

Abstract
Embryonic stem cells (ESC), derived from the early inner cell mass (ICM), are constituted of theoretically homogeneous pluripotent cells. Our study was designed to test this concept using experimental approaches that allowed characterization of progenies derived from single parental mouse ESC. Flow cytometry analysis showed that a fraction of ESC submitted to neural differentiation generates progenies that escape the desired phenotype. Live imaging of individual cells demonstrated significant variations in the capacity of parental ESC to generate neurons, raising the possibility of clonal diversity among ESC. To further substantiate this hypothesis, clonal sublines from ESC were generated by limit dilution. Transcriptome analysis of undifferentiated sublines showed marked differences in gene expression despite the fact that all clones expressed pluripotency markers. Sublines showed distinct differentiation potential, both in phenotypic differentiation assays and with respect to gene expression in embryoid bodies. Clones generated from another ESC line also showed individualities in their differentiation potential, demonstrating the wider applicability of these findings. Taken together, our observations demonstrate that pluripotent ESC consist of individual cell types with distinct differentiation potentials. These findings identify novel elements for the biological understanding of ESC and provide new tools with a major potential for their future in vitro and in vivo use.
Keywords: embryonic stem cell, cloning, differentiation, neural differentiation
Introduction
Embryonic stem cells (ESC) are cell lines derived from the inner cell mass (ICM) of the blastocyst. The ICM is generated through segregation from the trophectoderm, which represents the first differentiation event at early post-implantation embryonic stage. The trophectoderm is dedicated to the generation of extraembryonic tissues whereas the ICM further develops into the primitive endoderm and epiblast. The primitive endoderm generates additional extraembryonic tissues, whereas the epiblast gives rise to the three primordial germ layers and ultimately to the embryo proper.
ESC are frequently considered as a homogeneous cell population, but there are several reasons to doubt this assumption.
First, the ICM at E3.5 after fertilization is already heterogeneous. GATA-6, a transcription factor governing primitive endoderm formation, is expressed in the ICM in a random ‘salt-and-pepper’ fashion, but quite distinct from Nanog+ epiblast precursors [1]. As ESC derivation consists of the initial culture of the ICM, expanding cells could be already heterogeneous.
Secondly, several studies reported that some transcription factors associated with pluripotency are expressed in a heterogeneous fashion in ESC [2-4]. However, it has been suggested that this heterogeneity is not due to the coexistence of independent cell populations because the culture of isolated marker positive and negative fractions restored cells with the original expression pattern, thus implying that the two populations can convert into each other.
Thirdly, a recent study noted that the Stella gene is expressed in 30% of ESC [5]. Phenotypically, Stella-positive ESC resemble
ICM cells, whereas the Stella-negative part is closer to the epiblast. As observed for Nanog, when Stella-positive and Stella-negative ESC were isolated and placed in separate cultures, the original distribution of gene expression was restored. Despite their interchangeability, the two populations exhibited distinct differentiation potentials, and Stella-negative ESC differentiated more readily into cells expressing trophectodermal markers than Stella-positive cells, which were more prone to give rise to embryoid bodies.
Lastly, a recent study, using a reporter gene designed to detect the primitive endoderm lineage, a reversible early primitive endoderm-like cell subtype was identified in mouse ESC [6]. This finding supports the model in which ESC has trapped a set of interconvertible cell states reminiscent of the early stages in blastocyst.
Together, these data raise the possibility of subsets of cells within ESC populations, reflecting the presence of structures within the early embryo dedicated to formation of extraembryonic tissues. However, so far, there is little evidence for heterogeneity of early pluripotent stem cells derived from the ICM. Also, so far, it has been suggested that subpopulations within ESC lines are in a dynamic equilibrium. However, the question whether stable subpopulations of cells exist remains open.
In this study, experiments to explore in detail the differentiation potential of a single ESC were designed. We demonstrate diversity in the ESC-derived progenies, which is at least determined by intrinsic properties of the parental cell. Clonal ESC sublines showing bona fide markers of ICM and pluripotency were established. These lines displayed distinct differentiation potentials and were stable over time.
Materials and methods
Cell culture
The mouse CGR8 and D3 ESC lines were obtained from the European Collection of Cell Culture (ATCC); the stromal PA6 cell line was provided by the Riken BRC cell bank, Japan. The ESC lines were maintained in BHK-21 medium supplemented with 10% foetal calf serum, 2 mM L-glutamine, 1% non-essential amino acids, 1 mM sodium pyruvate, 1% penicillin/streptomycin (Gibco, Invitrogen, Grand Island, NY, USA; http://www.invitrogen.com), and leukaemia inhibitory factor. CGR8 were cultured on gelatin-coated dishes, D3 on a layer of irradiated mouse embryonic fibroblasts. PA6 stromal cell line was maintained in MEM-α medium supplemented with 10% foetal bovine serum (Gibco, Invitrogen).
Antibodies
The following primary antibodies were used: mouse anti-nestin, mouse anti-neuronal nuclei-specific protein (NeuN), rabbit anti-tyrosine hydroxylase, rat antidopamine transporter (DAT; Chemicon, Temecula, CA, USA; http://www.chemicon.com), mouse antityrosine hydroxylase (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA; http://www.scbt.com), mouse anti-β-III-tubulin (Sigma-Aldrich, St. Louis, MO, USA; http://www.sigmaaldrich.com), and rabbit anti-β-III-tubulin (Covance, Princeton, NJ, USA; http://www.covance.com). The following fluorochrome-labelled secondary antibodies were used: Alexa Fluor (555 or 488)-labelled antibodies from goat or donkey against mouse, goat or rabbit (Molecular Probes, Eugene, OR, USA; http://probes.invitrogen.com); Cy5-conjugated donkey against mouse IgG, PE-Cy5.5 goat against rabbit IgG (Jackson Immunoresearch Laboratories, PA, USA; http://www.jacksonimmuno.com).
