

Abstract
The major green tea polyphenol (-)-epigallocatechin-3-gallate (EGCG) has been shown to exhibit antitumour activities in several tumour models. One of the possible mechanisms by which EGCG can inhibit cancer progression is through the modulation of angiogenesis signalling cascade. The tumour cells’ ability to tightly adhere to endothelium is a very important process in the metastatic process, because once disseminated into the bloodstream the tumour cells must re-establish adhesive connections to endothelium in order to extravasate into the target tissues. In this study, we investigated the anti-angiogenic effects of EGCG treatment (10 μM) on human cervical tumour cells (HeLa) by evaluating the changes in the expression pattern of 84 genes known to be involved in the angiogenesis process. Transcriptional analysis revealed 11 genes to be differentially expressed and was further validated by measuring the induced biological effects. Our results show that EGCG treatment not only leads to the down-regulation of genes involved in the stimulation of proliferation, adhesion and motility as well as invasion processes, but also to the up-regulation of several genes known to have antagonist effects. We observed reduced proliferation rates, adhesion and spreading ability as well as invasiveness of HeLa tumour cells upon treatment, which suggest that EGCG might be an important anti-angiogenic therapeutic approach in cervical cancers.
Keywords: EGCG, gene expression, angiogenesis, HeLa cells
Introduction
In 2007, cervical cancer was reported to be the second most common female cancer worldwide, being the third cause of female cancer mortality annually [1]. Although in the developed countries cervical cancer incidence and mortality have decreased, this disease still remains a serious problem with high estimated incidence and mortality [2].
Cervical cancer can be prevented and if detected early it is generally curable [3]. However, the treatments for metastatic or recurrent cervical carcinoma are poorly effective and with serious adverse effects. There is a great need to identify and investigate new systemic acting agents for cervical cancer treatment.
Lately, the research has been focused on natural compounds; several dietary components show prevention and therapeutic proprieties in the pathogenesis of cancer. The use of dietary compounds for cancer prevention and therapy could be of major importance because in addition to the diverse biological effects, they have low toxicity and fewer adverse effects than traditional chemotherapeutic agents.
Green tea has received much attention, particularly its major component (-)-epigallocatechin-3-gallate (EGCG). EGCG has been shown to possess remarkable cancer chemopreventive and therapeutic potentials against various cancers, by modulating cell proliferation and apoptosis both in vitro and in vivo [4-8]. In addition, recent work revealed that EGCG could interfere with cell signalling pathways of angiogenesis, metastasis and migration in prostate, liver and breast cancer cells [9-11].
Angiogenesis is a central process in cervical carcinogenesis and progression [12]. Cervical tumour’s ability to develop vasculature in order to respond to the metabolic needs is essential to its progression. The antiproliferative effect of EGCG in cervical cancer has been intensively investigated [13-17], but little is known about its anti-angiogenic potential [18].
With these considerations, in this study we report the effect of EGCG on the angiogenic potential of HeLa cervical adenocarcinoma cells, specifically the molecular transcription pattern induced by EGCG treatment. Tumour invasiveness properties including cell morphology change, in vitro invasion, spreading and adhesion to extracellular matrix (ECM) and endothelium were evaluated in order to validate our findings.
Materials and methods
Cell culture
HeLa cells (ECACC, Salisbury, UK) were cultured in DMEM growth medium with 1000 mg/l glucose and Human Umbilical Vein Endothelial Cells (HUVEC) cells were cultured in RPMI-1640 medium at 37°C in 5% carbon dioxide–humidified air. Each culture medium was supplemented with 10% foetal bovine serum (FBS), 2 mM L-glutamine, 1% Non-essential amino acids (NEAA), 100 U/ml penicillin and 100 U/ml streptomycin. Cell culture reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA).
EGCG treatment
A total of 5 × 105 HeLa cells per well were seeded in 6-well plates in Opti-Mem medium (Gibco, Invitrogen, Carlsbad, CA) and treated with EGCG (Sigma-Aldrich) to a final concentration of 10 μM for 24 and 48 hrs. Control cells (UT) were treated with phosphate-buffered saline (PBS) instead of EGCG. All our experiments were conducted under serum starvation conditions in order to synchronize the cells and stimulate growth factor production.
Total RNA isolation
Total RNA was isolated with TriReagent (Sigma-Aldrich), purified with RNAeasy Mini Kit (Qiagen Inc., Valencia, CA, USA) and further analysed for quantity and quality with NanoDrop ND-1000 and Agilent Lab-on-a-chip Bioanalyzer 2100 (Agilent Technology Inc., Santa Clara, CA, USA). All the RNAs presented a RNA Integrity Number (RIN) between 9 and 10.
PCR array
Eighty-four genes involved in angiogenesis modulation were simultaneously evaluated using the Human Angiogenesis RT2 ProfilerTM PCR Array (SABiosciences, Frederick, MD, USA). One microgram of total RNA isolated after 24 hrs of treatment was reverse-transcribed with RT2 First Strand kit (SABiosciences), and subsequently amplified in a 96-well plate (The Human Angiogenesis RT2 ProfilerTM PCR Array) with RT2 Real-Timer SyBR Green (SABiosciences) using LightCycler 480 Detector System (Roche Diagnostics, Bucharest, Romania). The mRNA levels were analysed with The PCR Array Data Analysis Software using the DDCt method.
ELISA
Three of the differentially expressed genes were randomly selected and their protein levels were measured by standard sandwich ELISA method. Cell media were collected at 24 and 48 hrs after treatment and quantified for the expression levels of interleukin 1β (IL-1β, DLB50; R&D Systems, Inc., Minneapolis, MN, USA), platelet-derived growth factor α (PDGFA) (R&D, MAB1739, BAF221) and transforming growth factor-β2 (TGF-β2; R&D, MAB612, BAT302) proteins according to the manufacturer instructions and ELISA protocol. For IL-1β quantification the media were concentrated five times under speed vacuum.
Proliferation assay
A total of 2 × 104 HeLa cells were treated with EGCG (10 μM final concentration) in a 96-well plate and incubated for 24 and 48 hrs, respectively. After incubation, the cells were treated with 0.1% 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and incubated 1 hr for dye incorporation. Blue formazans were eluted in Dimethyl sulfoxide (DMSO) and quantified with Tecan Sunrise (Tecan Group Ltd., Männedorf, Switzerland) plate reader.
Attachment assay
Twenty-four and –48 hours after treatment, HeLa cells were harvested with 0.25% trypsin-ethylenediaminetetraacetic acid, followed by centrifugation at 1000 rpm for 5 min. at room temperature (RT). The cell pellets were suspended in serum-free DMEM medium at a density of 2.5 × 105 cells/ml. A total of 100 μl of cell suspension (25,000 cells/well) were seeded in a 96-well plate which was pre-treated with 15 μg/ml type IV collagen (Sigma-Aldrich) or laminin (Sigma-Aldrich) overnight at 4°C, rinsed with PBS, followed by blocking with 0.2% bovine serum albumin (BSA) at 37°C for 45 min. Cells were allowed to adhere for 20–30 min. in a cell culture incubator and non-attached cells were removed by gently washing with PBS. Attached cells were fixed with 4% formaldehyde for 30 min. at RT, followed by staining in 0.1% (w/v) crystal violet for 30 min. Stained cells were lysed in 1% SDS and the intensity of stain, which is proportional to the number of adherent cells, was quantitated by using a microtitre plate reader at the absorbance of 570 nm.
