

Abstract
Nuclear factor κB (NF-κB) is an inducible transcription factor that tightly regulates the expression of a large cohort of genes. As a key component of the cellular machinery NF-κB is involved in a wide range of biological processes including innate and adaptive immunity, inflammation, cellular stress responses, cell adhesion, apoptosis and proliferation. Appropriate regulation of NF-κB is critical for the proper function and survival of the cell. Aberrant NF-κB activity has now been implicated in the pathogenesis of several diseases ranging from inflammatory bowel disease to autoimmune conditions such as rheumatoid arthritis. Systems governing NF-κB activity are complex and there is an increased understanding of the importance of nuclear events in regulating NF-κB's activities as a transcription factor. A number of novel nuclear regulators of NF-κB such as IκB-ζ and PDZ and LIM domain 2 (PDLIM2) have now been identified, adding another layer to the mechanics of NF-κB regulation. Further insight into the functions of these molecules raises the prospect for better understanding and rational design of therapeutics for several important diseases.
Keywords: NF-κB, inflammation, nuclear regulation, immunity, cancer
Introduction
Nuclear factor κB (NF-κB) is an important transcription factor typically activated by pro-inflammatory cytokines and other specific stimuli and is involved in the regulation of a variety of biological responses. It was initially identified as a protein that binds to a specific DNA sequence within the intronic enhancer of the immunoglobulin κ light chain in mature B and plasma cells [1]. It has subsequently been shown to be a ubiquitously expressed transcription factor that plays a critical role in the regulation of inflammatory, apoptotic and immune processes. It achieves this by regulating the expression of proteins such as cytokines, chemokines, adhesion molecules and the cellular death cascade [2–4]. The critical role played by NF-κB is demonstrated by the fact that it has been implicated in the pathogenesis of many diseases with an immune or inflammatory component/mechanism, including H. pylori infection, α1 antitrypsin deficiency, acute respiratory distress syndrome, glomerulonephritis, septic shock and chronic diseases such as rheumatoid arthritis, inflammatory bowel disease, asthma, atherosclerosis and tumourigenesis [5–10].
NF-κB is constructed of homo- or heterodimers of a family of related proteins that share a common Rel homology domain. The family is made up of five proteins: p65, c-Rel, Rel B, p50/p105 and p52/p100. Both p105 and p100 are synthesized as precursor proteins that are processed to the smaller active forms of p50 and p52. The p65/p50 heterodimer is the most abundant form of the NF-κB protein followed by the p50/p50 and p65/p65 homodimer complexes [2, 11–13].
Activation of NF-κB
Activation of NF-κB is a complex process that is governed mainly by members of IκB kinase (IKK) complex that are upstream of another set of regulator proteins called Inhibitor of κB (IκBs). IκB proteins belong to a structurally and functionally distinct family whose members include IκB-α, IκB-β, IκB-ɛ, IκB-ζ, Bcl-3, p100 and p105, all important regulators of NF-κB [14]. In an unstimulated cell NF-κB is sequestered in a complex with IκB-α, IκB-β and IκB-ɛ. Activation and nuclear translocation of NF-κB in general requires the degradation of bound IκBs, leading to the unmasking of the nuclear localization domain of NF-κB. Degradation of IκB-α necessitates its phosphorylation by IKK-β with subsequent processing by the ubiquitin-proteasomal system. This releases NF-κB allowing it to translocate to the nucleus where it regulates gene expression [2].
Various different stimuli, including oxidative stress, inflammatory cytokines, radiation, viruses and bacteria activate NF-κB [15]. It has been shown that signal pathways from these various stimuli converge mainly upon the IKK complex. This complex consists of three proteins, two of which, IKK-α and IKK-β, are catalytic in activity while the third, IKK-γ/NF-κB essential modulator (NEMO) has a regulatory role serving as an adapter protein, connecting both the catalytic subunits with the upstream activators. In the well-characterized canonical pathway, the IKK-β and IKK-γ/NEMO containing complex phosphorylates two critical serine residues in IκB-α (Ser 32 and Ser 36) allowing targeting of the IκB-α for ubiquitination and degradation [15, 16]. However recent studies have also shown the existence of IKK independent activation of NF-κB. This alternative pathway involves selective activation of the [p100/p52]/RelB heterodimer complex, triggered by members of the TNF-α family and chemokines which result in IKK-α dimer formation. The importance of both of these pathways has been demonstrated by their ability to alter gene expression and end stage functional outcome [17, 18].
More recently it has become evident that basal NF-κB activity also plays a critical role in various physiological and pathophysiological conditions. For example, basal NF-κB activity has been shown to be required for the accumulation of HIF-1α, a transcription factor that mediates the response to hypoxic conditions. Absence of IKK-β-mediated NF-κB activity results in defective induction of HIF-1 α and its regulated genes including VEGF [19, 20], thereby providing a critical link between hypoxic and inflammatory responses. Therefore, while much progress has been made in understanding the regulation of induced NF-κB, the importance of basal NF-κB activity is only beginning to be elucidated.
Regulators of NF-κB activation
NF-κB is regulated at multiple levels. This includes the regulated synthesis of individual subunits of NF-κB coupled with subsequent post-translational modifications (e.g. phosphorylation and acetylation). The principle regulators of NF-κB, the IκB proteins, share a common domain essential for their function, called ankyrin repeats. Most members of the IκB family have six to seven ankyrin repeats (each being 33 amino acids in length). However, ankyrin repeat domains are widely found, in proteins as diverse as Cdk inhibitors, signal transduction and transcriptional regulator proteins (STATs), cytoskeletal organizers and developmental regulators. These motifs are known to facilitate protein-protein interactions but have no known enzymatic activity [21]. Another common characteristic of IκB proteins is the presence of the PEST peptide sequence. The PEST sequence (which is a region rich in the amino acids proline (P); glutamic acid (E); serine (S); or thre-onine (T)) present in members of the IκB family targets them for degradation via the 26 S proteasomal pathways although in the case of IκB-α, it has been reported that the PEST sequence is not essential for this process [22–24].
In an unstimulated cell, INF-κB is largely localized in the cytoplasm, in a complex with the IκBs. IκB-α plays a pivotal role in the regulation of NF-κB activity. This is illustrated by the observation that mice deficient in IκB-α have consititutive NF-κB activation. After phosphorylation, IκB-α is degraded, leading to the release of NF-κB permitting its translocation to the nucleus and its binding to target gene promoters. In the nucleus, NF-κB binds to the promoters of target genes - including IκB-α, leading to increased IκB-α protein expression. Newly synthesized IκB-α enters the nucleus and promotes the detachment of NF-κB from the DNA, causing it to be exported back to the cytoplasm. This feedback cycle is tightly regulated and is essential for terminating the action of NF-κB that would otherwise lead to a sustained inflammatory response.
Similarly to IκB-α, the other best-characterized inhibitor of NF-κB, IκB-β, predominantly associates with the p65/p50 and p50/cRel heterodimers. Unlike IκB-α however, IκB-β is only activated by a limited range of stimuli such as LPS, IL-1 or HTLV-1 Tax protein. It has been shown that IκB-β invokes a persistent state of NF-κB activation [25]. Moreover, it has been established that binding of NF-κB to the free form of IκB-β prevents it from binding to IκB-α thus triggering persistent activation of NF-κB [26].
Another cytoplasmic inhibitor of NF-κB, IκB-ɛ, was first identified by yeast two-hybrid screening [27]. IκB-ɛ is mainly expressed in T cells in the thymus, spleen, and lymph nodes. Importantly, enhanced up-regulation of IκB-α and IκB-β has been demonstrated in IκB-ɛ knockout mice, suggesting that IκB members may functionally compensate for each other [28].
More recently, p100, the precursor of the p52 subunit, was reported to be a bonafide fourth member of the IκB regulator family [29]. The authors identified that limited amounts of cytoplasmic p50/p65 was bound directly to p100 and that stimulation of the cells with LTβR led to an increase in NF-κB activity in a p100 dependent but IκB independent manner. Interestingly it has also been reported that p100 itself is an NF-κB target gene [29]. There are several reviews that provide extensive information about the function and mechanism of action of these classical regulators of NF-κB [4, 12].
Although there has been a remarkable advance in the understanding of NF-κB and its regulation, much remains to be elucidated regarding the exact mechanism of how it regulates the expression of so many genes, how this matrix of gene expression is dependent on tissue identity and temporal conditions, and crucially, the sequence of events leading to the termination of its activity. Furthermore, the mechanisms by which NF-κB is regulated in the nucleus are poorly described and only recently being explored in depth.
