

Abstract
Genes can maintain spatiotemporal expression patterns by long-range interactions between cis-acting elements. The cystic fibrosis transmembrane conductance regulator gene (CFTR) is expressed primarily in epithelial cells. An element located within a DNase I-hyper-sensitive site (DHS) 10 kb into the first intron was previously shown to augment CFTR promoter activity in a tissue-specific manner. Here, we reveal the mechanism by which this element influences CFTR transcription. We employed a high-resolution method of mapping DHS using tiled microarrays to accurately locate the intron 1 DHS. Transfection of promoter-reporter constructs demonstrated that the element displays classical tissue-specific enhancer properties and can independently recruit factors necessary for transcription initiation. In vitro DNase I footprinting analysis identified a protected region that corresponds to a conserved, predicted binding site for hepatocyte nuclear factor 1 (HNF1). We demonstrate by electromobility shift assays (EMSA) and chromatin immunoprecipitation (ChIP) that HNF1 binds to this element both in vitro and in vivo. Moreover, using chromosome conformation capture (3C) analysis, we show that this element interacts with the CFTR promoter in CFTR-expressing cells. These data provide the first insight into the three- dimensional (3D) structure of the CFTR locus and confirm the contribution of intronic cis-acting elements to the regulation of CFTR gene expression.
Keywords: CFTR, intronic enhancer, HNF1, enhancer-promoter interaction
Introduction
Structural and functional analysis of the human genome has revealed that cis-regulatory elements influencing transcription often exist some distance away from relevant basal promoters. These elements may include enhancers, silencers, insulators and locus control regions. Large-scale, functional genomics efforts such as the ENCyclopedia Of DNA Elements (ENCODE) project [1] have begun to annotate the broader context of the human genome by locating and defining these embedded regulatory elements. The cystic fibrosis transmembrane conductance regulator gene (CFTR), which when mutated causes the common genetic disease cystic fibrosis, is associated with a number of potential cis-regulatory sites. These elements often display specific changes in local chromatin structure, and we have previously evaluated the CFTR locus in many cell types for structural features including histone modifications, DNase I hypersensitivity and associated transcription factor binding [2–6]. Here, we use a high-resolution, tiled microarray-based assay, DNase-chip [7], to map DNase I-hypersensitive site (DHS) within 90 kb flanking the CFTRpromoter region. We detected a cell-type-specific DHS within the first intron of CFTR that corresponds to a regulatory element that we identified in earlier work [8, 9] and has been confirmed by others [10]. This element (known as 7/8 based on primer sets used to amplify the region [9]) was shown to positively regulate CFTR promoter activity specifically in intestinal cells both in vitro and in vivo. Removal of the element from a human CFTR yeast artificial chromosome (YAC) reduced expression levels of the human gene by about 60% in transgenic mice carrying the YAC but only within the epithelium of the small intestine [8]. Nonetheless, the mechanism of action of this key regulatory element has not yet been explored.
We now utilize this element in transient transfection experiments to show that it functions as a classical, tissue-specific enhancer and can also independently recruit general factors necessary for transcription initiation. To determine the nuclear factor(s) interacting with the regulatory sequence, we perform in vitro DNase I footprinting analysis, which reveals a significant protected sequence within which exists a conserved hepatocyte nuclear factor 1 (HNF1) binding site. Expression of HNF1a correlates with CFTR expression, and this transcription factor binds in vitro to another cluster of intronic DHS in CFTR[11]. In vivo, HNF1α contributes to the maintenance of normal mouse CFTR expression levels in the small intestine [11]. Here, we show that HNF1 binds to the intron 1 enhancer both in vitro, by elec-trophoretic mobility shift assay (EMSA), and in vivo, by chromatin immunoprecipitation (ChIP). Moreover, we use chromosome conformation capture (3C) analysis to show that this intronic enhancer interacts with the CFTR promoter in vivo, revealing a three-dimensional structure of the active CFTR locus.
Methods
Cell culture
The human colon carcinoma cell lines Caco2 [12] and HT29 [13], human bronchial epithelial cell line 16HBE14o- [14] and the human embryonic kidney cell line HEK293 [15] were grown in Dulbecco's Modified Eagle's Medium (DMEM) (Gibco, Carlsbad, CA, USA) supplemented with 5% fetal bovine serum (FBS) in a humidified atmosphere (37°C) of 5% CO2 and were passaged as necessary. Primary skin fibroblasts (ATCC: GM08333) were grown in MEM (Gibco) supplemented with 15% FBS.
Primer sequences
All primer sequences and locations used for DNase-chip, RT-PCR, plasmid cloning, mutagenesis, and 3C are listed in the Supporting Information.
DNase-chip
DNase-chip was performed as previously described, with modifications [7]. Briefly, 2–5 × 107 cells were lysed using 0.1% NP-40 buffer. Purified nuclei were exposed to increasing amounts of DNase I (0–30 U; NEB, Beverly, MA, USA), reactions were stopped with 0.1 M ethylenediaminetetraacetic acid (EDTA) and digested chromatin was embedded into InCert agarose plugs (Lonza, Walkersville, MD, USA). Chromatin digestion was determined by pulsed-field gel electrophoresis, and adequately digested samples were blunt-ended with T4 DNA polymerase (NEB). Chromatin was then extracted from agarose and the blunt ends were ligated to biotinylated linkers overnight. As a control, 25 μg of genomic DNA from the same cell type was ligated to linkers and processed in parallel. Chromatin was sonicated to generate 200–500 bp fragments and the biotinylated ends were captured with steptavidin Dynabeads (Invitrogen, Carlsbad, CA, USA). Sheared ends were blunted with T4 DNA polymerase and ligated to non-biotinylated linkers, and the samples were amplified with ligation-mediated PCR using primer oJW102C. PCR material was labelled with Cy5-dUTP and genomic DNA control was labelled with Cy3-dUTP and each was hybridized to ENCODE tiling arrays (NimbleGen, Madison, WI, USA; human genome build 17, May 2004). Hybridization data from three (Caco2 and fibroblasts) or two (16HBE14o- and HT29) experiments were analysed with ACME statistical software [16] using a window size of 500 bp and a threshold of 0.95.
Reverse transcriptase (RT)-PCR
RNA was isolated from Caco2, 16HBE14o–, HT29, HEK293 and primary skin fibroblasts using Trizol (Invitrogen), as per the manufacturer's instructions. cDNA was generated from 250 ng RNA using the Taqman® Reverse Transcription kit (Applied Biosystems). CFTR expression was assayed using E1 primer set [17]; as a control, (β-glucocerebrosidase (GenBank no. M16328) expression was assayed using primers GD67A and GD9B(11).
Plasmid construction
PCR amplification of the CFTR basal promoter and intron 1 putative regulatory elements (referred to as 7/8) was described previously [9]. Reporter constructs for transient expression were generated using the pGL3B vector (Promega, Madison, WI, USA). The CFTR basal promoter was cloned into Nhe I and Bgl II sites of the multiple cloning site, upstream of the luciferase reporter gene. The 7/8 fragment was digested out of the pCRII vector using BamH I, and one or two copies of this fragment were inserted in forward and reverse orientations into the enhancer site of the pGL3B CFTR promoter vector. All subcloned fragments were confirmed by sequencing.
Overlapping fragments of CFTR intron 1 were generated by PCR using cosmid F0424 [6] as a template. All primers included a 5′Mlu I restriction site, and all amplified products were ligated into the Mlu I site in the multiple cloning site of the promoter-less pGL3B vector upstream of the luciferase reporter gene. All cloned fragments were sequenced to confirm orientation and exclude PCR artefacts.
