ± 3.6 | 20.0 ± 6.9 | 9.2 ± 2.8 | 14.0 ± 3.8 | 11.4 ± 5.0 | 12.9 ± 4.7 | 9.9 ± 2.9 |
| Chemokine (C-X-C motif) ligand 11 | Cxcl11 | 5.2 ± 1.7 | 3.2 ± 1.1 | 6.0 ± 8.9 | 4.8 ± 1.7 | 2.8 ± 0.8 | 4.8 ± 1.3 | 2.3 ± 1.3 | 4.0 ± 1.1 |
| Aurora kinase B | Aurkb | 5.0 ± 2.0 | 3.7 ± 1.0 | 5.9 ± 1.7 | 5.4 ± 1.3 | 4.0 ± 1.6 | 5.0 ± 1.2 | 5.8 ± 1.7 | 4.8 ± 1.6 |
Data are expressed as fold-change ± S.E.
Discussion
Severance of fasciculated axonal bundles in mature rat neuronal clusters in culture has been used previously to study and characterize the cytoskeletal dynamics of regenerative sprouting after axonal injury [6]. Using the same in vitro model, we have applied a DNA microarray approach to gain mechanistic insight into the key molecular determinates that underlie neurite regeneration after axotomy at a transcriptomic level. Briefly, we found that regenerative sprouting is a complicated process that involves complex cytoskeleton dynamics and vigilant control of secondary processes such as oxidative stress, ion homeostasis and inflammation. These secondary processes have to be tightly regulated to ensure neurite regeneration without causing evident cell death. Physiologically, diffuse axonal injury is one of the many key pathological features associated with head trauma. Upon trauma, stretched axons become brittle and tear. This situation is immediately salvaged by the damaged neurons’ intrinsic capacity to regenerate. However, mechanisms are activated for cell demise when the injury is beyond redemption. Through transcriptomic studies, we may identify crucial cellular pathways that facilitate regeneration and manipulate them whenever necessary to lessen damage and avoid irreversible apoptosis.
Neurite cytoskeleton reorganization
In accordance with the study by Chuckowree and Vickers [6], we confirmed that adaptive sprouting of neurons after injury involves substantial cytoskeleton reorganization. For an axon to regenerate, its neurite arm, which is predominately composed of microtubules, needs to be extended accordingly. This involves microtubule formation and stabilization. Tubulins are fundamental for microtubule formation and this could explain their augmented gene expression. Stabilizing these newly formed microtubules is pivotal and this entails a concerted regulation of various microtubule destabilizing and stabilizing proteins. Elevated gene expression of Notch1, alongside decreased expression of Stmn, Cspg2 and Spg4, is consistent with this process. It has been found that microtubule stabilization could promote axon regeneration by preventing chondroitin sulphate proteoglycan (Cspg) accumulation at lesion site [20]. This stabilization could be mediated by Notch activation whereby it down-regulated spastin (Spg) expression, a microtubule severing protein, and deterred axonal degeneration in primary cortical neurons [21]. Stamins (Stmn) in general destabilize microtubules by preventing their assembly and promoting their disassembly.
The regenerating growth cone acts such as a molecular conveyor belt whereby actin bundles at the proximal end are constantly fragmented by actin-destabilizing or severing proteins. Released actin fragments are then retrogradely transported and reassembled at the leading edge with help from nucleation-promoting factors [22]. These changes were reflected by the down-regulated gene expression of actin regulators namely Pfn, Cdc42, Arp2/3 [23, 24]. In contrast, NWASP and Gas7 gene expression were up-regulated by an average of 3- and 1.5-fold, respectively, throughout [25, 26]. As such, it is imperative to account for the contrasting regulation of NWASP, Arp2/3 and Cdc42. NWASP and Arp2/3 are undeniably the master components of actin polymerization. During neurite regrowth, actin dynamics unlike microtubules steer towards instability, a phenomenon observed due to the complex processes governing polymerization and depolymerization of actin filament to support growth cone steering. Interestingly, Erin D.G and Matthew D.W critically reviewed that the way Arp2/3 affects actin polymerization is more dependent on its activity regulated by ATP rather than its expression [27]. It was mentioned that binding of WASP to ATP bound Arp2/3 and G actin, primes the complex and creates a nucleation point for daughter filaments formation. Subsequent ATP hydrolysis on Arp2 after nucleation has been temporally and functionally linked to actin branch disassembly. The authors also brought up the importance of recycling Arp2/3 itself for the formation of new actin processes, an event closely associated with actin recycling. With that, the possibility of regulating already expressed Arp2/3 proteins by ATP in neurons and the fact that it can be recycled could potentially explain the redundant need for its expression after axotomy. Unlike Arp2/3, NWASP is not directly regulated by ATP binding but its relevance is to elicit an active conformation of Arp2/3 for nucleation by physically binding to it. As such, our data illuminate an interesting questionnaire on how NWASP is being regulated in regards to neurite regeneration. Moreover, apart from NWASP physical interaction with Arp2/3 to direct actin polymerization, it also serves as focal points where transduced signals converge to orchestrate actin polymerization dynamics [28]. Hence, NWASP importance in connecting multiple signalling pathways to initiate actin assembly as well as how it is regulated could possibly account for its elevated expression in comparison to Arp2/3. As for Cdc42, unlike NWASP and Arp2/3, there has been evidence showing that NWASP recruitment of Arp2/3 that results in the formation of membrane protrusions and processes could occur via a Cdc42-independent manner, at least for neurite outgrowth of hippocampal neurons [26]. Because of the way actin proteins are recycled in the growth cone, this further explains the redundant need for newly synthesized actins and they were generally down-regulated transcriptionally following axonal injury. Recycling is essential to prevent excessive actin build-up that could thwart neuronal advances. This assumption is further supported by the heightened expression of myosin genes, which encode motor molecules that transport actins in a retrograde fashion. Using fluorescent speckle microscopy, it was found that inhibition of myosin II not only perturbed actin retrograde flow and affected neuronal growth but its contraction forces were also required to recycle actin fragments for growth cone formation [29]. Together, our gene profiling data showed that neurite regeneration is an event that involves stabilization of microtubules and dynamic instability of actin filaments [7].