Nuclear regulation of NF-κB
While IκB-α, IκB-β and IκB-ɛ are present in the cytoplasm, recent studies reveal that both IκB-αv and IκB-β, are also shuttled into the nucleus where they actively participate in the regulation of NF-κB. As described before, once in the nucleus, IκB-α binds with p65 and promotes its export back to the cytoplasm causing cessation of NF-κB activity and replenishment of the cytoplasmic IκB-α/p65 stockpile. Although the exact role of IκB-β in the nucleus is still unknown, there is evidence to suggest that it can bind to DNA-bound NF-κB [26]. Tergaonker et al., based on their elaborate experiments, reported that the three main regulators of NF-κB, namely IκB-α, IκB-β and IκB-ɛ, inhibit NF-κB in a redundant manner and that in cells lacking these proteins, NF-κB is still partially active [30]. Furthermore, nuclear roles for both IKK-α and IKK-γ/NEMO have been recently described. A novel function for IKK-α was demonstrated wherein it was shown to modulate the expression of NF-κB responsive genes by interacting with CBP ([C-Amp responsive element] CREB binding protein) and p65 bound to the promoter sequence of the target gene [31]. Interestingly IKK-α was also shown to cause phosphorylation of Histone H3 and increased transcription factor activity [31, 32]. Another member of the IKK complex, IKK-γ/NEMO has also been shown to shuttle between cytoplasm and nucleus [33]. Surprisingly NEMO does not possess the classical nuclear localization signal (NLS) suggesting that other domains/co-trans-porters might be involved in its translocation. IKK-γ/NEMO too binds to CBP and it has been suggested that it might influence histone deacetylase (HDAC) activity leading to decreased NF-κB activity. Thus while both IKK-α and IKK-γ/NEMO have nuclear roles, no such activity has been reported for IKK-β.
A number of nuclear specific regulators of NF-κB have also been described, including B-cell lymphoma 3 (Bcl-3), IκB-ζ, ubiquitously expressed transcript (UXT) and ZAS3 [34–36]. Identification of these nuclear localized, specific inhibitors of NF-κB is intriguing and may enable a greater understanding of how NF-κB activity is modulated in the nucleus. It has also been established that post-translational modification of NF-κB within the nucleus by processes such as phosphorylation, acetylation and deacetylation are important for the regulation of its activity (Table 1 and Fig. 1). These known nuclear regulators and their mechanism of action are elaborated upon in the following sections.
1.
Nuclear regulation of NF-κB. A number of proteins have been reported to regulate NF-κB activity in the nucleus. Some of these, such as Bcl-3 and UXT, bind directly to the members of the NF-κB complex, while other proteins catalyse post-translational modifications of NF-κB subunits. Bcl-3, B cell lymphoma-3; UXT, ubiquitously expressed transcript; HATs, histone acetyl transferases; HDACs, histone deacetylases.
| Name of the regulator | Mechanism of action |
|---|---|
| Bcl-3 | Binds to p50 and p65 subunits |
| IκB ζ | Binds to p50 and p65 subunits |
| UXT | Binds to p65 subunit |
| ZAS3 | Competes for IκB regulatory elements |
| HATs | Acetylates histones and transcription factors |
| HDACs | Deacetylates histones and transcription factors |
| Kinases | Phosphorylates NF-κB subunits |
1.
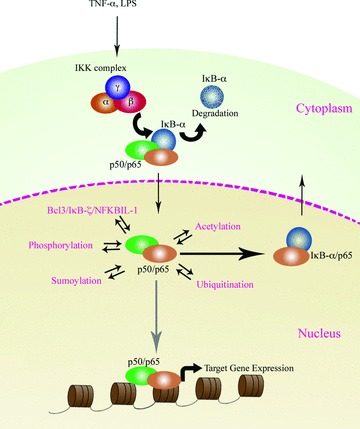
Regulation of NF-κB activity. When stimulated the active complex of p50/p65 subunit translocates to the nucleus and binds to the consensus DNA sequence. Different proteins and processes that regulate the nuclear activity of NF-κB are indicated by arrows. Post-translational modifications, including phosphorylation and acetylation, regulate binding of NF-κB to the target genes.
B cell lymphoma 3 (Bcl-3)
Bcl-3 is a nuclear protein that preferentially promotes NF-κB-dependent gene transcription [37]. Experiments with transgenic mice constitutively expressing Bcl-3 indicated that Bcl-3 associated with endogenous p50 and p52 [38]. Bcl-3 can cause derepression of transcription by removing p50 and p52 dimers, which are transcriptionally inactive, from the NF-κB sites, thus leading to the binding of an active complex consisting of p65, Rel-B or c-Rel [39]. While the ankyrin repeat domain of the Bcl-3 protein is essential for its activity as an IκB, its N- and C-terminal regions are not homologous to those of other IκB proteins, being very proline rich, which suggests a potential role as a transcriptional trans-activator. Zhang et al. demonstrated that Bcl-3 co-localizes with the NF-κB subunit p50 in a variety of punctate or speckled patterns strongly implying a interaction between these two proteins. This pattern of distribution, along with the proline rich sequence, corroborates the possible transactivator nature of the protein as does the formation of a ternary complex with DNA and p50 homodimers [40].
Bcl-3 is a crucial factor in the signal transduction pathways activated by a variety of pro-inflammatory ligands. For example, in cells stimulated with LPS, Bcl-3 facilitates production of TNF-α but not of IL-6 [41]. On the other hand, Bcl-3 inhibits IL-10 expression in macrophages in response to infection thus promoting activation of the innate immune system [42].
Bcl-3 knockout mice exhibit severe defects in humoural immune responses and protection from in vivo challenges [43]. Bcl-3 plays a critical role in sustaining immune responses and is crucial for optimal T-cell function. Over-expression of Bcl-3 results in increased T-cell survival whereas activated Bcl-3-null T cells die abnormally rapidly [44]. The pro-survival signal activated by Bcl-3 requires the blockade of the pro-apoptotic protein Bim and occurs in the presence of IL-12 [45].
The role of Bcl-3 in tumourigenesis first came to light when it was implicated in the 14: 19 chromosomal translocation in B-cell lymphomas. Kashatus et al. showed that Bcl-3 is induced by DNA damage and is an additional pathway for the induction of Hdm2 gene expression and suppression of p53. Indeed, constitutive expression of Bcl-3 suppressed DNA-damage induced p53 activity [46]. The contribution of Bcl-3 to tumour growth was further elaborated when it was reported that the cyclin D1 elevation seen in CYLD deubiquitinase gene knockout mice was mediated by increased activity of Bcl-3-associated p50 and p52 dimers [47]. CYLD is an important negative regulator of Bcl-3 (and another important mediator of NF-κB signals, TRAF2) causing Bcl-3 to be de-ubiquitinated, and therefore preventing its translocation to the nucleus. In addition, Bcl-3 expressing breast cancer cells have reduced duration of the G1 phase and increased phosphorylation of retinoblastoma protein thus promoting faster cell division rate [48]. Current evidence therefore suggests that Bcl-3 may play an important role in tumourigenesis via its regulation of NF-κB. Recent evidence also points towards a contribution of NF-κB-mediated inflammation in tumour progression [49, 50]. In this regard, Bcl-3 may be considered as a potential target for therapies centred on the NF-κB pathway.
IκB-ζ is another recently described regulator of NF-κB, which shares a high degree of homology with Bcl-3 [35, 51]. The expression of IκB-ζ is induced by IL-1β, LPS, peptidoglycan, bacterial lipoprotein, flagellin, and CpG DNA but not by TNF-α[51]. It has also been demonstrated that exogenous expression of IκB-ζ augmented the NF-κB mediated expression of beta defensins (hBD-2) and NGAL, whereas it inhibited that of E-selectin [52]. Consistent with the induction spectrum of IκB-ζ, both hBD-2 and NGAL are preferentially induced by IL-1p and not by TNF-α[52].
IκB-ζ is barely detected in resting cells but is strongly induced upon stimulation. Newly generated IκB-ζ subsequently translocates to the nucleus and associates with both the p50 and p65 subunits of NF-κB [53]. Studies have reported that one of the functions of IκB-ζ is likely to be in activating genes that play roles in the elimination of infectious bacteria as shown by its induction by microbial ligands but not by double-stranded RNA. IκB-ζ is essential for TLR and IL-1R -mediated induction of various inflammatory genes such as IL-6, the IL-12 p40 subunit and granulo-cyte-macrophage colony-stimulating factor [35]. In the case of IL-6, an IκB-ζ/NF-κB p50 complex is recruited to the IL-6 promoter leading to an increased expression of IL-6 in response to TLR or IL-1 signalling [51, 54].
