

Abstract
A prevalence of 1% in the general population and approximately 50% concordance rate in monozygotic twins was reported for schizophrenia, suggesting that genetic predisposition affecting neurodevelopmental processes might combine with environmental risk factors. A multitude of pathways seems to be involved in the aetiology and/or pathogenesis of schizophrenia, including dopaminergic, serotoninergic, muscarinic and glutamatergic signalling. The phosphoinositide signal transduction system and related phosphoinositide-specific phospholipase C (PI-PLC) enzymes seem to represent a point of convergence in these networking pathways during the development of selected brain regions. The existence of a susceptibility locus on the short arm of chromosome 20 moved us to analyse PLCB1, the gene codifying for PI-PLC β1 enzyme, which maps on 20p12. By using interphase fluorescent in situ hybridization methodology, we found deletions of PLCB1 in orbito-frontal cortex samples of schizophrenia-affected patients.
Keywords: PLCB1, schizophrenia, orbito-frontal cortex, PFC, genetics, signal transduction, I-FISH
Introduction
Schizophrenia (OMIM #181500) is a psychiatric disorder affecting cognitive functions such as attention, motivation, execution and emotion. In spite of many research efforts, the aetiopathogenesis remains largely unknown. The heterogeneous clinical expression reflects different etiological factors, such as altered expression of one or more genes, complications at birth, biochemical alterations including neurotransmitters imbalance, environmental factors and immunological abnormalities [1-3].
The complexity of the mechanisms underlying schizophrenia is witnessed by the involvement of a multitude of molecular pathways, including dopaminergic [4, 5], serotoninergic [6, 7], muscarinic [8-11] and glutamatergic signalling [12-14]. Remarkably, the phosphoinositide (PI) signalling, one of the major G-protein–linked signal transduction pathway operating in the human central nervous system, seems to be a point of convergence among the above pathways [15, 16]. Studies performed to assess the activity of the PI signal transduction system revealed that it is selectively impaired in specific brain regions of patients with neurological [17] and psychiatric disorders [18]. Further studies suggested that subtle alterations in the activity of the PI signal transduction system influence cognition, mood and behaviour associated with mental disorders, including schizophrenia [19]. Many findings corroborated this hypothesis. PI turnover in platelets from patients with schizophrenia was increased after thrombin stimulation, which activates platelet membrane Gqa [20]. PI hydrolysis was increased by a muscarinic acetylcholine receptor agonist, carbachol, but not by dopamine or serotonin [20]. By contrast, further studies reported increased PI hydrolysis by serotonin and a dopamine D1 agonist, SKF 38393 [21]. Dysregulation of the dopamine system in the prefrontal cortex (PFC) was also suspected to underlie the cognitive impairment observed in schizophrenia [22].
PI-PLC family of enzymes is crucial in PI signalling system, by regulating PI spatio-temporal balance. PI-PLC enzymes probably represent the bridge link existing between dopaminergic system and brain-derived neurotrophic factor (BDNF) levels [23]. The PLC-PKC-Src pathway was identified as the only required for surface expression and new synaptic insertion of N-methyl-D-aspartic (NMDA) receptors, which also seem to be involved in the pathogenesis of schizophrenia [24]. PI-PLC was also assumed to be a target of lithium, the first-line treatment for bipolar disorder [25]. Recently, D1 and D2 dopamine receptors were demonstrated to specifically activate the PI-PLC signalling and intracellular calcium release in the brain [26].
As a response to a wide panel of stimuli, PI-PLC cleaves the membrane phosphatydil-inositol bisphosphate (PIP2) into inositol trisphosphate (IP3) and diacylglycerol (DAG). IP3 diffuses rapidly into the cytoplasm inducing calcium release from the endoplasmic reticulum (ER) by binding to IP3-gated calcium-release channels [27, 28]. Calcium is a pleiotropic second messenger, mediator in many activities, both in cells and tissues. The increase in calcium levels induced by IP3 moves protein kinase C (PKC) to translocate from the cytoplasm to the plasma membrane, where it is activated to phosphorylate-specific serine or threonine residues on a number of target proteins [28]. DAG can be further cleaved to release arachidonic acid, which either acts as a messenger, is used in the synthesis of eicosanoids [27, 29] and activates a serine/threonine calcium-dependent PKC [30].
PI-PLC enzymes are classified on the basis of amino acid sequence, domain structure and mechanism of recruitment in response to activated receptors. Thirteen mammalian PI-PLC isoforms have been identified, divided into six sub-families: β(1–4), γ(1, 2), γ(1, 3, 4), (1), ζ(1) and η(1–2) [28]. The isoforms are strictly tissue specific, are codified by different genes and usually splicing variants exist [28]. Different expression of some isoforms was described in activated or pathological cells with respect to the corresponding quiescent counterparts [29, 31].
The PI-PLC β sub-family is regulated by the Gq family of GTP-binding proteins [32, 33]. With special regard to brain tissue, a modulation of PI-PLC β isoforms exists during the cortical development [28]. From post-natal stages onwards, PI-PLC β1 is abundant in selected areas of the brain such as cerebral cortex, hippocampus, amygdala, lateral septum and olfactory bulb [9, 34].
In adult mouse brain, Pi-plc β1 expression is evident in various grey matter regions, such as layers 2–6 of the cortex, pyramidal and granular layers of the hippocampus, mitral and granule layers of the olfactory bulb, amygdale, caudate, putamen and lateral septal nucleus [35]. The Pi-plc β1 up-regulation during critical periods of development marked by increased cortical plasticity, suggests that it might play a role in eliciting structural and functional adaptation [36, 37].
