

Abstract
The stromal interaction molecules STIM1 and STIM2 are Ca2+ sensors, mostly located in the endoplasmic reticulum, that detect changes in the intraluminal Ca2+ concentration and communicate this information to plasma membrane store-operated channels, including members of the Orai family, thus mediating store-operated Ca2+ entry (SOCE). Orai and STIM proteins are almost ubiquitously expressed in human cells, where SOCE has been reported to play a relevant functional role. The phenotype of patients bearing mutations in STIM and Orai proteins, together with models of STIM or Orai deficiency in mice, as well as other organisms such as Drosophila melanogaster, have provided compelling evidence on the relevant role of these proteins in cellular physiology and pathology. Orai1-deficient patients suffer from severe immunodeficiency, congenital myopathy, chronic pulmonary disease, anhydrotic ectodermal dysplasia and defective dental enamel calcification. STIM1-deficient patients showed similar abnormalities, as well as autoimmune disorders. This review summarizes the current evidence that identifies and explains diseases induced by disturbances in SOCE due to deficiencies or mutations in Orai and STIM proteins.
Keywords: STIM, Orai, store-operated Ca2+ entry, CRAC, immunodeficiency
Introduction
Changes in cytosolic-free Ca2+ concentration ([Ca2+]c) are a universal signal that regulates a diversity of cellular functions, from short-term responses, such as secretion, contraction or aggregation, to long-term responses, including cell proliferation [1]. Physiological agonists increase [Ca2+]c, which consist of two components: the release of Ca2+ from the intracellular organelles and Ca2+ entry through plasma membrane (PM) channels. Ca2+ release from intracellular Ca2+ stores is a mechanism regulated by agonist-generated second messengers, including the inositol 1,4,5-trisphosphate (IP3), cyclic ADP ribose, nicotinic acid adenine dinucleotide phosphate (NAADP) or sphingosine-1-phosphate [2-6]. However, Ca2+ release from finite intracellular Ca2+ stores is sometimes insufficient to induce full activation of cellular processes and, to maintain Ca2+ signals, as well as to refill intracellular stores, Ca2+ entry plays a relevant role. Ca2+ entry might be achieved by different mechanisms, including voltage-operated
Ca2+ entry through voltage-sensitive Ca2+ channels and receptor-operated Ca2+ entry following receptor occupation by means other than a change in membrane potential [7]. The latter may take several forms and is conducted by different types of channels: receptor-operated channels (ROC) formed by subunits of the receptor protein itself, second messenger-operated channels (SMOC) gated by a diffusible messenger generated as a consequence of receptor occupation and, finally, store-operated (or capacitative) Ca2+ channels (SOC/CRAC) activated when the luminal Ca2+ concentration in the intracellular Ca2+ stores is reduced as a result of receptor occupation and the subsequent generation of a Ca2+-mobilizing second messenger [8, 9]. In non-excitable cells, and also in certain excitable cells, store-operated Ca2+ entry (SOCE) is a major mechanism for Ca2+ influx [10].
It has been established that SOC channels group a family of Ca2+-permeable channels with different biophysical properties. The first identified store-operated current, ICRAC, was revealed in electrophysiological studies of mast cells [11]. The channel conducting ICRAC was found to be non-voltage activated, inwardly rectifying and highly Ca2+ selective [12]. In addition to ICRAC, other store-operated currents of greater conductance and lower Ca2+ selectivity, commonly named ISOC, have been described [10]. The nature of the SOC components that mediate ICRAC and ISOC has been an issue of intense debate for over the last decades. In 2006, Orai1 was presented as the candidate to mediate ICRAC [13-15]. Members of the Orai family (Orai1–3) are highly conversed Ca2+ channel-forming subunits consisting in four transmembrane (TM) domains located in the PM and both N- and C-termini located in the cytosol (Fig. 1; Refs. [15, 16]). The C- and N-terminal regions of Orai interact with STIM1. The N-terminal region is critical for STIM1-mediated gating [17-20], and also contains a putative calmodulin (CaM)-binding domain, suggesting a possible role of CaM as regulator of Orai channel function [17, 21].
Fig 1.
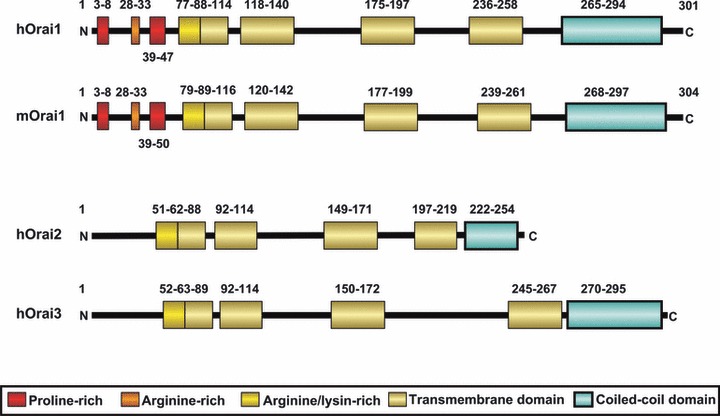
Orai protein family. Representation of domain organization of human (h) and mouse (m) Orai proteins. Mouse Orai1 shares a 90% identity with human Orai1 in the amino acid sequence according the pairwise alignment generated by BLAST (http://blast.ncbi.nlm.nih.gov/). Their domain structure is highly conserved. N and C represent the amino- and the carboxyl-terminus, respectively. Coloured boxes represent different domains. Numbers above and below the domains indicate their boundaries and the amino acid position. Boundaries of mouse Orai1 were predicted by clustal protein alignment (http://www.ebi.ac.uk/Tools/msa/clustalw2/). Modified version of the figure is taken from [148].
The role of Orai1 in ICRAC was identified by gene mapping in patients with hereditary severe combined immunodeficiency (SCID) attributed to the loss of ICRAC in T cells [22]. As described for ICRAC, Orai1 shows an extraordinarily high selectivity for Ca2+ over monovalent cations [23]. In addition to Orai1, its homologues Orai2 and Orai3 have been reported to be able to form SOC channels. Overexpression of all Orai homologues produced or enhanced SOCE, although with different efficiencies, being greater for Orai1 than for Orai2 and Orai3 [24].
In addition to Orai proteins, the mammalian homologues of Drosophila melanogaster transient receptor potential (TRP) channels have been presented as SOC candidates. The involvement of TRPs in SOC channel formation and the conduction cationic current ISOC remains controversial, with a number of laboratories providing evidence in favour or against this possibility. Particular attention has been paid to the canonical TRP (TRPC) subfamily members, which have been suggested to be activated by store depletion using different approaches, from overexpression of specific TRP proteins to knockdown of endogenous TRPs and pharmacological studies (for a review see Ref. [9]).
Intracellular Ca2+ stores and disease
Mechanisms of intracellular Ca2+ homeostasis
A variety of intracellular Ca2+ transporters and buffer systems modulate intracellular Ca2+ signals and maintain the low resting [Ca2+]c typically found in most cell types. At resting conditions, [Ca2+]c is maintained by Ca2+-ATPases, such as the secretory pathway Ca2+-ATPase (SPCA) in the Golgi compartments or the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) that pumps Ca2+ continuously towards the endoplasmic reticulum (ER) lumen opposing the Ca2+ leak that occurs through the ER membrane probably via the translocon [25]. During agonist stimulation, however, dramatic changes in [Ca2+]c occur due to opening of Ca2+ channels located in intracellular organelles, such as the IP3, ryanodine or NAADP receptors, which allow Ca2+ efflux from the intracellular stores, and Ca2+ entry through plasma membrane Ca2+-permeable channels. Once agonist-stimulation is terminated, [Ca2+]c is returned to the resting level through the collaborative work of Ca2+-ATPases and exchangers [26].
The advances in the understanding of Ca2+ signalling mostly occurred in parallel with the investigation of the intracellular Ca2+ stores, which are able to accumulate significant amounts of Ca2+. Although the resting [Ca2+]c is between 20 and 100 nM, depending on the cell type investigated, the Ca2+-concentration in the intracellular Ca2+-stores is within the micromolar range [27]. Intracellular Ca2+ stores include the ER, the mitochondria, the Golgi apparatus, the nuclear envelope and the acidic lysosomal-like organelles. The ER is the major source of the intracellular released Ca2+. The Ca2+ content in the ER is tightly regulated by SERCA that pumps Ca2+ back against a Ca2+ gradient across its membrane [28]. Ca2+ efflux from the intracellular stores has been reported to occur via occupation of the IP3 receptors (IP3R) or ryanodine receptors (RyR) [4]. Functional heterogeneity of the ER Ca2+ pool has been reported on the base of the heterogeneous expression of SERCA isoforms and the different sensitivity of ER Ca2+ compartments to distinct SERCA inhibitors. The presence of different ER Ca2+ compartments might have functional relevance, with function-specific Ca2+ compartments associated to discrete cellular mechanisms, although the occupation of different membrane receptors by agonists [29]. Intimately connected to the ER is the nuclear envelope, a small intracellular Ca2+ store with an intraluminal Ca2+ concentration of approximately 100 μM [30]. Ca2+ release from the nuclear envelope has been reported to be mediated by NAADP, as well as by IP3 and cyclic ADP-ribose [31], which act on specific Ca2+ release channels present in the inner nuclear membrane [30]; thus, leading to transient rises in the nucleoplasmic Ca2+ concentration, which could be important for the control of specific types of gene expression [30, 31].
Recently, particular attention has been paid on the acidic organelles including lysosomes and lysosomal-like organelles, such as secretory granules. These organelles show a proton-gradient across their membranes maintained by the vacuolar proton-ATPase (V-ATPase), which provides the driving force for Ca2+ uptake by a complex H+/Ca2+ exchange [32]. In human platelets, we have found that Ca2+ uptake in the acidic organelles involves the V-ATPase, which provides the driving force solely for the maintenance of stored Ca2+, and a SERCA3 isoform involved in Ca2+ store refilling [33].
Mitochondria initiate, transduce and modulate a variety of Ca2+ signals, regulating spatiotemporal dynamics of cellular Ca2+ signals [34]. This organelle might modulate Ca2+ signalling either directly, by Ca2+ uptake via the mitochondrial Ca2+ uniporter or by releasing accumulated Ca2+ into the cytosol by means of Na+/Ca2+ or H+/Ca2+ exchangers, or indirectly by regulating the concentration of ATP, NAD(P)H and reactive oxygen species, molecules that influence the activity of different pumps, exchangers and channels involved in the Ca2+ signalling machinery [35]. Among the roles of mitochondria in Ca2+ signalling, this organelle has been reported to play an essential role controlling the extent and duration of SOCE by buffering sub-plasmalemmal Ca2+ [36], and has also been found to contribute to ER Ca2+ refilling in the presence of IP3-generating agonists [37].
The Golgi apparatus has also been reported to act as an agonist-releasable intracellular Ca2+ store. Agonist-induced Ca2+ release from the Golgi apparatus was described in HeLa cells stably expressing targeted aequorin into this compartment and, as well as the ER, the Golgi apparatus was found to contribute to the rise in [Ca2+]c upon agonist stimulation [38]. In HEK293 cells, menthol causes Ca2+ release from both the ER and Golgi compartments [39]. Ca2+ transport into the Golgi pool is mediated by the secretory-pathway Ca2+-transport ATPases (SPCA), which supply the Golgi apparatus with both Ca2+ and Mn2+; thus, playing a relevant role in cellular Ca2+ and Mn2+ homeostasis [40-42].