Mutated plasmid was generated using the QuikChange® Mutagenesis Kit (Stratagene, LaJolla, CA, USA), according to the manufacturer's protocol.
Transient transfections
Cells were plated 1 day prior to transfection in 96-well plates at a confluence of 5000 cells per well. The cells were transfected with 50 ng of the luciferase reporter DNA and 1 ng of Renilla expression vector (Promega) per well using 0.25 μl/well of lipofectin (Invitrogen), according to the manufacturer's protocol. The luciferase assay was performed using dual luciferase reagent (DLR) kit (Promega), according to the manufacturer's protocol. All experiments were corrected for transfection efficiency by normalizing the firefly luciferase reporter gene activity to Renilla luciferase activity. Statistical significance of the results was calculated by unpaired t-tests.
In vitro DNase I footprinting
The intron 1 DHS 7/8 fragment was PCR-amplified using primers TSR7 and TSR8 and then cloned into pCRII (Invitrogen). Cleavage of this vector with Xho I or Sac I enabled Klenow DNA polymerase fill-in with [α-32P]-dCTP to label the sense or antisense strand, respectively. DNase I footprinting experiments were then performed as described previously [9].
In vitro transcription/translation of HNF1
The mouse HNF1α expression plasmid was made by PCR by amplifying the gene from the pBJ5-mHNF1α vector kindly donated by Dr. Gerald Crabtree (Stanford University, Stanford, CA, USA). Human HNF1β was cloned using Caco2 cDNA and specific primers. The mHNF1α gene was subcloned into pCRII (Invitrogen) and hHNF1β into pCR-Script (Stratagene); each sequence was verified and cloned into the pcDNA3.1 vector (Invitrogen) using Not I and Hind III (mHNF1α) or Xho I and Not I (hHNF1β).
Wild-type HNF1α and HNF1β proteins were produced using TNT® T7 Quick Coupled Reticulocyte System (Promega), as described in the manufacturer's protocol. To confirm the presence of translated protein, 5 μl of the reaction volume was resolved on a 10% SDS-PAGE gel and transferred onto nitrocellulose. Immunoblot analysis was performed by probing first with 0.25 μg/ml goat anti-HNF1α primary antibody (Santa Cruz Biotech, Santa Cruz, CA, USA; sc-6547) and anti-goat horseradish peroxidase (HRP)-conjugated secondary antibody and subsequently, after stripping (Restore Western Blot Stripping Buffer [Pierce, Rockford, IL, USA]), with rabbit anti-HNF1β primary antibody (Santa Cruz, sc-22840) and anti-rabbit HRP-conjugated secondary antibody (Sigma, St. Louis, MO, USA). Immunoblot results were visualized using enhanced chemiluminescence (ECL) detection reagent (Amersham, Piscataway, NJ, USA).
Electrophoretic mobility shift assays
Complementary single-stranded oligonucleotides (see Fig. 4 for sequences) were annealed and labelled with [α-32P]-dCTP by fill-in reactions with Klenow DNA polymerase, prior to purification with microspin G-25 columns (Amersham Biosciences). Labelled DNA fragments (∼100,000 cpm) were incubated for 20 min. with Caco2 nuclear extract (5 μg) or in vitro translated (IVT) proteins (5 μl) in a final reaction volume of 20 μl containing 20 mM HEPES pH 7.5, 100 mM KCl, 10 mM MgCl2, 1 mM EDTA, 12% (v/v) glycerol and 1 μg poly(dI-dC). For competition experiments, the proteins were pre-incubated with unlabelled oligonucleotide duplexes at 100-fold excess molar concentrations, for 20 min. at room temperature before addition of labelled DNA. For supershifts, a similar pre-incubation with nuclear extract or IVT proteins was included with addition of 1 μg of antibodies against HNF1α (Santa Cruz; sc-6547) or HNF1β (Santa Cruz; sc-7411). The samples were resolved on a 4% polyacrylamide gel at 4°C for 2 hrs at 300 V in a 0.5× TBE buffer (1 × TBE is 89 mM Tris, 89 mM boric acid and 2 mM EDTA). Following electrophoresis, the gels were dried and exposed to a Phosphormager screen (GE Healthcare, Piscataway, NJ, USA).
4.
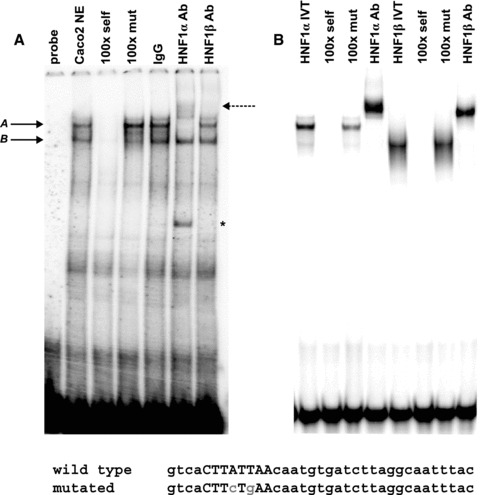
In vitro binding of HNF1 to 7/8 enhancer. EMSA using Caco2 nuclear extracts (A) or in vitro translated HNF1 proteins (B). (A) Two major protein complexes designated A and B with solid arrowheads are formed with a radiolabelled oligo -nucleotide spanning the 85 + 10 kb DHS region. Competition with 100-fold molar excess of unlabelled wild-type (100 × self) and mutant (100 × mut) probes is indicated. HNF1-specific antibodies (to either α or β) were used in supershift analysis, and free radiolabelled oligonucleotide (probe) and goat IgG were used as controls. Dashed arrow indicates HNF1α supershifted band in panel A; indicates non-specific band detected in HNF1α lane.
Chromatin immunoprecipitation (ChIP)
In total, 1 × 107 post-confluent Caco2 cells were trypsinized, resuspended in DMEM and cross-linked with 1% formaldehyde for 10 min. The cross-linking was stopped by the addition of glycine to 0.125 M. The cells were washed with cold PBS and lysed in 1 ml of 1% SDS, 10 mM EDTA, 50 mM Tris/HCl, pH 8.1 and 1 × protease inhibitor cocktail (Roche, Indianapolis, IN, USA). Chromatin was sonicated to an average size of 500 bp.
Immunoprecipitations were performed overnight at 4°C using 200 μl chromatin that was diluted 1:10 with ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl, pH 8.1 and 167 mM NaCl) and 10 μg of either rabbit anti-HNF1 antibody (Santa Cruz; sc-8986) or rabbit IgG (Santa Cruz; sc-2027). The complexes were collected using 60 μl Protein A/Salmon sperm agarose beads (Upstate, Billerica, MA, USA), washed several times according to the manufacturer's protocol and eluted with 1% SDS and 0.1 M NaHCO3. Cross-links were reversed at 65°C overnight, and the samples were treated with RNase (10 μg/ml) and proteinase K (40 μg/ml) before phenol/chloroform extraction and ethanol precipitation. The samples were resuspended in 0.5× TE and enrichment was analysed using Taqman® primer and probe sets and a Fast 7500 Real-Time PCR machine (Applied Biosystems).