Levels of PI-PLC β1 protein were found increased in the PFC and decreased in the superior temporal gyrus of schizophrenic patients [38]. This observation was supported by the evidence that Pi-Plc β1 knockout mice (KO) show abnormal cortical development, synaptogenesis and dendritic spine maturation [39, 40]. In adulthood, Pi-Plc β1 KO mice exhibit behavioural abnormalities, such as impaired pre-pulse inhibition of acoustic startle and hyperactivity in response to a novel environment, phenotypes currently considered representative of schizophrenia features [41, 42].
The activity of pathways involving PI-PLC β1 is modulated by the regulator of G-protein signalling 4 (RGS4; OMIM *602516), which is considered a candidate gene for schizophrenia [49-51].
An approximately 50% concordance rate in monozygotic twins and 1% prevalence in the general population was reported for schizophrenia [1]. These and other findings suggested that schizophrenia arises from a genetic predisposition affecting neurodevelopmental processes, combined with exposure to environmental risk factors [1].
Many susceptibility loci for schizophrenia were identified. Significance levels for linkage to schizophrenia were obtained for chromosome regions 6p24-p22 [52], 1q21-q22 [43], 13q32-q34 [53], 10p14 [54], 10q25.3-q26.3 [55], 8p22-p21 [53, 56], 6q21-q25 [57, 58], 22q11-q12 [59, 60] and 5q21-q33 [61, 62]. A further susceptibility locus was identified on the short arm of chromosome 20 (20p) [63, 64]. In particular, a smaller region was identified spanning 20p13-p12.2 [64, 65].
PLCB1 (OMIM *607120), the gene codifying for PI-PLC β1 enzyme, is constituted from 36 small exons and introns, and was located on the short arm of human chromosome 20 (20p12, nearby markers D20S917 and D20S177) [66].
The existence of a linkage with the 20p13.2-p12 region and the observation that PI-PLC β1 enzyme is a mediator and a point of convergence in the neurotransmitter pathways involved in schizophrenia, moved us to analyse the gene PLCB1.
Materials and methods
We analysed brain samples, furnished by the Stanley Brain Research Institute (Bethesda, MD, USA), of 30 anonymous individuals, divided into two groups constituted respectively of 15 normal controls (intended as donors that did not present with neurological nor psychiatric diseases) and 15 schizophrenia-affected patients. Age and sex distribution in the two groups were comparable. Samples were formaldehyde fixed and paraffin embedded biopsies of orbito-frontal cortex. Paraffin inclusion time ranged from 3 to 20 months. Ten 15-μm-thick slices for each sample were furnished. Interphase fluorescent in situ hybridization (I-FISH) analyses were conducted on a blind study. Subtelomeric short arm and painting FISH analyses of chromosome 20 were further conducted in the patients in which deletion of PLCB1 was identified and in four normal controls.
Nuclei extraction
Slices were deparaffinized in xylene, dehydrated in ethanol series, rinsed in ddH2O, mechanically detached from support slides and incubated at room temperature in 0.5 mg/ml pepsin in 0.2M HCl for 30 min. The digestion was stopped by washing the nuclei twice in PBS/bovine serum albumin (BSA). The supernatant was passed through a 40-μm nylon mesh and centrifuged. Collected nuclei were washed several times with PBS and pelleted by centrifugation. Supernatant was removed and replaced with Carnoy II fixative. Aliquots of the suspension were put on new slides and allowed to dry at room temperature.
Molecular cytogenetics analyses
For I-FISH procedure, we used the specific probe for PLCB1 (PAC clone HS881E24 from P. de Jong RPCI-5 PAC library), 115.611 bp long, spanning from exon 19 to 32. The probe was biotin labelled by nick translation following manufacturer's indications (Invitrogen, Carlsbad, CA, USA). Nuclei underwent I-FISH procedure as previously described [66]. Briefly, slides were rinsed several times in PBS, and dehydrated in ethanol series. Biotinylated PLCB1 probe was added to each slide. Co-denaturation was performed at 72–80°C and then slides were incubated in humidified chamber overnight at 37°C. Slides were washed in 50% formamide/2χ saline sodium citrate (SSC) buffer and several times in 2χ SSC. The hybridized probe was detected with Cy3-conjugated streptavidin (Sigma-Aldrich, St. Louis, MO, USA). From 95 to 100 nuclei were processed for each sample.
Subtelomeric FISH was performed with a specific FITC-conjugated probe for the short arm of chromosome 20 (20pter; Cytocell, Celbio, Italy) following manufacturer's indications. For each patient, bearing PLCB1 deletion from 45 to 50 nuclei and for each normal control from 42 to 50 nuclei were processed.
Chromosome painting FISH was performed with a specific FITC-conjugated probe for chromosome 20 (Oncor/Resnova, Italy) accordingly to manufacturer's indications. For each patient, bearing PLCB1 deletion from 47 to 50 nuclei and for each normal control from 45 to 50 nuclei were processed.
In every experiment, nuclei were then counterstained with DAPI (Oncor/Resnova) and slides were observed with Nikon Eclipse E1000 fluorescence microscope (Nikon Instruments, Florence, Italy), imaged with Genikon analyser system (Nikon Instruments).
Results
No deletions of PLCB1 were detected in the group of 15 normal controls at I-FISH analysis (Fig. 1). The 20pter and the painting FISH analyses of chromosome 20 performed in four normal controls did not detect abnormalities. All FISH results were homogeneous for each individual in all the analysed nuclei.
Fig 1.