Flow cytometry
Cells were labelled with 1.25 μmol/l 5,6-carboxy-fluorescein-succinimidyl-ester (CFSE, Sigma, Buchs, Switzerland) according to the manufacturer’s recommendations. The following antibodies used were against nestin and β-III-tubulin. For their intracellular detection, cells were fixed with paraformaldehyde 0.5% for 10 min. at room temperature under constant stirring before incubation (45 min.) with appropriate dilutions of antibodies in phosphate buffered saline (PBS) containing 0.2% Triton X-100 and 10% foetal bovine serum. Cells were rinsed twice with PBS, incubated for 45 min. with appropriate secondary antibodies and washed before fluorescent active cell sorting (FACS) analysis. Fluorescence was analysed with a FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA; http:www.bd.com) and the CellQuest software.
Immunofluorescence and microscopy
Glass cover slips containing cells were fixed with 2% paraformaldehyde in PBS for 15 min. at room temperature before permeabilization with Triton X-100 0.2% in PBS for an additional 30 min. After washing with PBS, cover slips were incubated overnight at +4°C with appropriate dilutions of primary antibodies in PBS containing 1% of foetal bovine serum. After washing in PBS, cover slips were incubated for 1 hr at room temperature with the appropriate dilution of secondary antibodies, washed again and incubated for 15 min. with 300 nM 4,6-diamidino-2-phenylindole. Cells were washed in PBS and rinsed with water before inclusion in FluorSave mounting medium (Calbiochem, San Diego, CA, USA; http://www.emdbiosciences.com). Automated imaging was performed with the imageXpress automated fluorescence microscope using the MetaXpress software (Molecular Devices, CA, USA).
Neural differentiation of ESC
ESC were washed with PBS before plating at low density (100 cell/cm2) on a confluent layer of irradiated (5000 rad) PA6. The medium for differentiation comprised: GMEM, 15% knockout serum replacement, 2 mM L-glutamine, 1 mM sodium pyruvate, 1 mM non-essential amino acids, 0.1 mM β-mercaptoethanol and 1% penicillin/streptomycin (Gibco, Invitrogen). In some experiments, cells ongoing neural dissociation were dissociated using trypsin/ethylenediaminetetraacetic acid (EDTA) 0.5% and re-plated on polyornithine-coated cell culture Petri dishes (0.001%) at the density of 5000 cells/cm2.
Generation of embryoid bodies
CGR8 was washed once with PBS and dissociated using trypsin/EDTA 5%. Cells were diluted in culture medium without leukaemia inhibitory factor and put on 20 μl drops of 500 cells on the cover of cell plates. Two days later, cells forming embryoid bodies were pooled in 10 ml ultralow attachment plates and left once again for 3 days in the incubator. On the day 5, embryoid bodies were plated on gelatin-coated dishes. First embryoid bodies started beating on day 7. Culture medium was changed every 2 days.
Lentivectors and ESC transduction
To generate entry vectors, the promoters [βIII-tubulin promoter (βIIIp) and Tα-1] and genes of interest (GFP and H2B-mRFP1) were cloned into pDONRP4-P1R and pDONR221, respectively, using the Gateway® BP clonase enzyme mix (Invitrogen). The resulting entry vectors were then recombined into 2K7bsd or 2K7neo lentivectors using the Gateway® LR plus clonase enzyme mix (Invitrogen). The lentivector particles were produced by transient transfection in 293T cells using calcium phosphate. The lentivector-containing supernatant was collected after 72 hrs, filtered through 0.45-μm pore-sized polyethersulfone membrane and concentrated 120-fold by ultracentrifugation (50,000 × g, for 90 min. at 4°C). The pellet was re-suspended in ESC culture medium and subsequently added to the target cells. Three days after transduction, blasticidin (7.5 μg/ml) or neomycin (400 μg/ml) was added to the culture medium and the selection was maintained for 6 (blasticidin) or 10 days (neomycin).
Microarrays
Total RNA was isolated with the RNA Mini Kit (Quiagen, Basel, Switzerland) and quality controlled for RNA integrity by capillary electrophoresis on Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). Five hundred nanograms of RNA was amplified and labelled using the Illumina TotalPrep RNA Amplification kit (Ambion, CA, USA). cRNA quality was assessed by capillary electrophoresis on Agilent 2100 Bioanalyzer. Hybridization on human expression arrays (Illumina, CA, USA) was carried out according to the manufacturer’s instructions.
Data were normalized and analysed using Illumina Beadstudio 3.1.3 (background correction and quantile normalization). Expression profiles of each sample were imported into GeneSpringGX 7.3.1 (Agilent Technologies). In addition to expression values, Illumina BeadStudio software computes a detection P value. Based on this, each probe was assigned a detection flag [P (present): P < 0.045; M (marginal): P between 0.050 and 0.045, A (absent): P > 0.05]. To identify differentially expressed transcripts, Student’s t-test and/or ANOVA and additional steps of filtering were carried out. Enrichment analysis for functional ontologies was made using MetaCore software (http://www.genego.com).
Cytogenetic and molecular analysis
ESC were treated with colcemid (Invitrogen) at 50 ng/ml for 4 hrs. Mitotically arrested cells were subjected to hypotonic treatment using KCl 0.075M for 5 min., fixed by changing the solution with Carnoy’s fixative (methanol:acetic acid = 3: 1, v/v) three times, after which the solution containing the cells was spread on a glass slide. Chromosomes were subsequently G-banded according to the standard procedure. Oligonucleotide array-comparative genomic hybridization (array-CGH) analyses were performed according to the manufacturer’s protocol using the Mouse Genome CGH Microarray Kit 244B (Agilent Technologies), covering the whole genome with a resolution of ∼20 kb. Data were analysed with Agilent CGH analytics 3.4 software, using the statistical algorithms z-score and ADAM-2 according to sensitivity threshold respectively at 2.5 and 6.0 and a moving average window of 0.2 Mb. Mapping data were analysed on the mouse genome sequence using the NCBI database Build 37 (http://www.ncbi.nlm.nih.gov).