Abnormal intracellular Ca2+ homeostasis and disease
Physiological agonists are known to induce typical Ca2+ signals to specifically regulate cellular functions, among the Ca2+ signals generated by agonist stimulation, Ca2+ oscillations play a relevant physiological role. Ca2+ oscillations consist of cyclical release and re-uptake of intracellularly stored Ca2+ and play an important role in the regulation of cellular functions. Current evidence suggests that Ca2+ influx across the PM plays a relevant role in the maintenance of Ca2+ oscillations, as well as in their localization within the cell [43]. Deregulation of cellular Ca2+ homeostasis leads to the development of a number of cellular dysfunctions that underlie a variety of disorders. An example for the pathophysiological relevance of intracellular Ca2+ signalling is cardiac diseases. In heart failure, the insufficient myocyte contraction is attributed to an insufficient increase in [Ca2+]c as a result of a reduced Ca2+ accumulation into the ER due to an abnormal expression of SERCA [44]. Abnormal ER Ca2+ homeostasis associated to presenilin-1 mutations has also been reported to contribute to the dysfunction and degeneration of neurons observed in Alzheimer’s disease [45]. Among other examples, SPCA mutations leading to loss of one functional copy of the human SPCA1 gene (ATP2C1) causes Hailey–Hailey disease, a rare skin disorder characterized by recurrent blisters and erosions in the flexural areas [46]. Specific mutations in RyRs results in enhanced sensitivity of RyR1 to activating Ca2+ concentrations and also to the exogenous and diagnostically used ligands caffeine and 4-chloro-m-cresol, thus leading to malignant hyperthermia, a skeletal myopathy where exposure to certain volatile anaesthetics and depolarizing muscle relaxants, commonly used in anaesthesia, trigger an abnormally high release of Ca2+ from the sarcoplasmic reticulum [47]. Finally, the discovery of a number of channelopathies has shed new light on the pathogenesis of a wide range of human diseases. Defects in L-type Ca2+ channels resulting in structural aberrations within their pore-forming region leads to a number of neurological disorders [48]. Homozygous expression of Orai1 bearing the R91W mutation results in impairment of SOCE leading to SCID [22]. Defects in cation permeable members of the TRP channel family have also been involved in human diseases such as hypomagnesemia with secondary hypocalcaemia, mucolipidosis type IV, autosomal-dominant polycystic kidney disease, familial focal segmental glomerulosclerosis or certain forms of cancer [49, 50]. The number of Ca2+ signalling dysfunctions underlying human diseases is growing, which highlights the key role that Ca2+ homeostasis plays in cellular physiopathology.
Sensing Ca2+ stores
Intracellular Ca2+ stores not only provide a source for agonist-induced Ca2+ mobilization but control a major Ca2+ influx pathway in non-excitable cells, SOCE, which is also present in excitable cells. SOCE was identified by Putney in 1986 as a mechanism by which the depletion of the intracellular Ca2+ stores activates Ca2+ entry through SOC channels [51]. The nature of the signal linking the Ca2+ content in the intracellular Ca2+ stores to the PM SOC channels has been a matter of intense investigation immediately after the discovery of SOCE. In 2005, Dr. Cahalan’s group reported that STIM1, a ubiquitously expressed protein in mammalian tissues, plays an essential role in SOCE and ICRAC, the best characterized capacitative current (Fig. 2; Refs. [52, 53]). The authors used an RNA interference (iRNA)-based screening to identify genes that impair Ca2+ entry in D. melanogaster S2 cells evoked by thapsigargin, a specific inhibitor of SERCA that stimulates SOCE. Among 170 screened genes, they found that ICRAC was suppressed in STIM knockdown S2 cells. Similarly, knockdown of the human homologue of D. melanogaster STIM1 significantly reduced ICRAC in Jurkat T cells and thapsigargin-evoked SOCE in HEK293 or SH-SY5Y cells [52]; thus, suggesting an essential role for STIM1 in the mechanism of activation of SOCE. Later on, the same group reported compelling evidence for a role of STIM1 as an ER Ca2+ sensor by using a STIM1 EF hand mutant that, being unable to sense intraluminal Ca2+ concentration, mimics Ca2+ store depletion, initiating translocation and activation of ICRAC [53]. Since 2005, a growing number of studies have provided evidence supporting a role for STIM1 as the ER Ca2+ sensor that communicates information concerning the Ca2+ content into the stores to PM SOC channels both in transiently or stably expressing cells and native cells [24, 54-57].
Fig 2.
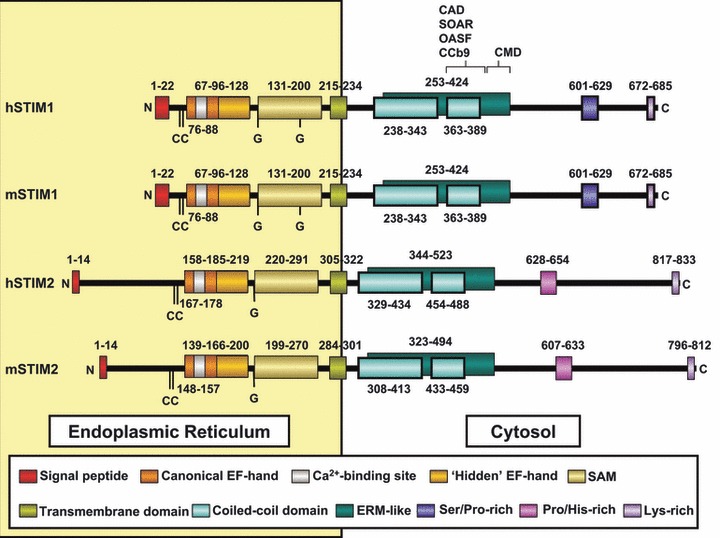
STIM protein family. Representation of domain organization of the human (h) and mouse (m) STIM proteins. Mouse STIM2 shares a 92% identity with human STIM2, while mouse STIM1 shares up to 97% identity in the amino acid sequence according the pairwise alignment generated by BLAST (http://blast.ncbi.nlm.nih.gov/). Their domain structure is also highly conserved. The amino- and the carboxyl-terminus are represented as N and C, respectively. Coloured boxes represent different domains. Numbers above and below the domains indicate their boundaries and the amino acid position. (CC) pair of highly conserved cysteines. (G) glycosylation sites. Boundaries of EF-hand and SAM motifs in hSTIM were biophysically characterized by [66,149,150], while transmembrane, coiled-coil and Ser/Pro/His/lys regions were predicted by computer models and clustal protein sequence alignment (http://www.ebi.ac.uk/Tools/msa/clustalw2/). Boundaries of mSTIM were predicted by clustal protein alignment.
STIM1 has a single TM domain with an EF-hand motif near the N-terminus, which is located in the lumen of the ER. In addition to the canonical EF-hand domain, the intraluminal N-terminus contains a hidden EF-hand motif and a sterile-a motif (SAM) that is important for STIM1 oligomerization (Fig. 2; Refs. [58, 59]). The cytosolic C-terminus includes two coiled-coil domains which overlap with an ezrin-radixin-moesin-like domain, a serine/proline-region and a lysine-rich region [60]. In addition, different research groups have identified a cytoplasmic STIM1 region essential for the activation of Orai1 and termed STIM1 Orai-activating region (SOAR; Ref. [61]), Orai-activating small fragment (OASF; Ref. [62]), CRAC-activating domain (CAD and CCb9; Refs. [63, 64], respectively). A decrease in ER luminal Ca2+ concentration results in dissociation of Ca2+ from the EF-hand motif, which, in turn, leads to STIM1 oligomerization and dissociation between the coiled-coil domain1 and SOAR, thus the positive charges located in the SOAR domain are free to interact with the acidic domain within the C-terminal domain of Orai1 and activate this channel (Fig. 2; Ref. [65]).
The STIM1 homologue STIM2 has a similar structure (Fig. 2). In the presence of Ca2+, STIM2 EF-SAM domain is monomeric and well-folded, as previously reported for STIM1 EF-SAM and, despite this domain shows similar Ca2+ -binding affinity in both STIMs, it is more stable in STIM2, which has been suggested to account for the different cellular functions of both proteins [66]. The function of STIM2 has not been completely elucidated. Early studies reported that, in contrast to the reported role of STIM1 in SOC activation, STIM2 suppressed this process, interfering with STIM1-mediated SOC activation, as a coordinated mechanism to regulate SOC-mediated Ca2+ entry [67]. Later on, STIM2 has been shown to maintain basal cytosolic and ER Ca2+ concentrations and to activate Ca2+ influx upon small changes in luminal ER Ca2+ content [68]. A role for STIM2 in the activation of SOC channels either in a store-operated mode activated through depletion of ER Ca2+ stores by IP3 or via a store-independent mechanism mediated by cell dialysis during whole-cell perfusion has been reported [69]. STIM2 has also been reported to play an essential role in SOCE in mouse neurons [70].
STIM isoforms have been widely recognized as the ER Ca2+ sensors [71]. However, we have recently reported that STIM1, and also STIM2, are located in the acidic Ca2+ stores. In human platelets, we detected STIM1 and STIM2 in isolated lysosomal compartments and dense granules. We have found association of STIM2 with STIM1, as well as between these proteins and Orai1, upon selective discharge of the acidic Ca2+ stores by using the vacuolar H+-ATPase inhibitor bafilomycin A1. Suppression of the association of STIM1 with Orai1 attenuates SOCE controlled by the acidic Ca2+ stores, thus suggesting a functional role for this interaction in SOCE in human platelets [72].
Importance of Orais and STIMs in tissues
Orai and STIM proteins are almost ubiquitously expressed in human and mouse tissues (Table 1; Refs. [73-76]). In humans, the strongest Orai1 expression has been found in a subset of cells located in primary and secondary lymphoid organs such as thymus and spleen, which is consistent with T cell expression. Other tissues that show Orai1 expression are endocrine and exocrine glands, hepatocytes, skeletal and cardiac muscle, skin, vascular endothelium, cells of the gastrointestinal tract, pneumocytes in the lung and kidney tubules [70, 73-75]. Interestingly, Orai1 staining is almost absent in brain, while Orai3 seems to be the only isoform that is strongly expressed in this organ, at least at RNA level [70, 73-75]. Orai3 transcripts are also widely expressed in human tissues, showing a minor abundance in spleen and colon [74, 75]. In contrast, Orai2 transcripts are prominently expressed in kidney, lung and spleen (Table 1; Refs. [74, 75]). In mouse, Orai transcripts exhibit similar expression pattern than in human (Table 1; Refs. [70, 74-76]). A weak expression of Orai1 transcripts was detected in murine testis and brain, while completely absent expression was observed in cortical neurons [70], indicating that other brain cells might express Orai1. Instead, a strong expression of Orai2 was detected in these cells compared with the weak expression of Orai3, indicating that Orai2 might be the predominant isoform in murine cortical neurons (Table 1; Ref. [70]).
Table 1.
Expression pattern of Orai and STIM isoforms in human and mouse tissues
| Human | Mouse | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Orai | STIM | Orai | STIM | |||||||||
| 1 | 2 | 3 | 1 | 2 | Refs. | 1 | 2 | 3 | 1 | 2 | Refs. | |
| Blood | ▴▴ | • | ▴▴ | − | − | [132] | ▴ | ▴▴ | ▴▴ | ▴▴ | ▾ | [70, 74, 100, 143] |
| Bone marrow | • | ▾ | ▴ | − | − | [132] | • | • | ▾ | − | − | [143] |
| Brain | ▾▾ | ▾ | • | • | ▴ | [68, 71, 132] | ▾ | ▴ | ▾ | ▾▾ | • | [70, 74, 79, 143] |
| Breast | − | − | − | − | − | • | ▾ | ▾ | − | − | [143] | |
| Heart | ▾ | ▾ | ▴ | • | ▴ | [68, 71, 132] | ▾ | ▾ | ▾ | • | • | [70, 74, 143] |
| Intestine | • | ▾ | ▾ | − | − | [68] | • | ▾ | • | − | − | [143] |
| Kidney | ▾ | • | ▴ | ▾ | ▾ | [68, 71, 132] | ▾ | ▾ | • | [70, 74, 143] | ||
| Lymph node | • | • | ▴ | − | − | [132] | • | ▴ | ▴ | ▴ | ▴ | [70, 143] |
| Liver | • | ▾ | ▴ | • | ▾ | [68, 71, 132] | ▾ | ▾ | ▾ | ▴ | ▴ | [70, 74, 143] |
| Lung | ▴ | • | ▴ | ▾ | • | [68, 71, 132] | • | • | • | ▾ | − | [74, 80, 143] |
| Pancreas | • | ▾ | ▾ | ▴ | ▴ | [68, 71, 132] | • | • | • | − | − | [143] |
| Placenta | • | • | ▴ | ▴ | • | [68, 71, 132] | • | ▾ | • | − | − | [143] |
| Skeletal muscle | ▴ | • | ▾ | • | ▾ | [68, 71, 132] | • | ▾ | ▾ | ▴▴ | ▴ | [70, 74, 143] |
| Skin | ▴ | • | • | − | − | [132] | • | • | • | − | − | [143] |
| Spleen | • | • | ▾ | − | − | [68] | • | • | • | ▴ | ▴ | [70, 74, 143] |
| Testis | • | ▴ | ▴ | − | − | [132] | ▾ | ▾ | ▾ | − | − | [74, 143] |
| Thymus | ▴ | ▾ | ▴ | − | − | [132] | • | ▴▴ | • | • | ▾ | [70, 74, 143] |
Expression levels referring to differences in mRNA or protein abundance in different tissues are not comparable among isoforms. Unknown abundance or unreported expression is represented as (−).▴: High; •: medium; ▾: low; [: absent; −: unknown.