Chromosome conformation capture (3C)
Chromosome conformation capture (3C) was performed as described previously [18, 19], with minor modifications. Briefly, 1 × 107 cells were fixed with 2% formaldehyde for 10 min. at room temperature. The cells were lysed in 5 ml cold lysis buffer (10 mM Tris pH 8, 10 mM NaCl, 0.2% NP-40 and 1 × protease inhibitor cocktail) and the nuclei were collected by centrifugation. Following extraction with 0.3% SDS, chromatin was digested overnight with 2000 U Hind III. Ligations were performed in a total reaction volume of 6.5 ml, using 100 U T4 DNA ligase (Roche), and incubated at 14°C for 4 hrs followed by 30 min. at room temperature. Cross-links were reversed by proteinase K treatment at 65°C overnight. The samples were purified by phenol/chloroform extraction followed by ethanol precipitation and then resuspended in 150 μl H2O. The concentration of each sample was determined by SYBR green quantitative PCR (qPCR), using the B13F/B13R primer set (amplicon found within a Hind III fragment; see Supporting Information), and each sample was compared with a genomic DNA reference of known concentration. The samples were subsequently diluted to a concentration of 100 ng/|jJ. A Taqman® probe and reverse primer was designed that was specific to the Hind III fragment encompassing the CFTR promoter. Multiple forward primers were then designed that were each specific to different Hind III fragments across the CFTR locus. Using a dilution series of digested/re-lig-ated bacterial artificial chromosome (BAC) DNA template, each forward primer was demonstrated to function with the ‘fixed’ Taqman® probe and reverse primer to amplify with approximately 100% efficiency. To quantify ligation events within 3C samples, 200 ng of 3C template were used per Taqman® qPCR reaction. The ligation efficiency at each site was corrected for the interaction between two Hind III fragments within the ubiquitously expressed excision repair cross-complementing rodent repair deficiency complementation group 3 (ERCC3) locus, which has been reported to adopt the same spatial conformation in different tissues [20–22].
Results
A tissue-specific DHS exists 10 kb into the first intron of CFTR
Previous DHS-mapping experiments using restriction enzyme digestion and Southern blotting with labelled CFTR probes revealed a prominent DHS approximately 10 kb into the first intron of CFTR at 185 + 10 kb (185 is the last base of exon 1), and weaker DHS at 185 + 12 kb and at + 14 kb. The DHS were only apparent in certain cell types, including the human colon carcinoma cell lines Caco2 and HT29, the human pancreatic adenocarcinoma cell line Capan1 and human primary male genital duct cells [9]. Each of these cell types express significant amounts of CFTR mRNA. However, in other cell types that express CFTR, in particular those derived from the airway epithelium, such as the human bronchial epithelial cell line 16HBE14o– (Harn's group, unpublished observations), no significant DHS was evident in this region. To more accurately define these intron 1 DHS, we used DNase-chip, a high-resolution assay for DHS using tiled NimbleGen microarrays, which can define DHS locations much more precisely than is possible by conventional methods. In Caco2 and HT29 cells, a single strong DHS was apparent 10 kb into the first intron (Fig. 1A).
1.
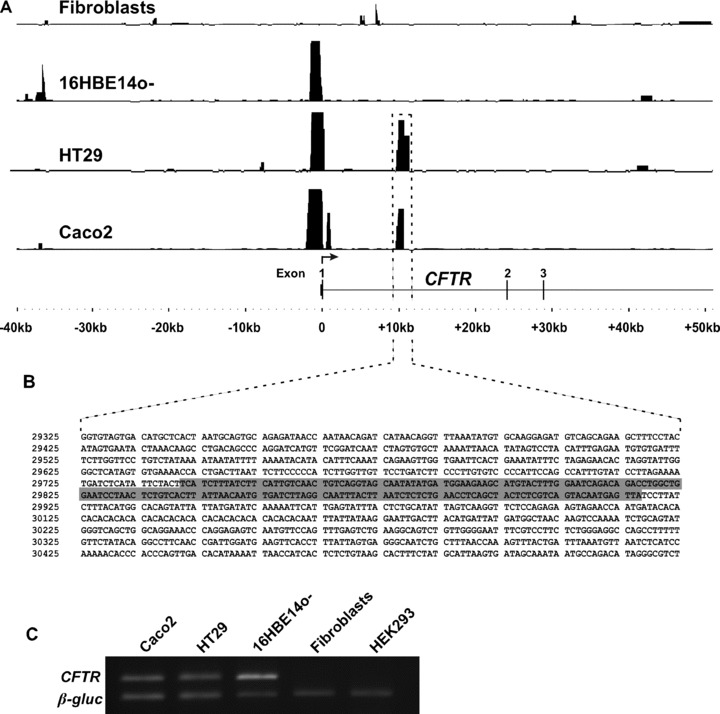
DNase-chip detects cell-type-specific DHS 10 kb into the first intron of CFTR. (A) Shown are DHS tracks in four different cell types: CFTR- primary human skin fibroblasts, CFTR+ human bronchial epithelial cell line 16HBE14o- and CFTR+ human colon carcinoma cell lines Caco2 and HT29. Significant peaks representing DHS are apparent in the 5′ promoter region of CFTR+ cells, whereas Caco2 and HT29 cells display another significant DHS within the first intron. Peak height is a measurement of –log10 (P-value) between 0 and 16, as determined by ACME (see Methods). The sequence associated with the identified intronic DHS is shown in panel B. Highlighted sequence corresponds to previously identified positive regulatory element 7/8 [9]. Sequence is numbered based on GenBank reference sequence AC000111. (Sequence corresponds to bases 116916908–116918007 of human genome build 17.) (C) CFTR expression in cell types used in this study measured by RT-PCR analysis compared with a β-glucocerebrosidase housekeeping gene control.
This DHS corresponds exactly to the previously defined 185 + 10 kb DHS and overlaps the 7/8 regulatory element [9] previously shown to positively regulate CFTR promoter activity (Fig. 1B). Interestingly, though a strong DHS is evident at the CFTR promoter in Caco2, HT29, and 16HBE14o- cells, the intron 1 DHS is only apparent in the intestinal cells. Moreover, the 185 + 10 kb DHS was the only significant DHS observed within the first intron, indicating that either this is the primary region within the first intron containing cis-regulatory elements or that the cells have evolved in culture and the +12 and +14 kb DHS are now less evident.
The 185 + 10 kb DHS contains a cell-type-specific enhancer of the CFTR promoter
We previously showed that the core of the cis-element within the 185 + 10 kb DHS lies within the 176 bp 7/8 fragment that augments the activity of the basal CFTR promoter in transient transfections of Caco2 cells [9]. These assays showed that this element increased promoter activity both alone and in the context of a larger cloned fragment, demonstrating its function in a position-independent manner. However, we did not determine whether this core had the classical enhancer property of functioning in each orientation. To assay for enhancer activity in transient transfection experiments, we generated a series of pGL3-derived constructs in which a 787 bp CFTR basal promoter fragment drives the expression of the firefly luciferase reporter gene. Each construct has a derivative of the 7/8 regulatory element cloned into the enhancer site of this vector. Single and double copies of 7/8 were cloned in both forward and reverse orientations. To determine tissue-specificity of 7/8 activity, these constructs were transfected into 16HBE14o- bronchial epithelial cells and Caco2 cells that both express CFTR and into human embryonic kidney cells (HEK293) that do not.