Totzke et al. further showed IκB-ζ to be differentially expressed in apoptosis-sensitive compared with resistant tumour cells [54]. They confirmed that in a stimulated cell, IκB-ζ is localized exclusively in the nucleus where it associates with both p65 and p50. Despite having a speckled pattern of distribution within the nucleus, IκB-ζ did not co-localize with either promyelocytic leukaemia protein (PML) or with SC-35, proteins that are commonly used as markers for nuclear speckles. However, it was shown to co-localize with the nuclear co-repressor silencing mediator of retinoid and thyroid receptors (SMRT), that constitutes part of the matrix-associated deacetylase complex along with HDAC5. This indicates that nuclear IκB-ζ is part of a larger complex with p50 and p65 and HDAC5, and could potentially function by modulating HDAC activity.
Induction of IκB-ζ gene expression depends on activation of NF-κB but efficient synthesis of IκB-ζ also requires stabilization of its transcript. Interestingly, stimulation with IL-17 causes a stabilization of IκB-ζ transcript without activation of NF-κB. The cis-element required for this stimulus-specific stabilization was localized to a 165 bp sequence within the 3’ untranslated region of the IκB-ζ and was not dependent on AU-rich elements [55].
κB-ζ is also likely to be critically involved in the transcriptional activation of a subset of genes that are elicited in B cells through stimulation of the antigen receptor. The important and selective role of IκB-ζ is demonstrated by the fact that IκB-ζ -deficient splenocytes exhibit defective proliferation in response to LPS [51]. This strongly suggests that B cells have the capacity to induce IκB-ζ and that the induced IκB-ζ plays an essential role in the LPS-mediated proliferation of B cells [56]. Since IL-6 is produced by antigen- or mitogen-stimulated B cells, IκB-ζ may be involved in the induction of IL-6 and therefore, in B-cell differentiation and haematopoietic cell growth [53]. These reports demonstrate that IκB-ζ is induced by both the innate and the adaptive immune system and is important in the proper functioning of both these critical responses.
UXT, ZAS3 and PDLIM2
Several novel proteins have been recently reported to regulate the activity of NF-κB. These proteins do not possess the characteristic sequences of known IκB family members, such as ankyrin repeats, and their identification as nuclear regulators of NF-κB significantly augments our understanding of the complexity of NF-κB regulation, while also enabling us to look at them as potential therapeutic targets. One of these proteins is UXT, which was first identified in 1999 [57] and was shown to be abundantly expressed in tumour tissue but not in normal tissue [57, 58]. More recently Sun et al. reported that UXT is an essential co-factor for NF-κB function and that it directly interacted with the p65 subunit of NF-κB inside the nucleus [36]. Indeed, the presence of NF-κB within the nucleus of stimulated or constitutively active cells was considerably diminished with decreased endogenous UXT levels. Furthermore, the authors reported that RNA interference knockdown of UXT led to impaired NF-κB activity dramatically attenuating the expression of NF-κB-dependent genes. Interestingly, in this study the UXT protein levels correlated with constitutive NF-κB activity in human prostate cancer cell lines suggesting a potential role for this protein in oncogenesis. The authors concluded that UXT was an integral component of the NF-κB enhanceosome and was essential for its nuclear function [36].
Hong et al. reported another novel protein, ZAS3 that inhibited NF-κB activity. ZAS3 is a large zinc finger protein that regulates IκB-mediated transcription and the TNF-α driven signal transduction pathway [34]. The ZAS genes encode transcriptional proteins that activate or repress the transcription of a variety of genes involved in growth, development and metastasis [59]. ZAS3 was shown to interfere with NF-κB mediated transcription by competing for IκB gene regulatory elements thus repressing transcription. Additionally ZAS3, or most probably an isoform of ZAS3, binds to TRAF2 (TNF receptor associated factor), inhibiting translocation of p65 [59]. It has been reported that NF-κB is constitutively active in a ZAS3-deficient cell line [34] and that ZAS3 deficiency leads to proliferation of cells leading to tumour formation in mice [60].
Regulation of NF-κB by ubiquitination has long been suggested to be an important factor in terminating its action. Indeed, in 2003 it was demonstrated that p65 protein stability is regulated by ubiquitin-mediated proteolysis and it was proposed that the cytokine signal inhibitor SOCS-1 was a putative p65 ubiquitin ligase [61]. However, more recently this process of NF-κB regulation by ubiquitination was attributed to the PDLIM2 protein [62]. PDLIM2 is a member of the PDZ and LIM domain containing protein family and some members of this family are implicated in diverse biological roles including cytoskeletal organization, neuronal signalling, cell lineage specification, organ development and oncogenesis [63]. However, unlike other members of this class, PDLIM2 itself was shown to inhibit NF-κB activity by facilitating the polyubiquitination of the p65 subunit of NF-κB in the nucleus [62]. For this activity, PDLIM2 functioned as an ubiquitin E3 ligase by means of its LIM domain, triggering the transfer of the soluble p65 fraction to the insoluble fraction within the promyelocytic leukaemia protein bodies in the nucleus and subsequently assisting the proteosomal degradation of p65 protein. Interestingly, PDLIM2 did not affect the cytoplasmic pool of p65, thus allowing for a critical pool of cytoplasmic p65 to remain enabling the cell to respond rapidly to subsequent stimulation.
Post-translational modification of NF-κB complex in the nucleus
Proteins within the NF-κB complex are subject to signal-induced post-translational modifications that influence their physiological functions. In addition to ubiquitination as discussed above, these include phosphorylation, hydroxylation and acetylation [64–66]. These modifications take place in the cytoplasm and/or the nucleus and within the nucleus their effects can range from tran-scriptional activation to repression.
Phosphorylation
Phosphorylation of proteins, a reversible process catalysed by kinases and phosphatases, enables various proteins to be regulated in a subtle manner. Of the Rel family members, phosphorylation of p65 has been most extensively analyzed. p65 is phosphorylated at nine different sites, including six serine and three threonine residues, with some occurring in the cytoplasm and others in the nucleus. The nine phosphorylated sites are Ser-536, Ser-535, Ser-529, Ser-468, Ser-311, Ser-276, Thr-435, Thr-505 and Thr-254 [65, 67]. Of these, phosphorylation of the two residues Ser-468 and Ser-276 are known to affect nuclear function of NF-κB. Phosphorylation of Ser-468 is mediated by glycogen synthase kinase -3 beta (GSK-3β) and results in the inhibition of the basal activity of NF-κB [65]. Ser-276, a target of mitogen- and stress-activated protein kinase 1 (MSK-1), is phosphorylated exclusively within the nucleus [68, 69]. One additional consequence of p65 phosphorylation is the increased recruitment of CBP/p300 leading to the export of repressor proteins such as HDACs [2, 70].
Other members of Rel family including Both RelB and C-Rel also undergo phosphorylation but while phosphorylation promotes RelB degradation, it enhances C-Rel transactivation [71, 72]. Of the described nuclear inhibitors of NF-κB, Bcl-3 is known to undergo GSK-3p mediated phosphorylation causing it to be degraded via the proteasomal pathway [73].
Acetylation and deacetylation
Transcription factors regulate gene expression by binding to a specific promoter sequence of the target gene [74] and it has become increasingly clear that their ability to access the DNA is a key regulatory nexus in the control of transcription factor activity. Histones form the fundamental part of the nucleosomal complex and regulate the manner in which the DNA is packaged. During activation of gene transcription in the presence of a compact nucleosomal complex, inaccessible DNA is made available to DNA binding proteins via modification of the nucleosomes. The arrangement of the nucleosomal complex is dependent on the post-translational modifications of histones, which include phosphorylation, methylation, acetylation among others [75]. It has been speculated that there is a ‘histone code’ which has, as its central tenet, the idea that specific patterns of post-translational modifications to histones act like a molecular ‘code’ which are recognized and used by non-histone proteins to regulate specific chromatin functions [76]. Of the various post-translational modifications, acetylation of core histone protein is probably the best understood. Enzymes such as histone acetyltransferases catalyse the addition of an acetyl moiety to the histone while HDACs proteins remove the acetyl group. The HDAC family members are classified into four groups; the main groups being: Class I HDACs that include HDAC 1, 2, 3 and 8 while Class II includes HDAC 4, 5, 6, 7, 9 and 10. Class I HDAC proteins, with the exception of HDAC3, are found exclusively in the nucleus, while Class II members shuttle in and out of the nucleus [77, 78]. In general, increased levels of histone acetylation are associated with increased transcription factor activity, whereas decreased levels of acetylation are associated with repression of gene expression [79].