STIMs are also ubiquitously expressed in human tissues (Table 1; Refs. [70, 77]). STIM1 transcripts are mainly expressed in lymphocytes [78], skeletal muscle, heart, brain, pancreas, placenta and almost absent in kidney and lung. STIM2 is strongly expressed in brain, pancreas, placenta, heart and almost absent in skeletal muscle, kidney, liver and lung (Table 1; Ref. [77]). Despite some variations concerning STIM isoform abundance in certain tissues, similar expression pattern was observed in mouse (Table 1; Refs. [56, 70, 76, 79–80]). STIM1 is mainly expressed in murine skeletal muscle, cerebellum, spleen, thymus, lymph nodes and additionally in platelets, while is almost absent in brain and completely absent in kidney. STIM2 is mainly expressed in skeletal muscle, liver, spleen and lymph nodes, while is completely absent in kidney (Table 1; Ref. [70]). Densitometric analysis of protein abundance revealed that STIM2 is the predominant isoform in murine brain, while the ratio of STIM1 to STIM2 abundance is reversed in T cells [70, 76, 79]. In this organ, STIM isoforms seems to be separately distributed to certain areas, such as cerebellum or hippocampus. Regarding the different properties exhibited by STIM1- or STIM2-mediated ICRAC currents and SOCE [81–82], these separated distribution suggested different mechanisms and requirements of SOCE in these brain areas [76, 79].
Participation of Orai and STIM in human diseases
Few papers published over the last 6 years reported patients carrying homozygous point mutations for Orai1 or STIM1 genes [22, 73, 78, 83–87]. These patients suffered from pathologies, which started early during infancy, due to absence of functional Orai1 or STIM1 proteins, indicating the participation of altered SOCE in certain human diseases. The prognosis of these patients was poor, with fatal consequences mainly due to immune response failure, unless treatment with haematopoietic stem cell transplantation, indicating a major role of Orai1- and STIM1-mediated SOCE in cells of the immune system. In contrast, heterozygous carriers of mutated alleles did not present any alteration affecting their normal lives [22, 73, 78, 88–90], indicating that the presence of a single wild-type allele is sufficient to sustain functional but reduced SOCE. The low frequency of such genetic alterations documented until now and the severity of their absence remark the importance of Orai1- and STIM1-mediated SOCE for normal life. Diseases caused by altered Orai2, Orai3 and STIM2 function have not been reported in humans yet. This section pretends to highlight the most relevant data taken from the study of these patients (Table 2), which were extensively summarized in the following excellent reviews [88, 89, 91].
Table 2.
Impact of Orai and STIM deficiency in human and mouse
| Orai | STIM | |||
|---|---|---|---|---|
| 1 | 3 | 1 | 2 | |
| Gene alteration | ||||
| Human | Single point mutations | iRNA | Mutation | None |
| Mouse | Knockout, knock-in | – | Knockout | Knockout |
| Orai/STIM function | Absent | Reduced | Absent | Absent |
| SOCE | Reduced | Reduced | Reduced | Reduced |
| Molecular alterations | ||||
| Defective TRC-mediated function [22,73,86,87,92–93,101,151] | Defective TCR-mediated function [78,99,100,113] | Defective TCR-mediated function [78,100,113] | ||
| Impaired NFAT nuclear translocation [22,93,100] | Defective FcRI-and FcγR-mediated function [112,152] | Defective cytokine production [100,113] | ||
| Defective production of several cytokines [22,73,86,87,92–94,100,101,151] | Impaired NFAT nuclear translocation [93,100,111] | Inability to retain NFAT in the nucleus [100] | ||
| Defective cytokine production [78,94,100,113] | ||||
| Cellular phenotype | ||||
| Human | ||||
| Defective T and NK cells [22,73,94] | Defective T and NK cells [78,94] | |||
| Predominance of muscle type I fibres and atrophic type II fibres [73] | Reduced number of CD4+CD25+ FoxP3+ T reg cells [78] | |||
| Impaired sweat gland cell function [73] | ||||
| Mouse | ||||
| Defective Th1, th2, th17, B cells [101,151] | Defective T, Th17, B, mast cells and macrophages [78,86,87,92] | Defective Th17 cell function [113] | ||
| Defective blood platelet function [106,107] | Lymphoproliferative disease [99] | Defective neuronal function [70] | ||
| Defective blood platelet function [108,117] | ||||
| Cancer cells | ||||
| Resistance to apoptosis in Pca cells [140] | Arrested MCF-7 cell cycle and proliferation [139] | |||
| Diabetes | ||||
| Impaired association of human STIM1 with Orai1, TRPC1 and TRPC6 [103] | ||||
| Main clinical phenotype | ||||
| Human | ||||
| SCID-like Immunodeficiency [22,83,84] | Immunodeficiency [78, 85] | |||
| Global muscular hypotonia [73] | Autoimmune thrombocytopenia [78] | |||
| Chronic pulmonary disease [73] | Lymphoproliferative disease [78] | |||
| Anhydrotic ectodermal dysplasia (impaired sweat production) [73] | Ectodermal dysplasia [78] | |||
| Mouse | ||||
| Perinatal death [101, 107, 151] | Perinatal death [56, 99, 100, 108] | Sudden death [70] | ||
| Immunodeficiency [101, 151] | Immunodeficiency [93, 94, 102] | Altered spatial memory [70] | ||
| Reduced procoagulant activity and thrombus formation [106, 107] | Reduced muscle cross-sectional area and mitochondriopathy [56] | |||
| Reduced procoagulant activity and thrombus formation [108,117] | ||||
| Reviewed in Refs. [88, 89, 91] |
Summary of the most important molecular and phenotipic alterations in absence of Orai/STIM functions in human and mice.
Orai1-deficient function and human disease
Different Orai1 mutated alleles were reported by Rao and Lewis groups in patients presenting a clinical phenotype characterized by an immunodeficiency similar to that observed in SCID patients (Table 2; Refs. [22, 83, 84]). Orai1-mutated alleles presented single point mutations which led to abrogated Orai1 function due to expression of defective Orai1 proteins (mutant R91W) [22] or to impaired Orai1 gene expression (mutant A88EfsX25, A103E and L194P; Refs. [73, 86, 87]). Orai1-deficient patients also suffered from congenital myopathy, chronic pulmonary disease, anhydrotic ectodermal dysplasia and a defect in dental enamel calcification, which, initially, were not a threat to the patient’s life [73, 88, 89, 91].
The most relevant phenotype in Orai1-deficient patients was the severely compromised immune response, similar to SCID patients, as consequence of abrogated SOCE in peripheral T cells, resulting in impaired T cell activation, cytokine production and absent proliferative responses in vitro (Table 2; Refs. [22, 73, 86–89, 91–93]). Impaired SOCE was observed also in B cells, natural killer (NK) cytotoxic cells and fibroblasts isolated from these patients [22, 73, 88, 89, 91, 94]. Normal immunoglobulin (Ig) levels were found in blood serum despite the absence of SOCE in these cells. However, Orai1-deficient patients failed to mount antigen-specific antibody responses upon vaccination or infection [89, 91]. In contrast to most of SCID patients, Orai1-deficient patients presented a normal number of B cells and CD4+ or CD8+ T cells in peripheral blood, indicating normal development of these cells in the absence of Orai1-mediated SOCE [88, 89, 91].
The absence of Orai1 also led to defects in the skeletal muscle characterized by global muscular hypotonia with decreased head control, delayed ambulation, reduced muscle strength and endurance (Table 2; Refs. [73, 88, 89]). Orai1-R91W mutation resulted in a predominance of type I fibres and atrophic type II fibres in these patients, suggesting a defect in fast twitch muscle fibre differentiation [73]. This defect might be explained by the requirement of SOCE during differentiation of human myoblasts, the precursors of adult skeletal muscle [95, 96]. Chronic pulmonary disease was also reported as a consequence of defective respiratory muscle function [73, 88, 89, 91]. The anhydrotic ectodermal dysplasia in Orai1-deficient patients was characterized by impaired sweat production, which results in dry skin and heat intolerance with recurrent fever [73, 88, 89, 91]. Ca2+ influx upon thapsigargin stimulation is required for secretion in sweat gland cells, indicating an important role of SOCE in sweat gland function [97, 98]. The absence of Orai1-mediated SOCE therefore, could alter the normal function of sweat glands in these patients.
In summary, the clinical phenotype associated with Orai1 deficiency in patients was limited to certain tissues and associated with impaired SOCE, indicating a predominant role of Orai1-mediated SOCE in a reduced number of cell types and tissues. Despite its severity, the limited clinical phenotype found in these patients contrasts with the wide Orai1 expression in a variety of cell types and tissues (Table 1). This could be explained by a minor relevance of SOCE in Ca2+ entry in those unaffected cell types and tissues, or by the presence of additional molecules which might compensate the lack of Orai1 or have a more relevant function in SOCE regulation [73, 88, 89, 91].
STIM1-deficient function and human disease
Different STIM1-mutated alleles were reported by Rao and Lewis groups in patients presenting a clinical phenotype very similar to those found in Orai1-deficient patients (Table 2; Ref. [78]), as expected, because both genes play their roles in the same signalling pathway according to data previously obtained in transgenic mouse [99-101] and in vitro models [6, 15, 102, 103]. Patients homozygous for single point mutations in STIM1 gene resulted in impaired STIM1 function due to a lack of STIM1 proteins (mutant E136X and mutant 1538-1G>A) [78, 85]. As a consequence, SOCE was severely impaired in cells from these patients.
The clinical phenotype was observed very early during infancy and was also limited to certain tissues similar to those observed in Orai1-deficient patients. It was characterized by immunodeficiency together with autoimmune disease, congenital myopathy and ectodermal dysplasia (Table 2; Refs. [78, 85, 88, 89, 91]). SOCE was absent in T, B and NK cells, which led to severely compromised T cell function, defective T cell proliferation and reduced cytokine production. However, a normal number of these cells were found in peripheral blood, indicating normal cell development in the absence of STIM1-mediated SOCE [78, 94]. Ig titres were normal for all subtypes in blood serum. In contrast, strongly reduced IgG titres were found in a patient due to nephrotic syndrome [78, 88, 89, 91].
STIM1-deficient patients also presented lymphoproliferative disease and an autoimmune response against blood platelets, which developed into thrombocytopenia (Table 2; Ref. [78]). The reduced number of CD4+CD25+FoxP3+ regulatory T cells (Tregs) found in peripheral blood might explain the immune thrombocytopenia observed in STIM1-deficient patients, because those cells regulate autoimmune responses [78, 88, 89, 91]. Taking together, the severe immunodeficiency observed in STIM1-deficient patients is very similar to that observed in Orai1-deficient patients with the exception of autoimmunity and reduced numbers of Treg cells.
