

Abstract
The fibrocytes are thought to serve as a source of newly deposited collagens I and III during reparative processes and in certain fibrotic disorders, but their matrix remodelling properties are incompletely understood. We evaluated their ability to produce several extracellular matrix (ECM) components, in comparison with fibroblasts, and to participate in collagen turnover. The collagen gene expression profile of fibrocytes differed from that of fibroblasts because fibrocytes constitutively expressed relatively high levels of the mRNA encoding collagen VI and significantly lower levels of the mRNA encoding collagens I, III and V. The proteoglycan (PG) gene expression profile was also different in fibrocytes and fibroblasts because fibrocytes constitutively expressed the mRNA encoding perlecan and versican at relatively high levels and the mRNA encoding biglycan and decorin at low and very low levels, respectively. Moreover, fibrocytes expressed the mRNA for hyaluronan synthase 2 at higher level than fibroblasts. Significant differences between the two cell populations were also demonstrated by metabolic labelling and analysis of the secreted collagenous proteins, PGs and hyaluronan. Fibrocytes constitutively expressed the scavenger receptors CD163 and CD204 as well as the mannose receptors CD206 and Endo180, and internalized and degraded collagen fragments through an Endo180-mediated mechanism. The results of this study demonstrate that human fibrocytes exhibit ECM remodelling properties previously unexplored, including the ability to participate in collagen turnover. The observed differences in collagen and PG expression profile between fibrocytes and fibroblasts suggest that fibrocytes may predominantly have a matrix-stabilizing function.
Keywords: cell adhesion, collagen receptor, Endo180, extracellular matrix, fibrocyte, fibrosis, matrix internalization, scavenger receptor
Introduction
The term ‘fibrocyte’ was coined in 1994 to define a proliferating cell with mixed leucocytic and mesenchymal features that appears in the wounded skin concurrently with circulating inflammatory cells and is present in cutaneous scars [1]. This cell was found to co-express the haematopoietic stem cell antigen CD34, the leucocyte common antigen CD45, the marker of the monocyte lineage CD11b, and fibroblast products such as collagens I and III [1]. It was subsequently demonstrated that the fibrocyte differentiates from a circulating bone marrow-derived precursor cell [2], which is present in the CD14+ fraction of peripheral blood mononuclear cells (PBMC) [3-7].
Several studies have suggested that fibrocytes may contribute to wound healing and may be involved in the pathogenesis of a number of pathological conditions characterized by excessive deposition of extracellular matrix (ECM) molecules [2, 8-10]. In particular, fibrocyte accumulation has been demonstrated in areas of ongoing subepithelial reticulin fibrosis (thickening of the lamina reticularis) in the bronchial mucosa of subjects with asthma [11–13] and in the fibrous dermal infiltrate present in the skin of patients with nephrogenic systemic fibrosis (NSF) [14-16]. The presence of high number of fibrocytes has also been observed in the fibrous cap of atherosclerotic lesions in human carotid arteries [17], in the pulmonary parenchyma of some individuals affected by idiopatic pulmonary fibrosis [18], and in the fibrous epiretinal membranes of patients with proliferative vitreoretinopathy [19].
In order to clarify what role fibrocytes may play in the pathogenesis of such diverse diseases, which have, however, in common alterations of the normally tightly regulated mechanisms of matrix deposition and collagen turnover, it is important to understand the matrix remodelling properties of these cells. Although it is widely recognized [2, 8-10] that fibrocytes can express to various extents collagen I, collagen III, fibronectin and some of the matrix metalloproteases (MMPs) commonly involved in ECM degradation, very little is known about their ability to produce other collagens and non-collagenous ECM components and to participate in collagen turnover. With this study we provide the first evidence that fibrocytes significantly differ from resident tissue fibroblasts in terms of level of expression of various collagen types, matrix proteoglycans (PGs) and hyaluronan. Moreover, we demonstrate that fibrocytes adhere to native collagenous proteins and internalize and degrade collagen fragments through an Endo180-mediated mechanism. Our findings suggest that fibrocytes may have a predominant matrix-stabilizing function, as opposed to the predominant matrix-building function of tissue fibroblasts.
Materials and methods
Generation and purification of human fibrocytes
Fibrocytes were generated in cultures of PBMC according to the originally described procedure [20], with some modifications. PBMC were isolated from human leukapheresis packs by density gradient centrifugation over Ficoll-Paque, washed and re-suspended in complete DMEM (Sigma-Aldrich, St. Louis, MO, USA): DMEM supplemented with 20% foetal calf serum (FCS) (HyClone Laboratories, Logan, UT, USA), 2 mM L-glutamine, 10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), 100 U/ml penicillin and 0.1 mg/ml streptomycin. PBMC were seeded into the fibronectin-coated wells of 6-well culture plates (Milllipore/Chemicon, Temecula, CA, USA) at the density of 5 × 106 cells/well and incubated for 72 hrs. The non-adherent cells were gently aspirated and the adherent cells were re-incubated in complete DMEM for additional 10 days. On the fifth day, 50% of the conditioned medium was replaced with fresh medium. At the end of the incubation period, cells were harvested and further purified by immunomagnetic depletion of macrophages, residual T lymphocytes and B lymphocytes as previously described [20]. The viability of the isolated cells was assessed by the trypan blue exclusion method and the purity of the isolated fibrocytes was checked by flow cytometry after staining with specific antibodies against CD45, CD34 and pro-collagen I as described below. By using these procedures, it was found that the isolated cells were ≥94.2% viable and that the viable cells were ≥96.8% CD45+CD34+pro-collagen I+ fibrocytes. In the experiments described below, the isolated fibrocytes were used without further passages unless otherwise specified.
Primary cultures of fibroblasts and fibroblast lines
In-house primary cultures of human lung fibroblasts were established using histologically normal fragments of resected pulmonary tissue from consenting patients undergoing lobectomy for lung cancer, as previously reported [21]. Cells were evaluated for the expression of CD90 (also known as Thy-1 and not expressed by fibrocytes and macrophages), CD45, CD34 and intracellular pro-collagen I by flow cytometry, as described below. Clonetics normal human dermal fibroblasts were used as positive controls for this analysis and for the subsequent phenotypic characterization by electron microscopy. Pure populations of CD45−CD34−CD90+pro-collagen I+ lung fibroblasts were re-plated and further expanded in serum-supplemented DMEM by serial passages at weekly intervals. Between passages 3 and 5, samples of each cell population were processed for ultrastructural analysis and aliquots of the remaining cells were frozen and stored in liquid nitrogen until use in the experiments described below.
Flow cytometry
For phenotypic analysis by flow cytometry, fibrocytes and fibroblasts (1 × 105) were suspended in a final volume of 100 μl flow cytometry buffer (BD Biosciences/Pharmingen, San Jose, CA, USA). To block non-specific binding of the primary antibodies via the Fc receptors, fibrocytes were pre-incubated with affinity-purified human FcγR-binding inhibitor (eBiosciences, San Diego, CA, USA) for 20 min. at 4°C prior to staining. For detection of surface antigens, including the extracellular domains of scavenger and mannose receptors, cells were incubated with one of the following primary antibodies or the isotype-matched control (2–5 μl) for 60 min. in the dark at 4°C: fluorescein isothiocyanate (FITC)-conjugated mouse anti-human CD14 (clone HDC14; BioLegend, San Diego, CA, USA); phycoerythrin (PE)-conjugated mouse anti-human CD45 (clone HI30; eBiosciences); peridinin chlorophyll protein-conjugated mouse anti-human CD45 (clone 2D1; BD Biosciences); PE-conjugated mouse anti-human CD34 (clone 8G12; BD Biosciences); PE-conjugated mouse anti-human CD90 (clone Thy-1A1; R&D Systems, Minneapolis, MN, USA); PE-conjugated mouse antibody against human integrin β1, also known as CD29, (clone P5D2; R&D Systems); unconjugated rabbit antibody against human Endo180, also known as urokinase-type plasminogen activator (uPA) receptor-associated protein, CD280 or mannose receptor C type 2 (ab70132; Abcam, Cambridge, UK); FITC-conjugated mouse anti-human uPA receptor (American Diagnostica, Stamford, CT, USA); PE-conjugated mouse anti-human CD163 (Clone RM3/1; BioLegend); PE-conjugated mouse antibody against the extracellular domain of CD204, also known as scavenger receptor AI, MSR1 or MSR-AI (clone 351615; R&D Systems); PE-conjugated mouse antibody against human discoidin domain receptor (DDR) 1 (Clone 48B3; Santa Cruz Biotechnology, Santa Cruz, CA, USA); unconjugated mouse antibody against the extracellular domain of DDR2 (clone 290804; IgG2B, R&D Systems) and unconjugated mouse antibody against CD206, also known as mannose receptor C type 1 or MRC1 (Clone 15–2, IgG1; BioLegend). Unbound antibody was removed by washing the cells twice in flow cytometry buffer. When unconjugated primary antibodies were used, antibody binding was revealed by incubation with the appropriate FITC-conjugated goat anti-rabbit (Abcam), PE-conjugated goat antimouse IgG2B (R&D Systems) or PE-conjugated goat antimouse IgG1 (BioLegend) secondary antibodies (1:250 to 1:500 dilution in flow cytometry buffer) for 60 min. in the dark at 4°C. For intracellular staining of the newly synthesized collagen I, the cells were fixed and permeabilized with Cytofix/ Cytoperm kit (BD Biosciences/Pharmingen) and incubated with 1 μl rat anti-human pro-collagen I, N-terminus (clone M58; Chemicon International, Temecula, CA, USA) or an isotype-matched control (R&D Systems) for 60 min. in the dark at 4°C. After washing, the cells were incubated with a FITC-conjugated goat anti-rat secondary antibody (Invitrogen, Carlsbad, CA, USA), diluted 1:500 in flow cytometry buffer, for 45 min. in the dark at 4°C. Analysis of the stained cells was performed on a FACSCalibur flow cytometry system (BD Biosciences), using the associated CellQuest software.