A potential role for acetylation in the regulation of NF-κB mediated transactivation was reported by Chen and Greene who showed that TSA enhances IκB-luciferase reporter gene expression in cells stimulated by TNF-α. p65 is acetylated by p300/CBP and deacetylated specifically by HDAC3. Acetylation of the 221 lysine residue of p65 was shown to enhance the DNA binding properties of p65. Additionally, they demonstrated that HDAC3 mediated deacetylation of p65 not only decreased the affinity of p65 for the DNA, but also potentiated the interaction of p65 with IκB-α, consequently leading to the export of the p65/κB-α complex to the cytoplasm. Based on this evidence, the authors suggested that reversible acetylation of intra-nuclear p65 regulated not only the duration of the NF-κB mediated transcriptional response but also contributed to the replenishment of the depleted cytoplasmic pool of latent NF-κB/IκB-α complexes, thereby preparing the cell for any further NF-κB inducing signal. In a series of interesting follow-on experiments the authors not only reported that acetylation of another residue -lysine 310 - was important for NF-κB activation, but they also explored the contribution of a combination of phosphorylation and acetylation on NF-κB and observed that prior phosphorylation of p65 at Ser 276 or Ser 536 facilitated the acetylation of p65 at lysine 310 [80]. These findings reveal how potentially different post-translational modifications can synergistically or antagonistically fine-tune the outcome to NF-κB activation.
Sumoylation
Sumolytion, another form of post-translational modification, is mediated by the small ubiquitin like modifier (SUMO) family of proteins [81, 82]. Sumoylation, like ubiquitination, requires four enzymatic components including SUMO protease, E1, E2 and E3 ligase and occurs on the consensus sequence ΨKXE (Ψ: hydrophobic, K: target lysine, X: any amino acid, E: glutamic acid) of the target protein. Current evidence indicates two different consequences to SUMO modification. In the first instance sumoylation effects protein stability while in the second, it promotes the nuclear translocation of the cytoplasmic protein [83, 84]. Within the nucleus, sumoylated proteins have been shown to accumulate as nuclear speckles. Indeed, the machinery required for sumoylation is generally found in the nucleus as are the majority of proteins known to be sumoylated [85]. Different families of proteins can undergo sumoylation, including transcription factors and co-factors such as CBP/p300 [82]. So far three proteins involved in the NF-κB signalling have been demonstrated to be sumoylated. Sumoylation prevents the stimulus induced ubiquitination and degradation of IκB-α[86]. However unlike ubiquitintion, this process has been shown to occur within the nucleus [85]. Interestingly sumoylation and ubiquitination target the same lysine residue indicating that stimulation specific post-translational modification of the critical lysine residue of IκB-α determines the fate of the protein [86]. PIAS1, an E3 ligase, has been shown to directly interact with the transactivation domain of the p65 subunit of NF-κB. This prevents the NF-κB -DNA binding reducing the expression of target genes including IκB-α[87]. Another SUMO E3 ligase, PIAS3, was shown to compete with CBP for binding with the Rel homology domain domain of p65 consequently suppressing NF-κB activity [88]. Finally IKKγ/NEMO, normally the non-catalytic member of the IKK complex, has also been reported to undergo sumoylation. However in this case NEMO is unbound to the IKK complex and sumoylation occurs in response to genotoxic stimuli such as heat shock and oxidative stress. The modified NEMO protein subsequently translocates to the nucleus where it is thought to repress NF-κB activity [33, 81]. It should be noted that the process of sumoylation is not only dynamic and reversible but also very subtle making it difficult to identify the effects of this post-translational modification.
Nuclear regulators of NF-κB: potential molecular chaperones
Transcription factors are critical cogs in the molecular machinery facilitating the transcription of genes in response to external stimuli. Crucially transcription factors need to respond dynamically and rapidly to variations in cellular stimulation. Since certain stimuli can induce the activation of several transcription factors at the same time, this would suggest that a common mechanism to rapidly terminate this activation must also exist. In addition, cells need to have a system in place to ‘sense’ such changes in the intensity of the stimuli and respond accordingly. Indeed, a critical transcription factor like NF-κB that can regulate the expression of a multitude of genes needs to respond to variations in the external stimuli and signal intensity.
Studies on the response of cells to changes in hormonal levels have provided additional insights into a possible process for the rapid termination of transcription factor activity and resetting of the cell for subsequent stimuli. In an interesting approach looking at how hormonal signals were modulated, Freeman and Yamamoto investigated the regulation of transcription factors including NF-κB [89]. They reported that over expression of two molecular chaperones, p23 and Hsp90, reduced the activity of AP-1 and NF-κB in cells stimulated with PMA and TNF-α respectively, apparently by triggering the release of the constituent transcription factor complexes from DNA. The authors suggested that transcription factor complexes undergo a continuous cycle of rapid assembly followed by disassembly from the DNA. This provides an attractive model which would enable regulators to sense and respond efficiently to fluctuating stimuli [89, 90].
The importance of molecular chaperones has been clearly outlined in circumstances of cellular stress where they are required for the trafficking of misfolded proteins [91]. Examples of molecular chaperones include heat shock protein family members such as Hsp27, Hsp90 and GRP78 (misfolded protein chaperone in the endoplasmic reticulum). Additional roles for such proteins came to light when it was shown that Hsp90 binds to the heat shock transcription factor and modulates its function independently of its chaperone activity [92, 93]. The additional functions of chaperone proteins were further corroborated when Virbasius et al. identified a novel protein, bZIP-enhancing factor (BEF), that not only acted as a molecular chaperone but also regulated the activity of nuclear transcription factors containing the bZIP DNA binding domain [94]. More recently, it was reported that Jun dimerization protein-2 (JDP-2), a component of the AP-1 transcription complex, was also a chaperone protein which could regulate histone acetylation and nucleosome formation. JDP-2 accomplished this by recruiting HDAC3 to the promoter region of its target gene [95]. These studies demonstrate that certain transcription factor complexes may routinely contain proteins displaying chaperone activities, and which may be integral to the correct functioning of these complexes.
In this context, some of the nuclear regulators of NF-κB and other transcription factors (such as JDP-2) can be considered as chaperone proteins. This provides a framework by which to understand their roles in the regulation of transcription factors. Several are documented as affecting more than one transcription factor. For example, the protein ZAS3 is known to regulate both NF-κB and AP-1 activity, while Bcl-3 regulates the transcriptional activity of not only NF-κB but also AP-1 and RXR [59, 96–98]. RXR belongs to a family of ligand-dependent transcriptional regulatory proteins and is an obligatory binding partner of several nuclear receptors including PXR/SXR [99]. In addition, Bcl-3 interacts with the basal transcription machinery including such members as TFIIA, TFIIB and TBP [96]. Bcl-3 has also been associated with other co-activators such as CBP/p300, TORC3 and SRC-1 and it is known to recruit HDACs to transcriptional complexes [100]. We have shown that another nuclear protein, NFκBIL1, inhibits several transcription factors including NF-κB and AP-1 (unpublished data). All these reports suggest that proteins such as Bcl-3, which have traditionally been classified strictly as regulators of NF-κB, may have a broader role in the regulation of several additional cellular processes and provides a mechanism allowing for extensive cross talk to occur between various transcription factors and their regulators while in the nucleus or bound to DNA.
Another mechanism for the regulation of transcription factor activity is likely to involve modulation of the availability of co-activators. For example, as mentioned above, Bcl-3 regulates NF-κB, AP-1 and RXR activity. Thus variability in the amount and the binding affinity of Bcl-3 protein would be expected to influence the activity of these transcription factors. There is evidence indicating that other regulators of NF-κB are also required transiently. For example, it has been demonstrated that IκB-ζ mRNA levels peak 1.5–2 hrs after a pulse of IL-1β stimulation and then rapidly declines, indicating that IκB-ζ synthesis is required only during assembly of a transcription complex on the target promoter (NGAL in this case) and at a later stage this regulator is dispensable [52]. Meanwhile, Allen et al. in 2007 reported altered NF-κB and AP-1 activity in ZAS3 knockout mice [98]. It is possible that in a cell and time specific manner, alterations in the spectrum of available co-regulators may regulate the transactivation potential of NF-κB or even enable one particular transcription factor to be more dominantly active than others. This concept has been reinforced by the observation that a single base difference in the IκB site of the MCP-1 gene was sufficient to alter the cofactor specificity of the NF-κB molecule bound to this site from IFN regulatory factor-3 (IRF-3) to Bcl-3 [101, 102]. This group of co-regulators may even contribute to the recently established oscillatory action of these transcription factors [103–105]. Sun et al. recently showed that TNF-α produces synchronous recruitment of AP-1 and NF-κB proteins to target promoters in an oscillatory manner [106, 107]. These findings suggest the existence of an intricate and dynamic role of co-regulators on the action of transcription factors including NF-κB, pointing to a possible ‘transcription factor co-regulator code’ that determines both the type of active transcription factor and the specificity of the target genes regulated downstream.