Further evidence of the dynamic response of the cytoskeleton to axotomy was the elevated expression of Tekt 1 and 2. These represent a group of cilia microtubule structural proteins, and supports an unexpected discovery that neuronal cilia might be vital for modulating signalling pathways to coordinate neuronal processes such as axonal guidance [30]. This highlights the need for further investigation on the roles of cilia-associated proteins in neurobiological events such as axotomy.
The importance of protein trafficking in axonal regeneration was indicated by the varied gene expression of motor proteins involved in cytoskeletal transport such as Dnc, Dna, Kif, Myh, Tpm and machinery factors linked to membrane vesicles such as Syt, Vamp, Stx. This clearly illustrated retrograde transport of injury signals from cut site to cell body which then evoked membrane expansion processes that involve cargo trafficking of cytoskeletal proteins and even neurotropic factors to promote axonal re-growth. Dynein-derived forces in particular, are able to oppose axon retraction and permit microtubules to advance [31].
Moreover, neurite regeneration is a process that involves reciprocal regulation of permissive and repulsive axon guidance cues. Neurotrophic factors, for instance, are capable of directing axonal regrowth in damaged neurons. Bdnf can sustain axon regeneration upon various nerve and brain injuries [32, 33] and overexpression of its receptor NtrkB was also found to elicit corticospinal axonal regeneration [34]. Here, transient gene expression of Bdnf and Ntrks at 8 hrs could highlight the probable importance of these molecules in the recovery process during the initial phase but not as much after specific projections had been formed. Most repulsive guidance molecules on the other hand, inhibit and deter axonal regrowth after injury. Sema3a is one such example. Chemically inhibiting Sema3a prevented its binding to neuropilin and enhanced neural regeneration in damage axons [35]. Eph/Ephrin signalling which controls axon guidance by contact repulsion also inhibits regeneration of axons following injury in neurons [36]. Similarly, our transcriptomic data concurred that these molecules and their receptors mainly Efn, Sema, Slit, Bmp, Nrp, Plxd and Eph were not favoured during neurite regrowth.
Notably, some neurite promoting factors including Apoe, Cntf and Lama5 were also significantly up-regulated. Apoe is required for lipid delivery for axon regrowth upon nerve crush injury [37] whereas Lama serves as a crucial extracellular matrix adhesion molecule along which axons would grow and thus permit regeneration in central neurons [38, 39]. Although Cntf is found mainly in astrocytes, it is also expressed in cortical neurons [40]. Being a cytokine-induced neurotrophic factor, it can potentiate axon regeneration in optic and spinal nerve injuries [41-43]. Overall, our results demonstrate that to facilitate axonal regrowth, the injured neurons themselves adapt to ensure that the cellular environment is permissive to regeneration via temporal generation of positive cues, concomitant restriction of negative cues and expression of neurotrophic factors.
Apart from the genes that regulate neurite cytoskeleton, our data have further shown that various genes commonly linked to secondary processes such as oxidative stress, apoptosis, cell cycle, calcium homeostasis and inflammation are also important in dictating the regenerative process of axonally cut neurons and their possible roles are briefly discussed as follow.
Oxidative stress
Reactive oxygen species (ROS) are inevitable harmful by-products of cellular activities. Oxidative stress, which could result in cell death, ensues when the build-up of ROS in cells overwhelm existing biological antioxidants under normal conditions. This occurs in neurons after axonal injury. To counteract such insult, our study showed that genes responsible for GSH synthesis and various antioxidant enzymes (Sods, Prdxs, Gpx) were elevated to alleviate the stress. This demonstrated that neurons have the transcriptional capability to activate these vital mechanisms for their fight against oxidative insult to prevent death and assist in regenerative sprouting.