Collagen production
Total collagen production was estimated by analysis of the incorporation of tritiated proline into collagenase-sensitive proteins (collagenous proteins) as previously described [22-25]. For this analysis, fibrocytes and fibroblasts were initially plated onto the uncoated wells of 24-well plates (5 × 104 cells/well) and cultured for 6 days in complete DMEM. On the third day, 50% of the conditioned medium was replaced with fresh medium. Cells were then extensively washed and re-incubated for 48 hrs in low-serum DMEM (DMEM supplemented with 0.4% FCS, 2 mM L-glutamine and 10 mM HEPES) and 50 μg/ml ascorbic acid (an activator of proline hydroxylation obtained from Sigma-Aldrich). After the first 24 hrs of the incubation period, 15 μCi/ml [3H]proline (L-[5–3H]–Proline, Amersham International, Little Chalfont, UK) and the inhibitor of lysyl oxidase β-aminopropionitrile (50 μg/ml, Sigma-Aldrich) were added to the culture medium. It was found in preliminary experiments that, under these conditions, fibrocytes and fibroblasts did not proliferate over the 48 hr period and that their viability remained unchanged. At the end of the incubation period, replicate cultures of fibrocytes and fibroblasts were either trypsinized to perform cell count with a Coulter counter or further processed as described in details elsewhere [22-25] and summarized below.
To measure the amounts of newly synthesized collagen that was produced in the form of soluble collagen over 24 hrs, aliquots of each conditioned medium were incubated for 4 hrs at 37°C in assay buffer (50 mM Tris–HCl, pH 7.5, 5 mM CaCl2 and 2.5 mM N-ethylmaleimide [NEM]), containing highly purified collagenase type VII (80 μg/100 μl) (Sigma-Aldrich). An ice-cold solution containing 10% trichloroacetic acid, 1 mM cold proline and 2% FCS was then added to each sample. After further incubation for 30 min. at 4°C, each sample was centrifuged (24,000 × g at 4°C) for 5 min. and the supernatant was analysed by liquid scintillation counting. The precipitate was hydrolysed in 1 ml 6 N HCl for 24 hrs at 70°C and subjected to liquid scintillation counting for measurement of the amounts of non-collagenous proteins released from cells. For analysis of the amounts of newly synthesized collagen that was deposited in the ECM over the same period of time, the cells were lysed and the matrix underlying each cell layer was fixed in 70% ethanol. The matrix was then incubated for 4 hrs at 37°C in assay buffer containing collagenase, as described above for the conditioned media. The supernatants were removed and residual matrix was solubilized by overnight incubation with 0.3 M NaOH in 1% SDS. Equal aliquots of supernatants and solubilized residual matrix were analysed by liquid scintillation counting. The contents of soluble and deposited collagenous proteins were expressed as disintegration per minute (dpm) of [3H]proline incorporated into collagenase-sensitive proteins in 24 hrs and as percentage of total soluble or deposited proteins [22-25]. Results were finally normalized to cell number.
Further experiments were subsequently performed to identify the types of collagenous proteins produced by fibrocytes and fibroblasts. Cells were labelled with tritiated proline for 24 hrs as described above. The conditioned media were aspirated and radio-labelled proteins were extracted from the cell layers. Non-collagenous proteins were digested with 20 μg/ml pepsin (Sigma-Aldrich) at 4°C overnight. Collagenous proteins were separated by SDS-PAGE, using equal fractions of radio-labelled material from fibrocytes and fibroblasts. The radio-labelled proteins were visualized by autoradiography. For Western blot analysis, the collagenous proteins were transferred to a Hybond ECL membrane (Amersham Biosciences, Piscataway, NJ, USA). The membrane was blocked and probed for 1 hr at 37°C with one of the following primary antibodies (1:800 to 1:5000 dilution): rabbit or goat polyclonal antibodies against human pro-collagen I (α1 chain), collagen III (α1 chain) and collagen V (α2 chain) from Santa Cruz Biotechnology; rabbit polyconal antibody against human collagen VI (α3 chain) from Millipore/Chemicon. After incubation with the appropriate horseradish peroxidase-conjugated secondary antibody, binding of the primary antibody to the target protein was visualized by an enzyme-linked chemoluminescence procedure, using ECL Western blotting detection reagents and Hyperfilm ECL (Amersham Biosciences).
Production of PGs
Fibrocytes and fibroblasts were grown for 6 days in complete DMEM as described above. Production of total PGs was then estimated by analysis of the incorporation of H235SO4 into their glycosaminoglycan (GAG) side chains according to a published protocol [26]. Briefly, cells were washed several times with low-sulphate DMEM (Gibco BRL, Paisley, UK), supplemented with 0.4% FCS and 2 mM L-glutamine, and re-incubated for 48 hrs in the same medium. After the first 24 hrs of incubation, 40 μCi/ml carrier-free [35S]sulphate (New England Nuclear Life Science Products, Boston, MA, USA) were added to each well. At the end of the incubation period, replicate cultures of fibrocytes and fibroblasts were either trypsinized to perform cell count with a Coulter counter or further processed as follows. The conditioned medium was harvested and each cell layer was washed twice with 100 μl phosphate-buffered saline (PBS). The two washes were added to the conditioned medium. After centrifugation at 1000 × g for 10 min., ultrapure urea (final concentration of 4 M) was added to the supernatant, which was stored at 4°C until further processing. Cell layers were rinsed and scraped sequentially twice with 2 ml solubilization buffer (pH 7.5) containing 4 M urea, 25 mM Tris base, 10 mM NEM, 20 mM ethylenediaminetetraacetic acid (EDTA), 5 mM benzamidine-HCl, 0.1 M ɛ-aminocaproic acid, 0.2 mM phenylmethylsulphonyl fluoride (PMSF) and 1% Triton X-100. The suspension was incubated for 60 min. on ice, vigorously shaken every 15 min. and centrifuged at 1000 × g for 10 min. at room temperature. To purify the soluble and cell layer-associated metabolically labelled PGs, the supernatant of the conditioned media and the supernatant of the solubilized cell layer were then subjected to anion-exchange chromatography on diethylaminoethyl (DEAE)-Sephacel, using 2 × 0.5 cm DEAE-Sephacel minicolumns (Pharmacia, Uppsala, Sweden). GAGs were eluted with DEAE elution buffer (4 M urea, 1.5 M NaCl, 50 mM sodium acetate, 10 mM NEM, 20 mM EDTA, 0.2 mM PMSF and 0.1% Triton X-100; pH 4.0) and the eluted material was analysed by liquid scintillation counting. Radioactivity in dpm was normalized to cell number.
Further experiments were separately performed to identify the PGs secreted by fibrocytes and fibroblasts. For metabolic labelling of newly synthesized proteins, cells were incubated for 24 hrs with 50 μCi/ml Tran35S-Label (ICN Biomedicals, Irvine, CA, USA) in methionine and cysteine-free DMEM medium. Cell samples were then processed for analysis of the PG core proteins as previously described [26, 27]. Briefly, radio-labelled PGs were extracted from the conditioned medium and the cell layers. The extracts were concentrated with DEAE-Sephacel minicolumns and precipitated with 1.3% potassium acetate in 95% ethanol. The precipitated PGs were separated on 0.75% agarose gel or by SDS-PAGE (with a 4–12% gradient slab gel and a 3% stacking gel), using equal fractions of radio-labelled material from fibrocytes and fibroblasts, with or without prior digestion of the GAG chains with both heparinase/heparitinase I (27.6 U/ml) and heparinase/heparitinase II (13.8 U/ml) (Sigma-Aldrich) or with chondroitinase ABC (1.67 U/ml, Seikagaku, Tokyo, Japan) [27]. The radio-labelled proteins were visualized by autoradiography. The core proteins obtained after digestion of the GAG chains were transferred from the gels to Hybond ECL membranes for Western blotting. The following primary antibodies were used to detect specific PG core proteins (1:500 to 1:1000 dilution): mouse anti-human biglycan (3E2; Santa Cruz Biotechnology); mouse anti-human perlecan (HSPG2; Zymed Laboratories, San Francisco, CA, USA); mouse antibody against the full length of human versican (Santa Cruz Biotechnology) and goat anti-human decorin (N-15; Santa Cruz Biotechnology).
Hyaluronan assay
To estimate the synthesis of hyaluronan in cultures of fibrocytes and fibroblasts, the cells were grown for 6 days in complete DMEM as reported above. The cells were then washed several times with low-serum DMEM and re-incubated for 24 hrs in the same medium. At the end of the incubation period, the conditioned media and cell layers were processed as previously described [28] and both soluble and cell-associated hyaluronan was quantified by using a commercially available assay kit (Hyaluronan DuoSet, R&D Systems) according to the manufacturer’s instructions.