Spreading assay
HeLa cells were harvested with 0.25% trypsin at 24 and 48 hrs after treatment, centrifuged at 1000 rpm for 5 min., and resuspended at a population density of 5 × 104 cells/ml in warm DMEM pre-equilibrated with 5% CO2. A total of 100 μl of cell suspension were plated onto a 96-well pre-treated plate coated with 15 μg/ml type IV collagen (Sigma-Aldrich) or laminin (Sigma-Aldrich) overnight at 4°C, washed with PBS and blocked with 0.2% BSA for 45 min. at 37°C. Cells were allowed to spread for 20 hrs at 37°C in a cell culture incubator. Cells were washed with PBS and fixed in 3% glutaraldehyde for 30 min. at RT. Cell images were taken by an Axiovert Zeiss (Carl Zeiss Instruments, Bucharest, Romania) microscope with 100× magnification. Spreading cells were defined as cells with extended processes, and non-spreading cells were defined as round cells. After image acquisition, the cells were stained for 30 min. with Giemsa solution, extensively washed and lysed in 1% SDS. The intensity of stain was quantified at 630 nm with a microtitre plate reader.
Adhesion to endothelium assay
Confluent monolayers of HUVEC cells were cultured on plastic cover slips which were pre-treated with 1% gelatine for 30 min. After 24 hrs of culture the medium was removed and 5 × 105 HeLa cells treated as previously described were added to each well and incubated for 50 min. at 37°C. After incubation the non-adherent HeLa cells were thoroughly washed with PBS–Alb, fixed in 4% paraformaldehyde for 15 min. at RT and stained with Giemsa solution for 5 min. The cover slips were washed with PBS, allowed to air dry and mounted on a microscope slide. Using light microscopy the number of cells adherent to the monolayer were counted in five fields of view per well selected at random.
In vitro invasion assay
The effect of EGCG treatment on the invasive properties of cervical cancer cells was determined using the matrigel transwell migration assay in co-culturing conditions. HeLa and HUVEC cells were trypsinized and resuspended in Opti-Mem and RPMI-1640 medium, respectively. Both cell types were treated with EGCG as described. A total of 1 × 105 HeLa cells were plated in the top chamber of the cell culture inserts (6.5 mm diameter insert, 3.0 μm pore size; Millipore, Milford, MA, USA) pre-treated with 1:10 diluted Matrigel BD (BD Biosciences, Franklin Lakes, NJ, USA) and 3 × 105 HUVEC cells were plated onto the bottom well of the chamber also pre-treated with matrigel. A total of 10% FBS was added in the bottom chamber as a chemoattractant. After incubation for 24 and 48 hrs, the cell inserts were removed from the plate and cells that did not migrate were mechanically removed with a cotton swab. Cells on the lower surface of the membrane were fixed in ice cold methanol; the membranes were cut from the housing insert and mounted on microscope slide with 4′,6-diamidino-2-phenylindole (DAPI) mounting medium. The migrated cells were examined using inverted phase fluorescence Zeiss Axiovert microscope and counted from six randomly selected fields in a blind way.
Statistical analysis
All experiments were performed in triplicates. All the data are presented as mean ± standard error of mean (S.E.M.). Differences were assessed by parametric and nonparametric tests according to data distribution using the statistical program SPSS 9.0 for Windows. P < 0.05 was considered to be statistically significant (*P < 0.05, **P < 0.01, ***P < 0.001).
Results
Effect of EGCG on the angiogenic potential of HeLa cells
The total RNA extracted from treated and untreated cells was profiled for 84 genes known to be involved in regulating the angiogenic process. The array includes growth factors and receptors, adhesion molecules, cytokines and chemokines, proteases and matrix proteins.
Five housekeeping genes (B2M, HPRT1, RPL13A, GAPDH and ACTB) were used for well to well normalization. Data analysis was done by DDCt method using the Superarray Data Analysis Web Portal. We have considered of interest all the genes with −1.5 ≤ fold regulation ≥ 1.5 in order to assess the genes involved in the early anti-angiogenic process. We found significant expression differences (P < 0.05) for 11 out of the 84 investigated genes. Four genes were up-regulated and seven down-regulated (Table 1).
Table 1.
Genes differentially expressed after 24 hrs of EGCG treatment versus control cells (UT)
| Gene | Fold regulation | P value |
|---|---|---|
| Angiopoietin-like 4 (ANGPTL4) | 2.22 | 0.0009 |
| Inhibitor of DNA binding 1 (ID1) | 1.98 | 0.001 |
| Interferon beta 1 (IFN-β1) | 2.49 | 0.01 |
| Interleukin 1 beta (IL-1β) | 1.77 | 0.0001 |
| Monocyte chemoattractant protein 1 (CCL2) | −2.117 | 0.0007 |
| Granulocyte chemotactic protein 2 (CXCL6) | −1.6958 | 0.001 |
| Ephrin A1 (EFNA1) | −1.7116 | 0.01 |
| Platelet-derived growth factor alpha (PDGFA) | −1.6958 | 0.003 |
| Transforming growth factor beta 2 (TGF-β2) | −1.6764 | 0.008 |
| Thrombospondin 1 (THBS-1) | −3.2013 | 0.0006 |
| Tumour necrosis factor alpha-induced protein 2 (TNFAIP2) | −1.5213 | 0.02 |
Three of these genes were validated at protein level by ELISA method. The results show the same expression pattern for the investigated proteins as their mRNA: we found down-regulated levels of PDGFA and TGF-β2 proteins and up-regulated levels of IL-1β (Fig. 1).
Fig 1.
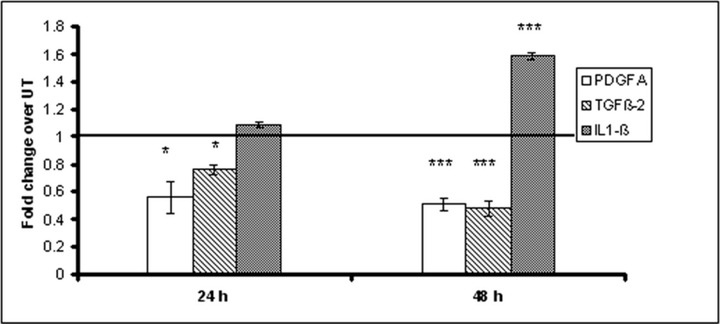
Fold change in protein expression upon EGCG treatment over untreated group. HeLa cells were incubated with 10 μM EGCG final concentration and the media were collected after 24 and 48 hrs and analysed by ELISA for PDGFA, TGF-β2 and IL-1β protein levels.
Effect of EGCG on HeLa cells proliferation
We examined the effect of EGCG on HeLa cells proliferation 24 and 48 hrs after treatment by MTT test. At both time-points, the cell proliferation was found to be significantly inhibited (Fig. 2). EGCG induced a 45% inhibition effect on the proliferation rate measured after 48 hrs of treatment compared to untreated cells; moreover, there was no significant difference between the number of cells evaluated at 24 hrs compared to cells treated for 48 hrs.
Fig 2.