Several heat shock proteins (Hsp) were also transcriptionally up-regulated. Mainly, Hsp act as molecular chaperones to refold misfolded proteins and prevent deleterious protein build-up. Intriguingly, small Hsp (12–43 kD) are viewed as vital neuroprotectants in several neurological disorders [44]. Hsp27 whose gene expression was up-regulated by ≥4-folds post-injury could suppress cytochrome c–mediated cell death [45]. Besides, it could interact, modulate and remodel neuronal cytoskeleton [46, 47]. As such, in addition to its protein refolding function, local gene expression of Hsp after axotomy might serve as anti-apoptotic and neurite regrowth signals.
Programmed cell death (PCD)
PCD is a process regulated by proto-oncogenes. It is logical that for an injured neuron to successfully regenerate, it must avoid axotomy-induced cell death. It is possible that apoptotic genes such as Apaf1, Bnips, Casp3, 11 and 12 were down-regulated in our study as an intrinsic protective mechanism to facilitate regeneration. This is supported by the fact that axotomized neuronal clusters in vitro did not result in evident cell death [6]. Similarly, studies have shown that overexpression of anti-apoptotic proteins could save neurons from axotomy-induced cell death [48, 49]. However, it is important to note that stringent control of apoptotic genes expression in denial of cell death in this case does not necessarily assist or speed up neurite regeneration, and probably indicates that neuron survival and neurite regeneration represent two distinct, although closely linked, processes within axotomized neurons.
Cell cycle
Cell cycle activation in response to DNA damage during extreme cellular stress could lead to the demise of neurons [50]. Interestingly, several cell cycle proteins were recently found to possess distinct, alternate neuronal functions that are independent from their cell cycle roles [19]. Plausibly, this mirrors our case of neurite regeneration in which certain cell cycle genes were elevated despite the down-regulation of several apoptotic genes consistent with cell death not being prominent. Aurkb, for example is a cell cycle kinase that remodels microtubule arrays for precise cell division [51, 52]. Its elevated gene expression could possibly reflect a function in neurite regrowth, a process akin to its involvement in cell division, because both processes involve complex cytoskeleton reorganization. However, functional biological studies are required to ascertain this speculation. Intriguingly, Aurora A kinase (Aurka), a sister kinase of Aurkb that is also known for its mitotic roles, had lately been reported by two separate groups to play a crucial role in the establishment of neuronal polarity [53, 54].
A more specific example involves Klf4, which was significantly down-regulated in our studies. Early studies show that Klf4 is a key cell cycle checkpoint transcription factor and it causes G1/S arrest by activating genes that are potent inhibitors of proliferation [55]. Recently, an alternate neuronal function of Klf4 was discovered whereby its overexpression in cortical neurons impeded neurite outgrowth while its knockout in retinal ganglion cells (RGCs) increased axon regeneration after crush injury [56]. Hence, beside cell cycle re-entry causing neuronal death, diverse gene expression of cell cycle proteins after axotomy could also signify alternate and unexpected functions for these proteins to regulate the cytoskeleton and promote axonal regeneration.
Calcium homeostasis
Calcium homeostasis after axotomy is vital as it determines survival or cell death onset. When the damage is too severe, excitotoxicity due to excessive influx of calcium ions causes neuronal cell death. However, in less damaging situations where calcium levels are appropriately controlled, this triggers proteolysis and membrane fusion/fission processes associated with axonal repair and cytoskeleton reorganization. First, local resealing is a critical early response for regrowth and this requires calcium activation of calpains to cleave the spectrin network and release membrane tension. This might explain why Capn3 was up-regulated, whereas Spnb gene expression was varied. Secondly, calcium is a fundamental co-factor for cytoskeletal remodelling as it is required for membrane vesicle fusion processes used in the delivery of constructive molecules and neurotrophic factors to axotomized ends for resealing purposes and growth cone formation [57]. Varied gene expression of calcium signalling associated genes, and the elevated gene expression of motor and machinery proteins involved in membrane vesicle transport and fusion mentioned earlier, further supported this supposition.
In addition, up-regulation of SNF1/AMPK, a calcium-activated kinase involve in metabolic stress, showed that energy level regulation after axotomy is important. This is accompanied by the concomitant elevated expression of its upstream activators such as Il-6 and Cntf [58, 59]. Research on the role of AMPK in neurons is new but recent studies have shown that AMPK activation in neurons is neuroprotective [60, 61]. More recently, plausible roles for AMPK in neuronal polarity and axon specification are indicated by its necessity for the maintenance of cell polarity in epithelial cells [62]. In the case of our study, it is possible that energy regulation in response to calcium ion levels could be crucial for the battle against metabolic insults and the re-establishment and/or maintenance of polarity in severed neurons.