There are an ever-increasing number of regulators of NF-κB being reported. Recently another novel protein, ribosomal protein S, was reported to be a part of a DNA-bound NF-κB complex causing its recruitment to selected NF-κB responsive genes [108]. The presence of so many regulators raises an obvious question: why does NF-κB require such complex control of its activity? From a cell-signalling perspective, the NF-κB pathway can be construed as an hourglass wherein, in order to generate signal specific responses, the complexity present at the level of the inducers of NF-κB and variation in the intensity of these inducing signals needs to be reflected at the level of the regulation of NF-κB and the consequent modulation of expression of the target genes (Fig. 2). It needs to be noted that these regulators can either be NF-κB specific or members of generic regulatory complexes.
2.

NF-κB signalling and regulation: hourglass phenomenon: signal pathways activated by several different stimuli converge at the NF-κB complex in the cytoplasm. However, in order to generate signal-specific responses, the complexity of signalling prior to NF-κB activation needs to be reflected by the intricacy of NF-κB regulation, both at the cytoplasmic and more critically at the nuclear level, and the consequent gene expression. UV: ultraviolet radiation; Ac: acetylation; P: phosphorylation; IκBs: inhibitors of NF-κB; Su: SUMOytion; Hy: hydroxylation; U: ubiquitination; Others: proteins including ZAS3, UXT, PDLIM2, etc.
Based on these novel findings we believe that nuclear regulators of NF-κB such as IκB-ζ, Bcl-3, UXT, PDLIM2, ZAS3 and NFκBIL1 may be viewed as nuclear chaperone proteins bringing together the NF-κB subunits or other transcription factor sub-units as required (Fig. 3). Likewise, upon reduction or cessation of the stimuli, these proteins chaperone the removal of NF-κB from DNA thus contributing to the termination of its action. While a majority of the NF-κB subunits might recycle back to the cytoplasm and prepare the cell for the next wave of stimuli, it may be beneficial that a critical amount of NF-κB remains constantly within the nucleus. In this context, it may be that these nuclear regulators bring together these transcription factors and their co-regulators together and maintain them as a single complex waiting for the next appropriate signal and when the cell has been induced, shuttle the transcription factor to the DNA.
3.
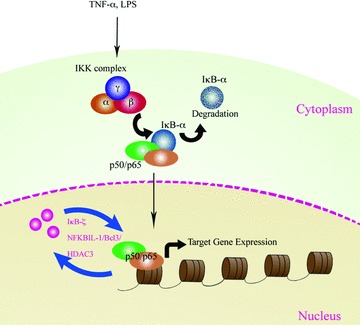
Nuclear regulators of NF-κB as potential chaperone proteins: nuclear regulators of NF-κB, including Bcl-3 and IκB-ζ, can potentially function as chaperone proteins facilitating the shuttling of transcription factor NF-κB between the promoter regions of target gene and the nuclear speckles.
Conclusion
Transcription factors regulate the expression of all genes and are essential components of the signalling process for maintaining cellular homeostasis. NF-κB is vital in regulating the expression of genes involved in immunity and inflammation. The action of NF-κB is critical for the cell and it needs to be regulated dynamically. Since NF-κB subunits undergo nucleo-cytoplasmic shuttling, its regulation in the nucleus could plausibly involve a mechanism whereby NF-κB is chaperoned to/from active sites within the nucleus. It is possible that, in a fashion similar to the cytoplasmic inhibitors of NF-κB, the nuclear inhibitors of NF-κB too sequester the subunits in inactive complexes.
A growing number of studies have implicated NF-κB dependent pathways in the development of various diseases such as inflammatory bowel disease, rheumatic arthritis, atherosclerosis and asthma and more recently dysregulated NF-κB activity has been identified as a contributing factor in tumourigenesis. Numerous novel therapeutic agents that inhibit either activation or function of NF-κB have been developed or are under development [109]. In the context of cancer it was recently reported that a combination of NF-κB inhibitors along with TRAIL inhibitors might decrease the rate of cancer progression [110]. Nevertheless, because of the presence of NF-κB in virtually all cells and its involvement in many different cellular pathways and functions, application of these drugs to inhibit NF-κB non-specifically may have side effects [111–114]. The nuclear regulators of NF-κB are downstream participants of the NF-κB signalling pathways and may enable a less blunt abolition of its activity. However, in spite of the intense focus on NF-κB and elucidation of its function and regulation, the road to the complete understanding of its regulation has only been partially explored. Further studies into the nuclear regulation of NF-κB are essential to providing a fuller understanding of the mechanism of action of this and other transcription factors in various diseases, with the ultimate prospect of identifying better and more specific therapeutic targets.
Acknowledgments
The authors wish to acknowledge support from Wellcome Trust/ HRB ‘New Blood’ Fellowship (RMM), Science Foundation Ireland and Higher Education Authority PRTLI. The authors wish to thank Serkan I˙smail Göktuna for the artwork.
References
- 1.Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986;46:705–16. doi: 10.1016/0092-8674(86)90346-6. [DOI] [PubMed] [Google Scholar]
- 2.Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell. 1998;1:661–71. doi: 10.1016/s1097-2765(00)80066-0. [DOI] [PubMed] [Google Scholar]
- 3.May MJ, Ghosh S. Signal transduction through NF-kappa B. Immunol Today. 1998;19:80–8. doi: 10.1016/s0167-5699(97)01197-3. [DOI] [PubMed] [Google Scholar]
- 4.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–62. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 5.Aud D, Peng SL. Mechanisms of disease: Transcription factors in inflammatory arthritis. Nature Clin Pract. 2006;2:434–42. doi: 10.1038/ncprheum0222. [DOI] [PubMed] [Google Scholar]
- 6.Abdel-Latif MM, Windle H, Terres A, Eidhin DN, Kelleher D, Reynolds JV. Helicobacter pylori extract induces nuclear factor-kappa B, activator protein-1, and cyclooxygenase-2 in esophageal epithelial cells. J Gastrointest Surg. 2006;10:551–62. doi: 10.1016/j.gassur.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 7.Lawless MW, Greene CM, Mulgrew A, Taggart CC, O’Neill SJ, McElvaney NG. Activation of endoplasmic reticulum-spe-cific stress responses associated with the conformational disease Z alpha 1-antit-rypsin deficiency. J Immunol. 2004;172:5722–6. doi: 10.4049/jimmunol.172.9.5722. [DOI] [PubMed] [Google Scholar]
- 8.Lawless MW, Mankan AK, Gray SG, Norris S. Endoplasmic reticulum stress-A double edged sword for Z alpha-1 antit-rypsin deficiency hepatoxicity. Int J Biochem Cell Biol. 2008;40:1403–14. doi: 10.1016/j.biocel.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 9.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Eng J Med. 2000;342:1334–49. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 10.Blaschke F, Bruemmer D, Law RE. Egr-1 is a major vascular pathogenic transcription factor in atherosclerosis and resteno-sis. Rev Endocr Metabol Disord. 2004;5:249–54. doi: 10.1023/B:REMD.0000032413.88756.ee. [DOI] [PubMed] [Google Scholar]
- 11.Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2:725–34. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 12.Moynagh PN. The NF-kappaB pathway. J Cell Sci. 2005;118:4589–92. doi: 10.1242/jcs.02579. [DOI] [PubMed] [Google Scholar]