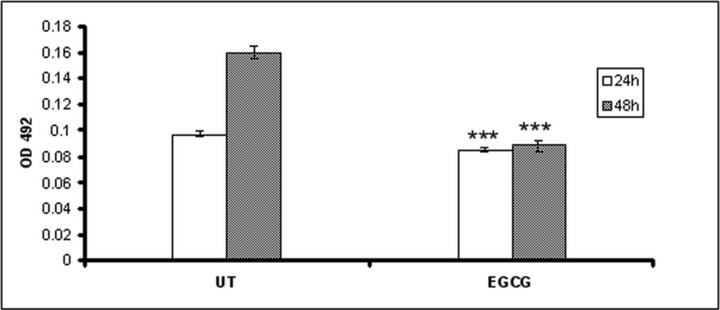
The effect of EGCG on HeLa cell proliferation. The HeLa cells were seeded in 96-well plates and treated with 10 μM EGCG final concentration in serum-free medium. After 24 and 48 hrs, the cells were treated with MTT and analysed for the number of live cells.
Effect of EGCG on cell attachment and spreading
We evaluated the influence of EGCG on cellular adhesion and motility on type IV collagen and laminin coated plates (Figs 3 and 5), two major components of ECM. Our results show (Fig. 3) that after 48 hrs of EGCG treatment the attachment of HeLa cells to type IV collagen was stimulated, but had no effect on the adhesion to laminin. There were no differences in cells attachment observed at 24 hrs after treatment (data not shown). In contrast, if the cells were allowed to attach for 20 hrs, the adhesion was dramatically inhibited (Fig. 4). Untreated cells presented multiple filopodia and lamellipodia, characteristic for spreading cells, whereas most of the EGCG treated cells maintained their round shape even after 20 hrs, indicating reduced spreading ability (Fig. 5).
Fig 3.
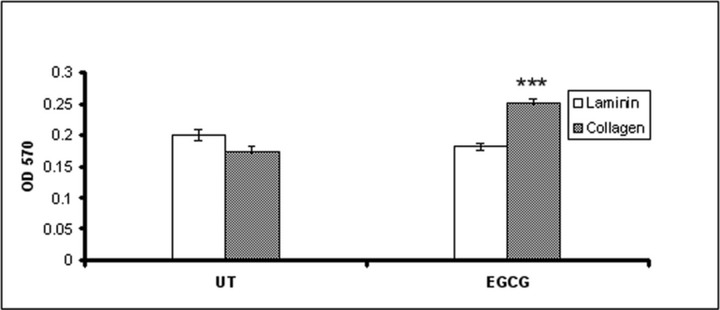
Adhesion of HeLa cells to collagen and laminin after EGCG treatment. Forty-eight hours after treatment, cells were harvested, resuspended and seeded in a 96-well plate which was pre-treated with 15 μg/ml type IV collagen or laminin. Cells were allowed to adhere for 20 min. and non-attached cells were removed by washing. Attached cells were fixed, stained with crystal violet, followed by measuring the absorbance at 570 nm.
Fig 5.
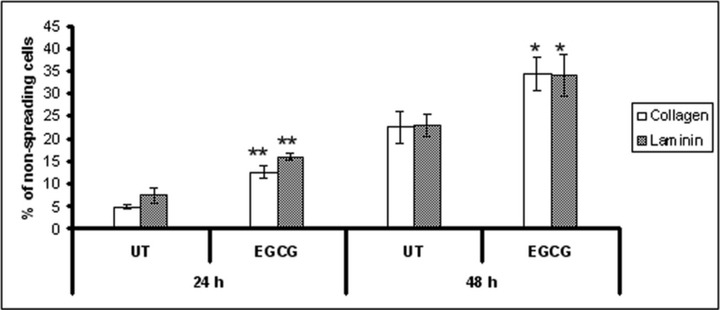
Effect of EGCG treatment on HeLa cells spreading. HeLa cells were treated with EGCG, and harvested at 24 and 48 hrs after treatment. Harvested cells were plated onto a 96-well plate which was coated with type IV collagen or laminin. Cells were allowed to spread for 20 hrs at 37°C. Spreading cells were defined as cells with extended processes, and un-spreading cells were defined as round cells. The cells were counted in nine different views.
Fig 4.
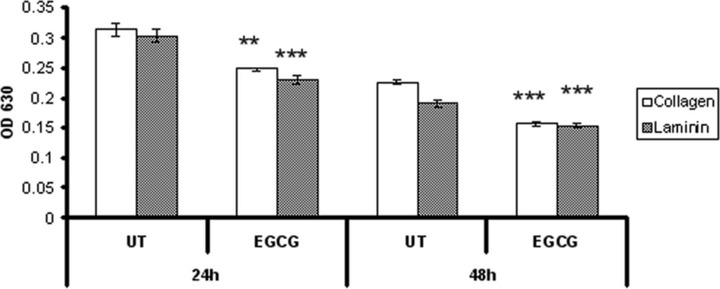
Adherent HeLa cells to collagen and laminin after 20 hrs of incubation. Twenty-four and forty-eight hours after treatment, cells were harvested, resuspended and seeded in a 96-well plate which was pre-treated with 15 μg/ml type IV collagen or laminin. Cells were allowed to adhere for 20 hrs and non-attached cells were removed by washing. Attached cells were fixed, stained with Giemsa solution, followed by measuring the absorbance at 630 nm.
Attachment to endothelium assay
In order to quantify the adhesion of the HeLa cells to endothelial HUVEC cells, HeLa cells were treated with EGCG and harvested at 24 and 48 hrs after treatment. The cells were seeded onto the HUVEC cell monolayers, and co-cultured for 50 min. After removing the non-adherent cells, the remaining adherent cells were stained and counted. As shown in Figure 6, the EGCG treatment reduced the adherence of HeLa cells to endothelial cells by 40%.
Inhibition of cell migration by EGCG
Migration towards a chemoattractant is a distinct cellular phenotype of metastatic tumour cells, and it is an essential step for tumour invasion and metastasis. Because cervical cancer is associated with more aggressive tumour phenotypes, we examined the effect of EGCG on the migration ability of HeLa cells using an in vitro migration assay, which simulates the in vivo metastatic process. As shown in Figure 7, there was a dramatic inhibition of HeLa cells’ migration ability after the treatment with EGCG. Compared with the untreated cells, EGCG caused an average of 48% and 68% reduction of migration ability after 24 and 48 hrs, respectively.
Discussions
Several studies describe the beneficial effects of green tea consumption, many of them pointing potential advantages compared to traditional cancer drugs like availability, and low toxicity to healthy cells. These observations led to the investigation of several compounds found in green tea in clinical trials. Currently, there are 51 ongoing clinical trials studying the effects of EGCG on different pathologies, including one on cervical cancer (information available at http://www.clinicaltrials.gov; accessed 12 March, 2011).
To date, the precise molecular mechanism of EGCG anticancer effects remain unclear [8]. In this study, we report the transcriptional mechanism that might modulate the anti-angiogenic and anti-metastatic effects of EGCG on HeLa cervical cells.
By profiling 84 genes involved in angiogenesis modulation we investigated the early molecular transcriptional mechanisms induced by EGCG treatment. The transcriptional analysis showed that EGCG treatment modulates the transcription of several genes involved in the angiogenic process (Table 1). These genes are known to mediate multiple mechanisms like endothelial cell proliferation (tumour necrosis factor α-induced protein 2 [TNFAIP2], ephrin A1 [EFNA1], PDGFA, chemotactic protein 2 [CXCL6], interferon β1 (IFN-β1), inhibitor of DNA binding 1 [ID1]), adhesion (thrombospondin 1 [THBS-1], CXCL6), migration (EFNA1, THBS-1, TGF-β2, chemoattractant protein 1 [CCL2]) and invasion (angiopoietin-like 4 [ANGPTL4], THBS-1, TGF-β2, IL-1β, ID1). The literature analysis relating the genes of interest showed that most of them act synergistically, on both vascular and tumour compartment. On the premises that every gene expression is associated with a biological effect, we further validated our findings by measuring the effects of EGCG treatment on tumour cell proliferation, adhesion, spreading and invasiveness.