Inflammation
Inflammation in response to axotomy as observed from the presence of Bf, C2, Nrf1 and Atrn could result in cell death if not properly regulated [63-65]. cAMP levels which are increased and limited by the activity of adenylate cyclase (Adcy) and phosphodiesterase (Pde), respectively, could mitigate inflammation [66, 67]. Elevated gene expression of Adcy, Adcyap and Gpr, but decreased expression of Pde, as per our study, clearly suggested that cAMP elevation was adopted to ease inflammation caused by axotomy. More important, increased levels of cAMP were found to promote axonal regeneration and recovery after CNS injury [68]. The likely mechanism for this is increasing the translocation of neurotrophic receptors, for example NtrkB, to the plasma membrane of neurons [69]. cAMP could also activate a series of kinases that augment growth promoting signalling pathways in brain and spinal cord injuries [70]. Our data confirm the importance of the role of cellular cAMP level to alleviate inflammation and to push regrowth mechanisms in response to injury. Henceforth, the roles of ATP in Arp2/3 regulation mentioned earlier, together with the significance of regulating secondary messengers to direct neurite regeneration, highlight the importance of secondary messenger precursors in the study of axonal injury.
Apart from the primary association of cytokines and chemokines with inflammation, they could also directly or indirectly modulate regeneration after injury. This was evident from their robust gene expression profiles. Through a knockout study, Il6, a cytokine, was found to promote re-growth of dorsal column axons after static nerve injury [71]. Changes in cytokine levels often alter expression of transcription factors involved in inflammation and regeneration. Stat3 is one such example and its activation in axons upon injury could act as a retrograde signalling factor to promote regeneration of sensory and motor neurons [72]. As for chemokines, Cxcr4 for instance, was implicated with neuronal regeneration because it could regulate axonal path finding in the CNS [73]. Notably, Il6r, Stat3 and Cxcr4 were all transcriptionally up-regulated in our study. These examples highlight the direct impact cytokines and chemokines can have upon regenerating neurons.
Physiologically however, cytokines and chemokines released upon neuroinflammation often have an indirect effect on neurons whereby they help to recruit immune-response cells to injured site. Most of these cells when recruited excessively to injured site could result in neuronal apoptosis but during regeneration, they could also secrete bioactive forms of neurotrophic factors that would potentially aid in neurons’ axon regrowth [74-76]. Supplementation of neurite promoting factors by immune cells could possibly explain the transient need for neurons to express their own, such as Ntrk and Bdnf found in our in vitro injury model. Intriguingly, a study showed that oncomodulin secreted by inflammation-activated macrophages could promote axon regeneration in RGCs only upon elevated cAMP levels [77]. It was speculated that increased cAMP caused translocation of specific receptors to the cell surface membrane. Even though this was observed in RGCs, the general idea may still apply for neurons in which they are probably programmed to modulate cellular second messenger levels, such as cAMP, during injury to elicit actions. For example, relocation of specific receptors to the cell membrane in anticipation of binding to neurite-promoting factors released by surrounding immune-response cells.
Conclusion
It is significant that a number of genes listed in our DNA microarray study were in accordance with findings from previous in vitro and in vivo regenerative studies reported by other groups in relation to spinal cord and brain injuries. This provides affirmation that our protocol of severing axonal bundles in vitro to study injury-induced neurite regeneration is a reasonable and valid experimental model. From a transcriptomic view, we have revealed that neurite regeneration is a complex process that mainly involves restructuring of neurite cytoskeleton, determined by intricate actin and microtubule dynamics, protein trafficking and appropriate control of both guidance cues and neurotrophic factors. We conclude that the molecular response of neurons to axotomy is not a simple process and evokes two distinct pathways; a cell survival response accompanied by activation of genes to protect against oxidative stress, inflammation and cellular ion imbalance and a regenerative response driven by modulation of the neuronal cytoskeleton. Gene expression profiles from our study demonstrated that injured neurons have an innate capability to survive axotomy by elevating biological antioxidants and molecular chaperones, regulating secondary messengers, suppressing apoptotic genes, controlling calcium ion-associated processes and possibly by expressing certain cell cycle proteins which might serve unique alternate neuronal functions. Genes involved in these highlighted protective and regenerative mechanics were summarized in Figure 3. Transcriptional analysis is a powerful and effective screening method to globally search for comprehensive clues to determine which biological pathways are involved in the intrinsic response of neurons to axotomy. Appropriate manipulation of selected biological targets involved in these pathways could then potentially facilitate the design and development of novel, effective therapeutics for axonal damage in CNS injury.
Fig 3.
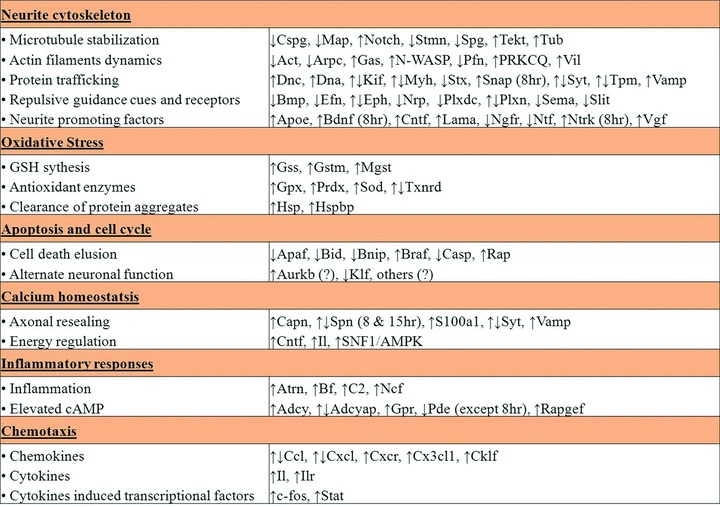
Summarized view on the regulation of selected genes (taken from gene profiles) that were involved in different major sub-processes associated with neurite regeneration after axonal injury in vitro as discussed.