STIM1-deficient patients also suffered from ectodermal dysplasia and congenital myopathy, similar to that observed in Orai1-deficient patients (Table 2). Myopathy was characterized by non-progressive global muscular hypotonia and partial iris hypoplasia. In contrast to Orai1-deficient patients, histological abnormalities were not observed in skeletal muscle of STIM1-deficient patients [78, 88, 89, 91].
Orai1 and STIM1 in human diabetic platelets
The contribution of SOCE to platelet activation and the nature of SOC channels in these cells have remained controversial. Bleeding times in Orai1- and STIM1-deficient patients were only moderately prolonged or normal and patients lacked signs of an enhanced bleeding diathesis [73, 78, 88, 89, 91]. Recently, we reported reduced SOCE in platelets from type 2 diabetic patients, which is likely mediated by impairment of the association of STIM1 with the channel subunits Orai1, but also with hTRPC1 and hTRPC6, and might be involved in the pathogenesis of the altered platelet responsiveness observed in diabetic patients [104].
In summary, the clinical phenotypes found in Orai1- and STIM1-deficient patients indicate that Orai1- and STIM1-mediated SOCE plays very important roles mainly in cells of the immune system, skeletal muscle and some ectodermal-derived tissues such as sweat glands. Orai2, Orai3 and STIM2 co-exists with Orai1 or STIM1 together with other non-SOCE elements of Ca2+ entry in many other tissues, which might compensate or minimize the lack of functional Orai1 and STIM1 in other unaffected tissues. So far, no Orai2-, Orai3- or STIM2-deficient patients have been identified yet. Further studies in Orai1- and STIM1-deficient clinical phenotype might give insights about additional roles of these proteins in other cell types or tissues.
Orai and STIM mutant mouse as models of disease
The mouse has shown to be an invaluable model organism to study mechanisms of human disease, because mouse is very similar to humans in both genetic and physiological aspects. Genetically engineered mice, which lack the function of known or unknown genes, are one of the most efficient ways to reveal their function in vivo.
Mice lacking Orai1, STIM1 and STIM2 expression have been generated over the last years by a number of laboratories [70, 100, 101, 105–108]. Comparison of both human and mouse Orai1- or STIM1-deficient phenotypes revealed interspecific similarities and discrepancies (Table 2; Refs. [88, 89, 91]). The analysis of these mouse models, together with previous abundant in vitro data, helped to elucidate the cellular function of these proteins and contributed to underhand the clinical phenotype in patients lacking these proteins. The data obtained in these mouse models can give a clue of further analysis to be done in other resembling human diseases to reveal underlying mechanisms of human pathogenesis.
Sudden and perinatal mortality
Mice lacking the expression of functional Orai1 and STIM1 proteins die at perinatal and early postnatal periods [56, 101, 105,107, 108]. Starting at 8 weeks after birth, sudden death of STIM2-deficient mice was observed, and only ∼10% of the animals reached the age of 30 weeks [70]. The precise cause of death is unclear in all cases. In contrast, spontaneous abortion, perinatal mortality or early neonatal death was not reported among families of Orai1- and STIM1-deficient patients [88, 89, 91]. However, the low number of patients identified until now makes not possible to determine the prevalence of perinatal mortality in these cases. Indeed, data obtained from these mouse models indicated that the altered function of Orai1, STIM1 and STIM2 could be a potential determinant of sudden, perinatal or early postnatal mortality in humans, which might be important to investigate.
Immunodeficiency
In line with the phenotype observed in STIM1- and Orai1-deficient patients, TCR-dependent and -independent T cell activation as well as B cell activation was severely impaired, while the number of T and B cells were normal in the blood stream of Orai1- and STIM1-deficient mice (Table 2; Refs. [78, 86–87, 92]). Analysis of primary and secondary lymphoid organs of mutant mice revealed a possible explanation. Normal numbers of T and B cells were found in murine thymus and bone marrow [99–101, 105], which indicates an unaltered T cell development. This finding was surprising, because TCR induced Ca2+ signals and SOCE has been considered necessary for T cell development [109–110]. Further analysis of T cell development in these murine models will be crucial to clarify this point. Expression of cytokines was substantially reduced in Orai1-deficient patients [92, 93], similar to the multiple cytokine expression defect found in T cells from Orai1- and STIM1-deficient mice, which involved a reduction in interleukin (IL)-2, interferon-γ (IF-γ), IL-4 and IL-10 production [100, 101]. Deeper analysis of these mice revealed as possible explanation an impaired SOCE-dependent nuclear translocation of the transcription factor NFAT, which, in turn, is necessary for cytokine production [89, 91, 93, 100, 111].
In addition, an impaired development of functional CD4+ Foxp3+ regulatory T cells was observed in STIM1-deficient patients [78] and double-deficient STIM1/STIM2 [78, 100]. The further analysis of mutant mice offered a potential explanation [100, 111]. The absence of both STIM1 and STIM2 in naive T cells abrogated the sustained Ca2+ influx required for nuclear translocation of NFAT, which, in turn, impaired NFAT-dependent induction of Foxp3 expression and formation of a NFAT/Foxp3/DNA-binding complex. This complex has been proposed to be important for the initiation of Treg differentiation and regulation of their function [89, 91, 100, 111].
Finally, STIM1-deficient patients and double-deficient STIM1/ STIM2 mice developed an autoimmune, lympho-myeloproliferative, phenotype characterized by hepatosplenomegaly and lymphadenopathy [78, 100]. Beyersdorf et al. also reported a lymphoproliferative disease in STIM1-deficient mice [99]. This phenotype was prevented when wild-type Treg cells were transferred into double deficient STIM1/STIM2 mice, indicating that the lympho-myeloproliferative disease is mainly caused by decreased regulatory Treg function. In agreement with this, Orai1-deficient human patients and mice showed normal numbers of Treg cells and signs of autoimmunity and lymphoproliferation were not observed [22, 101]. This might be explained by the residual SOCE detected in their T cells, which in turn could allow normal Treg differentiation and regulatory Treg function. This residual SOCE could be presumably mediated by other existing SOCE channels expressed in T cells such as Orai2 or Orai3 [22, 88, 89, 91, 101].
Autoimmune and inflammatory diseases
Human patients lacking STIM1 expression presented AIHA and thrombocytopenia [78], probably produced by an autoimmune response and functional macrophage-mediated phagocytosis of red blood cells and platelets. In contrast, STIM1-deficient mice injected with auto-antibodies against platelets or red blood cells were protected from thrombocytopenia and anaemia, which might be explained by the severely compromised Fc-gamma receptor (FcγR)-mediated SOCE and the abrogated function of STIM1-deficient macrophages and Kupffer cells observed in these mice [112]. These results indicate interspecific differences in STIM1 function in macrophages. Functional macrophages indicated that STIM1 does not seem to be essential for FcγR-mediated response in humans while the absence of STIM1 severely impairs macrophage function in mouse [88, 89, 91].
The analysis of Orai1 and STIM deficiency in murine models already evidenced additional potential roles of these proteins in autoimmune and inflammatory responses. A crucial function of STIM1 and STIM2 has been reported as regulator of autoreactive T cell activation in a murine model of myelin-oligodendrocyte glycoprotein (MOG(35–55))-induced experimental autoimmune encephalomyelitis (EAE) [113]. STIM1 deficiency significantly impaired autoimmune responses mediated by Th1/Th17 cells against neuronal tissue in vivo, resulting in complete protection from EAE. Instead, mice lacking STIM2 developed an ameliorated EAE disease. Deficiency of STIM2 was associated with a reduction of IF-γ/IL-17 production by neuroantigen-specific T cells, which might explain the reduced clinical peak at early stages of disease [113].
On the other hand, mast cells derived from Orai1-deficient mice showed severely impaired SOCE, degranulation and cytokine secretion upon Fc-epsilon receptor I (FcRI) stimulation and the allergic reactions elicited in vivo were inhibited in these mutant mice [105]. Taken together, these findings establish Orai1 and STIM as attractive new molecular therapeutic targets for the treatment of inflammatory and autoimmune disorders. Indeed, in addition to their roles in SOCE, the relevance of Orai and STIM proteins in autoimmune and inflammatory diseases could be discovered in the near future.
Skeletal muscle
The skeletal muscle defect in mice matches the congenital myopathy observed in Orai1- and STIM1-deficient patients (Table 2; Refs. [22, 78]). In addition, haematopoietic stem cell transplantation corrected immunodeficiency in surviving STIM1-deficient patients but still exhibited muscular hypotonia [78], suggesting that the myopathy is not a secondary effect to autoimmunity. STIM1-deficient mice showed reduced muscle cross-sectional area and mitochondriopathy [56]. The mechanism by which abrogated SOCE contributed to the pathogenesis of these myopathyes is unclear but most likely includes short term Ca2+ responses, such as muscle contraction, altered Ca2+-dependent signalling pathways leading to altered gene expression such as NFAT-dependent gene regulation, disorders of metabolism and adverse remodelling [56, 88, 89, 91]. Contraction of skeletal muscle fibres requires Ca2+ release from the sarco/endoplasmic reticulum (S/ER) through RyR (reviewed in Ref. [114]). Absence of STIM1 abrogated SOCE, impaired refilling of S/ER and conferred reduced tetanic force and increased susceptibility to fatigue in adult STIM1-deficient mice [56]. Thus, STIM1 was required to refill internal S/ER Ca2+ stores of myofibres subjected to repeated stimulation and increased motor nerve stimulation [56]. Although distinct mechanisms control myogenesis and muscle formation, additional studies reported that postnatal myogenesis critically relies on RyR-mediated store depletion [115] and Ca2+ influx through SOCE [95, 96, 116]. Therefore, STIM1 might be also required to refill internal S/ER Ca2+ stores in response to signals associated with muscle development and the absence of STIM1 function could lead to defective muscle differentiation [88, 89, 91].
Thrombosis and haemostasis
Studies in Orai1- and STIM1-deficient mice models showed that Orai1- and STIM1-mediated SOCE are essential for platelet activation, glycoprotein VI- and thrombin-dependent procoagulant activity in vitro and thrombus formation in vivo [106-108]. However, bleeding times were normal or moderately prolonged in Orai1- and STIM1-deficient patients, and they lacked signs of an enhanced bleeding diathesis [73, 78, 91, 117]. Similar results were observed in the Orai1 mutant R93W knock-in (similar to the mutant R91W Orai1 gene in humans), Orai1- and STIM1-deficient mice after mechanical injury [106–108]. However, murine Orai1- and STIM1-deficient platelets were unable to form stable thrombus in mice and failed to promote artery occlusion after chemical injury in arterial walls. These mutant mice were in turn significantly protected against ischaemic brain infarction or pulmonary thromboembolism [107, 108]. These results established therefore, an important role of STIM1 and Orai1 in mechanisms underlying arterial thrombosis, but not haemostasis upon mechanical injury in mice. The impairment in thrombus formation can be partially explained by the reduced glycoprotein VI- and thrombin-dependent surface exposure of phosphatidylserine (PS) observed in these mutant mice, which accomplishes platelet procoagulant activity [117]. Despite the fact that STIM2 is expressed in these cells, STIM2-deficient platelets did not show defects in SOCE, procoagulant activity or thrombus formation [117]. The absence of studies concerning procoagulant activity in Orai1- and STIM1-deficient patients makes impossible to confirm the presence of similar mechanistic differences in humans, but gives a clue for further investigation in human platelets. Taken together, the results obtained in mice establish STIM1 and Orai1 as an important mediator in the pathogenesis of ischaemic cardio- and cerebrovascular events and potential targets for the design of novel anti-thrombotic therapies.