Quantitative polymerase chain reaction
Total cellular RNA was extracted using RNeasy reagents (Qiagen, Valencia, CA, USA), and genomic DNA was removed by the DNA-Free product (Ambion, Austin, TX, USA). RNA was reverse transcribed using SuperScript III (Invitrogen), and specific transcripts were analysed by real-time polymerase chain reaction (PCR) using iQ SYBR green Supermix (Bio-Rad, Reinach, Switzerland). Gene expression was normalized to EEF1A1 and HPRT RNA expression. The sequences of the primers used were as follows:
Tal1 forward primer, ATAGCCTTAGCCAGCCGCTC; Tal1 reverse primer, GCCGCACTACTTTGGTGTGA; Myh7 forward primer, TTCTCCTGCTGTTTCCTTACTTGC; Myh7 reverse primer, TTCCTTTCTCGGAGCCACC; α-foeto protein forward primer, AGGAGAAATGGTCCGGCTGT; α-foeto protein reverse primer, GTCCAATGAAAATGTCGGCC; Insulin1 forward primer, TGACTCCTGAGAGGAAGGTTTATTC; Insulin1 reverse primer, TTGCTGTGACTCCCCTGCTA; Zic1 forward primer, GCCGGTAAATCCAGGACTGA; Zic1 reverse primer, AACCTAAAGGCTTGCTGCCTC; Actinin1 forward primer, TCAACCACTTTGACCGGGAT; Actinin1 reverse primer, TGAACTCTTCGGGACCCAAC.
Results
Differentiation of ESC generates proliferating cells that escape early to the neural fate
During induced differentiation of ESC, typically a fraction of cells do not acquire the desired cellular phenotype. Although this may be due to anisochronicity, that is, a delay of a subpopulation to progress in the maturation process, a subpopulation of cells might escape the desired differentiation because of inherent resistance to the differentiation protocol. To address this question, we developed a flow cytometry assay combining immunodetection of differentiation markers with an analysis of cell division.
Mouse CGR8 ESC were cultured for 5 days at low density on a confluent layer of irradiated stromal cells (PA6) [7] to induce early neural differentiation and, when indicated, dissociated and plated on polyornithin to progress towards more advanced neural differentiation. As expected, undifferentiated ESC were negative for nestin and β-III-tubulin (Fig. 1A, left panel). At day 5 of differentiation, a complex pattern of cellular expression of the two markers was observed (Fig. 1A, right panel) with β-III-tubulin-positive, nestin-negative neuronal cells, nestin-positive β-III-tubulin-negative precursor cells and a small population of double-positive transition cells. Of note, a sizeable population of double-negative cells was observed, but these were not due to a delayed exit of a subpopulation from the pluripotency state as SSEA-1 expression was abolished after 5 days of differentiation in both nestin-positive and nestin-negative populations (Fig. 1B).
Fig 1.
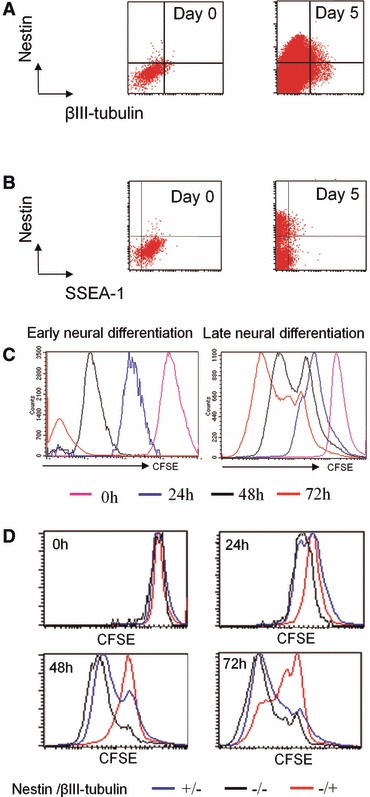
A subpopulation of ESC escapes neural differentiation. ESC were subjected to early neural differentiation by 5 days of coculture with PA6 stroma cells, then late differentiation was induced after cell dissociation and re-plating on polyornithine. (A) Flow cytometric analysis of nestin and βIII-tubulin expression during early differentiation. (B) Flow cytometric analysis of nestin and SSEA-1 expression during early differentiation. (C) Analysis of CFSE dilution by flow cytometry during early and late differentiation of ESC. (D) Combination of phenotypic and CFSE dilution analysis in differentiating ESC. The CFSE dilution was assessed at different time points for different subpopulations: nestin-positive/βIII-tubulin-negative (neuroepithelial cells), nestin-/ βIII-tubulin-positive (neuronal cells), nestin-/βIII-tubulin (non-neural cells).
The synchronicity of cell division was also monitored using the fluorescent probe CFSE [8]. CFSE is a stable and non-toxic fluorescent dye diluted by 50% in daughter cells after each cell division and thereby allows flow cytometry quantification of the number of mitotic events. A non-dividing cell maintains the initial level of CFSE fluorescence, whereas dividing cells lose fluorescence as a function of the number of divisions. As shown in the monophasic CFSE histograms, most cells divided during early neural differentiation (Fig. 1C, left panel). In contrast, the multiphasic CFSE histograms show that a subpopulation of cells slows down or stops division during late neural differentiation (Fig. 1C, right panel).
We then analysed cell proliferation as a function of cellular differentiation markers during late neural differentiation at 24, 48 and 72 hrs after plating on polyornithin (Fig. 1D). Two days after plating, the neuronal subpopulation (nestin-negative/β-III-tubulin-positive) slowed down its proliferation (higher CFSE intensity) in comparison to neuroepithelial (nestin-positive/β-III-tubulin-negative) and non-neural cells (nestin-negative/β-III-tubulin-negative) that actively proliferated (lower CFSE intensity). Three days after re-plating, the neuronal population included cells that had definitively stopped division, whereas non-neural and neuroepithelial cells continued division. Of note, several peaks corresponding to different CFSE intensities were observed within the three subpopulations, demonstrating also heterogeneity among them.
Taken together, these observations show that all ESC started a differentiation program, but some cells escaped to the neural fate at a very early stage and generated mixed cultures where post-mitotic neurons coexist with proliferating neural progenitors and non-neural cells. These non-neural cells could be very immature cells not still committed to other specific lineages. This escape response could be explained by heterogeneity among ESC submitted to neural differentiation.