Acknowledgments
This work was financially supported by Singapore Biomedical Research Council research grant R-184-000-093-305, Singapore National Medical Research Council research grant R-183-000-075-213, Strategic Initiative Funding (Menzies Research Institute), National Health and Medical Research Council (AUSTRALIA) research grant 544913, Australian Research Council research grants DP0984673 and DP0556630 and a research grant from the Motor Accident & Insurance Board of Tasmania.
Conflict of interest
There are no conflicts of interest.
References
- 1.David S, Aguayo AJ. Axonal regeneration after crush injury of rat central nervous system fibres innervating peripheral nerve grafts. J Neurocytol. 1985;14:1–12. doi: 10.1007/BF01150259. [DOI] [PubMed] [Google Scholar]
- 2.Sandvig A, Berry M, Barrett LB, et al. Myelin-, reactive glia-, and scar-derived CNS axon growth inhibitors: expression, receptor signaling, and correlation with axon regeneration. Glia. 2004;46:225–51. doi: 10.1002/glia.10315. [DOI] [PubMed] [Google Scholar]
- 3.Taylor AM, Blurton-Jones M, Rhee SW, et al. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat Methods. 2005;2:599–605. doi: 10.1038/nmeth777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taylor AM, Berchtold NC, Perreau VM, et al. Axonal mRNA in uninjured and regenerating cortical mammalian axons. J Neurosci. 2009;29:4697–707. doi: 10.1523/JNEUROSCI.6130-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dickson TC, Adlard PA, Vickers JC. Sequence of cellular changes following localized axotomy to cortical neurons in glia-free culture. J Neurotrauma. 2000;17:1095–103. doi: 10.1089/neu.2000.17.1095. [DOI] [PubMed] [Google Scholar]
- 6.Chuckowree JA, Vickers JC. Cytoskeletal and morphological alterations underlying axonal sprouting after localized transection of cortical neuron axons in vitro. J Neurosci. 2003;23:3715–25. doi: 10.1523/JNEUROSCI.23-09-03715.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sengottuvel V, Leibinger M, Pfreimer M, et al. Taxol facilitates axon regeneration in the mature CNS. J Neurosci. 2011;31:2688–99. doi: 10.1523/JNEUROSCI.4885-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Verma P, Chierzi S, Codd AM, et al. Axonal protein synthesis and degradation are necessary for efficient growth cone regeneration. J Neurosci. 2005;25:331–42. doi: 10.1523/JNEUROSCI.3073-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCurley AT, Callard GV. Time course analysis of gene expression patterns in zebrafish eye during optic nerve regeneration. J Exp Neurosci. 2010;2010:17–33. [PMC free article] [PubMed] [Google Scholar]
- 10.Veldman MB, Bemben MA, Thompson RC, et al. Gene expression analysis of zebrafish retinal ganglion cells during optic nerve regeneration identifies KLF6a and KLF7a as important regulators of axon regeneration. Dev Biol. 2007;312:596–612. doi: 10.1016/j.ydbio.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 11.Kuo LT, Tsai SY, Groves MJ, et al. Gene expression profile in rat dorsal root ganglion following sciatic nerve injury and systemic neurotrophin-3 administration. J Mol Neurosci. 2010;43:503–15. doi: 10.1007/s12031-010-9473-3. [DOI] [PubMed] [Google Scholar]
- 12.Vega-Avelaira D, Geranton SM, Fitzgerald M. Differential regulation of immune responses and macrophage/neuron interactions in the dorsal root ganglion in young and adult rats following nerve injury. Mol Pain. 2009;5:70. doi: 10.1186/1744-8069-5-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Giovanni S, Faden AI, Yakovlev A, et al. Neuronal plasticity after spinal cord injury: identification of a gene cluster driving neurite outgrowth. FASEB J. 2005;19:153–4. doi: 10.1096/fj.04-2694fje. [DOI] [PubMed] [Google Scholar]
- 14.Zai L, Ferrari C, Subbaiah S, et al. Inosine alters gene expression and axonal projections in neurons contralateral to a cortical infarct and improves skilled use of the impaired limb. J Neurosci. 2009;29:8187–97. doi: 10.1523/JNEUROSCI.0414-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chung RS, Vickers JC, Chuah MI, et al. Metallothionein-III inhibits initial neurite formation in developing neurons as well as postinjury, regenerative neurite sprouting. Exp Neurol. 2002;178:1–12. doi: 10.1006/exnr.2002.8017. [DOI] [PubMed] [Google Scholar]
- 16.Chung RS, Staal JA, McCormack GH, et al. Mild axonal stretch injury in vitro induces a progressive series of neurofilament alterations ultimately leading to delayed axotomy. J Neurotrauma. 2005;22:1081–91. doi: 10.1089/neu.2005.22.1081. [DOI] [PubMed] [Google Scholar]