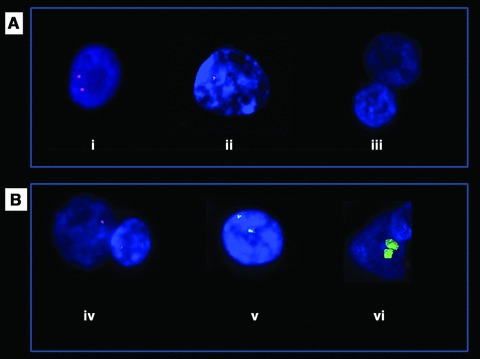
Molecular cytogenetic analyses of normal control and schizophrenia-affected patients (100χ). (A) I-FISH analysis with PLCB1 Cy3-conjugated probe: (A, i) normal control, (A, ii) schizophrenia-affected patient (#15) bearing mono-allelic deletion and (A, iii) schizophrenia-affected patient (#7) bearing bi-allelic deletion. (B) analyses in schizophrenia-affected patient (#10) bearing PLCB1 monoallelic deletion: (B, iv) mono-allelic deletion detected by I-FISH analysis with Cy3-conjugated PLCB1 probe, (B, v) Sub-telomeric FISH with 20pter FITC-conjugated probe and (B, vi) painting FISH with chromosome 20 FITC-conjugated probe.
Deletion of one allele of PLCB1 was detected in all the analysed nuclei of three out of 15 patients (two males and one female) affected with schizophrenia. Deletion of both alleles of PLCB1 was detected in the vast majority of nuclei of one patient affected with schizophrenia (patient 7#; Table 1; Fig. 1A, i and B, iv). In patient 7#, 100 nuclei were analysed; in 76 nuclei no PLCB1 signal was detected; one signal was detected in 24 nuclei.
Table 1.
Schizophrenia-affected patients: clinical data and results of I-FISH analysis performed with PLCB1 probe (right column)
| Patient no. | Gender | Age at onset | Survival | Age at death | Suicide | PLCβ1 (two alleles) | Nuclei |
|---|---|---|---|---|---|---|---|
| 1 | F | 22 | 8 | 30 | Yes | +/+ | 100/100 |
| 2 | M | 20 | 32 | 52 | No | +/+ | 95/95 |
| 3 | M | 13 | 17 | 30 | No | +/+ | 100/100 |
| 4 | F | 38 | 24 | 62 | No | +/+ | 95/95 |
| 5 | F | 15 | 45 | 60 | No | +/+ | 100/100 |
| 6 | M | 27 | 33 | 60 | No | +/+ | 100/100 |
| 7 | M | 27 | 5 | 32 | No | −/−; +/− | 76/100; 24/100 |
| 8 | M | 18 | 13 | 31 | Yes | +/− | 100/100 |
| 9 | F | 42 | 16 | 58 | No | +/+ | 100/100 |
| 10 | M | 20 | 5 | 25 | Yes | +/− | 98/98 |
| 11 | M | 17 | 27 | 44 | No | +/+ | 100/100 |
| 12 | M | 21 | 23 | 44 | No | +/+ | 100/100 |
| 13 | F | 24 | 32 | 56 | Yes | +/+ | 100/100 |
| 14 | M | 19 | 16 | 35 | No | +/+ | 100/100 |
| 15 | F | 25 | 24 | 49 | No | +/− | 100/100 |
Age at onset, survival and age at death must be intended in years. The last column indicates the number of analysed nuclei for patient. F: female patient; M: male patient; +: presence of PLCB1 allele signal; −: absence of PLCB1 allele signal.
The 20pter (Fig. 1B, v) and the painting FISH of chromosome 20 (Fig. 1B, vi), performed in the four patients bearing PLCB1 deletion, did not detect further abnormalities (Table 2). All the results were homogeneous for each patient in all the analysed nuclei.
Table 2.
Schizophrenia-affected patients bearing mono or bi-allelic deletion of PLCB1
| Patient# | Gender | PLCB1 I-FISH | 20pter | FISH | Chr. 20 painting | FISH | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| nuclei | nuclei | nuclei | ||||||||
| 7 | M | −/−; +/− | 76; 24 | +/+ | 50 | +/+ | 47 | |||
| 8 | M | +/− | 100 | +/+ | 48 | +/+ | 50 | |||
| 10 | M | +/− | 98 | +/+ | 50 | +/+ | 50 | |||
| 15 | F | +/− | 100 | +/+ | 45 | +/+ | 50 | |||
Comparison of the results of I-FISH, sub-telomeric 20pter FISH and chromosome 20 painting FISH analyses. F: female patient; M: male patient. I-FISH: +: presence of PLCB1 allele signal; −: absence of PLCB1 allele signal. Sub-telomeric 20pter FISH: ++: presence of both 20p telomere signals. Chromosome 20 painting FISH: +/+: presence of both chromosomes 20. Flanking columns indicate the number of analysed nuclei per patient.
Age at onset of the disease was comprised between 13 and 42 (mean 23.2) years in the total group, constituted of all 15 patients affected with schizophrenia, between 13 and 42 (mean 23.45) years in the patients with negative I-FISH analysis (with no PLCB1 deletion, as both the alleles were detected) and between 18 and 27 (mean 22.25) years in patients bearing the deletion of one or both PLCB1 alleles (Table 3).
Table 3.
Comparison of age at onset, age at death and survival in the analysed patients affected with schizophrenia and corresponding mean values (range in parentheses)
| Age at onset | Survival | Age at death | |
|---|---|---|---|
| Total group | 23.2 (13–42) | 21.33 (5–45) | 44.53 (25–62) |
| Patients with normal PLCB1 | 23.45 (13–42) | 24.9 (8–45) | 48.27 (30–62) |
| Patients bearing PLCB1 deletion | 22.25 (18–27) | 11.75 (5–24) | 34.25 (25–49) |
The survival rate, intended as the life time duration from diagnosis of schizophrenia to death, in the total group was comprised between 5 and 45 (mean 21.33) years, in the patients with no PLCB1 deletion was comprised between 8 and 45 (mean 24.9) years and in the patients bearing PLCB1 deletion between 5 and 24 (mean 11.75) years (Table 3).
The age at death in the total group was comprised between 25 and 62 (mean 44.53) years, in the patients with no PLCB1 deletion between 30 and 62 (mean 48.27) years, and in the patients bearing the deletion of PLCB1 between 25 and 49 (mean 34.25) years (Table 3).