Individual ESC submitted to neural differentiation generates a defined progeny
The hypothesis of heterogeneity among ESC was then investigated. We designed experimental conditions that allow characterization of progenies derived exclusively from one individual ESC. Neural differentiation was induced by plating ESC at very low density on PA6 stromal cells. Under these conditions, single ESC generates a colony with ongoing differentiation and the nature of each progeny can be monitored. After 3 days, ESC-derived colonies were heterogeneous. Some colonies included cells with a neural phenotype in majority (nestin-positive and βIII-tubulin-positive) whereas other colonies were mainly non-neural (double-negative; Fig. 2A). It is noteworthy that some colonies were a mixture of neural and non-neural cells (Fig. S1). This observation indicated strong variations in the nature of the progeny derived from one single ESC. Quantifications showed that: half of the colonies included βIII-tubulin-positive neuronal cells after 3 days; 25% of colonies included dopaminergic neurons with (tyrosine hydroxylase TH+); 10% of colonies included mature-stage neurons (NeuN+; Fig. 2B). Heterogeneity between colonies was confirmed using two other neural progenitor cells markers, Pax-6 and Sox-1 (data not shown). Heterogeneity between colonies for neural commitment was confirmed using a genetically modified ESC line expressing the green fluorescent protein (GFP) under the control of the early neural-specific promoter Tα1 [9]. ESC-Tα1-GFP submitted to neural differentiation in the same experimental colonies generated multiple colonies (Fig. 2C, upper panel), which expressed (48% ± 8.3) or not (52% ± 12.1) GFP (Fig. 2C, bottom panel), confirming the coexistence of neural and non-neural progenies. Thus, the capacity of some cells to escape to the neural fate is linked to the nature of the parental cell from which they derive.
Fig 2.
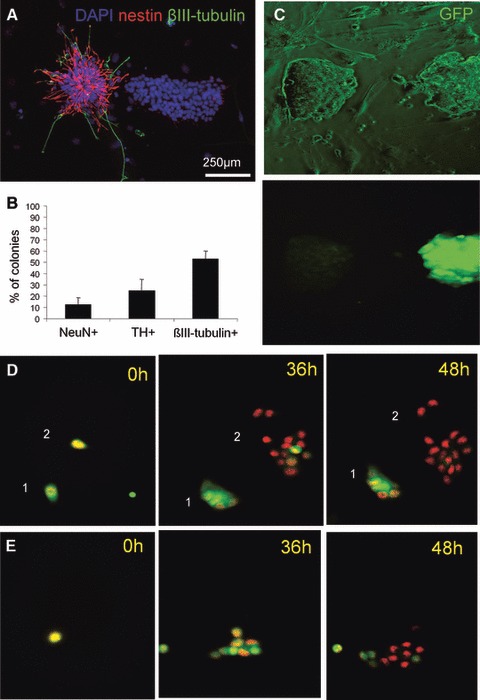
Variability of progenies derived from individual parental ESC. (A) In an experimental setup where one colony was derived from one parental ESC, cells were stained for nestin and βIII-tubulin after 72 hrs differentiation. (B) One hundred fifty ESC-derived colonies were analysed for the presence or not of NeuN (mature-stage neurons), TH (dopaminergic neurons) and βIII-tubulin (neuronal cells) cells. (C) ESC-Tα1-GFP were submitted to neural differentiation for 72 hrs and colonies were analysed for GFP expression. (D,E) ESC-H2B-mRFP1 were submitted to neural differentiation and monitored by live imaging during the first 2 days. The capacity of daughter cells to acquire a neural phenotype depended of the nature of parental ESC. Herein are presented three examples of different progenies.
The observed variability between the different progenies was analysed in greater detail using a promoter/reporter gene-based method. A genetically modified CGR8 ESC line was developed to express the GFP under the control of the βIIIp. ESC-βIIIp-GFP cells were cotransduced with a lentivector expressing the monomeric red fluoresecent protein (mRFP1) fused to the H2B histone for its targeting to cell nuclei. This allowed ESC-βIIIp-GFP-H2B-mRFP1 to be visualized by fluorescent microscopy because of their red fluorescent nuclei. The promoter for βIII-tubulin was constitutively active at a low level in undifferentiated ESC (data not shown). GFP expression was abolished in non-neural populations (nestin-negative; Fig. S2A), whereas it was increased in neuronal cells (βIII-tubulin-positive; Fig. S2B). ESC-βIIIp-GFP-H2B-mRFP1 were plated on PA6 for neural differentiation and several individual ESC were followed during 2 days using an automated high throughput imaging system (ImageXpress, Molecular Devices). Imaging confirmed that undifferentiated ESC-βIIIp-GFP-H2B-mRFP1 express a background level of GFP and divide rapidly after plating (data not shown). Six colonies were followed. In some colonies (1/6), GFP expression was maintained or increased in the ESC-derived progeny (Fig. 2D, colony 1). In other colonies, the GFP expression was rapidly abolished in all cells (1/6; Fig. 2D, colony 2), confirming that some individual ESC generate a non-neural progeny. Finally, colonies where only a fraction of cells switched off GFP expression were also observed (4/6; Fig. 2E) and indicated that the progeny derived from one individual ESC could be also a mix between neural and non-neural cells. The same analysis was also performed at late neural differentiation stage. Eight colonies were followed. In this case, plating on polyornithin which favour the survival and differentiation of neural cells generated six fully neural colonies and four mixed colonies.
Phylogenic trees including GFP expression were established from live imaging movies. None of all the analysed phylogenic trees was identical, thus confirming the uniqueness of progenies derived from individual ESC. For example, it can be observed that a part (Fig. S3a, b and d) or the totality (Fig. S3c) of the progeny switched off GFP expression. In other colonies, the promoter remained active in most cells after 2 days (Fig. S3d). Interestingly, cells that have switched off GFP expression were frequently derived exclusively from one of the two daughter cells generated by the first division. In contrast, those which kept GFP expression were derived from the other daughter cell, indicating that the first division produced two different daughter cells that generate respectively different progenies.