- 17.Dennis G, Jr, Sherman BT, Hosack DA, et al. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:R60. [PubMed] [Google Scholar]
- 18.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 19.Frank CL, Tsai LH. Alternative functions of core cell cycle regulators in neuronal migration, neuronal maturation, and synaptic plasticity. Neuron. 2009;62:312–26. doi: 10.1016/j.neuron.2009.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hellal F, Hurtado A, Ruschel J, et al. Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Science. 2011;331:928–31. doi: 10.1126/science.1201148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferrari-Toninelli G, Bonini SA, Bettinsoli P, et al. Microtubule stabilizing effect of notch activation in primary cortical neurons. Neuroscience. 2008;154:946–52. doi: 10.1016/j.neuroscience.2008.04.025. [DOI] [PubMed] [Google Scholar]
- 22.Lowery LA, Van Vactor D. The trip of the tip: understanding the growth cone machinery. Nat Rev Mol Cell Biol. 2009;10:332–43. doi: 10.1038/nrm2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Korobova F, Svitkina T. Arp2/3 complex is important for filopodia formation, growth cone motility, and neuritogenesis in neuronal cells. Mol Biol Cell. 2008;19:1561–74. doi: 10.1091/mbc.E07-09-0964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim YS, Furman S, Sink H, et al. Calmodulin and profilin coregulate axon outgrowth in Drosophila. J Neurobiol. 2001;47:26–38. doi: 10.1002/neu.1013. [DOI] [PubMed] [Google Scholar]
- 25.Pinyol R, Haeckel A, Ritter A, et al. Regulation of N-WASP and the Arp2/3 complex by Abp1 controls neuronal morphology. PLoS One. 2007;2:e400. doi: 10.1371/journal.pone.0000400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.You JJ, Lin-Chao S. Gas7 functions with N-WASP to regulate the neurite outgrowth of hippocampal neurons. J Biol Chem. 2010;285:11652–66. doi: 10.1074/jbc.M109.051094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goley ED, Welch MD. The ARP2/3 complex: an actin nucleator comes of age. Nat Rev Mol Cell Biol. 2006;7:713–26. doi: 10.1038/nrm2026. [DOI] [PubMed] [Google Scholar]
- 28.Fawcett J, Pawson T. Signal transduction. N-WASP regulation-the sting in the tail. Science. 2000;290:725–6. doi: 10.1126/science.290.5492.725. [DOI] [PubMed] [Google Scholar]
- 29.Medeiros NA, Burnette DT, Forscher P. Myosin II functions in actin-bundle turnover in neuronal growth cones. Nat Cell Biol. 2006;8:215–26. doi: 10.1038/ncb1367. [DOI] [PubMed] [Google Scholar]
- 30.Lee JH, Gleeson JG. The role of primary cilia in neuronal function. Neurobiol Dis. 2010;38:167–72. doi: 10.1016/j.nbd.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Myers KA, Tint I, Nadar CV, et al. Antagonistic forces generated by cytoplasmic dynein and myosin-II during growth cone turning and axonal retraction. Traffic. 2006;7:1333–51. doi: 10.1111/j.1600-0854.2006.00476.x. [DOI] [PubMed] [Google Scholar]
- 32.Song XY, Li F, Zhang FH, et al. Peripherally-derived BDNF promotes regeneration of ascending sensory neurons after spinal cord injury. PLoS One. 2008;3:e1707. doi: 10.1371/journal.pone.0001707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harris NG, Mironova YA, Hovda DA, et al. Pericontusion axon sprouting is spatially and temporally consistent with a growth-permissive environment after traumatic brain injury. J Neuropathol Exp Neurol. 2010;69:139–54. doi: 10.1097/NEN.0b013e3181cb5bee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hollis ER II, Jamshidi P, Low K, et al. Induction of corticospinal regeneration by lentiviral trkB-induced Erk activation. Proc Natl Acad Sci U S A. 2009;106:7215–20. doi: 10.1073/pnas.0810624106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Montolio M, Messeguer J, Masip I, et al. A semaphorin 3A inhibitor blocks axonal chemorepulsion and enhances axon regeneration. Chem Biol. 2009;16:691–701. doi: 10.1016/j.chembiol.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 36.Goldshmit Y, McLenachan S, Turnley A. Roles of Eph receptors and ephrins in the normal and damaged adult CNS. Brain Res Rev. 2006;52:327–45. doi: 10.1016/j.brainresrev.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 37.Boyles JK, Zoellner CD, Anderson LJ, et al. A role for apolipoprotein E, apolipoprotein A-I, and low density lipoprotein receptors in cholesterol transport during regeneration and remyelination of the rat sciatic nerve. J Clin Invest. 