The anti-proliferative effect of EGCG has been reported by several groups [13-17]. Cell proliferation and growth is promoted through various signalling pathways and EGCG seems to interfere with these pathways and lead to decreased proliferation rates in cervical cancer cells (Fig. 2).
PDGFA is a growth factor that upon binding to their receptors activates a variety of intracellular signalling molecules that control cell growth, proliferation and differentiation, leading to cancer progression. PDGF isoforms act mainly through a paracrine mechanism, their expression promoting the establishment of a well-vascularized and prominent stroma, which in turn provides growth factors and anti-apoptotic signals for tumour cells, support for tumour angiogenesis and facilitate invasion [19, 20]. Pietras et al. [21] have recently showed in a mouse model of cervical carcinoma that PDGFA stimulates the stromal components and impairment of the PDGF/PDGFR signalling with imatinib affected the angiogenic phenotype of both premalignant cervical neoplasias and invasive carcinomas; the treated lesions exhibited diminished blood vessel density and reduced pericyte coverage.
A considerable number of solid tumours [22, 23] including cervical cancer [24] have been showed to express PDGF receptors which suggest that these molecules also act by an autocrine mechanism leading to increased proliferation rates. We found that EGCG down-regulates the expression of both mRNA and protein levels of PDGFA and this could be one of the mechanisms by which EGCG decreases the proliferation rate as well as the angiogenic potential of cervical cancer cells.
IFN-β1 antitumour activity has been intensively investigated in human cancers. Studies have shown that its expression leads to immune stimulation [25, 26], tumour regression [27, 28] and angiogenesis inhibition by down-regulation of proangiogenic factors like bFGF in vivo [29-31] and blocking endothelial cell migration in vitro. Vannucchi et al. [32] reported that IFN-β1 induces a direct anti-proliferative effect on cervical tumour cells by inhibiting the cell cycle progression. This report seems to be in concordance with our study. We observed that the number of cells after 24 and 48 hrs of treatment remained the same and we assume that EGCG treatment decreases the proliferation rate by stopping the cell cycle progression through induced IFN-β1 expression.
ID genes are regulators of transcription being responsible for changes in gene expression that lead to increased growth, invasion and metastasis in several human carcinomas [33]. The precise role of ID1 in cervical cancer is little investigated; however, several studies correlated increased expression of this protein with higher tumour cell aggressiveness, poor prognosis and survival in early stages [34, 35]. Lyden et al. showed that ID proteins are required for the proliferative and invasive phenotype of endothelial cells during tumour-associated angiogenesis [33]; however, there was no association between ID1 expression and angiogenesis in cervical cancer, which suggest that ID1 expression on prognosis cannot be attributed to its proangiogenic effect alone [34]. Another study reported that ID genes are strongly responsive to signalling pathways coupled to growth factors [36]. ID proteins enhance cell cycle progression and their overexpression induces apoptosis in serum-deprived fibroblasts [37]. The exact mechanism of action of EGCG through ID1 gene up-regulation in HeLa cervical cells is still to be determined, ID1 overexpression might be induced as a resistance mechanism against EGCG treatment in order to progress the cell cycle or the serum withdrawal coupled with ID1 expression might lead to cell apoptosis as reported for fibroblasts.
IL-1β seems to engage different cellular signalling pathways depending on the cell type in a dose-dependent manner, leading to genotoxic damage, cell apoptosis or cell growth [38]. Breast cancer cells under genotoxic stress secreting moderate levels of IL-1β stimulated clonal expansion, whereas high levels of IL-1β engaged the apoptotic pathway. In angiogenesis, TNF-α and IL-1β induce increased TNFAIP2 mRNA levels [39, 40] during capillary tube formation in vitro [39]. In small cell lung cancer IL-1β up-regulates CXCL6 production and increases tumour cell proliferation [41]. CXCL6 overexpression has been associated with increased metastatic phenotype in small cell lung [41] and prostate [42] cancers. In prostate cancer, CXCL6 overexpression was also associated with increased cell adhesion to ECM and endothelium [42]. Roomi et al. reported recently that EGCG and IL-1β stimulation on HeLa cells leads to decreased matrix metalloproteinase 2 (MMP-2) secretion [43], a crucial matrix metaloprotease in cell invasion. Our results show that EGCG treatment seems to stimulate the transcription of IL-1β and decrease TNFAIP2 and CXCL6 levels.
In addition to decreased proliferation rate, EGCG treatment seems to also modulate the adhesion capability of HeLa cells to ECM and endothelium. The attachment test to ECM showed no effect on the rapid adhesion of HeLa cells to laminin at either time-points, but showed increased adhesion to type IV collagen after 48 hrs of treatment (Fig. 3). Cell motility is the key step in organ invasion by tumour cells and increased adherence to the ECM is the first process that indicates increased spreading ability and subsequently metastatic potential of a tumour cell. However, if the time of incubation was extended, the cells lost both their adherence as well as spreading potential (Fig. 4). We observed significant morphology change of the cervical cancer cells after the treatment with EGCG, indicating reduced motility (Fig. 5). These observations might be explained by the fact that even if cells establish initial focal complexes with the ECM, they fail to mature into stable focal adhesions, a process that seems to be impaired by the EGCG treatment. Moreover, we further investigated HeLa cells adherence properties to an endothelial cell monolayer. Re-establishment of adhesive connections to endothelium after entering the bloodstream is a key step in tumour metastasis [44]. Our results show that EGCG treatment reduces the adherence of HeLa cells to endothelial cells (Fig. 6).
Fig 6.
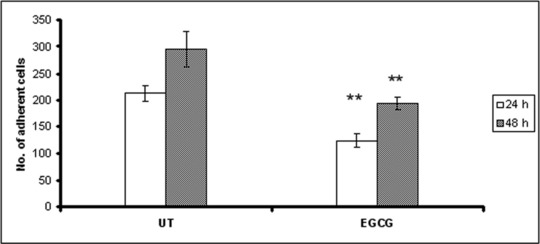
Adhesion of HeLa cells to endothelial cells monolayer after EGCG treatment. Twenty-four and 48 hours after treatment, cells were harvested, resuspended and seeded onto cover slips containing a HUVEC monolayer. Cells were allowed to adhere for 50 min. and non-attached cells were removed by washing. Attached cells were fixed and stained with Giemsa solution, followed by image acquisition and cell counting. Number of adherent HeLa cells were counted in five random views per well.
Adherence of tumour cells to ECM and endothelium is mainly mediated through integrin pathways. THBS-1 has been shown to be involved in modulating these pathways and affect tumour cell adhesion [45], migration and invasion [46]. THBS-1 has been proposed to have both pro- and anti-metastatic properties [46, 47] by regulating epithelial cell growth, motility [46], stromal/epithelial interactions and angiogenesis. THBS-1 has been shown to be a potent inhibitor of angiogenesis by multiple mechanisms, including direct interaction with vascular endothelial growth factor (VEGF) and inhibition of MMP-9 activation [47, 48]. However, recent studies reported different biological action in tumour progression and metastasis [46, 47]. Overall, the effects of THBS-1 on any given tumour’s metastatic potential probably depends on genetic and/or epigenetic changes in the tumour cells themselves, as well as its anti-angiogenic effects on endothelial cells [49].