Gene expression analysis
Gene expression analysis was performed by real-time RT-PCR. Fibrocytes and fibroblasts were grown for 6 days in complete DMEM as described above. Cell layers were then washed and re-incubated for 24 hrs in low-serum DMEM. Total RNA was isolated by lysing cells in Trizol (Invitrogen). Genomic DNA was removed by treatment with DNase-free DNA Treatment and Removal Agent (Ambion, Austin, TX, USA). Quantitation of total RNA was then performed with the RiboGreen RNA quantification assay (Invitrogen) and total RNA integrity was analysed by agarose gel electrophoresis. Purified RNA (50 ng) was reverse-transcribed into the corresponding complementary DNA by using the iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA, USA). Specific primers for the target genes and the reference gene, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), were synthesized on the basis of published sequences [29-31] and are listed in Table 1. Each real-time PCR was performed by using the Bio-Rad iQ SYBR Green SuperMix with forward and reverse primers for each target gene and for the reference gene at a final concentration of 300 and 100 nM, respectively, in a sample volume of 25 μl. PCR was conducted by employing the Bio-Rad iCycler thermal cycler and the iCycler iQ detection system (software version 3.1). Following PCR amplification, a melting curve analysis was performed to exclude the amplification of non-specific products. For each gene, cycle threshold (CT) values were calculated and normalized by subtraction of the CT value for GAPDH (ΔCT value). The relative expression level of each gene in fibrocytes versus fibroblasts was determined according to the comparative DDCT method [32, 33].
Table 1.
Primers used for real-time RT-PCR
| Gene symbol | Forward primer | Reverse primer | Amplicon length (bp) |
|---|---|---|---|
| COL1A1 | 5′-TACAGCGTCACTGTCGATGGC-3′ | 5′-TCAATCACTGTCTTGCCCCAG-3′ | 61 |
| COL3A1 | 5′-AATTTGGTGTGGACGTTGGC-3′ | 5′-TTGTCGGTCACTTGCACTGG-3′ | 130 |
| COL5A2 | 5′-GCACGCTTGCCCATCATAGA-3′ | 5′-CCAATTTCAACGCCGAATT-3′ | 74 |
| COL6A3 | 5′-GAGCAGCTTGACAACATTGCC-3′ | 5′-GCCCAGAGCACTTGCAGG-3′ | 61 |
| BGN | 5′-GCCAAGCTGACTGGCATCC-3 | 5′-AGTAGCGAAGCAGGTCCTCCA-3 | 106 |
| DCN | 5′-TGATGCAGCTAGCCTGAAAGG-3′ | 5′-AGGCGTGTTGGCCAGAGAG-3′ | 101 |
| HSPG2 | 5′-TGTGTCGAGATGGAATCAAAGGA-3′ | 5′-GTCGGACTCTGCTATGCCATGT-3′ | 174 |
| VCAN | 5′-GTGACTATGGCTGGCACAAATTCC-3′ | 5′-GGTTGGGTCTCCAATTCTCGTATTGC-3′ | 229 |
| HAS2 | 5′-TCGCAACAGGTAACGCAAT-3′ | 5′-ACTTCTGTTTTTCCACCCATTT-3′ | 77 |
| GAPDH | 5′-CCATGTTCGTCATGGGTGT-3′ | 5′-TGGTCATGAGTCCTTCCACGATA-3′ | 145 |
The target genes were collagen I, α1 chain (COL1A1); collagen III, α1 chain (COL3A1); collagen V, α2 chain (COL5A2); collagen VI, α3 chain (COL6A3); biglycan (BGN); decorin (DCN); perlecan (heparan sulphate PG2, HSPG2); versican (VCAN) and hyaluronan synthase 2 (HAS2). HAS2 is the hyaluronan synthase isoform more frequently expressed constitutively by cells that are able to produce hyaluronan [28, 31]. The primers used for analysis of the versican transcripts were chosen because they detect all known splice variants of the gene [30]. The reference gene was GAPDH.
Adhesion assay
For this assay, 96-well microtitre plates were coated for 1 hr at 37°C with the following matrix molecules (5–50 μg/ml in PBS at neutral pH) as previously described [34, 35]: human plasma fibronectin (Sigma-Aldrich), human collagen I, human collagen IV, human collagen V or human collagen VI (all from Calbiochem, San Diego, CA, USA). The coated wells were washed three times with Tris-buffered saline (TBS) to remove any unbound matrix molecule and residual binding sites were blocked by incubation with 0.2% bovine serum albumin (BSA) in TBS for 1 hr at 37°C. Following three washes with TBS at room temperature, fibrocytes were added to each coated well (2 × 104 cells per well) and were allowed to adhere for 1 hr at 37°C (humidified atmosphere, 5% CO2 in air) in serum-free DMEM, supplemented with 2 mM L-glutamine, 15 mg/ml BSA, 5 mM CaCl2 and 20 mM HEPES (pH 7.4). Unbound fibrocytes were removed by washing three times with TBS containing 5 mM CaCl2. To remove loosely adherent fibrocytes, the plates were inversely centrifuged in the swinging-bucket microplate holder of a centrifuge at 300 × g for 2 min. Quantitation of the firmly adherent cells was then performed as previously described [36]. Briefly, attached cells were fixed with 4% paraformaldehyde in PBS for 20 min. and then stained with crystal violet. Stained cells were lysed with 1% Triton X-100 in 10 mM Tris–Cl (pH 8.0) and absorbance was read at 595 nm on a Benchmark microtitre plate reader (Bio-Rad Laboratories). The number of adherent cells was estimated with the use of a standard curve, which was constructed in preliminary experiments by reading the absorbance of lysates of cell samples containing known numbers of stained cells (from 5 × 102 to 3 × 104). Non-specific adhesion was assessed using fibrocytes inoculated into BSA-coated wells and treated as described above.
Blocking of the cell binding to matrix molecules was performed as follows: fibrocytes were pre-incubated with medium containing 20 μg/ml of a blocking mouse antibody against human integrin β1 (clone P5D2, IgG1; R&D Systems) or the isotype-matched control, 25 μg/ml of the ligand of the macrophage scavenger receptors polyinosinic acid (poly[I]) (Calbiochem) [37] or 400 nM of the Endo180 ligand uPA [38, 39] (American Diagnostica) at 4°C for 30 min. with gentle rotation on a rocker platform to prevent cell re-attachment. The cells were then inoculated into the matrix-coated wells of microtitre plates and the adhesion assay was conducted as described above in presence of the blocking antibody, the isotype-matched control antibody or one of the receptor ligand, as appropriate.
Collagen uptake and degradation
Native trypsin-resistant collagen I from rat tail (BD Biosciences), human collagen VI (Calbiochem) or human plasma fibronectin (Sigma-Aldrich) were radio-labelled with Na[125I]iodide (Amersham) using the Iodogen procedure [40] and tubes pre-coated with the Pierce Iodination Reagent (Thermo Fisher Scientific, Rockford, IL, USA) as described elsewhere [39]. The labelled proteins were then separated from free iodide by gel filtration on a Sephadex G-25 column (Amersham). In some experiments, native collagen I was pre-cleaved by treatment with freshly activated recombinant human MMP13 (Calbiochem) at 25°C for 24 hrs under non-denaturing conditions, as previously described [41]. Complete cleavage of the native collagen I into one- and three-quarter fragments by this collagenase (collagenase 3) was confirmed by SDS-PAGE [41], and the cleaved collagen fragments were also radio-labelled with Na[125I]iodide. The specific activity of native [125I]collagens, cleaved [125I]collagen I and [125I]fibronectin ranged from 6 to 10 μCi/μg of protein.
Fibrocytes were plated onto the uncoated wells of 24-well culture plates (5 × 104 cells/well) and cultured for 6 days in complete DMEM as described above. Then, replicate cultures were either trypsinized to perform cell count with a Coulter counter or further processed as follows. The cell layers were washed three times with serum-free DMEM, supplemented with 15 mg/ml BSA, 2 mM L-glutamine, 5 mM CaCl2 and 20 mM HEPES (pH 7.4), and re-incubated for 12 hrs in the same medium. The medium was then replaced with fresh serum-free DMEM, supplemented as indicated above and containing 5 nM/105 cells/well of one of the 125I-labeled matrix proteins. The cells were incubated again at 37°C for 3 hrs. At the end of the incubation period, 100 μl aliquots of the conditioned media were retained for determination of the amounts of degraded radio-ligand (acid-soluble radioactivity) released from the cells, as previously described [39]. Briefly, the samples were treated with 10% trichloroacetic acid and 0.5% BSA and centrifuged at 1000 × g for 10 min. at 4°C. The acid-soluble radioactivity in the supernatants was counted in a gamma counter. The cell layers were washed twice with ice-cold PBS and incubated for 2 min. at 4°C with 50 μg/ml trypsin, 50 μg/ml proteinase K, and 0.53 mM EDTA, to release the radio-ligand bound to the cell surface and detach the cells [34]. The detached cells were centrifuged at 3000 × g for 5 min. at 4°C and the radioactivity in the supernatants and in the pellet was measured in the gamma counter to estimate the amount of radio-ligand released from the cell surface (bound radioactivity) and the amount of radio-ligand taken up by the cells (internalized radioactivity), respectively [34]. In all experiments, a control was included in which the amount of degraded radio-ligands generated in absence of cells was measured and subtracted to that generated in presence of fibrocytes. The internalized radioactivity and the released acid-soluble radioactivity were expressed as percentage of the initially added radioactivity.