Taken together, these observations confirm that individual ESC generate defined progenies and do not share the same potential for the generation of neural progenies.
Clonal diversity among ESC corresponding to the early pluripotent ICM
The observation that individual ESC in the same culture does not share the same neurogenic potential could be explained by heterogeneity between individuals or stochasticity in the decision triggering ESC neural differentiation. The hypothesis of individualities between ESC at the single-cell level was investigated. Seven clonal sublines were derived from ESC by a limit dilution method. The pluripotent phenotype of each clonal subline was first investigated. Most of the tested sublines expressed markers of pluripotent cells of the early ICM including rex-1, alkaline phosphatase, Oct-4, Nanog, Klf-4, Sox-2, Klf-2, Pecam-1 and Pramel-4 (Fig. 3A). It is noteworthy that Stella expression was found in most of clones, but with a variable expression level. One clonal line (clone 5) expressed primitive endoderm markers (data not shown), thus was excluded.
Fig 3.
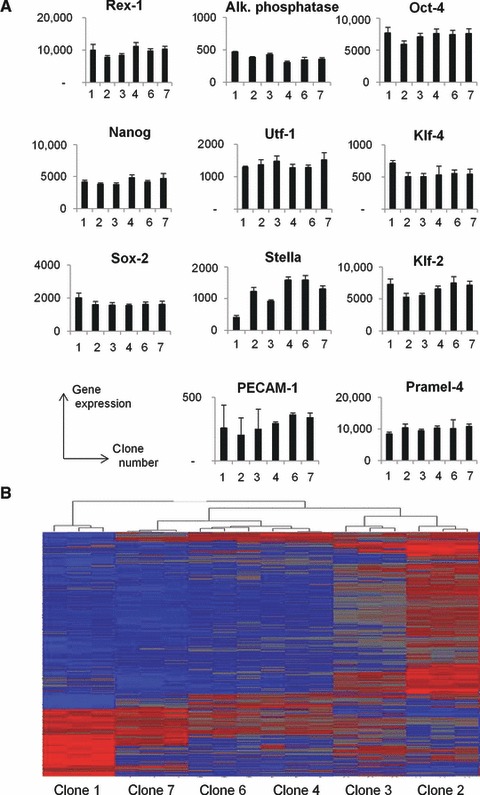
mRNA expression profile in ESC sublines. A total mRNA expression profile was performed on each clonal ESC subline. (A) The expression of mRNA associated to pluripotency and/or early inner cell mass was quantified. (B) The expression of 6800 genes varied significantly between ESC clones. Based on gene expression profile of each clone, a hierarchical cluster was established to classify ESC clones. The two most different clones were clones 1 and 2 whereas clones 4 and 6 were highly similar.
To exclude genomic abnormalities, clonal ESC sublines were submitted to an analysis of their genomic structure. Standard karyotyping (G-banding) of the different clones (clones 1–7) was performed. The chromosome number and the presence of chromosome abnormalities were evaluated at two culture time intervals (passages 10 and 16; Table 1). Most clones showed cell mosaicism, except clone 7 at passage 16. The normal 2n = 40 frequency value in clone 3 was 36% at passage 10 and up to 60% at passage 16 (Table 1). The analysis also revealed the presence of chromosomal abnormalities. Clones 1, 2, 6 and 7 showed an identical structural rearrangement by the presence of an unidentified derivative chromosome (der) present at both passages. This rearranged chromosome was present in the hyperploidic 41,XY preponderant population cell (Fig. S4). A high-resolution genomic analysis of clones was also performed by molecular karyotyping (array-CGH). This analysis revealed the presence of common partial deletion and duplication smaller than 1 Mb in all analysed clones (Table S1). It is noteworthy that duplication on chromosome X was present in clones 1 and 2, but absent in others. The abandoned clone 5 showed a different genomic profile typified by the lack of a region in 5qE1 (data not shown). Taken together, these results suggest that the genomic structure of the clones, with exception of clone 5, was similar and showed no major abnormalities.
Table 1.
Standard karyotyping of ESC clones by G-banding
| Clone_passage | Number of analysed metaphases | Karyotype | Results |
|---|---|---|---|
| Clone 1_10 | 15 | 2 | 41, XY, +der(?)[2]/40, XY, der(?)[13] |
| Clone 1_16 | 15 | 3 | 41, XY, +der(?)[8]/40, XY, der(?)[7] |
| Clone 2_10 | 14 | 2 | 41, XY, +der(?)[10]/40, XY, der(?)[4] |
| Clone 2_16 | 16 | 2 | 41, XY, +der(?)[3]/40, XY, der(?)[13] |
| Clone 3_10 | 11 | 2 | 42, XY[7]/40, XY[4] |
| Clone 3_16 | 15 | 1 | 42, XY[2]/41, XY[3]/40, XY[9] |
| Clone 4_10 | 10 | 2 | 42, XY[4]/41, XY[1]/40, XY[2] |
| Clone 4_16 | 12 | 4 | 42, XY[4]/41, XY[4]/40, XY[2] |
| Clone 6_10 | 10 | 1 | 42, XY, +der(?)[6]/41, XY, +der(?)[4] |
| Clone 6_16 | 10 | 2 | 41, XY, +der(?)[5]/40, XY, der(?)[4] |
| Clone 7_10 | 12 | 2 | 41, XY, +der(?)[2]/40, XY, der(?)[13] |
| Clone 7_16 | 12 | 2 | 40, XY, der(?)[9] |
Der(?): derivative.