- 13.Grossmann M, Nakamura Y, Grumont R, Gerondakis S. New insights into the roles of ReL/NF-kappa B transcription factors in immune function, hemopoiesis and human disease. Int J Biochem Cell Biol. 1999;31:1209–19. doi: 10.1016/s1357-2725(99)00068-0. [DOI] [PubMed] [Google Scholar]
- 14.Gerondakis S, Grossmann M, Nakamura Y, Pohl T, Grumont R. Genetic approaches in mice to understand Rel/NF-kappaB and IkappaB function: transgenics and knockouts. Oncogene. 1999;18:6888–95. doi: 10.1038/sj.onc.1203236. [DOI] [PubMed] [Google Scholar]
- 15.Karin M. How NF-kappaB is activated: the role of the IkappaB kinase (IKK) complex. Oncogene. 1999;18:6867–74. doi: 10.1038/sj.onc.1203219. [DOI] [PubMed] [Google Scholar]
- 16.Senftleben U, Karin M. The IKK/NF-kappaB pathway. Crit Care Med. 2002;30:S18–S26. [PubMed] [Google Scholar]
- 17.Shen G, Jeong WS, Hu R, Kong AN. Regulation of Nrf2, NF-kappaB, and AP-1 signaling pathways by chemopreventive agents. Antioxid Redox Signal. 2005;7:1648–63. doi: 10.1089/ars.2005.7.1648. [DOI] [PubMed] [Google Scholar]
- 18.Luo JL, Kamata H, Karin M. The anti-death machinery in IKK/NF-kappaB signaling. J Clin Immunol. 2005;25:541–50. doi: 10.1007/s10875-005-8217-6. [DOI] [PubMed] [Google Scholar]
- 19.Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG, Karin M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453:807–11. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taylor CT. Interdependent roles for hypoxia inducible factor and nuclear factor-kappaB in hypoxic inflammation. J Physiol. 2008;586:4055–9. doi: 10.1113/jphysiol.2008.157669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mosavi LK, Cammett TJ, Desrosiers DC, Peng ZY. The ankyrin repeat as molecular architecture for protein recognition. Protein Sci. 2004;13:1435–48. doi: 10.1110/ps.03554604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rogers S, Wells R, Rechsteiner M. Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science. 1986;234:364–8. doi: 10.1126/science.2876518. [DOI] [PubMed] [Google Scholar]
- 23.Shumway SD, Maki M, Miyamoto S. The PEST domain of IkappaBalpha is necessary and sufficient for in vitro degradation by mucalpain. J Biol Chem. 1999;274:30874–81. doi: 10.1074/jbc.274.43.30874. [DOI] [PubMed] [Google Scholar]
- 24.Wulczyn FG, Krappmann D, Scheidereit C. Signal-dependent degradation of IkappaBalpha is mediated by an inducible destruction box that can be transferred to NF-kappaB, bcl-3 or p53. Nucleic Acids Res. 1998;26:1724–30. doi: 10.1093/nar/26.7.1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bourke E, Kennedy EJ, Moynagh PN. Loss of Ikappa B-beta is associated with prolonged NF-kappa B activity in human glial cells. J Biol Chem. 2000;275:39996–40002. doi: 10.1074/jbc.M007693200. [DOI] [PubMed] [Google Scholar]
- 26.Suyang H, Phillips R, Douglas I, Ghosh S. Role of unphosphorylated, newly synthesized I kappa B beta in persistent activation of NF-kappa B. Mol Cell Biol. 1996;16:5444–9. doi: 10.1128/mcb.16.10.5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Whiteside ST, Epinat JC, Rice NR, Israel A. I kappa B epsilon, a novel member of the I kappa B family, controls RelA and cRel NF-kappa B activity. EMBO J. 1997;16:1413–26. doi: 10.1093/emboj/16.6.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mémet S, Laouini D, Epinat JC, Whiteside ST, Goudeau B, Philpott D, Kayal S, Sansonetti PJ, Berche P, Kanellopoulos J, Israël A. IkappaBepsilon-deficient mice: reduction of one T cell precursor subspecies and enhanced Ig isotype switching and cytokine synthesis. J Immunol. 1999;163:5994–6005. [PubMed] [Google Scholar]
- 29.Basak S, Kim H, Kearns JD, Tergaonkar V, O’Dea E, Werner SL, Benedict CA, Ware CF, Ghosh G, Verma IM, Hoffmann A. A fourth IkappaB protein within the NF-kappaB signaling module. Cell. 2007;128:369–81. doi: 10.1016/j.cell.2006.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tergaonkar V, Correa RG, Ikawa M, Verma IM. Distinct roles of IkappaB proteins in regulating constitutive NF-kappaB activity. Nat Cell Biol. 2005;7:921–3. doi: 10.1038/ncb1296. [DOI] [PubMed] [Google Scholar]
- 31.Anest V, Hanson JL, Cogswell PC, Steinbrecher KA, Strahl BD, Baldwin AS. A nucleosomal function for IkappaB kinase-alpha in NF-kappaB-dependent gene expression. Nature. 2003;423:659–63. doi: 10.1038/nature01648. [DOI] [PubMed] [Google Scholar]
- 32.Yamamoto Y, Gaynor RB. IkappaB kinases: key regulators of the NF-kappaB pathway. Trends Biochem Sci. 2004;29:72–9. doi: 10.1016/j.tibs.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 33.Verma UN, Yamamoto Y, Prajapati S, Gaynor RB. Nuclear role of I kappa B kinase-gamma/NF-kappa B essential modulator (IKK gamma/NEMO) in NF-kappa B-dependent gene expression. J Biol Chem. 2004;279:3509–15. doi: 10.1074/jbc.M309300200. [DOI] [PubMed] [Google Scholar]
- 34.Hong JW, Allen CE, Wu LC. Inhibition of NF-kappaB by ZAS3, a zinc-finger protein that also binds to the kappaB motif. Proc Natl Acad Sci USA. 2003;100:12301–6. doi: 10.1073/pnas.2133048100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamazaki S, Muta T, Takeshige K. A novel IkappaB protein, IkappaB-zeta, induced by proinflammatory stimuli, negatively regulates nuclear factor-kappaB in the nuclei. J Biol Chem. 2001;276:27657–62. doi: 10.1074/jbc.M103426200. [DOI] [PubMed] [Google Scholar]
- 36.Sun S, Tang Y, Lou X, Zhu L, Yang K, Zhang B, Shi H, Wang C. UXT is a novel and essential cofactor in the NF-{kappa}B transcriptional enhanceosome. J Cell Biol. 2007;178:231–44. doi: 10.1083/jcb.200611081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Q, Didonato JA, Karin M, McKeithan TW. BCL3 encodes a nuclear protein which can alter the subcellular location of NF-kappa B proteins. Mol Cell Biol. 1994;14:3915–26. doi: 10.1128/mcb.14.6.3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caamano JH, Perez P, Lira SA, Bravo R. Constitutive expression of Bc1–3 in thymocytes increases the DNA binding of NF-kappaB1 (p50) homodimers in vivo. Mol Cell Biol. 1996;16:1342–8. doi: 10.1128/mcb.16.4.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bundy DL, McKeithan TW. Diverse effects of BCL3 phosphorylation on its modulation of NF-kappaB p52 homodimer binding to DNA. J Biol Chem. 1997;272:33132–9. doi: 10.1074/jbc.272.52.33132. [DOI] [PubMed] [Google Scholar]
- 40.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 41.Kuwata H, Watanabe Y, Miyoshi H, Yamamoto M, Kaisho T, Takeda K, Akira S. IL-10-inducible Bcl-3 negatively regulates LPS-induced TNF-alpha production in macrophages. Blood. 2003;102:4123–9. doi: 10.1182/blood-2003-04-1228. [DOI] [PubMed] [Google Scholar]
- 42.Riemann M, Endres R, Liptay S, Pfeffer K, Schmid RM. The IkappaB protein Bcl-3 negatively regulates transcription of the IL-10 gene in macrophages. J Immunol. 2005;175:3560–8. doi: 10.4049/jimmunol.175.6.3560. [DOI] [PubMed] [Google Scholar]
- 43.Schwarz EM, Krimpenfort P, Berns A, Verma IM. Immunological defects in mice with a targeted disruption in Bcl-3. Genes Dev. 1997;11:187–97. doi: 10.1101/gad.11.2.187. [DOI] [PubMed] [Google Scholar]
- 44.Mitchell TC, Thompson BS, Trent JO, Casella CR. A short domain within Bcl-3 is responsible for its lymphocyte survival activity. Ann N Y Acad Sci. 2002;975:132–47. doi: 10.1111/j.1749-6632.2002.tb05947.x. [DOI] [PubMed] [Google Scholar]
- 45.Bauer A, Villunger A, Labi V, Fischer SF, Strasser A, Wagner H, Schmid RM, Häcker G. The NF-kappaB regulator Bcl-3 and the BH3-only proteins Bim and Puma control the death of activated T cells. Proc Natl Acad Sci USA. 2006;103:10979–84. doi: 10.1073/pnas.0603625103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kashatus D, Cogswell P, Baldwin AS. Expression of the Bcl-3 proto-oncogene suppresses p53 activation. Genes Dev. 2006;20:225–35. doi: 10.1101/gad.1352206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fassler R. Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-kappaB signaling. Cell. 2006;125:665–77. doi: 10.1016/j.cell.2006.03.041. [DOI] [PubMed] [Google Scholar]