Discussion
A large number of studies demonstrated that schizophrenia heritability is 0.70–0.85 and that risk in siblings of affected individuals is 10-fold increased [67]. These and other findings suggested that schizophrenia arises from a genetic predisposition affecting neurodevelopmental processes, combined with exposure to environmental risk factors [1]. This is not surprising, as the activity of the cerebral cortex, functionally altered in patients affected with schizophrenia, requires a strictly regulated genetic program for appropriate development [1, 68, 69]. The identification of candidate genes might open the way to improve the diagnosis, to evaluate and, in perspective, to limit the impact of environmental factors. It also might help to provide prognostic tools allowing to identify patients with different clinical outcome, and to personalize therapeutic approaches.
PI-PLC β1 is a rate-limiting enzyme involved in post-natal cortical development as well as in neuronal plasticity [36, 37] and regulates key G protein–coupled signalling pathways in the human cortex [18]. The involvement of PI-PLC β1 in schizophrenia was suggested, due to many evidence. PI-PLC β1 transduces intracellular signals from specific muscarinic, glutamate and serotonin receptors, all implicated in the pathogenesis of schizophrenia, therefore representing a point of convergence of their activities [70, 71].
A possible genetic association of PI-PLC β1 and schizophrenia was reported in linkage studies [63], and abnormal expression patterns in the brains of schizophrenia patients were reported [38, 71].
The outcome of disrupted PI-PLC β1 signalling was studied in Pi-plc β1 KO mice [38], which have abnormal cortical maturation including aberrant barrel formation in the somatosensory cortex3, abnormal synapse formation and dendritic spine dysmorphism [72]. Spatial memory deficits [73, 74] and behavioural abnormalities were also described [75], as well as deficit in working memory, altered fear conditioning, and motor/sensorimotor gating deficiencies, which all are considered features of relevance to schizophrenia [41, 73, 75]. Furthermore, during development a specific expression pattern for RGS4 was demonstrated, which resulted dramatically altered in the Pi-Plc β1 KO mice [75].
In this study, we analysed the orbito-frontal cortex of 30 patients, subdivided into two groups, comparable for age and sex distribution. Groups were respectively constituted by 15 donors, normal controls not presenting with neuropsychiatric disorders, and 15 schizophrenia-affected patients. No PLCB1 deletion was identified in the normal control group. Deletions of one or both alleles of PLCB1 were identified in four (three male and one female) out of 15 patients (26%) affected with schizophrenia. In the patients bearing the deletion of PLCB1, further analyses, such as 20pter and chromosome 20 painting FISH, did not detect other abnormalities, suggesting that the identified PLCB1 deletion was probably interstitial.
The observation that no PLCB1 deletions were found in normal controls, and was identified only in a limited number of schizophrenia-affected patients, suggests that the deletion of PLCB1 might be specifically associated to schizophrenia. Certainly, the number of individuals we analysed constitutes a small sample and further studies are required to enlarge the case record numbers. Moreover, the probe we used is 115.611 bp long. Therefore, this study identifies the presence of wide deletions of PLCB1, but not smaller abnormalities, such as single-point mutation or shorter deletions.
Interestingly, the disease outcome of patients bearing the deletion of PLCB1 significantly differed from the other schizophrenia patients we analysed. Differences were observed in the survival and in the death age, despite similar mean age at onset of schizophrenia.
Notwithstanding a wide range observed, the age at onset showed similar mean values in the total group, comprising all 15 patients affected with schizophrenia, in the patients bearing the deletion of one or both alleles of PLCB1, comprising four patients, and in the patients with no deletion, comprising the remaining 11 patients (Table 2). In fact, age at onset of the disease was comprised between 13 and 42 (mean 23.2) years in the total group, between 13 and 42 (mean 23.45) years in the patients with no deletion of PLCB1, and between 18 and 27 (mean 22.25) years in patients bearing the PLCB1 deletion (Table 3). The patients with the youngest age at onset (patient #3, #5 and #11) did not bear deletion of PLCB1 (Table 1).
The survival rate was significantly shorter in the patients bearing PLCB1 deletion (5–24; mean 11.75 years) with respect to the total group (5–45; mean 21.33 years) and to patients with no deletion (8–45; mean 24.9 years), suggesting that the deletion of PLCB1 might be related to a worse clinical outcome. Moreover, in patient #7 no alleles of PLCB1 were detected in 76% of the analysed nuclei (bi-allelic deletion; Fig. 1) and one allele was detected in the remaining 24% (Table 2), probably representing a high percentage mosaicism in the cortex tissue. The survival rate of this patient was the shortest (5 years) in the analysed group, suggesting that the bi-allelic deletion of PLCB1, although non-homogeneous, might be associated to a worse prognosis.
The age at death resulted significantly younger in the patients bearing the PLCB1 deletion (25–49; mean 34.25 years) with respect to the total group (25–62; mean 44.53 years) and to patients with no deletion (30–62; mean 48.27 years).
No remarkable differences were observed in the suicide incidence in the analysed group (Table 1).
Although no gross variations exist among the mean values of the onset age in our patients, nor in the incidence of suicide, differences exist both in the survival rate and in the age of death. Patients bearing the deletion of PLCB1 had a shorter survival (difference 13.15 years, reduced of about 47%) and died younger (difference 14 years, reduction of about 29%), although the age at onset was comparable (difference 1.25 years) with respect to patients that did not bear the same deletion. This finding suggests that the deletion of PLCB1 gene and the absence of PI-PLC β1 enzyme, which probably follows, might be associated to a worse clinical outcome and prognosis.