The TGF-β pathway is known to be one of THBS-1’s targets. High levels of TGF-β have been associated with various tumours of epithelial origin [50, 51] including cervical tumours [52, 53]. Many of these tumours show increased invasiveness [54], particularly those that overexpress TGF-β2 [55] and are correlated with CIN progression [56-58], higher metastatic phenotype and/or poor patient outcome [52].
Although generally known for its anti-proliferative effects, TGF-β is a well-characterized inducer of epithelial–mesenchymal transformation in tumour cells, resulting in enhanced cell migration and invasion. TGF-β controls the transcription of numerous integrins and although the down-regulation of integrin expression has been reported, in most cases TGF-β stimulates integrin expression in several cell types and tissues, as well as in various human cancers (rev. in [59]). We assume that EGCG treatment down-regulates the THBS-1 expression (fold regulation –3.2), which in turn down-regulates its downstream target, TGF-β2 (fold regulation –1.67) and subsequently integrin secretion leading to decreased tumour cell adhesion, motility and invasion (Fig. 7).
Fig 7.
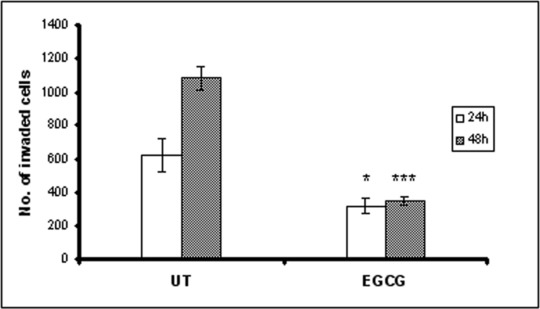
Inhibition of HeLa cells migration by EGCG. HeLa cells were trypsinized and plated in the top chamber of the transwell. HUVEC cells cultured in RPMI-1640 medium with 10% FBS was added in the lower chamber as a chemoattractant. After incubation for 24 and 48 hrs, cells that did not migrate through the pores were mechanically removed and cells on the lower surface of the membrane were fixed and stained. The number of migrated cells was counted from six randomly selected fields per well in a blind way.
It is well known that VEGF can regulate the proliferation and migration of both endothelial and tumour cells. EGCG has been shown to induce VEGF down-regulation in different cancers [60-63], including in HeLa cervical cancer cells [18]. Although we did not find decreased VEGF mRNA levels upon EGCG treatment (probably due to the different experimental conditions) we found decreased levels of EFNA1. EFNA1 is a prototypic ligand which bounds EphA2 receptor tyrosine kinase and triggers downstream signals that regulate cell growth [64, 65] and migration [66, 67]. EFNA1 and its receptor are known to be involved in the VEGF pathway by up-regulating VEGF expression in tumour cells and subsequently activate endothelial cells [68]. It seems that EGCG treatment down-regulates EFNA1 expression which might lead to VEGF inhibition as reported by Zhang et al. [18]. Concomitant increased expression of VEGF with EFNA1 was observed in patients with cervical cancer; following radiation therapy [69] and blocking EFNA1 signalling cascade decreased tumour-associated angiogenesis and consequently, tumour progression [70, 71].
Recently, Galaup et al. [72] showed that ANGPTL4 has an important role in the metastatic process; increased levels of ANGPTL4 inhibited both vascular activity and tumour cell motility and invasiveness. ANGPTL4’s role in angiogenesis is controversial suggesting a tissue specific activity, being induced in both endothelial cells [73, 74] and tumour cells [75]. Depending on the context, ANGPTL4 has been reported to exert complex biological function, acting as a proangiogenic factor [74, 76] by mediating endothelial cell survival [77] or having anti-angiogenic properties [78] by modulating endothelial cell adhesion [79]. It seems that in HeLa cells the ANGPTL4 up-regulation by EGCG treatment induce an anti-angiogenic and anti-metastatic effect.
EGCG treatment seems to mediate the angiogenic phenotype in HeLa cervical cells also through CCL2 inhibition. In human cervical cancer, CCL2 is abundantly expressed in the stroma and associated with macrophage infiltration [80]. CCL2 induces the migration and activation of monocytes, and it has been implicated in the recruitment of tumour-infiltrating macrophages (TIMs) in several tumour types [81, 82]. Previous studies showed that TIMs are a major source of MMP-9 [83] as well as a broad range of tumourigenic factors [84] promoting angiogenesis, tumour progression and metastasis. Zijlmans et al. [85] showed that cervical tumours that do not express CCL2 are characterized by a decreased number of TIMs, smaller tumour size and reduced vascular invasion as well as increased overall survival.
Conclusions
In this study, we explored the early transcriptional pattern of angiogenesis induced by EGCG treatment in cervical cancer cells. Our data show that EGCG treatment modulates the angiogenic pathways by interfering with the transcription pattern of several genes that act both on endothelial cells and tumour cells. EGCG treatment decreases the tumour cell proliferation rate, inhibits cell adhesion, motility and invasion potential of HeLa cells. Our results show that EGCG could be used as an anti-angiogenic agent in cervical adenocarcinomas treatment [86].
Acknowledgments
The authors acknowledge the financial support provided from programs co-financed by The Sectoral Operational Programme Human Resources Development, Contract POSDRU 6/1.5/S/3 – ‘Doctoral studies: through science towards society’ and National Grants Program – PNCDI2 (grant no. 42160/2008). We are grateful to Assoc. Prof. Marina Nechifor from the University of Bucharest, Romania for providing the HeLa cells.