Neuronal system
SOCE is a major mechanism for Ca2+ influx in non-electrically excitable cells. However, reports about the existence of SOCE, Orai and STIM function in electrically excitable cells such as skeletal muscle cells and neurons performed in genetically engineered mice and in vitro models offered an expanded view about the function of SOCE in cell physiology. Orai1- and STIM1-deficient patients did not show an altered cognitive or neuronal phenotype [22, 73, 78, 88, 89, 91] and matches with the low expression or specific localization of these proteins reported in human neuronal tissues (Table 1; Refs. [79, 118, 119]). This might indicate a minor function of Orai1- and STIM1-mediated SOCE in neuronal physiology. SOCE has also been observed in neuronal cells [10, 120, 121] and STIM2 is the predominant isoform in murine cortical neurons [70]. STIM2-deficient neurons isolated from mutant mice showed severely abrogated SOCE and decreased basal Ca2+ levels in the cytosol and intracellular Ca2+ stores [70]. In contrast to those observed in cells of the immune system, no significant changes in SOCE were reported in murine Orai1- and STIM1-deficient neurons [70]. This data suggested that STIM2 is the main mediator of SOCE in these cells. STIM2-deficient mice showed impaired spatial learning similar to that observed after blockade of NMDA ionotropic glutamate receptors [70, 122], which might be related with altered neurotransmitter release and synaptic plasticity [123]. Therefore, potentially altered STIM2 function might be expected in some patients showing familiar forms of mental disorders affecting cognitive functions, for instance memory processing. Moreover, STIM2 deficiency protected mice from neuronal damage after cerebral ischaemia, similar to those observed in STIM1 and Orai1-deficient mice [70]. However, while STIM1 and Orai1 deficiency conferred protection due to deficient platelet activation and impaired thrombus formation, which abrogated cerebral artery occlusion [107, 108], the lack of STIM2 conferred protection to neurons against ischaemic neuronal death, which prevented ischaemic brain damage [70]. Cytotoxic Ca2+ overload into the cell is considered the main factor of neuronal death during ischaemic conditions. The existing literature describes a reduction of SERCA re-uptake [124, 125] and active Ca2+ release from the ER through IP3R and RyR channels associated with the increased intracellular Ca2+ levels observed in these conditions. Such Ca2+ release from the ER is crucial for cellular Ca2+ damage as evidenced by protection of neurons against excitotoxic injury through blockade of IP3R or RyR [126, 127]. These events might lead to store depletion and Ca2+ accumulation in the cytosol, the earlier inducing an additional Ca2+ load into the cytosol via SOCE. SOCE may in turn, increase the release of glutamate and trigger an additional Ca2+ influx by activation of ionotropic glutamate receptors [128]. Both SOCE and glutamatergic Ca2+ entry might rapidly push the cytosolic Ca2+ concentration to damaging levels. In the same line, STIM2-deficient neurons might be less sensitive to apoptosis due to the absence of SOCE and the lower Ca2+ content observed in the cytosol and in the intracellular stores, which critically depends on a functional SOCE [10, 70]. The decreased store content could limit the initial Ca2+ release and might help to better utilize the remaining Ca2+ sequestration ability of SERCA during the ischaemic event. It is not clear why neurons use STIM2 instead of STIM1 to regulate SOCE, probably because different requirements in terms of Ca2+ influx dynamics which might depend of the cell type. Indeed, SOCE or ICRAC currents exhibit different properties depending on which STIM isoform regulate the process [81, 82]. STIM1 enhances both Orai1-mediated SOCE and constitutive coupling to activate Orai1 channels while STIM2 attenuates Orai1-mediated SOCE and drastically slows store-induced Orai1 channel activation. Additional studies reported a predominant function of STIM2 in other tissues [129, 130].
Different knockout models for other proteins related with SOCE suggested an important role of this mechanism in neuronal function. For instance, the absence of PLCβ1 led to epileptic-type seizures in mice [131], which indicated an involvement of PLCβ1 in the development and control of brain inhibitory pathways. IP3R type I null mice exhibited severe neurological symptoms, including ataxia and epilepsy [132]. This body of evidence, together with STIM2 function in neurons, suggests an unexpectedly important role of SOCE in electrically excitable cells such as neurons. These findings may serve as a basis for the development of novel neuroprotective agents for the treatment of ischaemic stroke and other neurodegenerative disorders in which disturbances in cellular Ca2+ homeostasis are considered a major pathophysiological component [133, 134].
In summary, despite certain discrepancies in Orai1- and STIM1-deficient phenotype among species, mouse models have demonstrated to be important for understanding Orai1 or STIM1 function in cell physiology and disease, being suitable to investigate novel therapies which seek to modulate SOCE for the treatment of disorders related with disturbances in cellular Ca2+ homeostasis. Certainly, more functions of SOCE will emerge from the study of Orai1- and STIM-deficient mice in the future.
Emerging studies of Orai and STIM in cancer and cell cycle
As mentioned earlier, SOCE mediated by STIM/Orai proteins is a ubiquitous pathway that controls a variety of important cell functions. Initial studies considered STIM1 as a molecule involved in growth arrest and degeneration in human G401 and RD cancer cell lines, suggesting a role in the pathogenesis of rhabdoid tumours [135, 136]. However, the discovery of its function as a Ca2+ sensor in the ER eclipsed further studies in these field. Current evidences support a role for STIMs and Orai1 in cell proliferation, with some differences depending on the cellular model investigated. In endothelial cells, knockdown of STIM1, STIM2 or Orai1 attenuated cell proliferation and induced cell cycle arrest at S and G2/M phase [54]. However, in HEK293 cells STIM1 has no role in cell proliferation, while silencing of Orai1 and STIM2 using siRNA resulted in SOCE inhibition and enhancement of cell population doubling time, thus suggesting that Orai1 and STIM2 are important for cell proliferation [82].
In addition to the involvement of SOCE in the regulation of cellular functions, emerging evidence suggests the involvement of the STIM/Orai pathway in certain types of cancer. A recent study showed that STIM1 gene expression is regulated by potential oncogenes such as Wilms tumour suppressor 1 (WT1) and early growth response (EGR) in human G401 rhabdoid tumour cells, thereby providing a molecular link between Ca2+ signalling and cancer [137]. WT1 and EGR1 protein can bind to putative regulatory elements located upstream of the STIM1 gene, and overexpressed WT1 or down-regulation of EGR1 induced both reduction of STIM1 expression and decreased SOCE [137]. Trebak’s group reported differences in SOCE and ICRAC in estrogen receptor-positive [ER(+)] and estrogen receptor-negative [ER(−)] breast cancer cell lines. In ER(+)-breast cancer cells, capacitative currents require STIM1/2 and Orai3 while SOCE in ER(−) breast cancer cells is mediated by the STIM1/Orai1 pathway [138]. In addition, isolated breast cancer tumours whose cells displayed higher STIM1/STIM2 rates had a significantly poorer prognosis [129]. The expression of Orai3 has been reported to be higher in breast cancer tissues and the MCF-7 breast cancer cell line than in normal tissues or mammary epithelial cell lines, which provide evidence for a significant effect of Orai3 on breast cancer cell growth [139]. In support of this hypothesis, down-regulation of Orai3 by siRNA has been reported to attenuate MCF-7 cell proliferation and arrest cell cycle at G1 phase [139]. Another study suggested that the resistance to apoptosis showed by human androgen-independent prostate cancer (Pca) cells is associated with their decreased Orai1 expression and SOCE [140]. Overexpressed Orai1 re-established SOCE and restored the normal rate of apoptosis in these cells, indicating a critical role of down-regulated Orai1 function in the establishment of an apoptotic resistance in Pca cells [140]. The involvement of components of the SOCE pathway in cancer highlights a possible role of STIM/Orai as therapeutic targets in cancer therapy.
Concluding remarks
A great advances in the understanding of SOCE has been done over the last years. The discovery of Orai and STIM isoforms as essential players of SOCE, where the participation of TRPC proteins and IP3R has also been described [Refs. [141, 142]; Fig. 3), helped to unravel the function of this mechanism in cell physiology. The phenotypic analysis of patients lacking these proteins showed a major function of Orai1- and STIM1-dependent SOCE in cells of the immune system, skeletal muscle some ectodermal-derived tissues such as sweat glands and teeth. Interestingly, cardiomyopathies were not reported in these patients, indicating a more prominent role of Orai1- and STIM1-dependent SOCE in skeletal muscle fibres than in cardiomyocytes. Studies in Orai1- and STIM1-deficient murine transgenic models performed in parallel complemented our knowledge of the mechanisms underlying disease in the absence of these proteins. The similar phenotype found in mouse and humans indicates that transgenic models could be suitable models to investigate novel therapies based on Orai, STIM and SOCE modulation. In addition, these models provided insights of new functions of SOCE in other tissues and pathological evens, such as ischaemic stroke and autoimmune diseases. The in vivo roles of their homologues Orai2, Orai3 and in a less extent STIM2 are still unclear, having overlapping functions which their respective isoforms in vitro. A major role for STIM1 and Orai1 in all tissues is unlikely, regarding the presence of SOCE in many cell types which did not show altered function in patients or mice lacking functional Orai1 and STIM1. Current evidence points out these molecules as new therapeutic targets, especially those related with immune disorders, severe T cell–dependent inflammatory diseases or cancer. Existing studies revealed that compared to current treatments (FK506, CsA and OKT3), Orai1 inhibitors could have a potential for higher efficacy without the need for expensive and side-effect-prone co-administration of additional immunosuppressants such as glucocorticoids (reviewed in Ref. [143]). However, the presence of immune-unrelated pathologies in the Orai1- or STIM1-deficient patients and the ubiquitous expression pattern of these molecules are issues that still have to be addressed for complete validation of these proteins as suitable therapeutic targets. In addition, members of the TRPC family were recently found to interact directly or indirectly to both STIM1 and Orai1 [55, 144] (reviewed in Ref. [145]), indicating that such TRPC members could participate as SOCE components, or that Orai and STIM1 could be involved in regulation of TRPC-dependent Ca2+ entry as well [144]. Because TRP channels are involved in a variety of physiological processes such as stress responses to noxious stimuli or thermo- and vasoregulation [146] (reviewed in Ref. [147]), the possibility that Orai or STIM inhibitors could elicit significant unwanted side effects by co-inhibition of other Orai- or STIM-interacting channels must be addressed as well [143].
Fig 3.
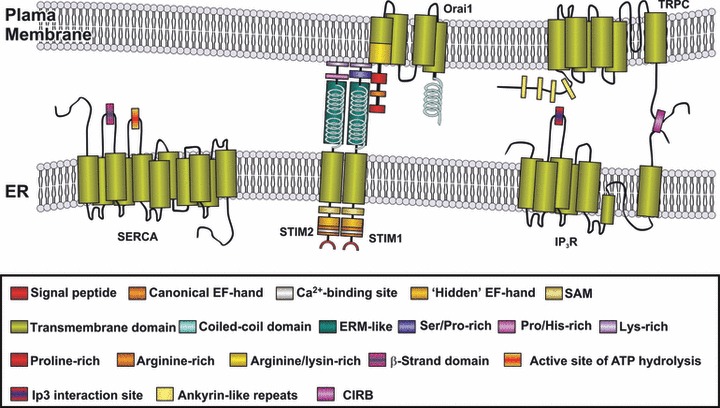
Overview of the major elements of SOCE. Discharge of the intracellular Ca2+ stores is detected by STIM proteins that communicate the filling state of the Ca2+ compartments to the store-operated channels in the plasma membrane, mostly consisting of Orai subunits and TRPC subfamily members. The latter have been reported to associate with IP3Rs, which regulates both Ca2+ release and entry [141, 142]. ER: endoplasmic reticulum; ERM: ezrin/radixin/moesin motif; SAM: sterile alpha motif; CIRB: calmodulin and IP3 receptor binding region.
Acknowledgments
This work was supported by MICINN grant BFU2010-21043-C02-01 and Junta de Extremadura-FEDER (GR10010).
Conflict of interest
The authors declare no conflicts of interest.