Blocking experiments were conducted as follows: fibrocytes were pre-incubated at 4°C for 30 min. with medium alone or with medium containing 400 nM of the Endo180 ligand uPA [38, 39] or 20 μM of the irreversible inhibitor of lysosomal cysteine proteases, E-64d (Calbiochem). The radio-ligands were then added to the media and the cells were re-incubated for a further 30 min. period at 37°C. To estimate the effects of the uPA and E-64d on the uptake and degradation of radio-ligands, the cell layers and conditioned media were processed as described above.
Statistical analysis
Each set of experiments was conducted using replicate cultures of fibrocyte from three to five donors and each experiment was repeated at least three times with fibrocytes from different donors. Statistical analysis of the data from at least five independent experiments was performed with the Wilcoxon rank sum test for two independent groups, the signed rank test for paired comparisons, or the Kruskal–Wallis one-way ANOVA by ranks, followed by the Wilcoxon rank sum test for post hoc comparison between groups, as appropriate [42]. These distribution-free non-parametric statistical tests were used because of the low number of observations for each experimental condition (n < 25) [42, 43]. A P value of <0.05 was considered statistically significant.
Results
Collagen production
Analysis of the incorporation of [3H]proline into collagenase-sensitive proteins revealed that fibrocytes were able to produce appreciable amounts of collagenous proteins (Fig. 1). However, the amount of newly synthesized collagens that was deposited in the ECM (Fig. 1A) over 24 hrs was significantly lower in cultured fibrocytes than in cultured fibroblasts, both in terms of absolute value and in terms of percentage of total deposited proteins. Considering the amounts of newly synthesized collagenous proteins present in the conditioned media, the differences between fibrocytes and fibroblasts were less marked, but also statistically significant (Fig. 1B).
Fig 1.
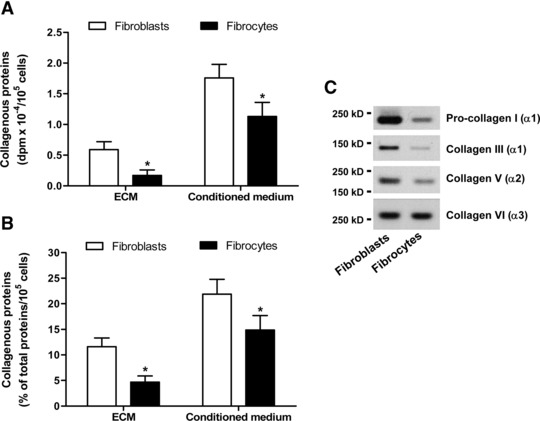
Comparison of collagen production in cultures of pulmonary fibroblasts and fibrocytes by metabolic labelling. (A) Absolute dpm values for the incorporation of [3H]proline into the insoluble collagenase-sensitive proteins (labelled collagenous proteins) deposited in the ECM and released in the conditioned medium over 24 hrs. (B) Labelled collagenous proteins expressed as percentage of the total labelled proteins deposited in the ECM and released in the conditioned medium over 24 hrs. Values are means ± S.D. from five independent experiments. The asterisk indicates statistically significant differences (P < 0.05) between the two cell populations. (C) Western blot analysis of the newly synthesized α chains of specific collagen types (intracellular precursor or mature proteins) extracted from the cell layers and separated by SDS-PAGE.
To examine the ability of fibrocytes to synthesize specific types of collagens, the relative levels of expression of the mRNA encoding the chains of collagens I, III, V and VI were evaluated by real-time RT-PCR, using specific primers for the target genes. These analyses showed that fibrocytes constitutively expressed the mRNA for all the collagen types tested, including collagens V and VI (Table 2). In comparison with fibroblasts, fibrocytes expressed the gene encoding collagen VI (COL6A3) at comparable levels and the genes encoding the chains of collagens I (COL1A1), III (COL3A1) and V (COL5A2) at significantly lower levels (Table 2). These differences between fibrocytes and fibroblasts were confirmed by Western blot analysis of the specific types of collagenous proteins produced by the two cell populations (Fig. 1C).
Table 2.
Relative expression of mRNAs encoding the chains of various collagen types, the core proteins of major PGs and hyaluronan synthase 2 in lung fibroblasts and fibrocytes by real-time RT-PCR
| Target gene | Gene symbol | Fibroblasts | Fibrocytes | Fold change (Mean) | ||
|---|---|---|---|---|---|---|
| CT (mean ± S.D.) | ΔCT (Mean) | CT (mean ± S.D.) | ΔCT (Mean) | |||
| Collagen I (α1) | COL1A1 | 15.3 ± 0.1 | 1.2 | 18.6 ± 0.3* | −2.5* | −13.0 |
| Collagen III (α1) | COL3A1 | 16.8 ± 0.2 | −0.5 | 23.1 ± 0.9* | −5.6* | −34.3 |
| Collagen V (α2) | COL5A2 | 15.9 ± 0.1 | 0.8 | 20.2 ± 0.3* | −2.3* | −8.6 |
| Collagen VI (α3) | COL6A3 | 18.1 ± 0.4 | −1.2 | 19.1 ± 0.7 | −1.9 | −1.4 |
| Biglycan | BGN | 19.0 ± 0.1 | −2.8 | 26.5 ± 0.6* | −9.3* | −90.5 |
| Decorin | dcn | 19.4 ± 0.3 | −2.5 | 28.7 ± 0.8* | −10.9* | −337.8 |
| Perlecan | HSPG2 | 22.2 ± 0.4 | −5.2 | 21.4 ± 0.6 | −3.9* | +2.5 |
| Versican | VCAN | 21.5 ± 0.6 | −5.1 | 19.4 ± 0.2* | −2.2* | +7.5 |
| Hyaluronan synthase 2 | HAS2 | 25.1 ± 0.2 | −8.3 | 23.6 ± 0.5* | −6.7* | +3.0 |
For each target gene, threshold cycle (CT) values were calculated and normalized by subtraction of the CT value for the reference gene GAPDH (ΔCT value). The relative expression level of each target was determined by subtracting the ΔCT value for fibrocytes from the ΔCT value for fibroblasts, and the fold increase (negative DDCT) or decrease (positive DDCT) in the level of expression of each gene in fibrocytes versus fibroblasts was calculated using the formula 2−[DDCT] or −(2[DDCT]), respectively. *Indicates statistically significant differences (P < 0.05) between the two cell populations.
Production of PGs and hyaluronan
Fibrocytes were able to synthesize appreciable amounts of PGs, as assessed by analysis of the incorporation of [35S]sulphate into the GAG side chains over a period of 24 hrs (Fig. 2A). The overall radioactivity incorporated into sulphated GAGs was significantly lower in cultured fibrocyte than in cultured fibroblast, but the fraction of metabolically labelled PGs that was associated to the cell layer was relatively high in fibrocytes (Fig. 2A). Fibrocytes were also able to synthesize appreciable levels of hyaluronan (Fig. 2B). The contents of newly synthesized hyaluronan were slightly lower in the conditioned media from fibrocytes than in the conditioned media from lung fibroblasts but, on the average, 18.6% of the hyaluronan synthesized by fibrocytes was membrane-associated compared with 7.9% of that synthesized by lung fibroblasts (Fig. 2B).
Fig 2.
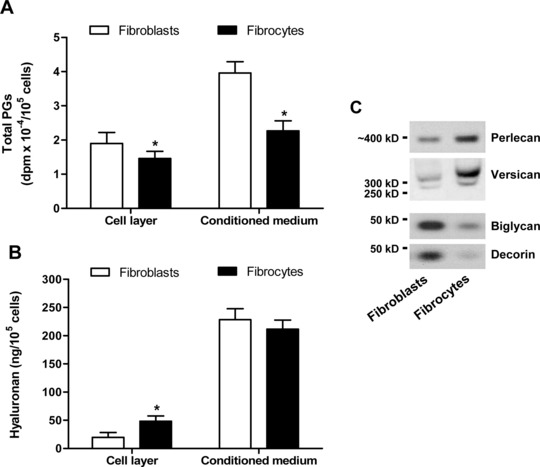
Comparison of PG and hyaluronan production in cultures of pulmonary fibroblasts and fibrocytes. (A) Incorporation of [35S]sulphate into the cell-associated fraction of PGs and in the fraction released into the conditioned media over 24 hrs. (B) Contents of hyaluronan in the cell layers and in the conditioned media after an incubation period of 24 hrs. Values are means ± S.D. from five independent experiments. The asterisk indicates statistically significant differences (P < 0.05) between the two cell populations. (C) Western blot analysis of the core proteins of PGs extracted from the cell layers (perlecan and versican) and conditioned media (biglycan and decorin) following labelling of the newly synthesized proteins with Tran35S-Label for 24 hrs. The core proteins were separated by agarose gel electrophoresis or SDS-PAGE after selective enzymatic degradation of the GAG chains attached to them.
Real-time RT-PCR analyses showed that fibrocytes expressed the mRNA for the PGs perlecan and versican and the mRNA encoding the hyaluronan synthase isoform 2 at high levels in comparison with fibroblasts (Table 2). By contrast, the levels of expression of the mRNA encoding the PGs biglycan and decorin in fibrocytes were on the average 90.5- and 337.8-fold lower, respectively, than those found in fibroblasts (Table 2). Western blot analysis of the core proteins of the cell-associated PGs (perlecan and versican) and PGs released into the conditioned media (biglycan and decorin) confirmed that fibrocytes produced predominantly perlecan and versican in comparison with fibroblasts (Fig. 2C).