- 48.Westerheide SD, Mayo MW, Anest V, Hanson JL, Baldwin AS., Jr The putative oncoprotein Bcl-3 induces cyclin D1 to stimulate G(1) transition. Mol Cell Biol. 2001;21:8428–36. doi: 10.1128/MCB.21.24.8428-8436.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–59. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 50.Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–6. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 51.Yamamoto M, Yamazaki S, Uematsu S, Sato S, Hemmi H, Hoshino K, Kaisho T, Kuwata H, Takeuchi O, Takeshige K, Saitoh T, Yamaoka S, Yamamoto N, Yamamoto S, Muta T, Takeda K, Akira S. Regulation of Toll/IL-1-receptor-mediated gene expression by the inducible nuclear protein IkappaBzeta. Nature. 2004;430:218–22. doi: 10.1038/nature02738. [DOI] [PubMed] [Google Scholar]
- 52.Cowland JBMT, Borregaard N. IL-1beta-specific up-regulation of neutrophil gelati-nase-associated lipocalin is controlled by IkappaB-zeta. J Immunol. 2006;176:5559–66. doi: 10.4049/jimmunol.176.9.5559. [DOI] [PubMed] [Google Scholar]
- 53.Muta T. IkappaB-zeta: an inducible regulator of nuclear factor-kappaB. Vitam Horm. 2006;74:301–16. doi: 10.1016/S0083-6729(06)74012-2. [DOI] [PubMed] [Google Scholar]
- 54.Totzke G, Essmann F, Pohlmann S, Lindenblatt C, Janicke RU, Schulze-Osthoff K. A novel member of the IkappaB family, human IkappaB-zeta, inhibits trans-activation of p65 and its DNA binding. J Biol Chem. 2006;281:12645–54. doi: 10.1074/jbc.M511956200. [DOI] [PubMed] [Google Scholar]
- 55.Watanabe STK, Muta T. A cis-element in the 3’-untranslated region of IkappaB-zeta mRNA governs its stimulus-specific expression. Biochem Biophys Res Commun. 2007;356:785–91. doi: 10.1016/j.bbrc.2007.03.044. [DOI] [PubMed] [Google Scholar]
- 56.Hijioka KMS, Eto-Kimura A, Takeshige K, Muta T. Induction of the nuclear IkappaB protein IkappaB-zeta upon stimulation of B cell antigen receptor. Biochem Biophys Res Commun. 2007;356:476–80. doi: 10.1016/j.bbrc.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 57.Schroer A, Schneider S, Ropers H, Nothwang H. Cloning and characterization of UXT, a novel gene in human Xp11, which is widely and abundantly expressed in tumor tissue. Genomics. 1999;56:340–3. doi: 10.1006/geno.1998.5712. [DOI] [PubMed] [Google Scholar]
- 58.Zhao H, Wang Q, Zhang H, Liu Q, Du X, Richter M, Greene MI. UXT is a novel centrosomal protein essential for cell viability. Mol Biol Cell. 2005;16:5857–65. doi: 10.1091/mbc.E05-08-0705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu LC. ZAS: C2H2 zinc finger proteins involved in growth and development. Gene Expr. 2002;10:137–52. doi: 10.3727/000000002783992479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Allen CE, Muthusamy N, Weisbrode SE, Hong JW, Wu LC. Developmental anomalies and neoplasia in animals and cells deficient in the large zinc finger protein KRC. Genes Chromosomes Cancer. 2002;35:287–98. doi: 10.1002/gcc.10128. [DOI] [PubMed] [Google Scholar]
- 61.Ryo A, Suizu F, Yoshida Y, Perrem K, Liou YC, Wulf G, Rottapel R, Yamaoka S, Lu KP. Regulation of NF-kappaB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol Cell. 2003;12:1413–26. doi: 10.1016/s1097-2765(03)00490-8. [DOI] [PubMed] [Google Scholar]
- 62.Tanaka T, Grusby MJ, Kaisho T. PDLIM2-mediated termination of transcription factor NF-kappaB activation by intranuclear sequestration and degradation of the p65 subunit. Nat Immunol. 2007;8:584–91. doi: 10.1038/ni1464. [DOI] [PubMed] [Google Scholar]
- 63.Te Velthuis AJ, Bagowski CP. PDZ and LIM domain-encoding genes: molecular interactions and their role in development. Sci World J. 2007;7:1470–92. doi: 10.1100/tsw.2007.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Perkins ND. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene. 2006;25:6717–30. doi: 10.1038/sj.onc.1209937. [DOI] [PubMed] [Google Scholar]
- 65.Neumann M, Naumann M. Beyond IkappaBs: alternative regulation of NF-kappaB activity. FASEB J. 2007;21:2642–54. doi: 10.1096/fj.06-7615rev. [DOI] [PubMed] [Google Scholar]
- 66.Cummins EP, Berra E, Comerford KM, Ginouves A, Fitzgerald KT, Seeballuck F, Godson C, Nielsen JE, Moynagh P, Pouyssegur J, Taylor CT. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci USA. 2006;103:18154–9. doi: 10.1073/pnas.0602235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005;30:43–52. doi: 10.1016/j.tibs.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 68.Vermeulen L, De Wilde G, Van Damme P, Vanden Berghe W, Haegeman G. Transcriptional activation of the NF-kappaB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1) EMBO J. 2003;22:1313–24. doi: 10.1093/emboj/cdg139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Duran A, Diaz-Meco MT, Moscat J. Essential role of RelA Ser311 phosphorylation by zetaPKC in NF-kappaB transcrip-tional activation. EMBO J. 2003;22:3910–8. doi: 10.1093/emboj/cdg370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhong H, May MJ, Jimi E, Ghosh S. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol Cell. 2002;9:625–36. doi: 10.1016/s1097-2765(02)00477-x. [DOI] [PubMed] [Google Scholar]
- 71.Marienfeld R, Berberich-Siebelt F, Berberich I, Denk A, Serfling E, Neumann M. Signal-specific and phosphorylation-dependent RelB degradation: a potential mechanism of NF-kappaB control. Oncogene. 2001;20:8142–7. doi: 10.1038/sj.onc.1204884. [DOI] [PubMed] [Google Scholar]
- 72.Maier HJ, Marienfeld R, Wirth T, Baumann B. Critical role of RelB serine 368 for dimerization and p100 stabilization. J Biol Chem. 2003;278:39242–50. doi: 10.1074/jbc.M301521200. [DOI] [PubMed] [Google Scholar]
- 73.Viatour P, Dejardin E, Warnier M, Lair F, Claudio E, Bureau F, Marine JC, Merville MP, Maurer U, Green D, Piette J, Siebenlist U, Bours V, Chariot A. GSK3-mediated BCL-3 phosphorylation modulates its degradation and its oncogenicity. Mol Cell. 2004;16:35–45. doi: 10.1016/j.molcel.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 74.Brivanlou AH, Darnell JE., Jr Signal transduction and the control of gene expression. Science. 2002;295:813–8. doi: 10.1126/science.1066355. [DOI] [PubMed] [Google Scholar]
- 75.Gray SG, Ekstrom TJ. The human histone deacetylase family. Exp Cell Res. 2001;262:75–83. doi: 10.1006/excr.2000.5080. [DOI] [PubMed] [Google Scholar]
- 76.Turner BM. Defining an epigenetic code. Nat Cell Biol. 2007;9:2–6. doi: 10.1038/ncb0107-2. [DOI] [PubMed] [Google Scholar]
- 77.De Ruijter AJ, Van Gennip AH, Caron HN, Kemp S, Van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–49. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cress WD, Seto E. Histone deacetylases, transcriptional control, and cancer. J Cell Physiol. 2000;184:1–16. doi: 10.1002/(SICI)1097-4652(200007)184:1<1::AID-JCP1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 79.Torchia J, Glass C, Rosenfeld MG. Co-activators and co-repressors in the integration of transcriptional responses. Curr Opin Cell Biol. 1998;10:373–83. doi: 10.1016/s0955-0674(98)80014-8. [DOI] [PubMed] [Google Scholar]
- 80.Chen LF, Williams SA, Mu Y, Nakano H, Duerr JM, Buckbinder L, Greene WC. NF-kappaB RelA phosphorylation regulates RelA acetylation. Mol Cell Biol. 2005;25:7966–75. doi: 10.1128/MCB.25.18.7966-7975.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mabb AM, Miyamoto S. SUMO and NF-kappaB ties. Cell Mol Life Sci. 2007;64:1979–96. doi: 10.1007/s00018-007-7005-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Seeler JS, Dejean A. Nuclear and unclear functions of SUMO. Nature reviews. 2003;4:690–9. doi: 10.1038/nrm1200. [DOI] [PubMed] [Google Scholar]