The clonal sublines without obvious genomic abnormalities (i.e. clones 1–4, 6 and 7) were submitted to a total mRNA expression analysis by microarray. The expression of 6800 genes varied significantly between clonal lines (variance analysis using ANOVA statistical test). Mathematical analysis of the expression profile of these 6800 genes for each clonal ESC allowed a hierarchical clustering (Fig. 3B). The most different clonal ESC were clones 1 and 2, which differed significantly in the expression of 315 genes. Clone 2 resembled more to clone 3 and clones 4–6. Variability in gene expression was confirmed with clone derived from another mouse ESC line (D3). In this case, the most important variability was observed between clones 3 and 5, which differed in the expression of 121 genes. Figure S5 summarizes families of genes that were differently expressed between clones 1 and 2 [from the public database GO process (Metacore software); http://www.genego.com]. Approximately, half of genes differently expressed between the two clones were classified in developmental processes, including the neuron generation. The nature of the most important changes between all clonal lines was also analysed. In Table S2, 30 genes showing the quantitatively most important differences in expression levels between different clones are listed. Notably, the list contains several groups of genes: (i) three guanylate binding proteins (Gbp 1, 2 and 3); (ii) three keratins (Krt 8, 18 and 19); (iii) two carbonic anhydrases (Car2 and 4). One of the potential interests in transcriptome analysis is the discovery of genes, which are predictive of the neurogenic potential. The neurogenic potential for each clonal line has been scored by the percentage of neural colonies indicated in Figure 4. Using the transcriptome database, the distribution of all gene expression levels among the different clones was statistically compared to the distribution of neurogenic scores. A correlation coefficient was calculated for all genes and they were classified according this value. Genes which were associated with the high or lower correlation coefficient were identified (Table S3). It shows that it is possible to find some candidate genes for a predictive strategy. However, further molecular studies are needed to further demonstrate causality between from a gene expression level to the neurogenic potential of clonal ESC.
Clonal ESC differ in their differentiation potential
We hypothesized that these differences in mRNA expression levels might confer a propensity for specific differentiation pathways to the different clones. To investigate variability at the functional level, clonal ESC were submitted to induced neural differentiation. All lines induced both neural and non-neural colonies. However, the ratio between neural and non-neural colonies differed between clones. Clones 2 and 6 generated a significantly higher percentage of colonies including neuroepithelial cells (nestin-positive) than clone 7 (Fig. 4A). This increased capacity of clones 2 and 6 was confirmed by neuronal βIII-tubulin staining (Fig. 4B). After 1-week differentiation and in accordance with these observations, clone 7 had a lower capacity to generate colonies with TH dopaminergic neurons (Fig. 4C). It is noteworthy that clone 1 induced a higher number of TH-positive colonies than other clones. Clones varied also in their cardiogenic potential (Fig. 4D). Clones 2 and 3 were significantly more efficient than clone 4 to produce beating cardiomyocytes. In contrast, clone 1 was not efficient to generate cardiac cells.
Fig 4.
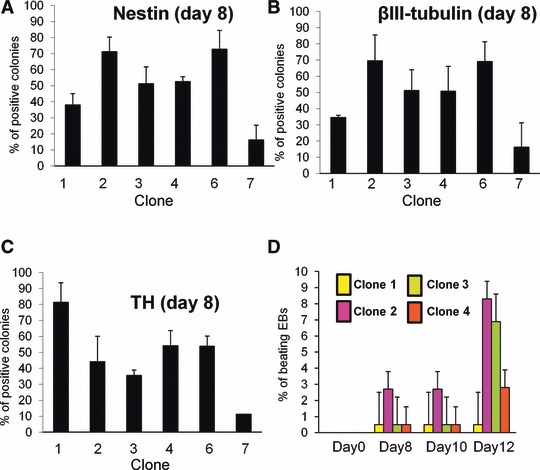
Clonal sublines do not share the neurogenic and cardiogenic potential. (A–C) Clonal ESC were submitted to neural differentiation by coculture on PA6 stromal cells. (A) The percentage of colonies including nestin-positive neuroepithelial cells was evaluated after 3 days. (B,C) The percentage of colonies including βIII-tubulin-positive neuronal cells (B) or (C) TH+ dopaminergic neurons was evaluated after 1 week. (D) ESC clones 1–4 were differentiated towards embryoid bodies. Cardiac differentiation was evaluated by the percentage of beating embryoid bodies at different time points. Statistical analysis was performed and all clonal ESC differed significantly from at least another one (Student’s t-test, P < 0.05).
Under appropriate conditions, all sublines had the capacity to generate floating embryoid bodies (data not shown). Expression of genes which are linked to different germ layers/cell types was then quantified in embryoid bodies after 2 weeks. First, a heterogeneous expression of the ectodermal Zic1, the endodermal Foxa2 and the mesodermal Tal1 was observed (data not shown), suggesting variability in the differentiation potential of ESC clones. Clones 6 induced higher levels of the blood markers (Fig. 5A), as well as bone (Fig. 5D) and pancreas markers (Fig. 5F). On the other hand, if clone number 7 was poorly neurogenic, it was more efficient to generate extraembryonic cells (Fig. 5B) and endothelial cells (Fig. 5E). Finally, clone 4 was suggested to generate more muscle cells (Fig. 5C).
Fig 5.
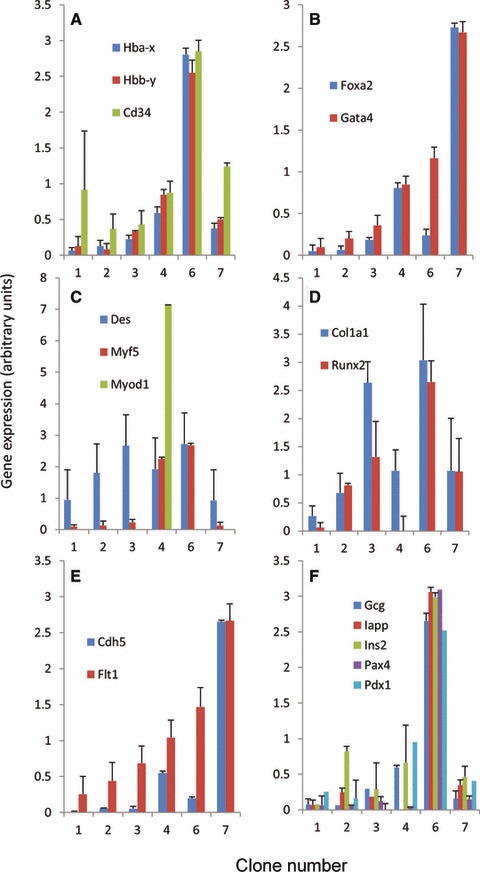
Clonal sublines do not share the same differentiation potential. ESC clones were differentiated in vitro towards embryoid bodies and assessed by quantitative PCR for the expression of genes specific for different lineages. Statistical analysis was performed and all clonal ESC differed significantly from at least another one (Student’s t-test, P < 0.05).