Moreover, remarkably, in a group of 15 patients affected with schizophrenia, I-FISH allowed us to identify a small sub-group whose mean survival was significantly shorter (difference 9.58 years, reduced of about 55%) than the mean value (21.33 years) obtained examining the raw clinical data. Moreover, in the sub-group identified by I-FISH, the age at death (34.25 years) was significantly younger (difference 10.28 years, reduced of about 24%) than the mean value (44.53 years) calculated using the raw clinical data of the total group (Table 2).
I-FISH analyses also allowed us to identify, within a group of 15 individuals affected with schizophrenia, two patients with the shortest survival time (patients #7 and #10), which both presented PLCB1 deletion (Table 1). Another patient (patient #8), also having short survival (eight years), did not bear PLCB1 deletion, although, due to the technical limitations of FISH methodology, it is not possible to exclude smaller abnormalities of the gene (Table 1).
However, I-FISH was performed on brain biopsies, so that findings might be different in tissues other than brain. Moreover, brain is not available to test, so that our results are far to offer a diagnostic and/or prognostic tool in alive patients, although they contribute to corroborate the hypothesis of an involvement of PLCB1 in the pathogenesis of schizophrenia.
Further studies are required to verify whether PLCB1 deletions are involved in the aetiology and/or pathogenesis of schizophrenia. Although significant progresses were achieved in studying the PI system and its alterations in human brain, many issues remain to be addressed with special regard to its relationship with schizophrenia. In particular, further studies are required both to refine the analysis of PLCB1 gene, the role of PI-PLC β1 protein and to extend the examination to other molecules involved in PI signalling.
Acknowledgments
The authors thank Professor Lucio Cocco, for providing the PLCB1 probe, and Mario De Meo, for technical assistance.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10:40–68. doi: 10.1038/sj.mp.4001558. [DOI] [PubMed] [Google Scholar]
- 2.Jarskog LF, Glantz LA, Gilmore JH, et al. Apoptotic mechanisms in the pathophysiology of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:846–58. doi: 10.1016/j.pnpbp.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 3.Pérez-Neri I, Ramìrez-Bermúdez J, Montes S, et al. Possible mechanisms of neurodegeneration in schizophrenia. Neurochem Res. 2006;31:1279–94. doi: 10.1007/s11064-006-9162-3. [DOI] [PubMed] [Google Scholar]
- 4.Seeman P, Lee T. Antipsychotic drugs: direct correlation between clinical potency and presynaptic action on dopamine neurons. Science. 1975;188:1217–9. doi: 10.1126/science.1145194. [DOI] [PubMed] [Google Scholar]
- 5.Creese I, Burt DR, Snyder SH. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science. 1976;192:481–3. doi: 10.1126/science.3854. [DOI] [PubMed] [Google Scholar]
- 6.Scarr E, Copolov DL, Dean B. A proposed pathological model in the hippocampus of subjects with schizophrenia. Clin Exp Pharmacol Physiol. 2001;28:70–3. doi: 10.1046/j.1440-1681.2001.03400.x. [DOI] [PubMed] [Google Scholar]
- 7.Lopez-Figueroa AL, Norton CS, Lopez-Figueroa MO, et al. Serotonin 5-HT1A, 5-HT1B, and 5-HT2A receptor mRNA expression in subjects with major depression, bipolar disorder, and schizophrenia. Biol Psychiatry. 2004;55:225–33. doi: 10.1016/j.biopsych.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 8.Bennett JP, Jr, Enna SJ, Bylund DB, et al. Neurotransmitter receptors in frontal cortex of schizophrenics. Arch Gen Psychiatry. 1979;36:927–34. doi: 10.1001/archpsyc.1979.01780090013001. [DOI] [PubMed] [Google Scholar]
- 9.Watanabe S, Nishikawa T, Takashima M, et al. Increased muscarinic cholinergic receptors in prefrontal cortices of medicated schizophrenics. Life Sci. 1983;33:2187–96. doi: 10.1016/0024-3205(83)90290-4. [DOI] [PubMed] [Google Scholar]
- 10.Dean B, Crook JM, Opeskin K, et al. The density of muscarinic M1 receptors is decreased in the caudateputamen of subjects with schizophrenia. Mol Psychiatry. 1996;1:54–8. [PubMed] [Google Scholar]
- 11.Raedler TJ, Bymaster FP, Tandon R, et al. Towards a muscarinic hypothesis of schizophrenia. Mol Psychiatry. 2006;12:232–46. doi: 10.1038/sj.mp.4001924. [DOI] [PubMed] [Google Scholar]
- 12.Noga JT, Hyde TM, Herman MM, et al. Glutamate receptors in the post-mortem striatum of schizophrenic, suicide, and control brains. Synapse. 1997;27:168–76. doi: 10.1002/(SICI)1098-2396(199711)27:3<168::AID-SYN2>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 13.Ohnuma T, Augood SJ, Arai H, et al. Expression of the human excitatory amino acid transporter 2 and metabotropic glutamate receptors 3 and 5 in the prefrontal cortex from normal individuals and patients with schizophrenia. Brain Res Mol Brain Res. 1998;56:207–17. doi: 10.1016/s0169-328x(98)00063-1. [DOI] [PubMed] [Google Scholar]