Oana Tudoran designed the research study, and wrote the paper, Olga Soritau and Piroska Virag performed the cell cultures, Oana Tudoran and Cornelia Braicu performed the research, Loredana Balacescu, Meda Rus and Claudia Gherman analysed the data, Ovidiu Balacescu, Florin Irimie and Ioana Berindan-Neagoe thoroughly discussed the results and revised the paper.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Garcia M, Jemal A, Ward EM, et al. Global cancer facts & figures 2007. Atlanta, GA: American Cancer Society; 2007. [Google Scholar]
- 2.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–49. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 3.Monk BJ, Herzog TJ. The evolution of cost-effective screening and prevention of cervical carcinoma: implications of the 2006 consensus guidelines and human papillomavirus vaccination. Am J Obstet Gynecol. 2007;197:337–9. doi: 10.1016/j.ajog.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 4.Yang CS, Lambert JD, Hou Z, et al. Molecular targets for the cancer preventive activity of tea polyphenols. Mol Carcinog. 2006;45:431–5. doi: 10.1002/mc.20228. [DOI] [PubMed] [Google Scholar]
- 5.Yang CS, Lambert JD, Ju J, et al. Tea and cancer prevention: molecular mechanisms and human relevance. Toxicol Appl Pharmacol. 2007;224:265–73. doi: 10.1016/j.taap.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Afaq F, Adhami VM, Ahmad N, et al. Inhibition of ultraviolet B-mediated activation of nuclear factor kappaB in normal human epidermal keratinocytes by green tea constituent (-)-epigallocatechin-3-gallate. Oncogene. 2003;22:1035–44. doi: 10.1038/sj.onc.1206206. [DOI] [PubMed] [Google Scholar]
- 7.Lambert JD, Yang CS. Mechanisms of cancer prevention by tea constituents. J Nutr. 2003;133:3262S–7S. doi: 10.1093/jn/133.10.3262S. [DOI] [PubMed] [Google Scholar]
- 8.Li WG, Li QH, Tan Z. Epigallocatechin gallate induces telomere fragmentation in HeLa and 293 but not in MRC-5 cells. Life Sci. 2005;76:1735–46. doi: 10.1016/j.lfs.2004.09.024. [DOI] [PubMed] [Google Scholar]
- 9.Jung YD, Ellis LM. Inhibition of tumour invasion and angiogenesis by epigallocatechin gallate (EGCG), a major component of green tea. Int J Exp Pathol. 2001;82:309–16. doi: 10.1046/j.1365-2613.2001.00205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khan N, Afaq F, Saleem M, et al. Targeting multiple signaling pathways by green tea polyphenol (-)-epigallocatechin-3-gallate. Cancer Res. 2006;66:2500–5. doi: 10.1158/0008-5472.CAN-05-3636. [DOI] [PubMed] [Google Scholar]
- 11.Fassina G, Vene R, Morini M, et al. Mechanisms of inhibition of tumour angiogenesis and vascular tumour growth by epigallocatechin-3-gallate. Clin Cancer Res. 2004;10:4865–73. doi: 10.1158/1078-0432.CCR-03-0672. [DOI] [PubMed] [Google Scholar]
- 12.Willmott LJ, Monk BJ. Cervical cancer therapy: current, future and anti-angiogensis targeted treatment. Expert Rev Anticancer Ther. 2009;9:895–903. doi: 10.1586/era.09.58. [DOI] [PubMed] [Google Scholar]
- 13.Qiao Y, Cao J, Xie L, et al. Cell growth inhibition and gene expression regulation by (-)-epigallocatechin-3-gallate in human cervical cancer cells. Arch Pharm Res. 2009;32:1309–15. doi: 10.1007/s12272-009-1917-3. [DOI] [PubMed] [Google Scholar]
- 14.Yokoyama M, Noguchi M, Nakao Y, et al. Antiproliferative effects of the major tea polyphenol, (-)-epigallocatechin gallate and retinoic acid in cervical adenocarcinoma. Gynecol Oncol. 2008;108:326–31. doi: 10.1016/j.ygyno.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 15.Yokoyama M, Noguchi M, Nakao Y, et al. The tea polyphenol, (-)-epigallocatechin gallate effects on growth, apoptosis, and telomerase activity in cervical cell lines. Gynecol Oncol. 2004;92:197–204. doi: 10.1016/j.ygyno.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 16.Noguchi M, Yokoyama M, Watanabe S, et al. Inhibitory effect of the tea polyphenol, (-)-epigallocatechin gallate, on growth of cervical adenocarcinoma cell lines. Cancer Lett. 2006;234:135–42. doi: 10.1016/j.canlet.2005.03.053. [DOI] [PubMed] [Google Scholar]
- 17.Ahn WS, Huh SW, Bae SM, et al. A major constituent of green tea, EGCG, inhibits the growth of a human cervical cancer cell line, CaSki cells, through apoptosis, G(1) arrest, and regulation of gene expression. DNA Cell Biol. 2003;22:217–24. doi: 10.1089/104454903321655846. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Q, Tang X, Lu Q, et al. Green tea extract and (-)-epigallocatechin-3-gallate inhibit hypoxia- and serum-induced HIF-1alpha protein accumulation and VEGF expression in human cervical carcinoma and hepatoma cells. Mol Cancer Ther. 2006;5:1227–38. doi: 10.1158/1535-7163.MCT-05-0490. [DOI] [PubMed] [Google Scholar]
- 19.Joyce JA. Therapeutic targeting of the tumour microenvironment. Cancer Cell. 2005;7:513–20. doi: 10.1016/j.ccr.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 20.Park CC, Bissell MJ, Barcellos-Hoff MH. The influence of the microenvironment on the malignant phenotype. Mol Med Today. 2000;6:324–9. doi: 10.1016/s1357-4310(00)01756-1. [DOI] [PubMed] [Google Scholar]
- 21.Pietras K, Pahler J, Bergers G, et al. Functions of paracrine PDGF signaling in the proangiogenic tumour stroma revealed by pharmacological targeting. PLoS Med. 2008;5:e19. doi: 10.1371/journal.pmed.0050019. , doi: 10.1371/journal.pmed.0050019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pietras K, Sjoblom T, Rubin K, et al. PDGF receptors as cancer drug targets. Cancer Cell. 2003;3:439–43. doi: 10.1016/s1535-6108(03)00089-8. [DOI] [PubMed] [Google Scholar]
- 23.Board R, Jayson GC. Platelet-derived growth factor receptor (PDGFR): a target for anticancer therapeutics. Drug Resist Updat. 2005;8:75–83. doi: 10.1016/j.drup.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 24.Taja-Chayeb L, Chavez-Blanco A, Martinez-Tlahuel J, et al. Expression of platelet derived growth factor family members and the potential role of imatinib mesylate for cervical cancer. Cancer Cell Int. 2006;6:22. doi: 10.1186/1475-2867-6-22. , doi: 10.1186/1476-4598-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tough DF, Borrow P, Sprent J. Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science. 1996;272:1947–50. doi: 10.1126/science.272.5270.1947. [DOI] [PubMed] [Google Scholar]
- 26.Rogge L, Barberis-Maino L, Biffi M, et al. Selective expression of an interleukin-12 receptor component by human T helper 1 cells. J Exp Med. 1997;185:825–31. doi: 10.1084/jem.185.5.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaido T, Bandu MT, Maury C, et al. IFN-alpha 1 gene transfection completely abolishes the tumourigenicity of murine B16 melanoma cells in allogeneic DBA/2 mice and decreases their tumourigenicity in syngeneic C57BL/6 mice. Int J Cancer. 1995;60:221–9. doi: 10.1002/ijc.2910600216. [DOI] [PubMed] [Google Scholar]
- 28.Sarkar S, Flores I, Ron Y, et al. Injection of Irradiated B16 Melanoma Genetically-Modified to Secrete Ifn-Alpha Causes Regression of an Established tumour. Int J Oncol. 1995;7:17–24. doi: 10.3892/ijo.7.1.17. [DOI] [PubMed] [Google Scholar]