Taken together, these data show that clonal lines do not share the same differentiation potential and confirm that individual pluripotent ESC in the same culture are functionally heterogeneous.
This verify wider applicability of the concept of clonal heterogeneity we investigated a second ESC line (D3). Sublines were generated from D3 by limit dilution and each clone was submitted to genomic analysis and total gene expression profile. As observed for the CGR8 line, there were no major genomic abnormalities among sublines (data not shown). Gene expression array was performed and all of the pluripotency markers corresponding to the early ICM were detected at comparable levels in the clones (Fig. 6A). However, D3 sublines differed significantly in their capacity to generate colonies containing βIII-tubulin-positive neurons, NeuN+ mature stage neurons and TH+ dopaminergic neurons (Fig. 6B).
Fig 6.
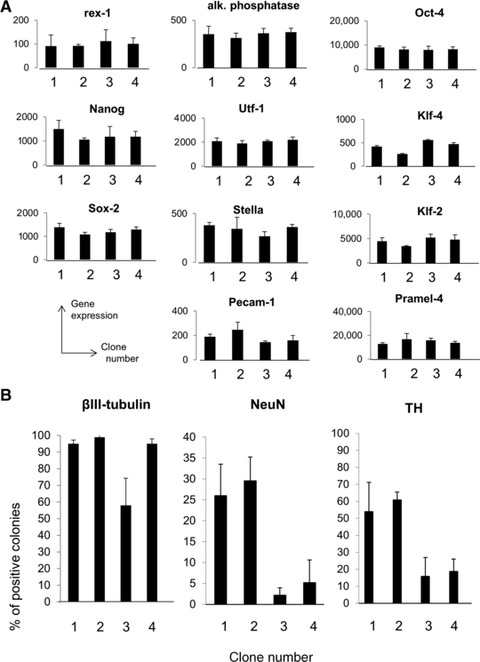
Clonal sublines from D3 confirms cellular diversity. (A) Total mRNA expression profile was performed on each clonal D3 subline. The expression of mRNA associated to pluripotency and/or early inner cell mass was established. (B) D3 sublines were submitted to neural differentiation by coculture on PA6 stromal cells. The percentage of colonies including βIII-tubulin-positive neuronal cells, NeuN+ mature neurons and TH+ dopaminergic neurons was evaluated after 1 week. Statistical analysis was performed and all clonal ESC differed significantly from at least another one (Student’s t-test, P < 0.05).
Discussion
In this study we report a hitherto not appreciated cause of heterogeneity of pluripotent ESC: namely, the existence of sublines within established ESC that show stable cellular individualities. These variations are not due to precursors of extraembryonic tissues, but represent cells expressing the typical marker profile for the early ICM. The individualities of the sublines are reflected in a distinct gene expression pattern in undifferentiated ESC, and a distinct cell fate potential upon differentiation. Pluripotency is defined as ‘the capacity of cells that may still differentiate into various types of specialized cells’. We have shown that all selected clonal ESC (i) express markers of the early ICM, (ii) have the capacity to be differentiated towards more than one cell type, (iii) have the capacity to generate embryoid bodies including cells of the three germ layers and (iv) are maintained in culture condition under leukaemia inhibiting factor exposure. These observations allow us to conclude that numerous individual ESC harbour pluripotent properties in culture. However, they differ significantly in their differentiation capacity with a proneness to more or less generate defined cell types.
What could be the origin of the observed cell heterogeneity within ESC? One possibility would be an artefact through the subcloning procedure. It is unlikely because as shown in Figures 1 and 2, there is ample evidence for such a cell heterogeneity within the parental cell line before subcloning. Genomic instability within ESC has been reported and could account at least in part for the observed heterogeneity. High-resolution karyotyping did not show noteworthy changes, except one clone which was excluded from the analysis. Thus, genomic changes could make some contribution to the observed individual cellular phenotypes, but it appears unlikely that they are the major cause behind it.
Could the observed heterogeneity reflect very early cell fate decisions in the early embryo? Early decisions determining different cell fates that occur in the early ICM can be suspected to generate such diversity. Cells positioned inside the embryo develop into the ICM, whereas outside cells develop into the first extraembryonic tissue, the trophectoderm, which will give rise to the placenta. The second decision determining cell fate distinguishes the pluripotent epiblast and the second extraembryonic primitive endoderm [10]. The possibility to derive several cell types from pre-implantation embryos has been reported, such as epiblast-like cells lines, trophoblast and extraembryonic endoderm-like cell lines [11,12]. From these findings, it cannot be excluded that some ESC lines could be contaminated by trophectodermal or primitive endoderm cells. Our study provides another level of cell heterogeneity. By focusing only on cells with a pluripotent ICM phenotype, we were able to observe that individual cells differ in their molecular profiling and differentiation potential. Because the early ICM show distinct precursors for the epiblast versus primitive endoderm [1], it is highly suggested that different pre-differentiation stages can be derived at the moment of ESC generation. Further studies on early ICM are still needed to formally demonstrate a causality link between ICM heterogeneity and the described ESC heterogeneity in culture.