1989;83:1015–31. doi: 10.1172/JCI113943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jang KJ, Kim MS, Feltrin D, et al. Two distinct filopodia populations at the growth cone allow to sense nanotopographical extracellular matrix cues to guide neurite outgrowth. PLoS One. 2010;5:e15966. doi: 10.1371/journal.pone.0015966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manthorpe M, Engvall E, Ruoslahti E, et al. Laminin promotes neuritic regeneration from cultured peripheral and central neurons. J Cell Biol. 1983;97:1882–90. doi: 10.1083/jcb.97.6.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dutta R, McDonough J, Chang A, et al. Activation of the ciliary neurotrophic factor (CNTF) signalling pathway in cortical neurons of multiple sclerosis patients. Brain. 2007;130:2566–76. doi: 10.1093/brain/awm206. [DOI] [PubMed] [Google Scholar]
- 41.Ye J, Cao L, Cui R, et al. The effects of ciliary neurotrophic factor on neurological function and glial activity following contusive spinal cord injury in the rats. Brain Res. 2004;997:30–9. doi: 10.1016/j.brainres.2003.10.036. [DOI] [PubMed] [Google Scholar]
- 42.Cui Q, Lu Q, So KF, et al. CNTF, not other trophic factors, promotes axonal regeneration of axotomized retinal ganglion cells in adult hamsters. Invest Ophthalmol Vis Sci. 1999;40:760–6. [PubMed] [Google Scholar]
- 43.Shuto T, Horie H, Hikawa N, et al. IL-6 up-regulates CNTF mRNA expression and enhances neurite regeneration. Neuroreport. 2001;12:1081–5. doi: 10.1097/00001756-200104170-00043. [DOI] [PubMed] [Google Scholar]
- 44.Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- 45.Bruey JM, Ducasse C, Bonniaud P, et al. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat Cell Biol. 2000;2:645–52. doi: 10.1038/35023595. [DOI] [PubMed] [Google Scholar]
- 46.Williams KL, Rahimtula M, Mearow KM. Hsp27 and axonal growth in adult sensory neurons in vitro. BMC Neurosci. 2005;6:24. doi: 10.1186/1471-2202-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Read DE, Gorman AM. Heat shock protein 27 in neuronal survival and neurite outgrowth. Biochem Biophys Res Commun. 2009;382:6–8. doi: 10.1016/j.bbrc.2009.02.114. [DOI] [PubMed] [Google Scholar]
- 48.Dubois-Dauphin M, Frankowski H, Tsujimoto Y, et al. Neonatal motoneurons overexpressing the bcl-2 protooncogene in transgenic mice are protected from axotomy-induced cell death. Proc Natl Acad Sci U S A. 1994;91:3309–13. doi: 10.1073/pnas.91.8.3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shibata M, Murray M, Tessler A, et al. Single injections of a DNA plasmid that contains the human Bcl-2 gene prevent loss and atrophy of distinct neuronal populations after spinal cord injury in adult rats. Neurorehabil Neural Repair. 2000;14:319–30. doi: 10.1177/154596830001400408. [DOI] [PubMed] [Google Scholar]
- 50.Kruman, Wersto RP, Cardozo-Pelaez F, et al. Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron. 2004;41:549–61. doi: 10.1016/s0896-6273(04)00017-0. [DOI] [PubMed] [Google Scholar]
- 51.Fuller BG, Lampson MA, Foley EA, et al. Midzone activation of aurora B in anaphase produces an intracellular phosphorylation gradient. Nature. 2008;453:1132–6. doi: 10.1038/nature06923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murata-Hori M, Wang YL. The kinase activity of aurora B is required for kinetochore-microtubule interactions during mitosis. Curr Biol. 2002;12:894–9. doi: 10.1016/s0960-9822(02)00848-5. [DOI] [PubMed] [Google Scholar]
- 53.Khazaei MR, Puschel AW. Phosphorylation of the par polarity complex protein Par3 at serine 962 is mediated by aurora a and regulates its function in neuronal polarity. J Biol Chem. 2009;284:33571–9. doi: 10.1074/jbc.M109.055897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamada M, Hirotsune S, Wynshaw-Boris A. The essential role of LIS1, NDEL1 and Aurora-A in polarity formation and microtubule organization during neurogensis. Cell Adh Migr. 2010;4:180–4. doi: 10.4161/cam.4.2.10715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rowland BD, Peeper DS. KLF4, p21 and context-dependent opposing forces in cancer. Nat Rev Cancer. 2006;6:11–23. doi: 10.1038/nrc1780. [DOI] [PubMed] [Google Scholar]