Adhesion to ECM molecules
Fibrocytes showed the ability to adhere to fibronectin and to immobilized collagen matrices within 1 hr, whereas their adhesion to BSA-coated surfaces over the same period of time was very low (Fig. 3). To investigate the mechanisms potentially involved in the adhesion process, the expression of receptors that are known to mediate the binding of monocytes or fibroblasts to fibronectin and to various types of native or denatured collagens [34–39, 41, 44–48] was investigated by flow cytometry. Fibrocytes were found to express high levels of integrin β1 and Endo180 immunoreactivity on the cell surface and most cells were specifically stained with the antibodies against these receptors (Fig. 4). Fibrocytes also showed significant immunoreactivity for CD163, CD204 and CD206 (Fig. 4), but did not appear to express appreciable levels of DDR1 or DDR2 immunoreactivity (Fig. 4). These results were confirmed by comparison of the normalized cumulative frequency distributions of the immunofluorescence test slopes and control slopes by the Kolmogorov–Smirnov two-sample test [49].
Fig 3.
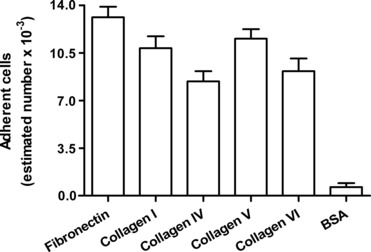
Adhesion of fibrocytes to human plasma fibronectin, human collagen I, human collagen IV, human collagen V or human collagen VI. Non-specific adhesion was assessed using BSA in place of the above-indicated ECM molecules for well coating. Values are means ± S.D. from three independent experiments.
Fig 4.
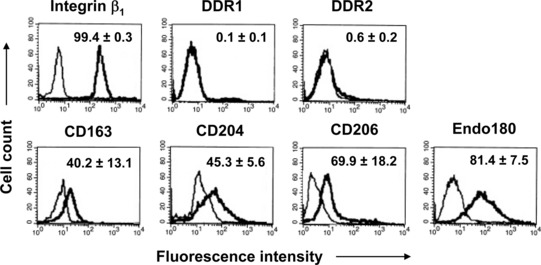
Expression of fibronectin- and collagen-binding receptors on the membrane of fibrocytes by flow cytometry. Each histogram shows staining with the antibody specific for the indicated marker (thick line) and the background non-specific staining with the irrelevant isotype-matched control (thin line), and is representative of three to four independent experiments. The numbers in the histograms are the percentages of positive cells, which are expressed as the means ± S.D.
On the basis of these results, blocking experiments were then conducted to evaluate the relative contribution of integrin β1, Endo180 and the scavenger receptors to the observed ECM binding activity of fibrocytes. Treatment with saturating concentrations of an antibody against integrin β1 specifically reduced fibrocyte adhesion to fibronectin by more than 90% and their adhesion to collagenous proteins by more than 70%, irrespective of the collagen type (Fig. 5). The Endo180 ligand uPA [38, 39] did not affect the adhesion of fibrocytes to fibronectin but significantly reduced the ability of these cells to adhere to the immobilized collagens, particularly to collagen V (Fig. 5). The ligand of the macrophage scavenger receptors, poly[I] [37], did not show any effect on fibrocyte adhesion to the ECM molecules tested (Fig. 5). The validity of the blocking experiments with uPA was confirmed by demonstrating the co-expression of Endo180 and the uPA receptor on the membrane of fibrocytes by flow cytometry. The mean percentage of cells that showed specific immunoreactivity for both surface markers was 79.4 (S.D. = 6.7).
Fig 5.
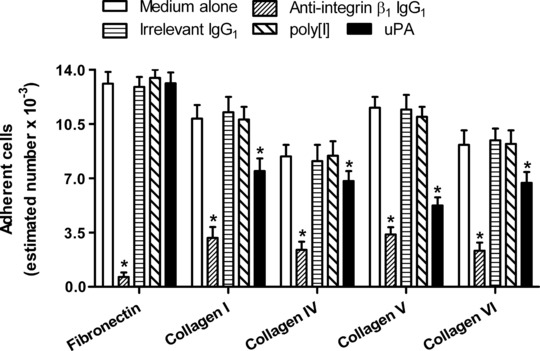
Effects of pre-incubation of fibrocytes with a specific antibody against integrin β1 or the irrelevant isotype-matched control, and with ligands of the scavenger receptors (poly[I]) and Endo180 (uPA) on cell adhesion to human plasma fibronectin, human collagen I, human collagen IV, human collagen V or human collagen VI. Values are means ± S.D. from five independent experiments. The asterisk indicates statistically significant differences (P < 0.05) versus medium alone.
Collagen uptake and degradation
Endo180 mediates cell binding to native or soluble collagens as well as the endocytosis and lysosomal degradation of collagen fragments resulting from collagenase-mediated cleavage [34, 36, 39, 41, 50]. Further experiments were therefore conducted to investigate the ability of fibrocytes to internalize and degrade radio-labelled intact collagens or collagen fragments under conditions that are known to be completely and exclusively dependent on the expression of a functional Endo180 [34, 41]. These experiments revealed that fibrocytes were able to take up both cleaved and uncleaved [125I]collagens while they did not internalize [125I]fibronectin (Fig. 6A). The internalized fraction of labelled collagenous protein was significantly higher when the added radio-ligand was the cleaved collagen I rather than the intact collagen I or collagen VI (Fig. 6A). The release of products derived from the intracellular degradation of internalized collagenous proteins was quantified by measuring the acid-soluble radioactivity in the conditioned media. This fraction of the initially added total radioactivity was also significantly higher when fibrocytes were incubated in the presence of collagen fragments than in presence of intact collagens (Fig. 6A). Taken together, these results suggested that fibrocytes internalized and degraded cleaved collagen I much more efficiently than intact collagens I and VI.
Fig 6.
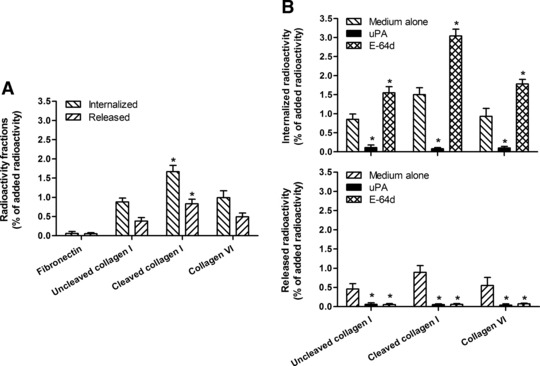
Internalization and degradation of radio-labelled uncleaved native collagen I, MMP13-generated fragments of native collagen I (cleaved collagen I) and intact collagen VI by fibrocytes in absence of any pre-treatment (A) and after pre-incubation of the cells with the Endo180 ligand uPA or the inhibitor of lysosomal cysteine proteases E-64d (B). The internalized radioactivity and the released acid-soluble radioactivity are expressed as the percentage of the initially added radioactivity. Values are means ± S.D. from five independent experiments. The asterisk indicates statistically significant differences (P < 0.05) versus uncleaved native collagen I and intact collagen VI (A) or versus medium alone (B).
In order to confirm the role of Endo180 in the uptake and degradation of collagens by fibrocytes, blocking experiments were conducted with the use of high concentrations of uPA, which has been shown to inhibit Endo180-mediated collagen internalization in other cell types [39]. Another series of blocking experiments were also performed with an inhibitor of lysosomal cysteine proteases (E-64d), which is known to block the lysosomal degradation of collagenous proteins mediated by Endo180 [39, 41]. In these experiments, uPA completely inhibited the internalization of both cleaved and uncleaved [125I]collagens and the release of acid-soluble degradation products from the cells (Fig. 6B). E-64d provoked a significant increase in the intracellular radioactivity, which was associated with a parallel decrease in the release of acid soluble degradation products from fibrocytes (Fig. 6B).
Discussion
In this study, we evaluated the production of several ECM molecules by cultured human fibrocytes, including the production of important ECM components for which no information is currently available. We found that fibrocytes are able to synthesize and release appreciable amounts of collagenous proteins and PGs, and that there are substantial differences between fibrocytes and fibroblasts in terms of gene expression profile and relative amounts of secreted proteins. In comparison with cultured human lung fibroblasts, fibrocytes constitutively expressed significantly lower levels of the mRNA encoding the chains of collagens I, III and V, but expressed comparable levels of the mRNA for collagen VI. The PG gene expression profile was also different in fibrocytes and fibroblasts because fibrocytes constitutively expressed relatively high levels the mRNA encoding perlecan and versican and much lower levels of the mRNA for the other tested PGs, particularly decorin. Moreover, fibrocytes expressed the mRNA for hyaluronan synthase 2 at higher level than fibroblasts. Significant differences between the two cell populations were also demonstrated by analysis of the secreted collagenous proteins, PGs and hyaluronan, and the results of these experiments were in keeping with the data obtained by gene expression analysis.