One of the starting points of this study was the observation that colonies derived from ESC often show all-or-none cell fate decision (Fig. 2A). However, while certain clones show a much higher propensity to form neuronal colonies, they are capable of forming non-neuronal colonies. Thus, additional elements are likely to intervene. Certain aspect of cell behaviour and cell fate decisions might be stochastic [13]. As a consequence, the nature of biological events varies even across isogenic populations and in individual cells over time. New experimental techniques allow gene expression to be followed in single cells over time and have revealed stochastic bursts of both mRNA and protein synthesis in many different types of cells. In an in vitro neural differentiation context, oscillations in the cell cycle, availability of transcription factors or in the gene expression could be responsible for the enormous flexibility and redundancy of cellular circuits required for a neural decision. This stochastic point of view could explain why certain individual cells among highly neurogenic clonal ESC lines still generate non-neural progenies. Thus, the decision for a neural fate could also depend on the cell machinery available at the moment of the decision.
ESC hold great promise for the study of early development, modelling disease as well as for cell therapy [14]. However, for all these applications, the heterogeneous behaviour of conventional ESC lines represents a limitation. Sublines with defined differentiation characteristics should have a major usefulness for in vitro and in vivo applications of ESC. A more homogenous starting material, obtained by ESC cloning and selection of the most neurogenic sublines should help to improve neural cell and tissue engineering. Clonal sublines can also be of interest for more fundamental science. Two questions are particularly relevant. First, which are the mechanisms, in particular epigenetic modifications that lead to a first pre-differentiation of ESC? Secondly, can these pre-differentiation steps be documented in the early embryo in vivo? The clonal sublines established in this study will be promising tools to address these questions.
Acknowledgments
The authors thank Graziella Chakroun-Ciglie for technical assistance. The work was supported by grants from the Clayton foundation (K.-H.K.), Parkinson Schweiz (M.D-.D., O.P-.S.) and the Swiss National Foundation (K.-H.K., O.P-.S.).
Conflict of interest
Authors declare that there are no potential conflicts of interest.
Supporting Information
Some individual ESC generate mixed colonies.
Specificity of the βIII-tubulin promoter.
Phylogeny of the progeny derived from individual parental ESC.
Presence of an unidentified derivative chromosome (der) in clones 1, 2, 6 and 7.
Fig. S5 Functional classification of genes which were differently expressed between ESC clones 1 and 2.
Genomic imbalances in clones
Classification of the 30 most important changes in mRNA expression between clones 1, 2, 3, 4, 6, and 7
Table S3 Identification of candidate genes that might positively or negatively predict the neurogenic potential of ESC clone
References
- 1.Chazaud C, Yamanaka Y, Pawson T, et al. Early lineage segregation between epiblast and primitive endoderm in mouse blastocysts through the Grb2-MAPK pathway. Dev Cell. 2006;10:615–24. doi: 10.1016/j.devcel.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 2.Chambers SM, Fasano CA, Papapetrou EP, et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27:275–80. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singh AM, Hamazaki T, Hankowski KE, et al. A heterogeneous expression pattern for Nanog in embryonic stem cells. Stem Cells. 2007;25:2534–42. doi: 10.1634/stemcells.2007-0126. [DOI] [PubMed] [Google Scholar]
- 4.Toyooka Y, Shimosato D, Murakami K, et al. Identification and characterization of subpopulations in undifferentiated ES cell culture. Development. 2008;135:909–18. doi: 10.1242/dev.017400. [DOI] [PubMed] [Google Scholar]
- 5.Hayashi K, Lopes SM, Tang F, et al. Dynamic equilibrium and heterogeneity of mouse pluripotent stem cells with distinct functional and epigenetic states. Cell Stem Cell. 2008;3:391–401. doi: 10.1016/j.stem.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Canham MA, Sharov AA, Ko MS, et al. Functional heterogeneity of embryonic stem cells revealed through translational amplification of an early endodermal transcript. PLoS Biol. 2010;8:e1000379. doi: 10.1371/journal.pbio.1000379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shintani A, Nakao N, Kakishita K, et al. Generation of dopamine neurons from embryonic stem cells in the presence of the neuralizing activity of bone marrow stromal cells derived from adult mice. J Neurosci Res. 2008;86:2829–38. doi: 10.1002/jnr.21748. [DOI] [PubMed] [Google Scholar]
- 8.Lyons AB. Analysing cell division in vivo and in vitro using flow cytometric measurement of CFSE dye dilution. J Immunol Methods. 2000;243:147–54. doi: 10.1016/s0022-1759(00)00231-3. [DOI] [PubMed] [Google Scholar]
- 9.Suter DM, Preynat-Seauve O, Tirefort D, et al. Phenazopyridine induces and synchronizes neuronal differentiation of embryonic stem cells. J Cell Mol Med. 2009;13:3517–27. doi: 10.1111/j.1582-4934.2009.00660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gardner RL. Investigation of cell lineage and differentiation in the extraembryonic endoderm of the mouse embryo. J Embryol Exp Morphol. 1982;68:175–98. [PubMed] [Google Scholar]
- 11.Chuykin I, Lapidus I, Popova E, et al. Characterization of trophoblast and extraembryonic endoderm cell lineages derived from rat preimplantation embryos. PLoS One. 2010;5:e9794. doi: 10.1371/journal.pone.0009794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tesar PJ, Chenoweth JG, Brook FA, et al. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature. 2007;448:196–9. doi: 10.1038/nature05972. [DOI] [PubMed] [Google Scholar]
- 13.Shahrezaei V, Swain PS. The stochastic nature of biochemical networks. Curr Opin Biotechnol. 2008;19:369–74. doi: 10.1016/j.copbio.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 14.Srivastava AS, Malhotra R, Sharp J, et al. Potentials of ES cell therapy in neurodegenerative diseases. Curr Pharm Des. 2008;14:3873–9. doi: 10.2174/138161208786898617. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Some individual ESC generate mixed colonies.
Specificity of the βIII-tubulin promoter.
Phylogeny of the progeny derived from individual parental ESC.
Presence of an unidentified derivative chromosome (der) in clones 1, 2, 6 and 7.
Fig. S5 Functional classification of genes which were differently expressed between ESC clones 1 and 2.
Genomic imbalances in clones
Classification of the 30 most important changes in mRNA expression between clones 1, 2, 3, 4, 6, and 7
Table S3 Identification of candidate genes that might positively or negatively predict the neurogenic potential of ESC clone