These luciferase assays show that one copy of the 7/8 element increases the firefly luciferase activity three-fold over the level of the CFTR promoter alone in Caco2 cells (Fig. 2A). These experiments were repeated in HT29 cells, with similar results (data not shown). This effect is cell-type-specific, as it is not observed in 16HBE14o- cells or HEK293 cells. Equivalent results were seen when the 7/8 fragment was cloned in a reverse orientation, suggesting that it is, indeed, a true enhancer element. Additionally two copies of the 7/8 enhancer in either orientation show a syner-gistic effect, increasing CFTR promoter activity by about 10-fold over the single copy element.
2.
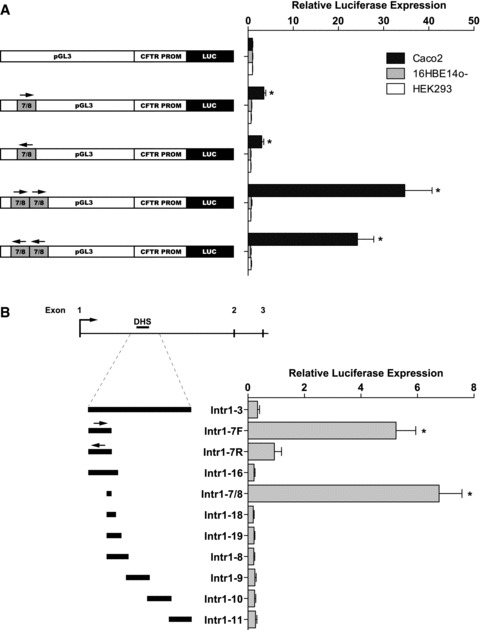
Transient transfections of pGL3 reporter vectors reveal enhancer and ‘promoter’ activity. (A) Caco2, 16HBE14o-, and CFTR- human embryonic kidney cell line HEK293 were transfected with constructs with 7/8 cloned into the enhancer site of the vector as a single copy or double copy and either in the forward or in the reverse orientation. A 787 bp frag-ment corresponding to the CFTR basal promoter used previously [9] was cloned into the promoter site of pGL3B. Renilla luciferase vector was used as a transfection control in all experiments; data shown are relative to the CFTR basal promoter vector without inserted enhancer sequence. (B) Intron 1 fragments cloned into the promoter site of pGL3B were transfected into Caco2 cells. Data shown are relative to CFTR basal promoter vector; error bars represent standard error of the mean (n= 12), *P< 0.01 using unpaired t-tests to analyse the difference from promoter-only vector.
The CFTR 7/8 element can recruit factors necessary for transcription initiation
Having confirmed the enhancer function of the 7/8 element, we next evaluated whether regions of the 185 + 10 kb DHS could recruit the necessary factors for transcriptional activation independent of the basal CFTR promoter. We cloned overlapping fragments of intron 1, including the 185 + 10 kb DHS region into the promoter-less pGL3B vector upstream of the firefly luciferase reporter gene. These plasmids were transiently transfected into Caco2 cells and assayed for their ability to activate reporter gene transcription (Fig. 2B). The construct containing one copy of the 7/8 sequence (pGL3B Intr1–7/8) showed strong ‘promoter’ activity, about seven-fold greater than that of the basal CFTR promoter. A construct containing 7/8 with an additional 882 bp of upstream sequence (pGL3B Intr1–7F) showed a similar level of activity. This activity was abolished when this fragment was cloned in a reverse orientation (pGL3B Intr1–7R), suggesting directional function of the 7/8 element. Constructs containing 7/8 with additional downstream sequences did not show ‘promoter’ activity, which may indicate that the inclusion of sequence downstream of 7/8 disrupts luciferase expression in the context of these transient assays, either by recruiting repressive factors or by inhibiting translation of the luciferase transcript.
HNF1 binds an element within the CFTR intron 1 DHS in vitro and in vivo
Previous studies demonstrated that the 185 + 10 kb DHS element binds specific protein complexes in Caco2 cells, though the identity of the factors was not revealed. To identify potential transcription factor-binding sites across the DHS 185 + 10 kb region, in vitro DNase I footprinting was performed (Fig. 3A). Two protected regions were evident upon binding of Caco2 nuclear extracts to a radiolabelled probe spanning the sense strand of 7/8. The protected regions encompass 30 (footprint 1, FP1) and 23 (footprint 2, FP2) nucleotide sequences and suggest a complex pattern of DNA-protein interactions within this site. These data are consistent with footprints identified in our previous analysis of this region, utilizing, in addition to Caco2 cells, nuclear extracts from primary male genital duct epithelial cells [9]. At the time of the original analysis, we were unable to predict the candidate transcription factors involved at this site due to the relative paucity of defined binding sites in available databases. We again performed in silico analysis for putative transcription factor-binding sites using MatInspector (http://www.genomatix.de). Within FP1, a strong predicted binding site for HNF1, which recognizes the consensus sequence GTTAATNATTANC [23], was identified on the antisense strand and it resides within a region of high sequence conservation between a number of species (Fig. 3B), suggesting the presence of a functional element.
3.
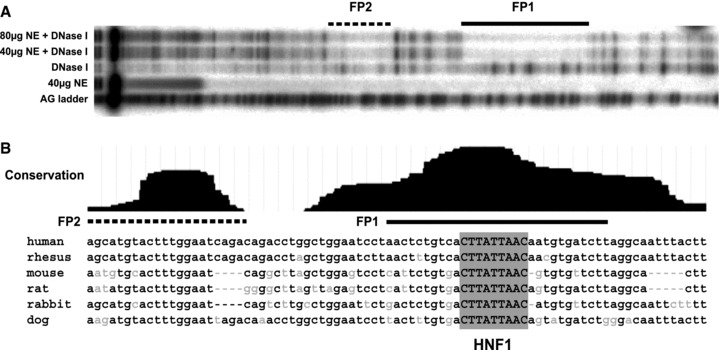
In vitro DNase I footprinting of 7/8 enhancer reveals two distinct protected regions. (A) A/G ladder used as reference (first lane); samples incubated with increasing amounts of Caco2 nuclear extracts (NE) and radiolabelled sense strand of 7/8 sequence. Lanes with NE alone and DNase I alone were used as controls. (B) Sequence corresponding to the two protected regions. Conservation track based on phastCons MultiZ alignment of 17 vertebrate species, available on ENCODE browser (http://www.genome.ucsc.edu/ENCODE). In silicoanalysis revealed a highly conserved HNF1 site on the antisense strand within FP1.
To determine the ability of HNF1 to bind at the 185 + 10 kb DHS, EMSAs were carried out using nuclear extracts from Caco2 cells. The nuclear extracts were incubated with an α-32P-labelled double-stranded DNA probe that spans the consensus HNF1-binding sequence, and the mobility of DNA-protein complexes was examined. Two major DNA-protein complexes (A and B) were observed, as indicated by retardation in gel mobility, suggesting that protein-DNA interactions occur at this site (Fig. 4A). These interactions were specific, as the binding of protein complexes was effectively competed with a 100-fold molar excess of the corresponding unlabelled probe.