Like fibroblasts, fibrocytes were able to incorporate part of the collagenous proteins and PGs they actively produced into the matrix underlying or surrounding the cells, as demonstrated by metabolic labelling. The membrane-associated fraction of newly synthesized hyaluronan and PGs was relatively high in fibrocyte cultures as compared to fibroblast cultures. These findings may be explained by the observed differences between the two cell types in terms of synthesis and secretion of hyaluronan and specific types of PGs. In comparison with fibroblasts, fibrocytes predominantly produced two PGs commonly associated to cell membranes, perlecan and versican, rather than biglycan and decorin [51]. Moreover, versican and hyaluronan aggregate together in the pericellular matrix and on the cell surface [51, 52] by binding to fibronectin and to the cell receptors integrin β1 and CD44 [52], which are also highly expressed on the cell surface of fibrocytes.
Collagens I, III and V are all fibrillar collagens [53], but their fibrils can be assembled differently and constitute distinct structural fibrous components of tissue ECM [54]. Interstitial collagen is predominantly composed of thick collagen I fibres, whereas the reticular sheet of fine fibrils which typically underlies epithelial basement membranes (lamina reticularis) is predominantly composed of collagens III and V [54]. Collagen VI is a non-fibrillar collagen that forms a network of beaded filaments [53]. This collagen, hyaluronan, perlecan and versican are all ECM molecules mainly involved in the modulation of cell-to-cell interaction and cell–matrix interaction [52, 55-58]. Perlecan binds a number of growth factors [59] and promotes angiogenesis [60]. It regulates the permeability of the basement membranes and interacts with other matrix components (laminin, fibronectin and collagen IV) to maintain tissue integrity by regulating stress transfer between the ECM, the cell membranes and the cytoskeletons [58, 61]. Increased production of perlecan and versican has been demonstrated in airway walls subjected to mechanical strain, and such response seems to represent part of a feedback mechanism through which the system adjusts to altered mechanical requirements by changing the visco-elastic properties of the lung tissue [61]. The ability of fibrocytes to produce relatively high levels of collagen VI, perlecan, versican and hyaluronan, in comparison with other ECM components, supports the hypothesis that these cells may be primarily implicated in the modulation of inflammatory responses and in tissue stabilization during wound healing or in chronic diseases characterized by impaired tissue repair [2, 8]. For example, fibrocytes may contribute to the thickening of the reticular basement membrane in asthma by producing the excessive amounts of non-interstitial collagens, versican, perlecan and hyaluronan that have been described in this condition [62–64]. If the progressive accumulation of fibrocytes in the subepithelial zone may have several detrimental effects on lung function [2, 11–13], it may also serve to enhance the resistance to mechanical stress of a bronchial wall weakened by the presence of a chronic inflammatory infiltrate [2]. Through similar mechanisms, the fibrocytes that preferentially localize to the fibrous cap of atherosclerotic lesions [17] might stabilize the plaques and prevent their fracture in the vessel zones mostly exposed to mechanical stress.
In this study, fibrocytes showed the ability to adhere to fibronectin and to immobilized collagen matrices within 1 hr. The adhesion to fibronectin was completely inhibited by blocking the binding of integrin β1 on the cell surface, whereas the same procedure significantly reduced but did not abolish the adhesion of fibrocytes to collagenous molecules. A phenotypic analysis of fibrocytes by flow cytometry revealed that these cells expressed various known collagen receptors, in addition to integrin β1, and the results of a series of blocking experiments indicated that the additional receptor involved in collagen adhesion was the mannose receptor Endo180. The same data also confirmed that uPA is competitive ligand for the collagen binding sites of Endo180 on the surface of cells that, like fibrocytes, express both the uPA receptor and the uPA receptor-associated protein Endo180 [38]. This receptor not only promotes cell binding to various collagen types but also mediates the uptake of collagen fragments and soluble or denatured collagens by some fibroblast-like cells, alternatively activated macrophages and hepatic stellate cells [34, 38, 39, 41, 50]. Following collagen binding, Endo180 is routed to the early endosomal compartment in the perinuclear zone for collagen delivery and the unoccupied receptor is immediately recycled to the cell surface. The initially bound collagen is rapidly transported to the late endosomal compartment and then routed to the lysosomes, where it is completely degraded primarily by cysteine proteases. In the present study, fibrocytes were able to internalize collagen I fragments more efficiently than uncleaved native collagen I or intact collagen VI, and collagen uptake was inhibited by excess amounts of uPA. Moreover, products of intracellularly degraded collagens could be recovered in the fibrocyte conditioned media, and inhibition of the activity of the lysosomal cysteine proteases blocked the release of such degraded products. These results are consistent with previous observations on Endo180-mediated collagen endocytosis in other cell types [34, 36, 39, 41] and all experiments were conducted under conditions that are known to be unaffected by the MMPs released by the cells themselves [34, 41]. Therefore, our findings collectively suggest that fibrocytes express functional Endo180 receptors on their cell surface and may be involved in collagen turnover.
As demonstrated in this study, collagen that has been pre-cleaved by a fibroblast-derived collagenase (MMP13) is easily internalized and degraded by fibrocytes. Considering that the initial cleavage of collagens at tissue sites is indeed initiated by MMP13 and other fibroblast-derived collagenases (MMP1 and MMP14) [65], it is conceivable to believe that collagen degradation by fibrocytes can occur in vivo and that any impairment of the Endo180-mediated collagen uptake in these cells may affect the outcome of inflammatory conditions and repair processes. This concept may help understand how fibrocytes contribute to the pathogenesis of NSF [16, 66], a fibrotic disorder that develops in certain patients with advanced kidney disease who have been exposed to gadolinium-based contrast agents [66-68]. Predisposing conditions such as vascular injury or inflammatory states are necessary for the development of NSF [67, 68] and all these conditions also favour the recruitment of fibrocyte precursor cells and the accumulation of fibrocyte at tissue sites [2, 8]. Histological analysis of the typical skin lesions shows the presence of thick collagen bundles in the dermis and subcutaneous tissue, and a dermal infiltrate almost exclusively composed of macrophages and CD45+CD34+fibrocytes co-expressing collagen I mRNA or protein [14-16]. These fibrocytes can form a complex dendritic network, and their dendritic processes encircle the collagen bundles [16, 66]. Numerous deposits of gadolinium with co-associated elements, such as phosphorus and calcium, are present in the extracellular compartment and within the cytoplasm of macrophages and fibrocytes [69].
The spreading of fibrocytes on collagen bundles and the close contact between their dendritic process and the collagen fibres are peculiar histological findings in NSF [66] and are in keeping with our observations that fibrocytes firmly adhere to and bind intact collagenous matrices in vitro. In our study, fibrocytes were also found to express some of the scavenger receptors normally expressed at high levels by certain subpopulations of macrophages, particularly alternatively activated macrophages [70]. CD204 and the other known scavenger receptors for unopsonized particles bind a broad spectrum of ligands, and their main function is the clearance of chemically modified host molecules such as oxidized and acetylated lipoproteins, apoptotic cells, pathogens and inorganic particles [71-73]. It is therefore possible that they are also implicated in the phagocytosis of gadolinium-containing particles by macrophages and fibrocytes. In the dermal infiltrate of NSF patients, the intracellular deposits of gadolinium appear to be localized to the lysosomal compartment of fibrocytes [69], where degradation of internalized collagens also takes place. The engulfment of fibrocytes with these inorganic particles may interfere with their ability to degrade collagenous proteins through the Endo180-mediated pathway. Such an impairment of collagen turnover would result in a net increase in the amounts of collagenous proteins deposited in the pericellular matrix by resident fibroblasts and fibrocytes themselves, and might substantially contribute to the development and progression of this fibrotic disorder. Additional mechanisms potentially involved in the pathogenesis of NSF include a progressive increase in the total number of collagen-producing cells at tissue sites and an up-regulation of the synthesis of ECM components in these cells. Recent in vitro studies have in fact demonstrated that gadolinium-containing contrast agents promote fibroblast growth [74] as well as the development of fibrocytes from their monocyte-like precursor cells [75]. Moreover, it has been reported that the fibroblasts and fibroblast-like cells isolated from NSF lesions spontaneously synthesize excessive levels of collagens and hyaluronan in vitro, in comparison with normal tissue fibroblasts [74].
In conclusion, the results of this study demonstrate that human fibrocytes exhibit specific ECM remodelling properties previously unexplored, including the ability to participate in collagen turnover. The observed differences between fibrocytes and normal tissue fibroblasts in terms of collagen and PG gene expression profile may reflect different functions of the two cell populations in wound healing and in inflammatory conditions associated with extensive ECM remodelling, such as asthma and atherosclerosis. In particular, fibrocytes seem to have a predominant matrix-stabilizing function, which is at variance with the predominant matrix-building function of most fibroblasts. The results of this study also suggest that interference with the ability of fibrocytes to degrade collagenous proteins through an Endo180-mediated pathway may represent one of the possible mechanisms involved in the pathogenesis of NSF.
Conflict of interest
This work was supported in part through funds from the Böhler-Thiele Foundation and the European Initiative for the Advancement of Regenerative Medicine. S.M. is a founding shareholder and board member of the company Avail GmbH, Basel, Switzerland. The other authors do not have conflicts of interest to declare.