- 56.Moore DL, Blackmore MG, Hu Y, et al. KLF family members regulate intrinsic axon regeneration ability. Science. 2009;326:298–301. doi: 10.1126/science.1175737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hay JC. Calcium: a fundamental regulator of intracellular membrane fusion. EMBO Rep. 2007;8:236–40. doi: 10.1038/sj.embor.7400921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kelly M, Gauthier MS, Saha AK, et al. Activation of AMP-activated protein kinase by interleukin-6 in rat skeletal muscle: association with changes in cAMP, energy state, and endogenous fuel mobilization. Diabetes. 2009;58:1953–60. doi: 10.2337/db08-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Escartin C, Pierre K, Colin A, et al. Activation of astrocytes by CNTF induces metabolic plasticity and increases resistance to metabolic insults. J Neurosci. 2007;27:7094–104. doi: 10.1523/JNEUROSCI.0174-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dasgupta B, Milbrandt J. Resveratrol stimulates AMP kinase activity in neurons. Proc Natl Acad Sci U S A. 2007;104:7217–22. doi: 10.1073/pnas.0610068104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Spasic MR, Callaerts P, Norga KK. AMP-activated protein kinase (AMPK) molecular crossroad for metabolic control and survival of neurons. Neuroscientist. 2009;15:309–16. doi: 10.1177/1073858408327805. [DOI] [PubMed] [Google Scholar]
- 62.Williams T, Brenman JE. LKB1 and AMPK in cell polarity and division. Trends Cell Biol. 2008;18:193–8. doi: 10.1016/j.tcb.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 63.Zipfel PF, Lauer N, Skerka C. The role of complement in AMD. Adv Exp Med Biol. 2010;703:9–24. doi: 10.1007/978-1-4419-5635-4_2. [DOI] [PubMed] [Google Scholar]
- 64.Gupta-Bansal R, Parent JB, Brunden KR. Inhibition of complement alternative pathway function with anti-properdin monoclonal antibodies. Mol Immunol. 2000;37:191–201. doi: 10.1016/s0161-5890(00)00047-x. [DOI] [PubMed] [Google Scholar]
- 65.Hultqvist M, Sareila O, Vilhardt F, et al. Positioning of a polymorphic quantitative trait nucleotide in the Ncf1 gene controlling oxidative burst response and arthritis severity in rats. Antioxid Redox Signal. 2011;14:2373–83. doi: 10.1089/ars.2010.3440. [DOI] [PubMed] [Google Scholar]
- 66.Silveira MS, Linden R. Neuroprotection by cAMP: another brick in the wall. Adv Exp Med Biol. 2006;557:164–76. doi: 10.1007/0-387-30128-3_10. [DOI] [PubMed] [Google Scholar]
- 67.Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414:916–20. doi: 10.1038/414916a. [DOI] [PubMed] [Google Scholar]
- 68.Spencer T, Filbin MT. A role for cAMP in regeneration of the adult mammalian CNS. J Anat. 2004;204:49–55. doi: 10.1111/j.1469-7580.2004.00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Meyer-Franke A, Wilkinson GA, Kruttgen A, et al. Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron. 1998;21:681–93. doi: 10.1016/s0896-6273(00)80586-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cui Q, So KF. Involvement of cAMP in neuronal survival and axonal regeneration. Anat Sci Int. 2004;79:209–12. doi: 10.1111/j.1447-073x.2004.00089.x. [DOI] [PubMed] [Google Scholar]
- 71.Cafferty WB, Gardiner NJ, Das P, et al. Conditioning injury-induced spinal axon regeneration fails in interleukin-6 knock-out mice. J Neurosci. 2004;24:4432–43. doi: 10.1523/JNEUROSCI.2245-02.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee N, Neitzel KL, Devlin BK, et al. STAT3 phosphorylation in injured axons before sensory and motor neuron nuclei: potential role for STAT3 as a retrograde signaling transcription factor. J Comp Neurol. 2004;474:535–45. doi: 10.1002/cne.20140. [DOI] [PubMed] [Google Scholar]
- 73.Stumm R, Hollt V. CXC chemokine receptor 4 regulates neuronal migration and axonal pathfinding in the developing nervous system: implications for neuronal regeneration in the adult brain. J Mol Endocrinol. 2007;38:377–82. doi: 10.1677/JME-06-0032. [DOI] [PubMed] [Google Scholar]
- 74.Elkabes S, DiCicco-Bloom EM, Black IB. Brain microglia/macrophages express neurotrophins that selectively regulate microglial proliferation and function. J Neurosci. 1996;16:2508–21. doi: 10.1523/JNEUROSCI.16-08-02508.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kerschensteiner M, Gallmeier E, Behrens L, et al. Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: a neuroprotective role of inflammation. J Exp Med. 1999;189:865–70. doi: 10.1084/jem.189.5.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nakajima K, Honda S, Tohyama Y, et al. Neurotrophin secretion from cultured microglia. J Neurosci Res. 2001;65:322–31. doi: 10.1002/jnr.1157. [DOI] [PubMed] [Google Scholar]
- 77.Yin Y, Henzl MT, Lorber B, et al. Oncomodulin is a macrophage-derived signal for axon regeneration in retinal ganglion cells. Nat Neurosci. 2006;9:843–52. doi: 10.1038/nn1701. [DOI] [PubMed] [Google Scholar]