- 83.Huang TT, Wuerzberger-Davis SM, Wu ZH, Miyamoto S. Sequential modification of NEMO/IKKgamma by SUMO-1 and ubiquitin mediates NF-kappaB activation by genotoxic stress. Cell. 2003;115:565–76. doi: 10.1016/s0092-8674(03)00895-x. [DOI] [PubMed] [Google Scholar]
- 84.Agbor TA, Taylor CT. SUMO, hypoxia and the regulation of metabolism. Biochem Soc Trans. 2008;36:445–8. doi: 10.1042/BST0360445. [DOI] [PubMed] [Google Scholar]
- 85.Rodriguez MS, Dargemont C, Hay RT. SUMO-1 conjugation in vivo requires both a consensus modification motif and nuclear targeting. J Biol Chem. 2001;276:12654–9. doi: 10.1074/jbc.M009476200. [DOI] [PubMed] [Google Scholar]
- 86.Desterro JM, Rodriguez MS, Hay RT. SUMO-1 modification of IkappaBalpha inhibits NF-kappaB activation. Mol Cell. 1998;2:233–9. doi: 10.1016/s1097-2765(00)80133-1. [DOI] [PubMed] [Google Scholar]
- 87.Liu B, Yang R, Wong KA, Getman C, Stein N, Teitell MA, Cheng G, Wu H, Shuai K. Negative regulation of NF-kappaB signaling by PIAS1. Mol Cell Biol. 2005;25:1113–23. doi: 10.1128/MCB.25.3.1113-1123.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jang HD, Yoon K, Shin YJ, Kim J, Lee SY. PIAS3 suppresses NF-kappaB-mediated transcription by interacting with the p65/RelA subunit. J Biol Chem. 2004;279:24873–80. doi: 10.1074/jbc.M313018200. [DOI] [PubMed] [Google Scholar]
- 89.Freeman BC, Yamamoto KR. Disassembly of transcriptional regulatory complexes by molecular chaperones. Science. 2002;296:2232–5. doi: 10.1126/science.1073051. [DOI] [PubMed] [Google Scholar]
- 90.Freeman BC, Yamamoto KR. Continuous recycling: a mechanism for modulatory signal transduction. Trends Biochem Sci. 2001;26:285–90. doi: 10.1016/s0968-0004(01)01834-5. [DOI] [PubMed] [Google Scholar]
- 91.Loyola A, Reinberg D. Histone deposition and chromatin assembly by RSF. Methods. 2003;31:96–103. doi: 10.1016/s1046-2023(03)00093-8. [DOI] [PubMed] [Google Scholar]
- 92.Giardina C, Lis JT. Dynamic protein-DNA architecture of a yeast heat shock promoter. Mol Cell Biol. 1995;15:2737–44. doi: 10.1128/mcb.15.5.2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Blake MJ, Buckley AR, Zhang M, Buckley DJ, Lavoi KP. A novel heat shock response in prolactin-dependent Nb2 node lymphoma cells. J Biol Chem. 1995;270:29614–20. doi: 10.1074/jbc.270.49.29614. [DOI] [PubMed] [Google Scholar]
- 94.Virbasius CM, Wagner S, Green MR. A human nuclear-localized chaperone that regulates dimerization, DNA binding, and transcriptional activity of bZIP proteins. Mol Cell. 1999;4:219–28. doi: 10.1016/s1097-2765(00)80369-x. [DOI] [PubMed] [Google Scholar]
- 95.Jin C, Kato K, Chimura T, Yamasaki T, Nakade K, Murata T, Li H, Pan J, Zhao M, Sun K, Chiu R, Ito T, Nagata K, Horikoshi M, Yokoyama KK. Regulation of histone acetylation and nucleosome assembly by transcription factor JDP2. Nat Struct Mol Biol. 2006;13:331–8. doi: 10.1038/nsmb1063. [DOI] [PubMed] [Google Scholar]
- 96.Na SY, Choi HS, Kim JW, Na DS, Lee JW. Bcl3, an IkappaB protein, as a novel transcription coactivator of the retinoid X receptor. J Biol Chem. 1998;273:30933–8. doi: 10.1074/jbc.273.47.30933. [DOI] [PubMed] [Google Scholar]
- 97.Na SY, Choi JE, Kim HJ, Jhun BH, Lee YC, Lee JW. Bcl3, an IkappaB protein, stimulates activating protein-1 transactivation and cellular proliferation. J Biol Chem. 1999;274:28491–6. doi: 10.1074/jbc.274.40.28491. [DOI] [PubMed] [Google Scholar]
- 98.Allen CE, Richards J, Muthusamy N, Auer H, Liu Y, Robinson ML, Barnard JA, Wu LC. Disruption of ZAS3 in mice alters NF-kappaB and AP-1 DNA binding and T-cell development. Gene Expr. 2007;14:83–100. doi: 10.3727/105221607783417574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schütz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–9. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hishiki T, Ohshima T, Ego T, Shimotohno K. BCL3 acts as a negative regulator of transcription from the HTLV-1 LTR through interactions with TORC3. J Biol Chem. 2007;282:28335–43. doi: 10.1074/jbc.M702656200. [DOI] [PubMed] [Google Scholar]
- 101.Leung TH, Hoffmann A, Baltimore D. One nucleotide in a kappaB site can determine cofactor specificity for NF-kappaB dimers. Cell. 2004;118:453–64. doi: 10.1016/j.cell.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 102.Natoli G. Little things that count in transcriptional regulation. Cell. 2004;118:406–8. doi: 10.1016/j.cell.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 103.Kangaspeska S, Stride B, Métivier R, Polycarpou-Schwarz M, Ibberson D, Carmouche RP, Benes V, Gannon F, Reid G. Transient cyclical methylation of promoter DNA. Nature. 2008;452:112–5. doi: 10.1038/nature06640. [DOI] [PubMed] [Google Scholar]
- 104.Métivier R, Gallais R, Tiffoche C, Le Péron C, Jurkowska RZ, Carmouche RP, Ibberson D, Barath P, Demay F, Reid G, Benes V, Jeltsch A, Gannon F, Salbert G. Cyclical DNA methylation of a transcriptionally active promoter. Nature. 2008;452:45–50. doi: 10.1038/nature06544. [DOI] [PubMed] [Google Scholar]
- 105.Nelson DE, Ihekwaba AE, Elliott M, Johnson JR, Gibney CA, Foreman BE, Nelson G, See V, Horton CA, Spiller DG, Edwards SW, McDowell HP, Unitt JF, Sullivan E, Grimley R, Benson N, Broomhead D, Kell DB, White MR. Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science. 2004;306:704–8. doi: 10.1126/science.1099962. [DOI] [PubMed] [Google Scholar]
- 106.Sun L, Yang G, Zaidi M, Iqbal J. TNF-induced gene expression oscillates in time. Biochem Biophys Res Commun. 2008;371:900–5. doi: 10.1016/j.bbrc.2008.03.114. [DOI] [PubMed] [Google Scholar]
- 107.Sun L, Yang G, Zaidi M, Iqbal J. TNF-induced oscillations in combinatorial transcription factor binding. Biochem Biophys Res Commun. 2008;371:912–6. doi: 10.1016/j.bbrc.2008.03.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wan F, Anderson DE, Barnitz RA, Snow A, Bidere N, Zheng L, Hegde V, Lam LT, Staudt LM, Levens D, Deutsch WA, Lenardo MJ. Ribosomal protein S3: a KH domain subunit in NF-kappaB complexes that mediates selective gene regulation. Cell. 2007;131:927–39. doi: 10.1016/j.cell.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 109.Jain MK, Ridker PM. Anti-inflammatory effects of statins: clinical evidence and basic mechanisms. Nat Rev Drug Discov. 2005;4:977–87. doi: 10.1038/nrd1901. [DOI] [PubMed] [Google Scholar]
- 110.Malhi H, Gores GJ. TRAIL resistance results in cancer progression: a TRAIL to perdition? Oncogene. 2006;25:7333–5. doi: 10.1038/sj.onc.1209765. [DOI] [PubMed] [Google Scholar]
- 111.Greten FR, Arkan MC, Bollrath J, Hsu LC, Goode J, Miething C, Göktuna SI, Neuenhahn M, Fierer J, Paxian S, Van Rooijen N, Xu Y, O’Cain T, Jaffee BB, Busch DH, Duyster J, Schmid RM, Eckmann L, Karin M. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell. 2007;130:918–31. doi: 10.1016/j.cell.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107:7–11. doi: 10.1172/JCI11830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yamamoto Y, Gaynor RB. Therapeutic potential of inhibition of the NF-kappaB pathway in the treatment of inflammation and cancer. J Clin Invest. 2001;107:135–42. doi: 10.1172/JCI11914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Karin M, Yamamoto Y, Wang QM. The IKK NF-kappa B system: a treasure trove for drug development. Nat Rev Drug Discov. 2004;3:17–26. doi: 10.1038/nrd1279. [DOI] [PubMed] [Google Scholar]