References
- 1.Bucala R, Spiegel LA, Chesney J, et al. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med. 1994;1:71–81. [PMC free article] [PubMed] [Google Scholar]
- 2.Mattoli S, Bellini A, Schmidt M. The role of a human hematopoietic mesenchymal progenitor in wound healing and fibrotic diseases and implications for therapy. Curr Stem Cell Res Ther. 2009;4:266–80. doi: 10.2174/157488809789649232. [DOI] [PubMed] [Google Scholar]
- 3.Abe R, Donnelly SC, Peng T, et al. Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J Immunol. 2001;166:7556–62. doi: 10.4049/jimmunol.166.12.7556. [DOI] [PubMed] [Google Scholar]
- 4.Yang L, Scott PG, Giuffre J, et al. Peripheral blood fibrocytes from burn patients: identification and quantification of fibrocytes in adherent cells cultured from peripheral blood mononuclear cells. Lab Invest. 2002;82:1183–92. doi: 10.1097/01.lab.0000027841.50269.61. [DOI] [PubMed] [Google Scholar]
- 5.Shao DD, Suresh R, Vakil V, et al. Pivotal advance: Th-1 cytokines inhibit, and Th-2 cytokines promote fibrocyte differentiation. J Leukoc Biol. 2008;83:1323–33. doi: 10.1189/jlb.1107782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Curnow SJ, Fairclough M, Schmutz C, et al. Distinct types of fibrocyte can differentiate from mononuclear cells in the presence and absence of serum. PLoS One. 2010;5:e9730. doi: 10.1371/journal.pone.0009730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.GarcÌa-de-Alba C, Becerril C, Ruiz V, et al. Expression of matrix metalloproteases by fibrocytes. Possible role in migration and homing. Am J Respir Crit Care Med. 2010;182:1144–52. doi: 10.1164/rccm.201001-0028OC. [DOI] [PubMed] [Google Scholar]
- 8.Bellini A, Mattoli S. The role of the fibrocyte, a bone marrow-derived mesenchymal progenitor, in reactive and reparative fibroses. Lab Invest. 2007;87:858–70. doi: 10.1038/labinvest.3700654. [DOI] [PubMed] [Google Scholar]
- 9.Keeley EC, Mehrad B, Strieter RM. The role of circulating mesenchymal progenitors (fibrocytes) in the pathogenesis of fibrotic disorders. Thromb Haemost. 2009;101:613–8. [PMC free article] [PubMed] [Google Scholar]
- 10.Herzog EL, Bucala R. Fibrocytes in health and disease. Exp Hematol. 2010;38:548–56. doi: 10.1016/j.exphem.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmidt M, Sun G, Stacey MA, et al. Identification of circulating fibrocytes as precursors of bronchial myofibroblasts in asthma. J Immunol. 2003;171:380–9. doi: 10.4049/jimmunol.171.1.380. [DOI] [PubMed] [Google Scholar]
- 12.Nihlberg K, Larsen K, Hultgardh-Nilsson A, et al. Tissue fibrocytes in patients with mild asthma: a possible link to thickness of reticular basement membrane. Respir Res. 2006;7:50. doi: 10.1186/1465-9921-7-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saunders R, Siddiqui S, Kaur D, et al. Fibrocyte localization to the airway smooth muscle is a feature of asthma. J Allergy Clin Immunol. 2009;123:376–84. doi: 10.1016/j.jaci.2008.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cowper SE, Bucala R. Nephrogenic fibrosing dermopathy: suspect identified, motive unclear. Am J Dermatopathol. 2003;25:358. doi: 10.1097/00000372-200308000-00017. [DOI] [PubMed] [Google Scholar]
- 15.Ortonne N, Lipsker D, Chantrel F, et al. Presence of CD45RO+ CD34+ cells with collagen synthesis activity in nephrogenic fibrosing dermopathy: a new pathogenetic hypothesis. Br J Dermatol. 2004;150:1050–2. doi: 10.1111/j.1365-2133.2004.05900.x. [DOI] [PubMed] [Google Scholar]
- 16.Cowper SE, Rabach M, Girardi M. Clinical and histological findings in nephrogenic systemic fibrosis. Eur J Radiol. 2008;66:191–9. doi: 10.1016/j.ejrad.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 17.Medbury HJ, Tarran SLS, Guiffre AK, et al. Monocytes contribute to the atherosclerotic cap by transformation into fibrocytes. Int Angiol. 2008;27:114–23. [PubMed] [Google Scholar]
- 18.Andersson-Sjöland A, de Alba CG, Nihlberg K, et al. Fibrocytes are a potential source of lung fibroblasts in idiopatic pulmonary fibrosis. Int J Biochem Cell Biol. 2008;40:2129–40. doi: 10.1016/j.biocel.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 19.Abu El-Asrar AM, Struyf S, Van Damme J, et al. Circulating fibrocytes contribute to the myofibroblast population in proliferative vitreoretinopathy epiretinal membranes. Br J Ophthalmol. 2008;92:699–704. doi: 10.1136/bjo.2007.134346. [DOI] [PubMed] [Google Scholar]
- 20.Chesney J, Metz C, Stavitsky AB, et al. Regulated production of type I collagen and inflammatory cytokines by peripheral blood fibrocytes. J Immunol. 1998;160:419–25. [PubMed] [Google Scholar]
- 21.Sun G, Stacey MA, Bellini A, et al. Endothelin-1 induces bronchial myofibroblast differentiation. Peptides. 1997;18:1449–51. doi: 10.1016/s0196-9781(97)00194-0. [DOI] [PubMed] [Google Scholar]
- 22.Peterkofsky B, Diegelmann R. Use of a mixture of proteinase-free collagenases for the specific assay of radioactive collagen in presence of other proteins. Biochemistry. 1971;10:988–94. doi: 10.1021/bi00782a009. [DOI] [PubMed] [Google Scholar]
- 23.Peterkofsky B. Estimation of total collagen production by collagenase digestion. In: Haralson MA, Hassel JL, editors. Extracellular matrix. London: Oxford University Press; 1995. pp. 31–8. [Google Scholar]
- 24.Köhler E, Bertschin S, Woodtly T, et al. Does aldosterone-induced cardiac fibrosis involve direct effects on cardiac fibroblasts. J Vasc Res. 1996;33:315–26. doi: 10.1159/000159159. [DOI] [PubMed] [Google Scholar]
- 25.Eickelberg O, Köhler E, Reichenberger F, et al. Extracellular matrix deposition by primary human lung fibroblasts in response to TGF-β1 and TGF-β3. Am J Physiol Lung Cell Mol Physiol. 1999;276:L814–24. doi: 10.1152/ajplung.1999.276.5.L814. [DOI] [PubMed] [Google Scholar]
- 26.Woods A, Couchman JR. Proteoglycan isolation and analysis. In: Bonifacino JS, Dasso M, Harford JB, Lippincott-Schwartz J, Yamada KM, editors. Current protocols in cell biology. Hoboken, NJ: Wiley; 2001. pp. 10.7.1–19. [Google Scholar]
- 27.Yamamoto C, Shimada S, Fujiwara Y, et al. Proteoglycans released from cultured bovine aortic endothelial cell layers by sodium spirulan are both perlecan and biglycan. Biol Pharm Bull. 2005;28:32–6. doi: 10.1248/bpb.28.32. [DOI] [PubMed] [Google Scholar]
- 28.Jacobson A, Brink J, Briskin MJ, et al. Expression of human hyaluronan synthases in response to external stimuli. Biochem J. 2000;348:29–35. [PMC free article] [PubMed] [Google Scholar]
- 29.Schnoor M, Cullen P, Lorkowski J, et al. Production of type VI collagen by human macrophages: a new dimension in macrophage functional heterogeneity. J Immunol. 2008;180:5707–19. doi: 10.4049/jimmunol.180.8.5707. [DOI] [PubMed] [Google Scholar]
- 30.Arslan F, Bosserhoff A-K, Nickl-Jockschat T, et al. The role of versican isoforms V0/V1 in glioma migration mediated by transforming growth factor-β2. Br J Cancer. 2007;96:1560–8. doi: 10.1038/sj.bjc.6603766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li L, Asteriou T, Bernert B, et al. Growth factor regulation of hyaluronan synthesis and degradation in human dermal fibroblasts: importance of hyaluronan for the mitogenic response of PDGF-BB. Biochem J. 2007;404:327–36. doi: 10.1042/BJ20061757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 33.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–8. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 34.Engelholm LH, List K, Netzel-Arnett S, et al. uPARAP/Endo 180 is essential for cellular uptake of collagen and promotes fibroblast collagen adhesion. J Cell Biol. 2003;160:1009–15. doi: 10.1083/jcb.200211091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gowen BB, Borg TK, Ghaffar A, et al. The collagenous domain of class A scavenger receptors is involved in macrophage adhesion to collagens. J Leukoc Biol. 2001;69:575–82. [PubMed] [Google Scholar]
- 36.Thomas EK, Nakamura M, Wienke D, et al. Endo180 binds to the C-terminal region of type I collagen. J Biol Chem. 2005;280:22596–605. doi: 10.1074/jbc.M501155200. [DOI] [PubMed] [Google Scholar]
- 37.El Khoury J, Thomas CA, Loike JD, et al. Macrophages adhere to glucose-modified basement membrane collagen IV via their scavenger receptors. J Biol Chem. 1994;269:10197–200. [PubMed] [Google Scholar]