- 29.Sidky YA, Borden EC. Inhibition of angiogenesis by interferons: effects on tumour- and lymphocyte-induced vascular responses. Cancer Res. 1987;47:5155–61. [PubMed] [Google Scholar]
- 30.Dinney CP, Bielenberg DR, Perrotte P, et al. Inhibition of basic fibroblast growth factor expression, angiogenesis, and growth of human bladder carcinoma in mice by systemic interferon-alpha administration. Cancer Res. 1998;58:808–14. [PubMed] [Google Scholar]
- 31.Dong Z, Greene G, Pettaway C, et al. Suppression of angiogenesis, tumourigenicity, and metastasis by human prostate cancer cells engineered to produce interferon-beta. Cancer Res. 1999;59:872–9. [PubMed] [Google Scholar]
- 32.Vannucchi S, Percario ZA, Chiantore MV, et al. Interferon-beta induces S phase slowing via up-regulated expression of PML in squamous carcinoma cells. Oncogene. 2000;19:5041–53. doi: 10.1038/sj.onc.1203883. [DOI] [PubMed] [Google Scholar]
- 33.Lyden D, Young AZ, Zagzag D, et al. Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumour xenografts. Nature. 1999;401:670–7. doi: 10.1038/44334. [DOI] [PubMed] [Google Scholar]
- 34.Schindl M, Oberhuber G, Obermair A, et al. Overexpression of Id-1 protein is a marker for unfavorable prognosis in early-stage cervical cancer. Cancer Res. 2001;61:5703–6. [PubMed] [Google Scholar]
- 35.Darnel AD, Wang D, Ghabreau L, et al. Correlation between the presence of high-risk human papillomaviruses and Id gene expression in Syrian women with cervical cancer. Clin Microbiol Infect. 2010;16:262–6. doi: 10.1111/j.1469-0691.2009.02774.x. [DOI] [PubMed] [Google Scholar]
- 36.Christy BA, Sanders LK, Lau LF, et al. An Id-related helix-loop-helix protein encoded by a growth factor-inducible gene. Proc Natl Acad Sci USA. 1991;88:1815–9. doi: 10.1073/pnas.88.5.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Norton JD, Atherton GT. Coupling of cell growth control and apoptosis functions of Id proteins. Mol Cell Biol. 1998;18:2371–81. doi: 10.1128/mcb.18.4.2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roy D, Sarkar S, Felty Q. Levels of IL-1 beta control stimulatory/inhibitory growth of cancer cells. Front Biosci. 2006;11:889–98. doi: 10.2741/1845. [DOI] [PubMed] [Google Scholar]
- 39.Sarma V, Wolf FW, Marks RM, et al. Cloning of a novel tumour necrosis factor-alpha-inducible primary response gene that is differentially expressed in development and capillary tube-like formation in vitro. J Immunol. 1992;148:3302–12. [PubMed] [Google Scholar]
- 40.Wolf FW, Sarma V, Seldin M, et al. B94, a primary response gene inducible by tumour necrosis factor-alpha, is expressed in developing hematopoietic tissues and the sperm acrosome. J Biol Chem. 1994;269:3633–40. [PubMed] [Google Scholar]
- 41.Zhu YM, Bagstaff SM, Woll PJ. Production and upregulation of granulocyte chemotactic protein-2/CXCL6 by IL-1beta and hypoxia in small cell lung cancer. Br J Cancer. 2006;94:1936–41. doi: 10.1038/sj.bjc.6603177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Engl T, Relja B, Blumenberg C, et al. Prostate tumour CXC-chemokine profile correlates with cell adhesion to endothelium and extracellular matrix. Life Sci. 2006;78:1784–93. doi: 10.1016/j.lfs.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 43.Roomi MW, Monterrey JC, Kalinovsky T, et al. In vitro modulation of MMP-2 and MMP-9 in human cervical and ovarian cancer cell lines by cytokines, inducers and inhibitors. Oncol Rep. 2010;23:605–14. doi: 10.3892/or_00000675. [DOI] [PubMed] [Google Scholar]
- 44.Eccles SA, Welch DR. Metastasis: recent discoveries and novel treatment strategies. Lancet. 2007;369:1742–57. doi: 10.1016/S0140-6736(07)60781-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilson KE, Li Z, Kara M, et al. Beta 1 integrin- and proteoglycan-mediated stimulation of T lymphoma cell adhesion and mitogen-activated protein kinase signaling by thrombospondin-1 and thrombospondin-1 peptides. J Immunol. 1999;163:3621–8. [PubMed] [Google Scholar]
- 46.Yee KO, Connolly CM, Duquette M, et al. The effect of thrombospondin-1 on breast cancer metastasis. Breast Cancer Res Treat. 2009;114:85–96. doi: 10.1007/s10549-008-9992-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kazerounian S, Yee KO, Lawler J. Thrombospondins in cancer. Cell Mol Life Sci. 2008;65:700–12. doi: 10.1007/s00018-007-7486-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Folkman J. The role of angiogenesis in tumour growth. Semin Cancer Biol. 1992;3:65–71. [PubMed] [Google Scholar]
- 49.Nucera C, Porrello A, Antonello ZA, et al. B-Raf(V600E) and thrombospondin-1 promote thyroid cancer progression. Proc Natl Acad Sci USA. 2010;107:10649–54. doi: 10.1073/pnas.1004934107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Derynck R, Jarrett JA, Chen EY, et al. Human transforming growth factor-beta complementary DNA sequence and expression in normal and transformed cells. Nature. 1985;316:701–5. doi: 10.1038/316701a0. [DOI] [PubMed] [Google Scholar]
- 51.Pasche B. Role of transforming growth factor beta in cancer. J Cell Physiol. 2001;186:153–68. doi: 10.1002/1097-4652(200002)186:2<153::AID-JCP1016>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 52.Hazelbag S, Kenter GG, Gorter A, et al. Overexpression of the alpha v beta 6 integrin in cervical squamous cell carcinoma is a prognostic factor for decreased survival. J Pathol. 2007;212:316–24. doi: 10.1002/path.2168. [DOI] [PubMed] [Google Scholar]
- 53.Diaz-Chavez J, Hernandez-Pando R, Lambert PF, et al. Down-regulation of transforming growth factor-beta type II receptor (TGF-betaRII) protein and mRNA expression in cervical cancer. Mol Cancer. 2008;7:3. doi: 10.1186/1476-4598-7-3. , doi: 10.1186/1476-4598-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Welch DR, Fabra A, Nakajima M. Transforming growth factor beta stimulates mammary adenocarcinoma cell invasion and metastatic potential. Proc Natl Acad Sci USA. 1990;87:7678–82. doi: 10.1073/pnas.87.19.7678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gold LI, Jussila T, Fusenig NE, et al. TGF-beta isoforms are differentially expressed in increasing malignant grades of HaCaT keratinocytes, suggesting separate roles in skin carcinogenesis. J Pathol. 2000;190:579–88. doi: 10.1002/(SICI)1096-9896(200004)190:5<579::AID-PATH548>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 56.Baritaki S, Sifakis S, Huerta-Yepez S, et al. Overexpression of VEGF and TGF-beta1 mRNA in Pap smears correlates with progression of cervical intraepithelial neoplasia to cancer: implication of YY1 in cervical tumourigenesis and HPV infection. Int J Oncol. 2007;31:69–79. [PubMed] [Google Scholar]