- 14.Nudmamud-Thanoi S, Reynolds GP. The NR1 subunit of the glutamate/NMDA receptor in the superior temporal cortex in schizophrenia and affective disorders. Neurosci Lett. 2004;372:173–7. doi: 10.1016/j.neulet.2004.09.035. [DOI] [PubMed] [Google Scholar]
- 15.Berridge MJ. Inositol trisphosphate and calcium signaling. Nature. 1993;361:315–25. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- 16.Fisher SK. Homologous and heterologous regulation of receptor-stimulated phosphoinositide hydrolysis. Eur J Pharmacol. 1995;288:231–50. doi: 10.1016/0922-4106(95)90035-7. [DOI] [PubMed] [Google Scholar]
- 17.Wallace MA, Claro E. Transmembrane signaling through phospholipase C in human cortical membranes. Neurochem Res. 1993;18:139–45. doi: 10.1007/BF01474676. [DOI] [PubMed] [Google Scholar]
- 18.Pacheco MA, Jope RS. Phosphoinositide signaling in human brain. Prog Neurobiol. 1996;50:255–73. doi: 10.1016/s0301-0082(96)00035-4. [DOI] [PubMed] [Google Scholar]
- 19.Jope RS, Song L, Li PP, et al. The phosphoinositide signal transduction system is impaired in bipolar affective disorder brain. J Neurochem. 1996;66:2402–19. doi: 10.1046/j.1471-4159.1996.66062402.x. [DOI] [PubMed] [Google Scholar]
- 20.Kaiya H, Nishida A, Imai A, et al. Accumulation of diacyl-glycerol in platelet phosphoinositide turnover in schizophrenia: a biological marker of good prognosis? Biol Psychiatry. 1989;26:669–76. doi: 10.1016/0006-3223(89)90101-7. [DOI] [PubMed] [Google Scholar]
- 21.Jope RS, Song L, Grimes CA, et al. Selective increases in phosphoinositide signaling activity and G protein levels in postmortem brain from subjects with schizophrenia or alcohol dependence. J Neurochem. 1998;70:763–71. doi: 10.1046/j.1471-4159.1998.70020763.x. [DOI] [PubMed] [Google Scholar]
- 22.Zhang F, St Clair D, Liu X, et al. Association analysis of the RGS4 gene in Han Chinese and Scottish populations with schizophrenia. Genes Brain Behav. 2005;4:444–8. doi: 10.1111/j.1601-183X.2005.00167.x. [DOI] [PubMed] [Google Scholar]
- 23.Hasbi A, Fan T, Alijaniaram M, et al. Calcium signalling cascade links dopamine D1-D2 receptor heteromer to striatal BDNF roduction and neuronal growth. Proc Natl Acad Sci USA. 2009;106:21377–82. doi: 10.1073/pnas.0903676106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li YC, Liu G, Hu JL, et al. Dopamine D1 receptor-mediated enhancement of NMDA receptor trafficking requires rapid PKC-dependent synaptic insertion in the prefrontal neurons. J Neurochem. 2010;114:62–73. doi: 10.1111/j.1471-4159.2010.06720.x. [DOI] [PubMed] [Google Scholar]
- 25.Alda M. Pharmacogenetics of lithium response in bipolar disorder. J Psychiatry Neurosci. 1999;4:154–8. [PMC free article] [PubMed] [Google Scholar]
- 26.Rashid AJ, So CH, Kong MM, et al. D1-D2 dopamine receptor heterooligomers with unique pharmacology are coupled to rapid activation of Gq/11 in the striatum. Proc Natl Acad Sci USA. 2007;104:654–9. doi: 10.1073/pnas.0604049104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suh PG, Park J, Manzoli L, et al. Multiple roles of phosphoinositide-specific phospholipase C isozymes. BMB Rep. 2008;41:415–34. doi: 10.5483/bmbrep.2008.41.6.415. [DOI] [PubMed] [Google Scholar]
- 28.Hisatsune C, Nakamura K, Kuroda Y, et al. Amplification of Ca2+ signalling by diacylglycerol-mediated inositol 1,4,5-trisphosphate production. J Biol Chem. 2005;280:11723–30. doi: 10.1074/jbc.M409535200. [DOI] [PubMed] [Google Scholar]
- 29.Lo Vasco VR, Fabrizi C, Fumagalli L, et al. Expression of phosphoinositide specific phospholipase C isoenzymes in cultured astrocytes activated after stimulation with Lipopolysaccharide. J Cell Biochem. 2010;109:1006–12. doi: 10.1002/jcb.22480. [DOI] [PubMed] [Google Scholar]
- 30.Noh DY, Shin SH, Rhee SG. Phosphoinositide-specific phospholipase C and mitogenic signalling. Biochim Biophys Acta. 1995;1242:99–114. doi: 10.1016/0304-419x(95)00006-0. [DOI] [PubMed] [Google Scholar]
- 31.Lo Vasco VR, Fabrizi C, Artico M, et al. Expression of phosphoinositide-specific phospholipase C isoenzymes in cultured astrocytes. J Cell Biochem. 2007;100:952–9. doi: 10.1002/jcb.21048. [DOI] [PubMed] [Google Scholar]
- 32.Hepler JR, Kozasa T, Smrcka AV, et al. Purification from Sf9 cells and characterization of recombinant Gqa and G11a. J Biol Chem. 1993;268:14367–75. [PubMed] [Google Scholar]
- 33.Jope RS, Song L, Powers R. [3H]PtdIns hydrolysis in postmortem human brain membranes is mediated by the G-protein Gq/11 and phospholipase C-b. Biochem J. 1994;304:655–9. doi: 10.1042/bj3040655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takenawa T, Homma Y, Emori Y. Properties of phospholipase C isozymes. Methods Enzymol. 1991;197:511–8. doi: 10.1016/0076-6879(91)97177-z. [DOI] [PubMed] [Google Scholar]
- 35.Watanabe M, Nakamura M, Sato K, et al. Patterns of expression for the mRNA corresponding to the four isoforms of phospholipase C beta in mouse brain. Eur J Neurosci. 1998;10:2016–25. doi: 10.1046/j.1460-9568.1998.00213.x. [DOI] [PubMed] [Google Scholar]