To determine whether HNF1 proteins are present within the complexes bound to DNA, we performed supershift analysis using antibodies directed against HNF1α and HNF1β. Both factors bind to the same consensus DNA sequence and can either homodimer-ize or heterodimerize [23]. Both HNF1α and HNF1β are produced in Caco2 cells, although the a form is produced at higher levels than the β form (data not shown). The migration of complex A was retarded upon pre-incubation of the nuclear extracts with an antibody specific for HNF1α alone, but not with one specific for HNF1β, suggesting both that HNF1α is a dominant factor present within the Caco2 protein complexes that are formed within the 7/8 enhancer and that other factors are also present (Fig. 4A). The specificity of DNA–protein interactions with HNF1 was confirmed using a probe with a 2 bp mutation within the core of the HNF1-binding site. Pre-incubation of the nuclear extracts with the mutated unlabelled probe competitor had no effect on complex A, suggesting that binding of these factors to DNA requires an intact HNF1-binding motif (Fig. 4A). However, the faster migrating complex B may include other DNA-binding proteins. No clear consensus site was identified in FP2, although in silico analysis with the Alibaba 2.1 (http://www.gene-regulation.com) transcrip tion factor search engine, but not with MatInspector (http://www.genomatix.de), predicted a CCAAT enhancer binding protein (C/EBP)-binding site. EMSA analysis of FP2 confirmed the formation of a DNA-protein complex, yet it was not supershifted with a C/EBP-specific antibody (data not shown). Moreover, no protein complexes were observed when an oligonucleotide probe spanning both FP1 and FP2 with the HNF1 site mutated was radio-labelled and used in EMSA experiments with Caco2 nuclear extracts (not shown), suggesting that destroying the HNF1-binding site eliminates possible binding of any other transcription factors in this region.
To further confirm the identity of the proteins and their direct DNA binding at 7/8, EMSA experiments were performed using IVT HNF1α and HNF1β proteins (Fig. 4B). In this case, both factors were able to bind and form complexes with the DNA probe. Pre-incubation of IVT proteins with specific antibodies also resulted in the supershift of both HNF1α- and HNF1 β-containing complexes. This suggests that the recombinant HNF1β protein in addition to HNF1α can bind directly to this DNA element, although not in nuclear extracts where other factors are present.
To demonstrate in vivo binding of HNF1 to this element, ChIP was performed using an antibody that recognizes an epitope present on both forms of HNF1, as the specific antibodies used for EMSA were not suitable for ChIP analysis. Several regions of CFTRwere analysed for HNF1 binding in Caco2 cells, including 2 kb upstream of the translation start site, the CFTR basal promoter, +5.7 kb into the first intron, the 185 + 10 kb DHS region and + 15 kb into the first intron (Fig. 5). The a1-antitrypsin (AAT) promoter provided a positive control for HNF1 binding [24, 25]. The intronic enhancer element located in the 185 + 10 kb DHS is significantly enriched in HNF1 immunoprecipitation of Caco2 chromatin over an isotype-matched IgG control, establishing in vivo binding of HNF1 to this element. It is also apparent that the basal promoter region of CFTR is enriched at about the same level as the AAT promoter control. Although at low stringency, the AliBaba 2.1 program (http://www.gene-regulation.com) predicts three potential HNF1-binding sites within 1 kb of the CFTR basal promoter, whereas the MatInspector (http://www.genomatix.de) program does not identify these sites. Moreover, none of these sites bound HNF1 in vitro when evaluated by EMSA (data not shown). Thus, this enrichment could be a result of a direct interaction between the CFTR basal promoter and the distal elements that directly bind HNF1, such as the intronic enhancer at 185 + 10 kb. HNF1 ChIP assays on 16HBE14o- and skin fibroblast chromatin showed no enrichment within the intronic DHS (data not shown).
5.
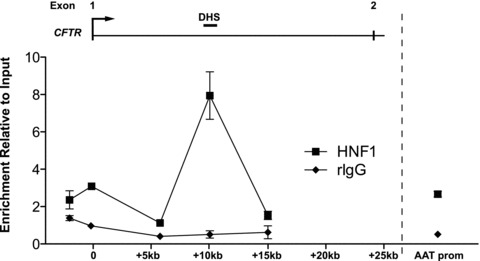
In vivo binding of HNF1 to 7/8 enhancer. ChIP analysis using rabbit HNF1 antibody or rabbit IgG as control and cross-linked Caco2 chromatin, followed by realtime PCR using Taqman® primer and probe sets at the indicated positions of the CFTR locus. A primer/probe set at the a1-antit-rypsin (AAT) promoter was used as a positive control for HNF1 binding. Each PCR was normalized to 18s rRNA levels and shown relative to an input control. Error bars represent standard error of the mean (n= 4). Lines are used to connect assayed sites and do not necessarily indicate enrichment levels between the sites.
Destroying the HNF1-binding site abrogates enhancer and ‘promoter’ activity of 7/8
In Fig. 4, we showed that the alteration of two bases in the consensus binding site for HNF1 abolished protein-DNA interactions in EMSA experiments. Next, we mutated the same two bases within the HNF1-binding site in fragment 7/8 cloned into either the enhancer or the promoter site of the pGL3 reporter vector and assayed them for activity (Fig. 6). Figure 6 shows that destruction of the HNF1 site completely abolishes both the enhancer (Fig. 6A) and the ‘promoter’ (Fig. 6B) activity.
6.
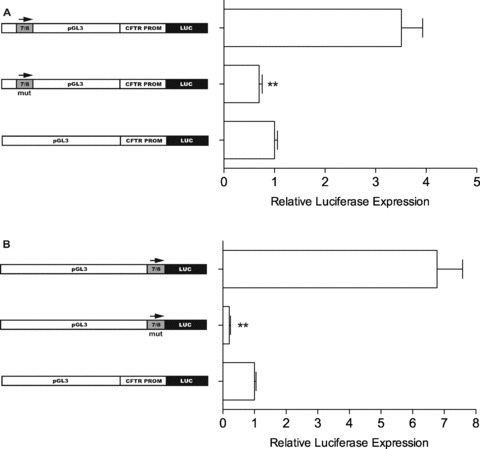
Mutations in 7/8 abolish enhancer and ‘promoter’ activity. (A) Transient transfections of Caco2 cells using pGL3 reporter vector with inserted CFTR basal promoter and 7/8 in enhancer site. Middle bar represents construct with two bases mutated within HNF1 site (same bases changed with mutated EMSA probe). (B) 7/8 cloned as a ‘promoter’ (top bar) and with HNF1 site mutated (middle). Renilla luciferase vector used as transfection control in all experiments; normalized luciferase activity shown relative to pGL3 with CFTR basal promoter alone. Error bars represent standard error of the mean (n= 12), **P < 0.01 using unpaired t-tests comparing mutated and wild-type vectors.
The CFTR intronic enhancer directly interacts with the promoter
As our ChIP analysis suggested that the HNF1-containing complex forming at the 7/8 enhancer might also be interacting with the CFTR promoter, we performed 3C using a Taqman® reverse primer and probe within a Hind III fragment encompassing the basal CFTR promoter and forward primers on a 5′ fragment and fragments within the first, third and seventh introns (Fig. 7). Using 3C, we measured the relative interaction frequency between the CFTR basal promoter and both 5′ and 3′ distal regions using real-time PCR in Caco2 (intestinal) and 16HBE14o- (airway) cells that express CFTR and in skin fibrob-lasts that do not. In 16HBE14o- cells and fibroblasts, in which no interaction between the first intron and the promoter is expected (because the intron 1 DHS is absent), the interaction frequency gradually decreases with Hind III fragments that are further from the promoter fragment, as the chance of random interactions decreases. However, in Caco2 cells in which the 7/8 enhancer is active, interaction frequencies between the promoter and the intron 1 fragments are all considerably elevated, and the typical gradual decrease in interaction frequency does not occur, revealing that, in vivo, these fragments are directly interacting with the promoter. The fragment with a Hind III site closest to the observed intron 1 DHS (see DHS bar above Fig. 7, panel A) displayed a significantly higher interaction frequency than a fragment lying equidistant to the basal promoter on the opposite side from intron 1, at approximately 10 kb 5′. This demonstrates that the observed interaction frequency of the first intron is not a result of chance random interactions.