- 57.Soufla G, Sifakis S, Baritaki S, et al. VEGF, FGF2, TGFB1 and TGFBR1 mRNA expression levels correlate with the malignant transformation of the uterine cervix. Cancer Lett. 2005;221:105–18. doi: 10.1016/j.canlet.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 58.Tervahauta A, Syrjanen S, Yliskoski M, et al. Expression of transforming growth factor-beta 1 and -beta 2 in human papillomavirus (HPV)-associated lesions of the uterine cervix. Gynecol Oncol. 1994;54:349–56. doi: 10.1006/gyno.1994.1222. [DOI] [PubMed] [Google Scholar]
- 59.Margadant C, Sonnenberg A. Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 2010;11:97–105. doi: 10.1038/embor.2009.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhu BH, Zhan WH, Li ZR, et al. (-)-Epigallocatechin-3-gallate inhibits growth of gastric cancer by reducing VEGF production and angiogenesis. World J Gastroenterol. 2007;13:1162–9. doi: 10.3748/wjg.v13.i8.1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rodriguez SK, Guo W, Liu L, et al. Green tea catechin, epigallocatechin-3-gallate, inhibits vascular endothelial growth factor angiogenic signaling by disrupting the formation of a receptor complex. Int J Cancer. 2006;118:1635–44. doi: 10.1002/ijc.21545. [DOI] [PubMed] [Google Scholar]
- 62.Shimizu M, Shirakami Y, Sakai H, et al. (-)-Epigallocatechin gallate inhibits growth and activation of the VEGF/VEGFR axis in human colorectal cancer cells. Chem Biol Interact. 2010;185:247–52. doi: 10.1016/j.cbi.2010.03.036. [DOI] [PubMed] [Google Scholar]
- 63.Dann JM, Sykes PH, Mason DR, et al. Regulation of vascular endothelial growth factor in endometrial tumour cells by resveratrol and EGCG. Gynecol Oncol. 2009;113:374–8. doi: 10.1016/j.ygyno.2009.02.014. [DOI] [PubMed] [Google Scholar]
- 64.Easty DJ, Guthrie BA, Maung K, et al. Protein B61 as a new growth factor: expression of B61 and up-regulation of its receptor epithelial cell kinase during melanoma progression. Cancer Res. 1995;55:2528–32. [PubMed] [Google Scholar]
- 65.Potla L, Boghaert ER, Armellino D, et al. Reduced expression of EphrinA1 (EFNA1) inhibits three-dimensional growth of HT29 colon carcinoma cells. Cancer Lett. 2002;175:187–95. doi: 10.1016/s0304-3835(01)00613-9. [DOI] [PubMed] [Google Scholar]
- 66.Pratt RL, Kinch MS. Activation of the EphA2 tyrosine kinase stimulates the MAP/ERK kinase signaling cascade. Oncogene. 2002;21:7690–9. doi: 10.1038/sj.onc.1205758. [DOI] [PubMed] [Google Scholar]
- 67.Walker-Daniels J, Riese DJ, 2nd, Kinch MS. c-Cbl-dependent EphA2 protein degradation is induced by ligand binding. Mol Cancer Res. 2002;1:79–87. [PubMed] [Google Scholar]
- 68.Brantley-Sieders DM, Fang WB, Hwang Y, et al. Ephrin-A1 facilitates mammary tumour metastasis through an angiogenesis-dependent mechanism mediated by EphA receptor and vascular endothelial growth factor in mice. Cancer Res. 2006;66:10315–24. doi: 10.1158/0008-5472.CAN-06-1560. [DOI] [PubMed] [Google Scholar]
- 69.Nojiri K, Iwakawa M, Ichikawa Y, et al. The proangiogenic factor ephrin-A1 is up-regulated in radioresistant murine tumour by irradiation. Exp Biol Med. 2009;234:112–22. doi: 10.3181/0806-RM-189. [DOI] [PubMed] [Google Scholar]
- 70.Brantley DM, Cheng N, Thompson EJ, et al. Soluble Eph A receptors inhibit tumour angiogenesis and progression in vivo. Oncogene. 2002;21:7011–26. doi: 10.1038/sj.onc.1205679. [DOI] [PubMed] [Google Scholar]
- 71.Dobrzanski P, Hunter K, Jones-Bolin S, et al. Antiangiogenic and antitumour efficacy of EphA2 receptor antagonist. Cancer Res. 2004;64:910–9. doi: 10.1158/0008-5472.can-3430-2. [DOI] [PubMed] [Google Scholar]
- 72.Galaup A, Cazes A, Le Jan S, et al. Angiopoietin-like 4 prevents metastasis through inhibition of vascular permeability and tumour cell motility and invasiveness. Proc Natl Acad Sci USA. 2006;103:18721–6. doi: 10.1073/pnas.0609025103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Le Jan S, Le Meur N, Cazes A, et al. Characterization of the expression of the hypoxia-induced genes neuritin, TXNIP and IGFBP3 in cancer. FEBS Lett. 2006;580:3395–400. doi: 10.1016/j.febslet.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 74.Le Jan S, Amy C, Cazes A, et al. Angiopoietin-like 4 is a proangiogenic factor produced during ischemia and in conventional renal cell carcinoma. Am J Pathol. 2003;162:1521–8. doi: 10.1016/S0002-9440(10)64285-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lal A, Peters H, St Croix B, et al. Transcriptional response to hypoxia in human tumours. J Natl Cancer Inst. 2001;93:1337–43. doi: 10.1093/jnci/93.17.1337. [DOI] [PubMed] [Google Scholar]
- 76.Hermann LM, Pinkerton M, Jennings K, et al. Angiopoietin-like-4 is a potential angiogenic mediator in arthritis. Clin Immunol. 2005;115:93–101. doi: 10.1016/j.clim.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 77.Kim I, Kim HG, Kim H, et al. Hepatic expression, synthesis and secretion of a novel fibrinogen/angiopoietin-related protein that prevents endothelial-cell apoptosis. Biochem J. 2000;346:603–10. [PMC free article] [PubMed] [Google Scholar]
- 78.Ito Y, Oike Y, Yasunaga K, et al. Inhibition of angiogenesis and vascular leakiness by angiopoietin-related protein 4. Cancer Res. 2003;63:6651–7. [PubMed] [Google Scholar]
- 79.Cazes A, Galaup A, Chomel C, et al. Extracellular matrix-bound angiopoietin-like 4 inhibits endothelial cell adhesion, migration, and sprouting and alters actin cytoskeleton. Circ Res. 2006;99:1207–15. doi: 10.1161/01.RES.0000250758.63358.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kleine-Lowinski K, Gillitzer R, Kuhne-Heid R, et al. Monocyte-chemo-attractant-protein-1 (MCP-1)-gene expression in cervical intra-epithelial neoplasias and cervical carcinomas. Int J Cancer. 1999;82:6–11. doi: 10.1002/(sici)1097-0215(19990702)82:1<6::aid-ijc2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 81.Koide N, Nishio A, Sato T, et al. Significance of macrophage chemoattractant protein-1 expression and macrophage infiltration in squamous cell carcinoma of the esophagus. Am J Gastroenterol. 2004;99:1667–74. doi: 10.1111/j.1572-0241.2004.30733.x. [DOI] [PubMed] [Google Scholar]
- 82.Ueno T, Toi M, Saji H, et al. Significance of macrophage chemoattractant protein-1 in macrophage recruitment, angiogenesis, and survival in human breast cancer. Clin Cancer Res. 2000;6:3282–9. [PubMed] [Google Scholar]
- 83.Giraudo E, Inoue M, Hanahan D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J Clin Invest. 2004;114:623–33. doi: 10.1172/JCI22087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lewis CE, Pollard JW. Distinct role of macrophages in different tumour microenvironments. Cancer Res. 2006;66:605–12. doi: 10.1158/0008-5472.CAN-05-4005. [DOI] [PubMed] [Google Scholar]
- 85.Zijlmans HJ, Fleuren GJ, Baelde HJ, et al. The absence of CCL2 expression in cervical carcinoma is associated with increased survival and loss of heterozygosity at 17q11.2. J Pathol. 2006;208:507–17. doi: 10.1002/path.1918. [DOI] [PubMed] [Google Scholar]
- 86.Sommer AP, Zhu D, Scharnweber T. Extraordinary anticancer effect of green tea and red light. Photomed Laser Surg. 2010;28:429–30. doi: 10.1089/pho.2009.2706. [DOI] [PubMed] [Google Scholar]