- 36.Kind PC, Kelly GM, Fryer HJ, et al. Phospholipase C-beta1 is present in the botrysome, an intermediate compartment-like organelle, and is regulated by visual experience in cat visual cortex. J Neurosci. 1997;17:1471–80. doi: 10.1523/JNEUROSCI.17-04-01471.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hannan AJ, Kind PC, Blakemore C. Phospholipase C-beta1 expression correlates with neuronal differentiation and synaptic plasticity in rat somatosensory cortex. Neuropharmacology. 1998;37:593–605. doi: 10.1016/s0028-3908(98)00056-2. [DOI] [PubMed] [Google Scholar]
- 38.Lin XH, Kitamura N, Hashimoto T, et al. Opposite changes in phosphoinositide-specific phospholipase C immunoreactivity in the left prefrontal and superior temporal cortex of patients with chronic schizophrenia. Biol Psychiatry. 1999;46:1665–71. doi: 10.1016/s0006-3223(99)00036-0. [DOI] [PubMed] [Google Scholar]
- 39.Hannan AJ, Blakemore C, Katsnelson A, et al. PLC-beta1, activated via mGluRs, mediates activity-dependent differentiation in cerebral cortex. Nat Neurosci. 2001;4:282–8. doi: 10.1038/85132. [DOI] [PubMed] [Google Scholar]
- 40.Spires TL, Molnar Z, Kind PC, et al. Activity-dependent regulation of synapse and dendritic spine morphology in developing barrel cortex requires phospholipase C-beta1 signalling. Cereb Cortex. 2005;15:385–93. doi: 10.1093/cercor/bhh141. [DOI] [PubMed] [Google Scholar]
- 41.McOmish CE, Hannan AJ. Enviromimetics: exploring gene environment interactions to identify therapeutic targets for brain disorders. Expert Opin Ther Targets. 2007;11:899–913. doi: 10.1517/14728222.11.7.899. [DOI] [PubMed] [Google Scholar]
- 42.van den Buuse M, Garner B, et al. Importance of animal models in schizophrenia research. Aust N Z J Psychiatry. 2005;39:550–7. doi: 10.1080/j.1440-1614.2005.01626.x. [DOI] [PubMed] [Google Scholar]
- 43.Brzustowicz LM, Hodgkinson KA, Chow EW, et al. Location of a major susceptibility locus for familial schizophrenia on chromosome 1q21-q22. Science. 2000;288:678–82. doi: 10.1126/science.288.5466.678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mirnics K, Middleton FA, Stanwood GD, et al. Disease-specific changes in regulator of G-protein signaling 4 (RGS4) expression in schizophrenia. Mol Psychiatry. 2001;6:293–301. doi: 10.1038/sj.mp.4000866. [DOI] [PubMed] [Google Scholar]
- 45.Chowdari KV, Mirnics K, Semwal P, et al. Association and linkage analyses of RGS4 polymorphisms in schizophrenia. Hum Mol Genet. 2002;11:1373–80. doi: 10.1093/hmg/11.12.1373. [DOI] [PubMed] [Google Scholar]
- 46.O'Donovan MC, Williams NM, Owen MJ. Recent advances in the genetics of schizophrenia. Hum Mol Genet. 2003;2:R125–33. doi: 10.1093/hmg/ddg302. [DOI] [PubMed] [Google Scholar]
- 47.Traynor JR, Neubig RR. Regulators of G protein signaling & drugs of abuse. Mol Interv. 2005;5:30–41. doi: 10.1124/mi.5.1.7. [DOI] [PubMed] [Google Scholar]
- 48.Gold SJ, Ni YG, Dohlman HG, et al. Regulators of G protein signaling (RGS) proteins: region-specific expression of nine subtypes in rat brain. J Neurosci. 1997;17:8024–37. doi: 10.1523/JNEUROSCI.17-20-08024.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nomoto S, Adachi K, Yang LX, et al. Distribution of RGS4 mRNA in mouse brain shown by in situ hybridization. Biochem Biophys Res Commun. 1997;241:281–7. doi: 10.1006/bbrc.1997.7802. [DOI] [PubMed] [Google Scholar]
- 50.Druey KM, Sullivan BM, Brown D, et al. Expression of GTPase-deficient Gialpha2 results in translocation of cytoplasmic RGS4 to the plasma membrane. J Biol Chem. 1998;273:18405–10. doi: 10.1074/jbc.273.29.18405. [DOI] [PubMed] [Google Scholar]
- 51.Dowal L, Elliott J, Popov S, et al. Determination of the contact energies between a regulator of G protein signaling and G protein subunits and phospholipase C beta 1. Biochemistry. 2001;40:414–21. doi: 10.1021/bi001923+. [DOI] [PubMed] [Google Scholar]
- 52.Straub RE, MacLean CJ, O'Neill FA, et al. A potential vulnerability locus for schizophrenia on chromosome 6p24-22: evidence for genetic heterogeneity. Nat Genet. 1995;11:287–93. doi: 10.1038/ng1195-287. [DOI] [PubMed] [Google Scholar]
- 53.Blouin JL, Dombroski BA, Nath SK, et al. Schizophrenia susceptibility loci on chromosomes 13q32 and 8p21. Nat Genet. 1998;20:70–3. doi: 10.1038/1734. [DOI] [PubMed] [Google Scholar]
- 54.DeLisi LE, Shaw SH, Crow TJ, et al. A genome-wide scan for linkage to chromosomal regions in 382 sibling pairs with schizophrenia or schizoaffective disorder. Am J Psychiatry. 2002;159:803–12. doi: 10.1176/appi.ajp.159.5.803. [DOI] [PubMed] [Google Scholar]
- 55.Williams NM, Rees MI, Holmans P, et al. A two-stage genome scan for schizophrenia susceptibility genes in 196 affected sibling pairs. Hum Mol Genet. 1999;8:1729–39. doi: 10.1093/hmg/8.9.1729. [DOI] [PubMed] [Google Scholar]
- 56.Kendler KS, MacLean CJ, O'Neill FA, et al. Evidence for a schizophrenia vulnerability locus on chromosome 8p in the Irish Study of high-density schizophrenia families. Am J Psychiatry. 1996;153:1534–40. doi: 10.1176/ajp.153.12.1534. [DOI] [PubMed] [Google Scholar]