7.
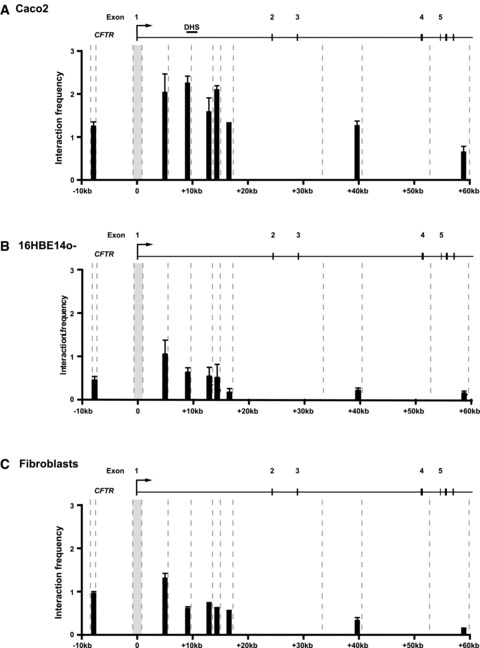
The 7/8 enhancer directly interacts with the CFTR promoter. Chromosome conformation analysis of CFTR+ Caco2 (A), CFTR+ 16HBE14o- (B) and CFTR- primary skin fibroblasts (C) show higher interaction of the first intron with the promoter in cells with the intron 1 DHS. Cross-linked chro-matin from each cell type was digested with Hind III (sites indicated by dashed lines) and re-ligated. The interaction frequency between a fixed Hind III fragment at the CFTR promoter and Hind III fragments 5′ and within the first, third and seventh introns was measured by Taqman® quantitative PCR. Each reaction was normalized to a control region in the ERCC3gene, as previously described [31]. The amplification efficiencies of all primer sets were verified using Hind III-digested CFTR and ERCC3 BACs. Each experiment was repeated at least twice; a single representative experiment is shown with PCR reactions performed in triplicate. Error bars represent standard error of the mean (n= 3).
Discussion
Our data suggest a model whereby HNF1 binds to the intronic enhancer at 185 + 10 kb and mediates a loop that functions to augment the recruitment of RNA polymerase II and other general transcription factors to the CFTR promoter, thus increasing CFTR transcription in intestinal epithelial cells. Chromatin loop structures have been shown to play a role in the differential expression of genes at a number of loci and in diverse cellular model systems including erythroid differentiation and T-cell development [26–31]. These loops contain cis-regulatory elements that often exist at large genomic distances from the interacting promoter. Several nuclear factors have been implicated in mediating these higher-order chromatin structures, including factors that are expressed both ubiquitously and in a tissue-specific manner. The expression of CFTR is, with a few exceptions, limited to the chloride-secreting epithelia of various tissues [17, 32–35], yet extensive analysis of the greater CFTR promoter region has not revealed the elements that confer this spatial expression pattern [36–38]. The promoter, which lacks a TATA box, contains potential regulatory elements such as AP-1- and Sp1-binding sites [38], a cAMP response element (CRE) that binds CRE-binding protein (CREB) upon protein kinase A activation [39, 40] and an inverted CCAAT element (Y box) that binds CCAAT enhancer binding protein (C/EBP) [41]. Additionally, NF-κB has been shown to bind about 1 kb upstream of the transcriptional start site and positively regulate transcription [42], and a polymorphic YY1 site exists within the core promoter that regulates transcription, possibly via interaction with serum response factor (SRF) [43]. Yet none of these defined transcriptional control mechanisms are responsible for the tissue-specific expression pattern of the gene. Thus, it is likely that cis-regulatory elements exist outside the core promoter, and these can often be detected by locating the DHS within and around the gene. We have consistently observed a DHS 10 kb into the first intron of CFTR, first by conventional Southern blot analysis [9], and now by the higher-resolution DNase-chip method shown in Fig. 1. This DHS is significant as it is only apparent in epithelial cell types of intestinal, pancreatic duct or genital duct origin, all three of which transcribe the gene at high levels. The first introns of many genes have been shown to play a role in influencing promoter activity, including several regulated by HNF1 [44, 45]. Although the mechanisms accounting for this have not been shown, DNA looping and physical interaction of cis-elements bound by HNF1 have been shown to occur [46]. Our data demonstrate that HNF1 mediates a chromatin loop structure between the CFTR promoter and an intronic enhancer, and although it is not definitive from our ChIP data which form of HNF1 is directly binding to the enhancer, it is generally accepted that HNF1α homod-imers have a greater ability to positively regulate transcriptional activity [47]. Moreover, we have previously demonstrated an important role for HNF1α in CFTR expression in the intestinal epithelium using HNF1α knockout mice [11]. HNF1 has also been shown to play a role in remodelling chromatin structure by altering nucleosome occupancy at specific sites [48]. Thus, HNF1 binding to the CFTR intronic enhancer may be playing several roles: recruiting factors responsible for modifying the chromatin structure of the enhancer region so that other nuclear factors may bind, and/or directly recruiting general transcription factors to the CFTR promoter to increase transcriptional activity. Evidence for HNF1 playing the latter role is seen in the ability of the 7/8 enhancer to enable transcription initiation when cloned into the ‘promoter’ site in transient transfection assays. There are, however, no data to suggest that this intronic element acts as a classical promoter within the context of chromatin. Thus, we suggest that the in vitro activity is a specific feature of certain enhancers that coordinate transcription with endogenous promoters. Indeed, other known regulatory elements within the CFTR locus also exhibit this activity in transient assays (data not shown). Several co-factors have been shown to associate with HNF1α, including the histone acetyltransferase complex CREB-binding protein CBP/p300 and the CBP-associated factor P/CAF [49], and these may also play a role in CFTR promoter activity. In a recent study, we measured the levels of histone acetylation and methylation at the CFTR promoter and found enrichment of histone marks associated with active chromatin in both Caco2 and primary male genital duct cells [2]. Our in vitro footprinting analysis of the intron 1 enhancer suggests that other nuclear factors are associated with this region, yet at this time, we have not identified which factors are cooperating with HNF1.
Thus, the intron 1 enhancer element is likely the focus for a complex configuration of multiple factors including HNF1. An additional regulatory element lying 2 kb 3′ to the site that we characterize here was recently also shown to bind HNF1 in Caco2 cells [50]. This element likely corresponds to the weak DHS that we observed previously at 185 + 12 kb [9]. Yet, our current analysis utilizing tiled microarrays clearly shows that the 185 + 10 kb site is a much stronger DHS and is thus most likely the site of greatest regulatory activity in the first intron.
It is also probable that other cis-regulatory elements exist elsewhere within the CFTR locus that play a coordinating role in regulating transcription with the 7/8 enhancer. Our previous analysis of the CFTR locus revealed a number of putative cis-regulatory elements, some of which also bind to HNF1 [11]. Hence, it is likely that, although the 185 + 10 kb intronic enhancer plays a key role in augmenting the recruitment of general transcription factors and RNA polymerase II to the promoter in the intestinal epithelium, other distal elements also physically interact in a complex multiple-loop configuration that includes the CFTR promoter in a tran-scriptionally active chromatin hub. Understanding the mechanism of action of CFTR enhancer elements and associated nuclear factors may provide us with novel tools or methods to modulate CFTR gene expression for therapeutical benefit.