References
- 1.Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 2.Berridge MJ, Irvine RF. Inositol phosphates and cell signalling. Nature. 1989;341:197–205. doi: 10.1038/341197a0. [DOI] [PubMed] [Google Scholar]
- 3.Cheek TR, Berridge MJ, Moreton RB, et al. Quantal Ca2+ mobilization by ryanodine receptors is due to all-or-none release from functionally discrete intracellular stores. Biochem J. 1994;301:879–83. doi: 10.1042/bj3010879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cancela JM, Churchill GC, Galione A. Coordination of agonist-induced Ca2+-signalling patterns by NAADP in pancreatic acinar cells. Nature. 1999;398:74–6. doi: 10.1038/18032. [DOI] [PubMed] [Google Scholar]
- 5.Churchill GC, Okada Y, Thomas JM, et al. NAADP mobilizes Ca(2+) from reserve granules, lysosome-related organelles, in sea urchin eggs. Cell. 2002;111:703–8. doi: 10.1016/s0092-8674(02)01082-6. [DOI] [PubMed] [Google Scholar]
- 6.Xu SZ, Muraki K, Zeng F, et al. A sphingosine-1-phosphate-activated calcium channel controlling vascular smooth muscle cell motility. Circ Res. 2006;98:1381–9. doi: 10.1161/01.RES.0000225284.36490.a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sage SO. The Wellcome Prize Lecture. Calcium entry mechanisms in human platelets. Exp Physiol. 1997;82:807–23. doi: 10.1113/expphysiol.1997.sp004066. [DOI] [PubMed] [Google Scholar]
- 8.Sage SO. Three routes for receptor-mediated Ca2+ entry. Curr Biol. 1992;2:312–4. doi: 10.1016/0960-9822(92)90885-e. [DOI] [PubMed] [Google Scholar]
- 9.Salido GM, Sage SO, Rosado JA. TRPC channels and store-operated Ca(2+) entry. Biochim Biophys Acta. 2009;1793:223–30. doi: 10.1016/j.bbamcr.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 10.Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 11.Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–6. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- 12.Zweifach A, Lewis RS. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc Natl Acad Sci USA. 1993;90:6295–9. doi: 10.1073/pnas.90.13.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vig M, Peinelt C, Beck A, et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006;312:1220–3. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peinelt C, Vig M, Koomoa DL, et al. Amplification of CRAC current by STIM1 and CRACM1 (Orai1) Nat Cell Biol. 2006;8:771–3. doi: 10.1038/ncb1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prakriya M, Feske S, Gwack Y, et al. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443:230–3. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- 16.Prakriya M. The molecular physiology of CRAC channels. Immunol Rev. 2009;231:88–98. doi: 10.1111/j.1600-065X.2009.00820.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lis A, Zierler S, Peinelt C, et al. A single lysine in the N-terminal region of store-operated channels is critical for STIM1-mediated gating. J Gen Physiol. 2010;136:673–86. doi: 10.1085/jgp.201010484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takahashi Y, Murakami M, Watanabe H, et al. Essential role of the N-terminus of murine Orai1 in store-operated Ca2+ entry. Biochem Biophys Res Commun. 2007;356:45–52. doi: 10.1016/j.bbrc.2007.02.107. [DOI] [PubMed] [Google Scholar]
- 19.Muik M, Frischauf I, Derler I, et al. Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J Biol Chem. 2008;283:8014–22. doi: 10.1074/jbc.M708898200. [DOI] [PubMed] [Google Scholar]
- 20.Li Z, Lu J, Xu P, et al. Mapping the interacting domains of STIM1 and Orai1 in Ca2+ release-activated Ca2+ channel activation. J Biol Chem. 2007;282:29448–56. doi: 10.1074/jbc.M703573200. [DOI] [PubMed] [Google Scholar]
- 21.Mullins FM, Park CY, Dolmetsch RE, et al. STIM1 and calmodulin interact with Orai1 to induce Ca2+-dependent inactivation of CRAC channels. Proc Natl Acad Sci USA. 2009;106:15495–500. doi: 10.1073/pnas.0906781106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feske S, Gwack Y, Prakriya M, et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–85. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 23.Yamashita M, Navarro-Borelly L, McNally BA, et al. Orai1 mutations alter ion permeation and Ca2+-dependent fast inactivation of CRAC channels: evidence for coupling of permeation and gating. J Gen Physiol. 2007;130:525–40. doi: 10.1085/jgp.200709872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mercer JC, Dehaven WI, Smyth JT, et al. Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. J Biol Chem. 2006;281:24979–90. doi: 10.1074/jbc.M604589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flourakis M, Van Coppenolle F, Lehen’kyi V, et al. Passive calcium leak via translocon is a first step for iPLA2-pathway regulated store operated channels activation. FASEB J. 2006;20:1215–7. doi: 10.1096/fj.05-5254fje. [DOI] [PubMed] [Google Scholar]
- 26.Rosado JA, Sage SO. Platelet signalling: calcium. In: Gresele P, Page CP, Fuster V, Vermylen J, editors. Platelets in thrombotic and non-thrombotic disorders pathophysiology, pharmacology and therapeutics. Cambridge: Cambridge University Press; 2000. pp. 260–71. [Google Scholar]
- 27.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–29. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 28.Vangheluwe P, Raeymaekers L, Dode L, et al. Modulating sarco(endo)plasmic reticulum Ca2+ ATPase 2 (SERCA2) activity: cell biological implications. Cell Calcium. 2005;38:291–302. doi: 10.1016/j.ceca.2005.06.033. [DOI] [PubMed] [Google Scholar]
- 29.Aulestia FJ, Redondo PC, Rodriguez-Garcia A, et al. Two distinct calcium pools in the endoplasmic reticulum of HEK-293T cells. Biochem J. 2011;435:227–35. doi: 10.1042/BJ20101427. [DOI] [PubMed] [Google Scholar]
- 30.Petersen OH, Gerasimenko OV, Gerasimenko JV, et al. The calcium store in the nuclear envelope. Cell Calcium. 1998;23:87–90. doi: 10.1016/s0143-4160(98)90106-3. [DOI] [PubMed] [Google Scholar]
- 31.Gerasimenko JV, Maruyama Y, Yano K, et al. NAADP mobilizes Ca2+ from a thapsigargin-sensitive store in the nuclear envelope by activating ryanodine receptors. J Cell Biol. 2003;163:271–82. doi: 10.1083/jcb.200306134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patel S, Docampo R. Acidic calcium stores open for business: expanding the potential for intracellular Ca2+ signaling. Trends Cell Biol. 2010;20:277–86. doi: 10.1016/j.tcb.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lopez JJ, Camello-Almaraz C, Pariente JA, et al. Ca2+ accumulation into acidic organelles mediated by Ca2+- and vacuolar H+-ATPases in human platelets. Biochem J. 2005;390:243–52. doi: 10.1042/BJ20050168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jacobson J, Duchen MR. Interplay between mitochondria and cellular calcium signalling. Mol Cell Biochem. 2004:256–257. doi: 10.1023/b:mcbi.0000009869.29827.df. : 209–18. [DOI] [PubMed] [Google Scholar]
- 35.Walsh C, Barrow S, Voronina S, et al. Modulation of calcium signalling by mitochondria. Biochim Biophys Acta. 2009;1787:1374–82. doi: 10.1016/j.bbabio.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 36.Parekh AB. Store-operated Ca2+ entry: dynamic interplay between endoplasmic reticulum, mitochondria and plasma membrane. J Physiol. 2003;547:333–48. doi: 10.1113/jphysiol.2002.034140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malli R, Frieden M, Trenker M, et al. The role of mitochondria for Ca2+ refilling of the endoplasmic reticulum. J Biol Chem. 2005;280:12114–22. doi: 10.1074/jbc.M409353200. [DOI] [PubMed] [Google Scholar]
- 38.Missiaen L, Van Acker K, Van Baelen K, et al. Calcium release from the Golgi apparatus and the endoplasmic reticulum in HeLa cells stably expressing targeted aequorin to these compartments. Cell Calcium. 2004;36:479–87. doi: 10.1016/j.ceca.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 39.Mahieu F, Owsianik G, Verbert L, et al. TRPM8-independent menthol-induced Ca2+ release from endoplasmic reticulum and Golgi. J Biol Chem. 2007;282:3325–36. doi: 10.1074/jbc.M605213200. [DOI] [PubMed] [Google Scholar]
- 40.Van Baelen K, Dode L, Vanoevelen J, et al. The Ca2+/Mn2+ pumps in the Golgi apparatus. Biochim Biophys Acta. 2004;1742:103–12. doi: 10.1016/j.bbamcr.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 41.Van Baelen K, Vanoevelen J, Callewaert G, et al. The contribution of the SPCA1 Ca2+ pump to the Ca2+ accumulation in the Golgi apparatus of HeLa cells assessed via RNA-mediated interference. Biochem Biophys Res Commun. 2003;306:430–6. doi: 10.1016/s0006-291x(03)00977-x. [DOI] [PubMed] [Google Scholar]
- 42.Wuytack F, Raeymaekers L, Missiaen L. PMR1/SPCA Ca2+ pumps and the role of the Golgi apparatus as a Ca2+ store. Pflugers Arch. 2003;446:148–53. doi: 10.1007/s00424-003-1011-5. [DOI] [PubMed] [Google Scholar]
- 43.Putney JW, Bird GS. Cytoplasmic calcium oscillations and store-operated calcium influx. J Physiol. 2008;586:3055–9. doi: 10.1113/jphysiol.2008.153221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.del Monte F, Hajjar RJ, Harding SE. Overwhelming evidence of the beneficial effects of SERCA gene transfer in heart failure. Circ Res. 2001;88:E66–7. doi: 10.1161/hh1101.092004. [DOI] [PubMed] [Google Scholar]
- 45.Auffret A, Gautheron V, Mattson MP, et al. Progressive age-related impairment of the late long-term potentiation in Alzheimer’s disease presenilin-1 mutant knock-in mice. J Alzheimers Dis. 2010;19:1021–33. doi: 10.3233/JAD-2010-1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vanoevelen J, Dode L, Raeymaekers L, et al. Diseases involving the Golgi calcium pump. Subcell Biochem. 2007;45:385–404. doi: 10.1007/978-1-4020-6191-2_14. [DOI] [PubMed] [Google Scholar]
- 47.Richter M, Schleithoff L, Deufel T, et al. Functional characterization of a distinct ryanodine receptor mutation in human malignant hyperthermia-susceptible muscle. J Biol Chem. 1997;272:5256–60. doi: 10.1074/jbc.272.8.5256. [DOI] [PubMed] [Google Scholar]
- 48.Striessnig J, Bolz HJ, Koschak A. Channelopathies in Cav1.1, Cav1.3, and Cav1.4 voltage-gated L-type Ca2+ channels. Pflugers Arch. 2010;460:361–74. doi: 10.1007/s00424-010-0800-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kiselyov K, Soyombo A, Muallem S. TRPpathies. J Physiol. 2007;578:641–53. doi: 10.1113/jphysiol.2006.119024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nilius B, Owsianik G, Voets T, et al. Transient receptor potential cation channels in disease. Physiol Rev. 2007;87:165–217. doi: 10.1152/physrev.00021.2006. [DOI] [PubMed] [Google Scholar]
- 51.Putney JW., Jr A model for receptor-regulated calcium entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- 52.Roos J, DiGregorio PJ, Yeromin AV, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–45. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang SL, Yu Y, Roos J, et al. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–5. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abdullaev IF, Bisaillon JM, Potier M, et al. Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ Res. 2008;103:1289–99. doi: 10.1161/01.RES.0000338496.95579.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lopez JJ, Salido GM, Pariente JA, et al. Interaction of STIM1 with endogenously expressed human canonical TRP1 upon depletion of intracellular Ca2+ stores. J Biol Chem. 2006;281:28254–64. doi: 10.1074/jbc.M604272200. [DOI] [PubMed] [Google Scholar]