- 38.Behrendt N, Jensen ON, Engelholm LH, et al. A urokinase receptor-associated protein with specific collagen binding properties. J Biol Chem. 2000;275:1993–2002. doi: 10.1074/jbc.275.3.1993. [DOI] [PubMed] [Google Scholar]
- 39.Mousavi SA, Sato M, Sporstøl M, et al. Uptake of denatured collagen into hepatic stellate cells: evidence for the involvement of urokinase plasminogen activator receptor-associated protein/Endo180. Biochem J. 2005;387:39–46. doi: 10.1042/BJ20040966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fraker PJ, Speck JC. Protein and cell membrane iodinations with a sparingly soluble chloroamide,1,3,4,6-tetrachloro-3a,6a-diphrenylglucoluril. Biochem Biophys Res Commun. 1978;80:849–57. doi: 10.1016/0006-291x(78)91322-0. [DOI] [PubMed] [Google Scholar]
- 41.Madsen DH, Engelholm LH, Ingvarsen S, et al. Extracellular collagenases and the endocytic receptor, urokinase plasminogen activator receptor-associated protein/ Endo180, cooperate in fibroblast-mediated collagen degradation. J Biol Chem. 2007;282:27037–45. doi: 10.1074/jbc.M701088200. [DOI] [PubMed] [Google Scholar]
- 42.Dawson B, Trapp RG. Basic & clinical biostatistics. New York: Lange Medical Books/McGraw-Hill; 2001. [Google Scholar]
- 43.Kitchen CMR. Nonparametric versus parametric tests of location in biomedical research. Am J Ophtalmol. 2009;147:571–2. doi: 10.1016/j.ajo.2008.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Staatz WD, Walsh JJ, Pexton T, et al. The alpha 2 beta 1 integrin cell surface collagen receptor binds to the alpha 1 (I)-CB3 peptide of collagen. J Biol Chem. 1990;265:4778–81. [PubMed] [Google Scholar]
- 45.Staatz WD, Fok KF, Zutter MM, et al. Identification of the tetrapeptide recognition sequence for the alpha 2 beta 1 integrin in collagen. J Biol Chem. 1991;266:7363–7. [PubMed] [Google Scholar]
- 46.Danen EH, Sonnenberg A. Integrins in the regulation of tissue development and function. J Pathol. 2003;201:632–41. doi: 10.1002/path.1472. [DOI] [PubMed] [Google Scholar]
- 47.Vogel W, Gish GD, Alves F, et al. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol Cell. 1997;1:13–23. doi: 10.1016/s1097-2765(00)80003-9. [DOI] [PubMed] [Google Scholar]
- 48.Shrivastava A, Radziejewski C, Campbell E, et al. An orphan receptor tyrosine kinase family whose members serve as nonintegrin collagen receptors. Mol Cell. 1997;1:25–34. doi: 10.1016/s1097-2765(00)80004-0. [DOI] [PubMed] [Google Scholar]
- 49.Watson JV. Flow cytometry data analysis: basic concept and statistics. Cambridge, UK: Cambridge University Press; 2005. [Google Scholar]
- 50.Wienke D, MacFadyen JR, Isacke CM. Identification and characterization of the endocytic transmembrane glycoprotein Endo180 as a novel collagen receptor. Mol Biol Cell. 2003;14:3592–604. doi: 10.1091/mbc.E02-12-0814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kähäri V-M, Larjava H, Uitto J. Differential regulation of extracellular matrix proteoglycans (PG) gene expression. Transforming growth factor-β1 upregulates biglycan (PGI), and versican (large fibroblast PG) but down-regulates decorin (PGII) mRNA levels in human fibroblasts in culture. J Biol Chem. 1991;266:10608–16. [PubMed] [Google Scholar]
- 52.Wu YJ, La Pierre DP, Wu J, et al. The interaction of versican with its binding partners. Cell Res. 2005;15:483–94. doi: 10.1038/sj.cr.7290318. [DOI] [PubMed] [Google Scholar]
- 53.Myllyharju J, Kivirikko KI. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 2004;20:33–43. doi: 10.1016/j.tig.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 54.Ushiki T. Collagen fibers, reticular fibers and elastic fibers. A comprehensive understanding from a morphological viewpoint. Arch Histol Cytol. 2002;65:109–26. doi: 10.1679/aohc.65.109. [DOI] [PubMed] [Google Scholar]
- 55.Hirose J, Kawashima H, Yoshie O, et al. Versican interacts with chemokines and modulate cellular responses. J Biol Chem. 2001;276:5228–34. doi: 10.1074/jbc.M007542200. [DOI] [PubMed] [Google Scholar]
- 56.Howell SJ, Doane KJ. Type VI collagen increases cell survival and prevents anti-β1 integrin-mediated apoptosis. Exp Cell Res. 1998;241:230–41. doi: 10.1006/excr.1998.4051. [DOI] [PubMed] [Google Scholar]
- 57.Myllyharju J, Kivirikko KI. Collagens and collagen-related diseases. Ann Med. 2001;33:7–21. doi: 10.3109/07853890109002055. [DOI] [PubMed] [Google Scholar]
- 58.Iozzo RV. Matrix proteoglycans: from molecular design to cellular function. Annu Rev Biochem. 1998;67:609–52. doi: 10.1146/annurev.biochem.67.1.609. [DOI] [PubMed] [Google Scholar]
- 59.Ruoslahti E, Yamaguchi Y. Proteoglycans as modulators of growth factor activities. Cell. 1991;64:867–9. doi: 10.1016/0092-8674(91)90308-l. [DOI] [PubMed] [Google Scholar]
- 60.Zhang W, Chuang YJ, Swanson R, et al. Antiangiogenic antithrombin downregulates the expression of the pro-angiogenic heparan sulfate proteoglycan, perlecan, in endothelial cells. Blood. 2004;103:1185–91. doi: 10.1182/blood-2003-08-2920. [DOI] [PubMed] [Google Scholar]
- 61.Al-Jamal R, Ludwig MS. Changes in proteoglycans and lung tissue mechanics during excessive mechanical ventilation. Am J Physiol Lung Cell Mol Physiol. 2001;281:L1078–87. doi: 10.1152/ajplung.2001.281.5.L1078. [DOI] [PubMed] [Google Scholar]
- 62.Wilson JW, Li X. The measurement of reticular basement membrane and submucosal collagen in the asthmatic airway. Clin Exp Allergy. 1997;27:363–71. [PubMed] [Google Scholar]
- 63.Roberts CR. Is asthma a fibrotic disease. Chest. 1995;107:111S–7S. doi: 10.1378/chest.107.3_supplement.111s. [DOI] [PubMed] [Google Scholar]
- 64.Pini L, Hamid Q, Shannon J, et al. Differences in proteoglycans deposition in the airways of moderate and severe asthmatics. Eur Respir J. 2007;29:71–7. doi: 10.1183/09031936.00047905. [DOI] [PubMed] [Google Scholar]
- 65.Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92:827–39. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- 66.Galan A, Cowper SE, Bucala R. Nephrogenic systemic fibrosis (nephrogenic fibrosing dermopathy) Curr Opin Rheumatol. 2006;18:614–7. doi: 10.1097/01.bor.0000245725.94887.8d. [DOI] [PubMed] [Google Scholar]
- 67.Agarwal R, Brunelli SM, Williams K, et al. Gadolinium-based contrast agents and nephrogenic systemic fibrosis: a systematic review and meta-analysis. Nephrol Dial Transplant. 2009;24:856–63. doi: 10.1093/ndt/gfn593. [DOI] [PubMed] [Google Scholar]
- 68.Perazella MA. Tissue deposition of gadolinium and development of NSF: a convergence of factors. Semin Dial. 2008;21:150–4. doi: 10.1111/j.1525-139X.2007.00403.x. [DOI] [PubMed] [Google Scholar]
- 69.Thakral C, Abraham JL. Gadolinium-induced nephrogenic systemic fibrosis is associated with insoluble Gd deposits in tissues: in vivo trasmetallation confirmed by microanalysis. J Cutan Pathol. 2009;36:1244–54. doi: 10.1111/j.1600-0560.2009.01283.x. [DOI] [PubMed] [Google Scholar]
- 70.Martinez FO, Gordon S, Locati M, et al. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and pattern of gene expression. J Immunol. 2006;177:7303–11. doi: 10.4049/jimmunol.177.10.7303. [DOI] [PubMed] [Google Scholar]
- 71.Murphy JE, Tedbury PR, Homer-Vanniasinkam S, et al. Biochemistry and cell biology of mammalian scavenger receptors. Atherosclerosis. 2005;182:1–15. doi: 10.1016/j.atherosclerosis.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 72.Arredouani MS, Yang Z, Imrich A, et al. The macrophage scavenger receptor SR-AI/II and lung defense against pneumococci and particles. Am J Respir Cell Mol Biol. 2006;35:474–8. doi: 10.1165/rcmb.2006-0128OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thakur SA, Hamilton R, Pikkarainen T, et al. Differential binding of inorganic particles to MARCO. Toxicol Sci. 2009;107:238–46. doi: 10.1093/toxsci/kfn210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Edward M, Quinn JA, Mukherjee S, et al. Gadodiamide contrast agent ‘activates’ fibroblasts: a possible cause of nephrogenic systemic fibrosis. J Pathol. 2008;214:584–93. doi: 10.1002/path.2311. [DOI] [PubMed] [Google Scholar]
- 75.Vakil V, Sung JJ, Piecychna M, et al. Gadolinium-containing magnetic resonance image contrast agent promotes fibrocyte differentiation. J Magn Reson Imaging. 2009;30:1284–8. doi: 10.1002/jmri.21800. [DOI] [PMC free article] [PubMed] [Google Scholar]