Acknowledgments
This work was supported in part by a Cystic Fibrosis Foundation post-doctoral fellowship (MS), the Cystic Fibrosis Foundation (Harris07P0), a Northwestern University Alumnae award (CJO) and also the Children's Memorial Research Center. We thank Dr. Gerald Crabtree (Stanford University, Stanford, CA) for the HNF1α expression plasmid and Jenny Kerschner for her contribution.
Supporting Information
Primers
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.The ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–40. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- 2.Blackledge NP, Carter EJ, Evans JR, Lawson V, Rowntree RK, Harris A. CTCF mediates insulator function at the CFTR locus. Biochem J. 2007;408:267–75. doi: 10.1042/BJ20070429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nuthall HN, Moulin DS, Huxley C, Harris A. Analysis of DNase-I-hypersensitive sites at the 3′ end of the cystic fibrosis trans-membrane conductance regulator gene (CFTR) Biochem J. 1999;341:601–11. [PMC free article] [PubMed] [Google Scholar]
- 4.Phylactides M, Rowntree R, Nuthall H, Ussery D, Wheeler A, Harris A. Evaluation of potential regulatory elements identified as DNase I hypersensitive sites in the CFTR gene. Eur J Biochem. 2002;269:553–9. doi: 10.1046/j.0014-2956.2001.02679.x. [DOI] [PubMed] [Google Scholar]
- 5.Smith AN, Wardle CJ, Harris A. Characterization of DNASE I hypersensitive sites in the 120 kb 5′ to the CFTR gene. Biochem Biophys Res Commun. 1995;211:274–81. doi: 10.1006/bbrc.1995.1807. [DOI] [PubMed] [Google Scholar]
- 6.Smith DJ, Nuthall HN, Majetti ME, Harris A. Multiple potential intragenic regulatory elements in the CFTR gene. Genomics. 2000;64:90–6. doi: 10.1006/geno.1999.6086. [DOI] [PubMed] [Google Scholar]
- 7.Crawford GE, Davis S, Scacheri PC, Renaud G, Halawi MJ, Erdos MR, Green R, Meltzer PS, Wolfsberg TG, Collins FS. DNase-chip: a high-resolution method to identify DNase I hypersensitive sites using tiled microarrays. Nat Methods. 2006;3:503–9. doi: 10.1038/NMETH888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rowntree RK, Vassaux G, McDowell TL, Howe S, McGuigan A, Phylactides M, Huxley C, Harris A. An element in intron 1 of the CFTR gene augments intestinal expression in vivo. Hum Mol Genet. 2001;10:1455–64. doi: 10.1093/hmg/10.14.1455. [DOI] [PubMed] [Google Scholar]
- 9.Smith AN, Barth ML, McDowell TL, Moulin DS, Nuthall HN, Hollingsworth MA, Harris A. A regulatory element in intron 1 of the cystic fibrosis transmem-brane conductance regulator gene. J Biol Chem. 1996;271:9947–54. doi: 10.1074/jbc.271.17.9947. [DOI] [PubMed] [Google Scholar]
- 10.Mogayzel PJ, Ashlock MA. CFTR intron 1 increases luciferase expression driven by CFTR 5′-flanking DNA in a yeast artificial chromosome. Genomics. 2000;64:211–5. doi: 10.1006/geno.2000.6119. [DOI] [PubMed] [Google Scholar]
- 11.Mouchel N, Henstra SA, McCarthy VA, Williams SH, Phylactides M, Harris A. HNF1alpha is involved in tissue-specific regulation of CFTR gene expression. Biochem J. 2003;378:909–18. doi: 10.1042/BJ20031157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fogh J, Wright WC, Loveless JD. Absence of HeLa cell contamination in 169 cell lines derived from human tumors. J Natl Cancer Inst. 1977;58:209–14. doi: 10.1093/jnci/58.2.209. [DOI] [PubMed] [Google Scholar]
- 13.Huet C, Sahuquillo-Merino C, Coudrier E, Louvard D. Absorbtive and mucus-secreting subclones isolated from a multipotent intestinal cell line (HT-29) provide new models for cell polarity and terminal differentiation. J Cell Biol. 1987;105:345–57. doi: 10.1083/jcb.105.1.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cozens AL, Yezzi MJ, Kunzelmann K, Ohrui T, Chin L, Eng K, Finkbeiner WE, Widdicombe JH, Gruenert DC. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am J Respir Cell Mol Biol. 1994;10:38–47. doi: 10.1165/ajrcmb.10.1.7507342. [DOI] [PubMed] [Google Scholar]
- 15.Simmons NL. A cultured human renal epithelioid cell line responsive to vasoac-tive intestinal peptide. Exp Physiol. 1990;75:309–19. doi: 10.1113/expphysiol.1990.sp003406. [DOI] [PubMed] [Google Scholar]
- 16.Scacheri PC, Crawford GE, Davis S. Statistics for ChIP-chip and DNase hyper-sensitivity experiments on NimbleGen arrays. Meth Enzymol. 2006;411:270–82. doi: 10.1016/S0076-6879(06)11014-9. [DOI] [PubMed] [Google Scholar]
- 17.Harris A, Chalkley G, Goodman S, Coleman L. Expression of the cystic fibro-sis gene in human development. Development. 1991;113:305–10. doi: 10.1242/dev.113.1.305. [DOI] [PubMed] [Google Scholar]
- 18.Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–11. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 19.Hagège H, Klous P, Braem C, Splinter E, Dekker J, Cathala G, De Laat W, Forné T. Quantitative analysis of chromosome conformation capture assays (3C-qPCR) Nat Prot. 2007;2:1722–33. doi: 10.1038/nprot.2007.243. [DOI] [PubMed] [Google Scholar]
- 20.De Laat W, Grosveld F. Spatial organization of gene expression: the active chro-matin hub. Chromosome Res. 2003;11:447–59. doi: 10.1023/a:1024922626726. [DOI] [PubMed] [Google Scholar]
- 21.Drissen R, Palstra RJ, Gillemans N, Splinter E, Grosveld F, Philipsen S, De Laat W. The active spatial organization of the beta-globin locus requires the transcription factor EKLF. Genes Dev. 2004;18:2485–90. doi: 10.1101/gad.317004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palstra RJ, Tolhuis B, Splinter E, Nijmeijer R, Grosveld F, De Laat W. The beta-globin nuclear compartment in development and erythroid differentiation. Nat Genet. 2003;35:190–4. doi: 10.1038/ng1244. [DOI] [PubMed] [Google Scholar]
- 23.Ryffel GU. Mutations in the human genes encoding the transcription factors of the hepatocyte nuclear factor (HNF)1 and HNF4 families: functional and pathological consequences. J Mol Endocrinol. 2001;27:11–29. doi: 10.1677/jme.0.0270011. [DOI] [PubMed] [Google Scholar]
- 24.Courtois G, Morgan JG, Campbell LA, Fourel G, Crabtree GR. Interaction of a liver-specific nuclear factor with the fib-rinogen and alpha 1-antitrypsin promoters. Science. 1987;238:688–92. doi: 10.1126/science.3499668. [DOI] [PubMed] [Google Scholar]
- 25.Hu C, Perlmutter DH. Regulation of alpha1-antitrypsin gene expression in human intestinal epithelial cell line Caco-2 by HNF-1alpha and HNF-4. Am J Physiol. 1999;276:1181–94. doi: 10.1152/ajpgi.1999.276.5.G1181. [DOI] [PubMed] [Google Scholar]