- 56.Stiber J, Hawkins A, Zhang ZS, et al. STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat Cell Biol. 2008;10:688–97. doi: 10.1038/ncb1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Frischauf I, Muik M, Derler I, et al. Molecular determinants of the coupling between STIM1 and Orai channels: differential activation of Orai1–3 channels by a STIM1 coiled-coil mutant. J Biol Chem. 2009;284:21696–706. doi: 10.1074/jbc.M109.018408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stathopulos PB, Li GY, Plevin MJ, et al. Stored Ca2+ depletion-induced oligomerization of stromal interaction molecule 1 (STIM1) via the EF-SAM region: an initiation mechanism for capacitive Ca2+ entry. J Biol Chem. 2006;281:35855–62. doi: 10.1074/jbc.M608247200. [DOI] [PubMed] [Google Scholar]
- 59.Zheng L, Stathopulos PB, Schindl R, et al. Auto-inhibitory role of the EF-SAM domain of STIM proteins in store-operated calcium entry. Proc Natl Acad Sci USA. 2011;108:1337–42. doi: 10.1073/pnas.1015125108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schindl R, Muik M, Fahrner M, et al. Recent progress on STIM1 domains controlling Orai activation. Cell Calcium. 2009;46:227–32. doi: 10.1016/j.ceca.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 61.Yuan JP, Zeng W, Dorwart MR, et al. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol. 2009;11:337–43. doi: 10.1038/ncb1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Muik M, Fahrner M, Derler I, et al. A cytosolic homomerization and a modulatory domain within STIM1 C terminus determine coupling to ORAI1 channels. J Biol Chem. 2009;284:8421–6. doi: 10.1074/jbc.C800229200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Park CY, Hoover PJ, Mullins FM, et al. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. 2009;136:876–90. doi: 10.1016/j.cell.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kawasaki T, Lange I, Feske S. A minimal regulatory domain in the C terminus of STIM1 binds to and activates ORAI1 CRAC channels. Biochem Biophys Res Commun. 2009;385:49–54. doi: 10.1016/j.bbrc.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang Y, Deng X, Gill DL. Calcium signaling by STIM and Orai: intimate coupling details revealed. Sci Signal. 2010;3:pe42. doi: 10.1126/scisignal.3148pe42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zheng L, Stathopulos PB, Li GY, et al. Biophysical characterization of the EF-hand and SAM domain containing Ca2+ sensory region of STIM1 and STIM2. Biochem Biophys Res Commun. 2008;369:240–6. doi: 10.1016/j.bbrc.2007.12.129. [DOI] [PubMed] [Google Scholar]
- 67.Soboloff J, Spassova MA, Hewavitharana T, et al. STIM2 is an inhibitor of STIM1-mediated store-operated Ca2+ entry. Curr Biol. 2006;16:1465–70. doi: 10.1016/j.cub.2006.05.051. [DOI] [PubMed] [Google Scholar]
- 68.Brandman O, Liou J, Park WS, et al. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell. 2007;131:1327–39. doi: 10.1016/j.cell.2007.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Parvez S, Beck A, Peinelt C, et al. STIM2 protein mediates distinct store-dependent and store-independent modes of CRAC channel activation. FASEB J. 2008;22:752–61. doi: 10.1096/fj.07-9449com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Berna-Erro A, Braun A, Kraft R, et al. STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Sci Signal. 2009;2:ra67. doi: 10.1126/scisignal.2000522. [DOI] [PubMed] [Google Scholar]
- 71.Jousset H, Frieden M, Demaurex N. STIM1 knockdown reveals that store-operated Ca2+ channels located close to sarco/endoplasmic Ca2+ ATPases (SERCA) pumps silently refill the endoplasmic reticulum. J Biol Chem. 2007;282:11456–64. doi: 10.1074/jbc.M609551200. [DOI] [PubMed] [Google Scholar]
- 72.Zbidi H, Jardin I, Woodard GE, et al. STIM1 and STIM2 are located in the acidic Ca2+ stores and associates with Orai1 upon depletion of the acidic stores in human platelets. J Biol Chem. 2011;286:12257–70. doi: 10.1074/jbc.M110.190694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McCarl CA, Picard C, Khalil S, et al. ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia. J Allergy Clin Immunol. 2009;124:1311–8. doi: 10.1016/j.jaci.2009.10.007. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gwack Y, Srikanth S, Feske S, et al. Biochemical and functional characterization of Orai proteins. J Biol Chem. 2007;282:16232–43. doi: 10.1074/jbc.M609630200. [DOI] [PubMed] [Google Scholar]
- 75.Su AI, Wiltshire T, Batalov S, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci USA. 2004;101:6062–7. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wissenbach U, Philipp SE, Gross SA, et al. Primary structure, chromosomal localization and expression in immune cells of the murine ORAI and STIM genes. Cell Calcium. 2007;42:439–46. doi: 10.1016/j.ceca.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 77.Williams RT, Manji SS, Parker NJ, et al. Identification and characterization of the STIM (stromal interaction molecule) gene family: coding for a novel class of transmembrane proteins. Biochem J. 2001;357:673–85. doi: 10.1042/0264-6021:3570673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Picard C, McCarl CA, Papolos A, et al. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N Engl J Med. 2009;360:1971–80. doi: 10.1056/NEJMoa0900082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Skibinska-Kijek A, Wisniewska MB, Gruszczynska-Biegala J, et al. Immunolocalization of STIM1 in the mouse brain. Acta Neurobiol Exp (Wars) 2009;69:413–28. doi: 10.55782/ane-2009-1753. [DOI] [PubMed] [Google Scholar]
- 80.Overall ML, Parker NJ, Scarcella DL, et al. Murine Stim1 maps to distal chromosome 7 and is not imprinted. Mamm Genome. 1998;9:657–9. doi: 10.1007/s003359900839. [DOI] [PubMed] [Google Scholar]
- 81.Zhou Y, Mancarella S, Wang Y, et al. The short N-terminal domains of STIM1 and STIM2 control the activation kinetics of Orai1 channels. J Biol Chem. 2009;284:19164–8. doi: 10.1074/jbc.C109.010900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.El Boustany C, Katsogiannou M, Delcourt P, et al. Differential roles of STIM1, STIM2 and Orai1 in the control of cell proliferation and SOCE amplitude in HEK293 cells. Cell Calcium. 2010;47:350–9. doi: 10.1016/j.ceca.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 83.Feske S, Giltnane J, Dolmetsch R, et al. Gene regulation mediated by calcium signals in T lymphocytes. Nat Immunol. 2001;2:316–24. doi: 10.1038/86318. [DOI] [PubMed] [Google Scholar]
- 84.Feske S, Prakriya M, Rao A, et al. A severe defect in CRAC Ca2+ channel activation and altered K+ channel gating in T cells from immunodeficient patients. J Exp Med. 2005;202:651–62. doi: 10.1084/jem.20050687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Byun M, Abhyankar A, Lelarge V, et al. Whole-exome sequencing-based discovery of STIM1 deficiency in a child with fatal classic Kaposi sarcoma. J Exp Med. 2010;207:2307–12. doi: 10.1084/jem.20101597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Le Deist F, Hivroz C, Partiseti M, et al. A primary T-cell immunodeficiency associated with defective transmembrane calcium influx. Blood. 1995;85:1053–62. [PubMed] [Google Scholar]
- 87.Partiseti M, Le Deist F, Hivroz C, et al. The calcium current activated by T cell receptor and store depletion in human lymphocytes is absent in a primary immunodeficiency. J Biol Chem. 1994;269:32327–35. [PubMed] [Google Scholar]
- 88.Feske S, Picard C, Fischer A. Immunodeficiency due to mutations in ORAI1 and STIM1. Clin Immunol. 2010;135:169–82. doi: 10.1016/j.clim.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Feske S. ORAI1 and STIM1 deficiency in human and mice: roles of store-operated Ca2+ entry in the immune system and beyond. Immunol Rev. 2009;231:189–209. doi: 10.1111/j.1600-065X.2009.00818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thompson JL, Mignen O, Shuttleworth TJ. The Orai1 severe combined immune deficiency mutation and calcium release-activated Ca2+ channel function in the heterozygous condition. J Biol Chem. 2009;284:6620–6. doi: 10.1074/jbc.M808346200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Feske S. CRAC channelopathies. Pflugers Arch. 2010;460:417–35. doi: 10.1007/s00424-009-0777-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Feske S, Muller JM, Graf D, et al. Severe combined immunodeficiency due to defective binding of the nuclear factor of activated T cells in T lymphocytes of two male siblings. Eur J Immunol. 1996;26:2119–26. doi: 10.1002/eji.1830260924. [DOI] [PubMed] [Google Scholar]
- 93.Feske S, Draeger R, Peter HH, et al. The duration of nuclear residence of NFAT determines the pattern of cytokine expression in human SCID T cells. J Immunol. 2000;165:297–305. doi: 10.4049/jimmunol.165.1.297. [DOI] [PubMed] [Google Scholar]
- 94.Maul-Pavicic A, Chiang SC, Rensing-Ehl A, et al. ORAI1-mediated calcium influx is required for human cytotoxic lymphocyte degranulation and target cell lysis. Proc Natl Acad Sci USA. 2011;108:3324–9. doi: 10.1073/pnas.1013285108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Darbellay B, Arnaudeau S, Konig S, et al. STIM1- and Orai1-dependent store-operated calcium entry regulates human myoblast differentiation. J Biol Chem. 2009;284:5370–80. doi: 10.1074/jbc.M806726200. [DOI] [PubMed] [Google Scholar]
- 96.Darbellay B, Arnaudeau S, Ceroni D, et al. Human muscle economy myoblast differentiation and excitation-contraction coupling use the same molecular partners, STIM1 and STIM2. J Biol Chem. 2010;285:22437–47. doi: 10.1074/jbc.M110.118984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bovell DL, Clunes MT, Elder HY, et al. Nucleotide-evoked ion transport and [Ca2+)](i) changes in normal and hyperhidrotic human sweat gland cells. Eur J Pharmacol. 2000;403:45–8. doi: 10.1016/s0014-2999(00)00570-7. [DOI] [PubMed] [Google Scholar]
- 98.Ko WH, Chan HC, Wong PY. Anion secretion induced by capacitative Ca2+ entry through apical and basolateral membranes of cultured equine sweat gland epithelium. J Physiol. 1996;497:19–29. doi: 10.1113/jphysiol.1996.sp021746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Beyersdorf N, Braun A, Vogtle T, et al. STIM1-independent T cell development and effector function in vivo. J Immunol. 2009;182:3390–7. doi: 10.4049/jimmunol.0802888. [DOI] [PubMed] [Google Scholar]
- 100.Oh-Hora M, Yamashita M, Hogan PG, et al. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat Immunol. 2008;9:432–43. doi: 10.1038/ni1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gwack Y, Srikanth S, Oh-Hora M, et al. Hair loss and defective T- and B-cell function in mice lacking ORAI1. Mol Cell Biol. 2008;28:5209–22. doi: 10.1128/MCB.00360-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xu P, Lu J, Li Z, et al. Aggregation of STIM1 underneath the plasma membrane induces clustering of Orai1. Biochem Biophys Res Commun. 2006;350:969–76. doi: 10.1016/j.bbrc.2006.09.134. [DOI] [PubMed] [Google Scholar]
- 103.Soboloff J, Spassova MA, Tang XD, et al. Orai1 and STIM reconstitute store-operated calcium channel function. J Biol Chem. 2006;281:20661–5. doi: 10.1074/jbc.C600126200. [DOI] [PubMed] [Google Scholar]