- 57.Cao Q, Martinez M, Zhang J, et al. Suggestive evidence for a schizophrenia susceptibility locus on chromosome 6q and a confirmation in an independent series of pedigrees. Genomics. 1997;43:1–8. doi: 10.1006/geno.1997.4815. [DOI] [PubMed] [Google Scholar]
- 58.Lindholm E, Ekholm B, Shaw S, et al. A schizophrenia-susceptibility locus at 6q25, in one of the world's largest reported pedigrees. Am J Hum Genet. 2001;69:96–105. doi: 10.1086/321288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pulver AE, Karayiorgou M, Wolyniec PS, et al. Sequential strategy to identify a susceptibility gene for schizophrenia: report of potential linkage on chromosome 22q12-q13.1: Part 1. Am J Med Genet. 1994;54:36–43. doi: 10.1002/ajmg.1320540108. [DOI] [PubMed] [Google Scholar]
- 60.Schizophrenia Linkage Collaborative Group for Chromosomes 3, 6 and 8. Additional support for schizophrenia linkage on chromosomes 6 and 8: a multicenter study. Am J Med Genet. 1996;67:580–94. doi: 10.1002/(SICI)1096-8628(19961122)67:6<580::AID-AJMG11>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 61.Bassett AS. Chromosomal aberrations and schizophrenia. Autosomes. Br J Psychiatry. 1992;161:323–34. doi: 10.1192/bjp.161.3.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Paunio T, Ekelund J, Varilo T, et al. Genome-wide scan in a nationwide study sample of schizophrenia families in Finland reveals susceptibility loci on chromosomes 2q and 5q. Hum Mol Genet. 2001;10:3037–48. doi: 10.1093/hmg/10.26.3037. [DOI] [PubMed] [Google Scholar]
- 63.Arinami T, Ohtsuki T, Ishiguro H, et al. Japanese Schizophrenia Sib-Pair Linkage Group. Genomewide high-density SNP linkage analysis of 236 Japanese families supports the existence of schizophrenia susceptibility loci on chromosomes 1p, 14q, and 20p. Am J Hum Genet. 2005;77:937–44. doi: 10.1086/498122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fanous AH, Neale MC, Webb BT, et al. Novel linkage to chromosome 20p using latent classes of psychotic illness in 270 Irish high-density families. Biol Psychiatry. 2008;64:121–7. doi: 10.1016/j.biopsych.2007.11.023. [DOI] [PubMed] [Google Scholar]
- 65.Hovatta I, Lichtermann D, Juvonen H, et al. Linkage analysis of putative schizophrenia gene candidate regions on chromosomes 3p, 5q, 6p, 8p, 20p and 22q in a population-based sampled Finnish family set. Mol Psychiatry. 1998;3:452–7. doi: 10.1038/sj.mp.4000443. [DOI] [PubMed] [Google Scholar]
- 66.Peruzzi D, Calabrese G, Faenza I, et al. Identification and chromosomal localisation by fluorescence in situ hybridisation of human gene of phosphoinositide-specific phospholipase Cb1. Biochim Biophys Acta. 2000;484:175–82. doi: 10.1016/s1388-1981(00)00012-3. [DOI] [PubMed] [Google Scholar]
- 67.Levinson DF, Mowry BJ, Escamilla MA, et al. The lifetime dimensions of psychosis scale (LDPS): description and interrater reliability. Schizophr Bull. 2002;28:683–95. doi: 10.1093/oxfordjournals.schbul.a006972. [DOI] [PubMed] [Google Scholar]
- 68.Harrison PJ. The neuropathology of schizophrenia. A critical review of the data and their interpretation. Brain. 1999;122:593–624. doi: 10.1093/brain/122.4.593. [DOI] [PubMed] [Google Scholar]
- 69.Lewis DA, Moghaddam B. Cognitive dysfunction in schizophrenia: convergence of gamma-aminobutyric acid and glutamate alterations. Arch Neurol. 2006;63:1372–6. doi: 10.1001/archneur.63.10.1372. [DOI] [PubMed] [Google Scholar]
- 70.Bunney TD, Katan M. PLC regulation: emerging pictures for molecular mechanisms. Trends Biochem Sci. 2011;36:88–96. doi: 10.1016/j.tibs.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 71.Shirakawa O, Kitamura N, Lin XH, et al. Abnormal neurochemical asymmetry in the temporal lobe of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2001;25:867–77. doi: 10.1016/s0278-5846(01)00149-x. [DOI] [PubMed] [Google Scholar]
- 72.Spires TL, Molnar Z, Kind PC, et al. Activity-dependent regulation of synapse and dendritic spine morphology in developing barrel cortex requires phospholipase C-beta1 signalling. Cereb Cortex. 2005;15:385–93. doi: 10.1093/cercor/bhh141. [DOI] [PubMed] [Google Scholar]
- 73.Bohm D, Schwegler H, Kotthaus L, et al. Disruption of PLC-beta 1-mediated signal transduction in mutant mice causes age-dependent hippocampal mossy fiber sprouting and neurodegeneration. Mol Cell Neurosci. 2002;21:584–601. doi: 10.1006/mcne.2002.1199. [DOI] [PubMed] [Google Scholar]
- 74.Shin J, Kim D, Bianchi R, et al. Genetic dissection of theta rhythm heterogeneity in mice. Proc Natl Acad Sci USA. 2005;102:18165–70. doi: 10.1073/pnas.0505498102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McOmish CE, Burrows EL, Howard M, et al. PLC-b1 knockout mice as a model of disrupted cortical development and plasticity: behavioral endophenotypes and dysregulation of RGS4 gene expression. Hippocampus. 2008;18:824–34. doi: 10.1002/hipo.20443. [DOI] [PubMed] [Google Scholar]