- 26.Cai S, Lee CC, Kohwi-Shigematsu T. SATB1 packages densely looped, tran-scriptionally active chromatin for coordinated expression of cytokine genes. Nat Genet. 2006;38:1278–88. doi: 10.1038/ng1913. [DOI] [PubMed] [Google Scholar]
- 27.Jing H, Vakoc CR, Ying L, Mandat S, Wang H, Zheng X, Blobel GA. Exchange of GATA factors mediates transitions in looped chromatin organization at a devel-opmentally regulated gene locus. Mol Cell. 2008;29:232–42. doi: 10.1016/j.molcel.2007.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song SH, Hou C, Dean A. A positive role for NLI/Ldb1 in long-range beta-globin locus control region function. Mol Cell. 2007;28:810–22. doi: 10.1016/j.molcel.2007.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spilianakis CG, Lalioti MD, Town T, Lee GR, Flavell RA. Interchromosomal associations between alternatively expressed loci. Nature. 2005;435:637–45. doi: 10.1038/nature03574. [DOI] [PubMed] [Google Scholar]
- 30.Splinter E, Heath H, Kooren J, Palstra RJ, Klous P, Grosveld F, Galjart N, De Laat W. CTCF mediates long-range chro-matin looping and local histone modification in the beta-globin locus. Genes Dev. 2006;20:2349–54. doi: 10.1101/gad.399506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vernimmen D, Gobbi MD, Sloane-Stanley JA, Wood WG, Higgs DR. Long-range chromosomal interactions regulate the timing of the transition between poised and active gene expression. EMBO J. 2007;26:2041–51. doi: 10.1038/sj.emboj.7601654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crawford I, Maloney PC, Zeitlin PL, Guggino WB, Hyde SC, Turley H, Gatter KC, Harris A, Higgins CF. Immunocytochemical localization of the cystic fibrosis gene product CFTR. Proc Natl Acad Sci USA. 1991;88:9262–6. doi: 10.1073/pnas.88.20.9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Engelhardt JF, Zepeda M, Cohn JA, Yankaskas JR, Wilson JM. Expression of the cystic fibrosis gene in adult human lung. J Clin Invest. 1994;93:737–49. doi: 10.1172/JCI117028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hyde K, Reid CJ, Tebbutt SJ, Weide L, Hollingsworth MA, Harris A. The cystic fibrosis transmembrane conductance regulator as a marker of human pancreatic duct development. Gastroenterology. 1997;113:914–9. doi: 10.1016/s0016-5085(97)70187-2. [DOI] [PubMed] [Google Scholar]
- 35.Strong TV, Boehm K, Collins FS. Localization of cystic fibrosis transmem-brane conductance regulator mRNA in the human gastrointestinal tract by in situ hybridization. J Clin Invest. 1994;93:347–54. doi: 10.1172/JCI116966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chou JL, Rozmahel R, Tsui LC. Characterization of the promoter region of the cystic fibrosis transmembrane conductance regulator gene. J Biol Chem. 1991;266:24471–6. [PubMed] [Google Scholar]
- 37.Koh J, Sferra TJ, Collins FS. Characterization of the cystic fibrosis transmembrane conductance regulator promoter region: chromatin context and tissue-specificity. J Biol Chem. 1993;268:15912–21. [PubMed] [Google Scholar]
- 38.Yoshimura K, Nakamura H, Trapnell BC, Dalemans W, Pavirani A, Lecocq JP, Crystal RG. The cystic fibrosis gene has a “housekeeping”-type promoter and is expressed at low levels in cells of epithelial origin. J Biol Chem. 1991;266:9140–4. [PubMed] [Google Scholar]
- 39.Matthews RP, McKnight GS. Characterization of the cAMP response element of the cystic fibrosis transmem-brane conductance regulator gene promoter. J Biol Chem. 1996;271:31869–77. doi: 10.1074/jbc.271.50.31869. [DOI] [PubMed] [Google Scholar]
- 40.McDonald RA, Matthews RP, Idzerda RL, McKnight GS. Basal expression of the cystic fibrosis transmembrane conductance regulator gene is dependent on protein kinase A activity. Proc Natl Acad Sci USA. 1995;92:7560–4. doi: 10.1073/pnas.92.16.7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pittman N, Shue G, LeLeiko NS, Walsh MJ. Transcription of cystic fibrosis trans-membrane conductance regulator requires a CCAAT-like element for both basal and cAMP-mediated regulation. J Biol Chem. 1995;270:28848–57. doi: 10.1074/jbc.270.48.28848. [DOI] [PubMed] [Google Scholar]
- 42.Cafferata EG, Guerrico AM, Pivetta OH, Santa-Coloma TA. NF-kappaB activation is involved in regulation of cystic fibrosis transmembrane conductance regulator (CFTR) by interleukin-1beta. J Biol Chem. 2001;276:15441–4. doi: 10.1074/jbc.M010061200. [DOI] [PubMed] [Google Scholar]
- 43.René C, Taulan M, Iral F, Doudement J, L’Honoré A, Gerbon C, Demaille J, Claustres M, Romey MC. Binding of serum response factor to cystic fibrosis transmembrane conductance regulator CArG-like elements, as a new potential CFTR transcriptional regulation pathway. Nucl Acids Res. 2005;33:5271–90. doi: 10.1093/nar/gki837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gregori C, Porteu A, Lopez S, Kahn A, Pichard AL. Characterization of the aldolase B intronic enhancer. J Biol Chem. 1998;273:25237–43. doi: 10.1074/jbc.273.39.25237. [DOI] [PubMed] [Google Scholar]
- 45.Shah RN, Ibbitt JC, Alitalo K, Hurst HC. FGFR4 overexpression in pancreatic cancer is mediated by an intronic enhancer activated by HNF1alpha. Oncogene. 2002;21:8251–61. doi: 10.1038/sj.onc.1206020. [DOI] [PubMed] [Google Scholar]
- 46.Su JS, Tsai TF, Chang HM, Chao KM, Su TS, Tsai SF. Distant HNF1 site as a master control for the human class I alcohol dehy-drogenase gene expression. J Biol Chem. 2006;281:19809–21. doi: 10.1074/jbc.M603638200. [DOI] [PubMed] [Google Scholar]
- 47.Rey-Campos J, Chouard T, Yaniv M, Cereghini S. vHNF1 is a homeoprotein that activates transcription and forms het-erodimers with HNF1. EMBO J. 1991;10:1445–57. doi: 10.1002/j.1460-2075.1991.tb07665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pontoglio M, Faust DM, Doyen A, Yaniv M, Weiss MC. Hepatocyte nuclear factor 1alpha gene inactivation impairs chro-matin remodeling and demethylation of the phenylalanine hydroxylase gene. Mol Cell Biol. 1997;17:4948–56. doi: 10.1128/mcb.17.9.4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Soutoglou E, Viollet B, Vaxillaire M, Yaniv M, Pontoglio M, Talianidis I. Transcription factor-dependent regulation of CBP and P/CAF histone acetyltransferase activity. EMBO J. 2001;20:1984–92. doi: 10.1093/emboj/20.8.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paul T, Li S, Khurana S, Leleiko NS, Walsh MJ. The epigenetic signature of CFTR expression is coordinated via chro-matin acetylation through a complex intronic element. Biochem J. 2007;408:317–26. doi: 10.1042/BJ20070282. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primers