- 104.Jardin I, Lopez JJ, Zbidi H, et al. Attenuated store-operated divalent cation entry and association between STIM1, Orai1, hTRPC1 and hTRPC6 in platelets from type 2 diabetic patients. Blood Cells Mol Dis. 2011;46:252–60. doi: 10.1016/j.bcmd.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 105.Vig M, DeHaven WI, Bird GS, et al. Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels. Nat Immunol. 2008;9:89–96. doi: 10.1038/ni1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bergmeier W, Oh-Hora M, McCarl CA, et al. R93W mutation in Orai1 causes impaired calcium influx in platelets. Blood. 2009;113:675–8. doi: 10.1182/blood-2008-08-174516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Braun A, Varga-Szabo D, Kleinschnitz C, et al. Orai1 (CRACM1) is the platelet SOC channel and essential for pathological thrombus formation. Blood. 2009;113:2056–63. doi: 10.1182/blood-2008-07-171611. [DOI] [PubMed] [Google Scholar]
- 108.Varga-Szabo D, Braun A, Kleinschnitz C, et al. The calcium sensor STIM1 is an essential mediator of arterial thrombosis and ischemic brain infarction. J Exp Med. 2008;205:1583–91. doi: 10.1084/jem.20080302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Aifantis I, Gounari F, Scorrano L, et al. Constitutive pre-TCR signaling promotes differentiation through Ca2+ mobilization and activation of NF-kappaB and NFAT. Nat Immunol. 2001;2:403–9. doi: 10.1038/87704. [DOI] [PubMed] [Google Scholar]
- 110.von Boehmer H, Fehling HJ. Structure and function of the pre-T cell receptor. Annu Rev Immunol. 1997;15:433–52. doi: 10.1146/annurev.immunol.15.1.433. [DOI] [PubMed] [Google Scholar]
- 111.Wu Y, Borde M, Heissmeyer V, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126:375–87. doi: 10.1016/j.cell.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 112.Braun A, Gessner JE, Varga-Szabo D, et al. STIM1 is essential for Fcgamma receptor activation and autoimmune inflammation. Blood. 2009;113:1097–104. doi: 10.1182/blood-2008-05-158477. [DOI] [PubMed] [Google Scholar]
- 113.Schuhmann MK, Stegner D, Berna-Erro A, et al. Stromal interaction molecules 1 and 2 are key regulators of autoreactive T cell activation in murine autoimmune central nervous system inflammation. J Immunol. 2010;184:1536–42. doi: 10.4049/jimmunol.0902161. [DOI] [PubMed] [Google Scholar]
- 114.Franzini-Armstrong C. The sarcoplasmic reticulum and the control of muscle contraction. FASEB J. 1999;13:S266–70. doi: 10.1096/fasebj.13.9002.s266. [DOI] [PubMed] [Google Scholar]
- 115.Pisaniello A, Serra C, Rossi D, et al. The block of ryanodine receptors selective. J Cell Sci. 2003;116:1589–97. doi: 10.1242/jcs.00358. [DOI] [PubMed] [Google Scholar]
- 116.Arnaudeau S, Holzer N, Konig S, et al. Calcium sources used by post-natal human myoblasts during initial differentiation. J Cell Physiol. 2006;208:435–45. doi: 10.1002/jcp.20679. [DOI] [PubMed] [Google Scholar]
- 117.Gilio K, van Kruchten R, Braun A, et al. Roles of platelet STIM1 and Orai1 in glycoprotein VI- and thrombin-dependent procoagulant activity and thrombus formation. J Biol Chem. 2010;285:23629–38. doi: 10.1074/jbc.M110.108696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Keil JM, Shen Z, Briggs SP, et al. Regulation of STIM1 and SOCE by the ubiquitin-proteasome system (UPS) PLoS One. 2010;5:e13465. doi: 10.1371/journal.pone.0013465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gasperini R, Choi-Lundberg D, Thompson MJ, et al. Homer regulates calcium signalling in growth cone turning. Neural Dev. 2009;4:29. doi: 10.1186/1749-8104-4-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Danoff SK, Ross CA. The inositol trisphosphate receptor gene family: implications for normal and abnormal brain function. Prog Neuropsychopharmacol Biol Psychiatry. 1994;18:1–16. doi: 10.1016/0278-5846(94)90021-3. [DOI] [PubMed] [Google Scholar]
- 121.Putney JW., Jr Capacitative calcium entry in the nervous system. Cell Calcium. 2003;34:339–44. doi: 10.1016/s0143-4160(03)00143-x. [DOI] [PubMed] [Google Scholar]
- 122.Morris RG, Anderson E, Lynch GS, et al. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5. Nature. 1986;319:774–6. doi: 10.1038/319774a0. [DOI] [PubMed] [Google Scholar]
- 123.Emptage NJ, Reid CA, Fine A. Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron. 2001;29:197–208. doi: 10.1016/s0896-6273(01)00190-8. [DOI] [PubMed] [Google Scholar]
- 124.Larsen GA, Skjellegrind HK, Moe MC, et al. Endoplasmic reticulum dysfunction and Ca2+ deregulation in isolated CA1 neurons during oxygen and glucose deprivation. Neurochem Res. 2005;30:651–9. doi: 10.1007/s11064-005-2753-6. [DOI] [PubMed] [Google Scholar]
- 125.Henrich M, Buckler KJ. Effects of anoxia and aglycemia on cytosolic calcium regulation in rat sensory neurons. J Neurophysiol. 2008;100:456–73. doi: 10.1152/jn.01380.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Frandsen A, Schousboe A. Dantrolene prevents glutamate cytotoxicity and Ca2+ release from intracellular stores in cultured cerebral cortical neurons. J Neurochem. 1991;56:1075–8. doi: 10.1111/j.1471-4159.1991.tb02031.x. [DOI] [PubMed] [Google Scholar]
- 127.Mattson MP, Zhu H, Yu J, et al. Presenilin-1 mutation increases neuronal vulnerability to focal ischemia in vivo and to hypoxia and glucose deprivation in cell culture: involvement of perturbed calcium homeostasis. J Neurosci. 2000;20:1358–64. doi: 10.1523/JNEUROSCI.20-04-01358.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Arundine M, Tymianski M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell Mol Life Sci. 2004;61:657–68. doi: 10.1007/s00018-003-3319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.McAndrew D, Grice DM, Peters AA, et al. ORAI1-mediated calcium influx in lactation and in breast cancer. Mol Cancer Ther. 2011;10:448–60. doi: 10.1158/1535-7163.MCT-10-0923. [DOI] [PubMed] [Google Scholar]
- 130.Bandyopadhyay BC, Pingle SC, Ahern GP. Store-operated Ca2+ signaling in dendritic cells occurs independently of STIM1. J Leukoc Biol. 2011;89:57–62. doi: 10.1189/jlb.0610381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kim D, Jun KS, Lee SB, et al. Phospholipase C isozymes selectively couple to specific neurotransmitter receptors. Nature. 1997;389:290–3. doi: 10.1038/38508. [DOI] [PubMed] [Google Scholar]
- 132.Matsumoto M, Nagata E. Type 1 inositol 1,4,5-trisphosphate receptor knock-out mice: their phenotypes and their meaning in neuroscience and clinical practice. J Mol Med. 1999;77:406–11. doi: 10.1007/s001090050370. [DOI] [PubMed] [Google Scholar]
- 133.Wojda U, Salinska E, Kuznicki J. Calcium ions in neuronal degeneration. IUBMB Life. 2008;60:575–90. doi: 10.1002/iub.91. [DOI] [PubMed] [Google Scholar]
- 134.Mattson MP. Calcium and neurodegeneration. Aging Cell. 2007;6:337–50. doi: 10.1111/j.1474-9726.2007.00275.x. [DOI] [PubMed] [Google Scholar]
- 135.Sabbioni S, Veronese A, Trubia M, et al. Exon structure and promoter identification of STIM1 (alias GOK), a human gene causing growth arrest of the human tumour cell lines G401 and RD. Cytogenet Cell Genet. 1999;86:214–8. doi: 10.1159/000015341. [DOI] [PubMed] [Google Scholar]
- 136.Sabbioni S, Barbanti-Brodano G, Croce CM, et al. GOK: a gene at 11p15 involved in rhabdomyosarcoma and rhabdoid tumour development. Cancer Res. 1997;57:4493–7. [PubMed] [Google Scholar]
- 137.Ritchie MF, Yue C, Zhou Y, et al. Wilms tumour suppressor 1 (WT1) and early growth response 1 (EGR1) are regulators of STIM1 expression. J Biol Chem. 2010;285:10591–6. doi: 10.1074/jbc.M109.083493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Motiani RK, Abdullaev IF, Trebak M. A novel native store-operated calcium channel encoded by Orai3: selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J Biol Chem. 2010;285:19173–83. doi: 10.1074/jbc.M110.102582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Faouzi M, Hague F, Potier M, et al. Down-regulation of Orai3 arrests cell-cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. J Cell Physiol. 2011;226:542–51. doi: 10.1002/jcp.22363. [DOI] [PubMed] [Google Scholar]
- 140.Flourakis M, Lehen’kyi V, Beck B, et al. Orai1 contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells. Cell Death Dis. 2010;1:e75. doi: 10.1038/cddis.2010.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Jardin I, Lopez JJ, Salido GM, et al. Functional relevance of the de novo coupling between hTRPC1 and type II IP3 receptor in store-operated Ca2+ entry in human platelets. Cell Signal. 2008;20:737–47. doi: 10.1016/j.cellsig.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 142.Woodard GE, Lopez JJ, Jardin I, et al. TRPC3 regulates agonist-stimulated Ca2+ mobilization by mediating the interaction between type I inositol 1,4,5-trisphosphate receptor, RACK1, and Orai1. J Biol Chem. 2010;285:8045–53. doi: 10.1074/jbc.M109.033605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Huang YH, Hoebe K, Sauer K. New therapeutic targets in immune disorders: ItpkB, Orai1 and UNC93B. Expert Opin Ther Targets. 2008;12:391–413. doi: 10.1517/14728222.12.4.391. [DOI] [PubMed] [Google Scholar]
- 144.Ong HL, Cheng KT, Liu X, et al. Dynamic assembly of TRPC1-STIM1-Orai1 ternary complex is involved in store-operated calcium influx. Evidence for similarities in store-operated and calcium release-activated calcium channel components. J Biol Chem. 2007;282:9105–16. doi: 10.1074/jbc.M608942200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Salido GM, Jardin I, Rosado JA. The TRPC ion channels: association with Orai1 and STIM1 proteins and participation in capacitative and non-capacitative calcium entry. Adv Exp Med Biol. 2011;704:413–33. doi: 10.1007/978-94-007-0265-3_23. [DOI] [PubMed] [Google Scholar]
- 146.Hicks GA. TRP channels as therapeutic targets: hot property, or time to cool down. Neurogastroenterol Motil. 2006;18:590–4. doi: 10.1111/j.1365-2982.2006.00823.x. [DOI] [PubMed] [Google Scholar]
- 147.Abramowitz J, Birnbaumer L. Physiology and pathophysiology of canonical transient receptor potential channels. FASEB J. 2009;23:297–328. doi: 10.1096/fj.08-119495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Fahrner M, Muik M, Derler I, et al. Mechanistic view on domains mediating STIM1-Orai coupling. Immunol Rev. 2009;231:99–112. doi: 10.1111/j.1600-065X.2009.00815.x. [DOI] [PubMed] [Google Scholar]
- 149.Stathopulos PB, Zheng L, Ikura M. Stromal interaction molecule (STIM) 1 and STIM2 calcium sensing regions exhibit distinct unfolding and oligomerization kinetics. J Biol Chem. 2009;284:728–32. doi: 10.1074/jbc.C800178200. [DOI] [PubMed] [Google Scholar]
- 150.Stathopulos PB, Zheng L, Li GY, et al. Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell. 2008;135:110–22. doi: 10.1016/j.cell.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 151.McCarl CA, Khalil S, Ma J, et al. Store-operated Ca2+ entry through ORAI1 is critical for T cell-mediated autoimmunity and allograft rejection. J Immunol. 2010;185:5845–58. doi: 10.4049/jimmunol.1001796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Baba Y, Nishida K, Fujii Y, et al. Essential function for the calcium sensor STIM1 in mast cell activation and anaphylactic responses. Nat Immunol. 2008;9:81–8. doi: 10.1038/ni1546. [DOI] [PubMed] [Google Scholar]