

Abstract
Treatment of high-risk neuroblastoma (NB) represents a major challenge in paediatric oncology. Alternative therapeutic strategies include antibodies targeting the disialoganglioside GD2, which is expressed at high levels on NB cells, and infusion of donor-derived natural killer (NK) cells. To combine specific antibody-mediated recognition of NB cells with the potent cytotoxic activity of NK cells, here we generated clonal derivatives of the clinically applicable human NK cell line NK-92 that stably express a GD2-specific chimeric antigen receptor (CAR) comprising an anti-GD2 ch14.18 single chain Fv antibody fusion protein with CD3-ζ chain as a signalling moiety. CAR expression by gene-modified NK cells facilitated effective recognition and elimination of established GD2 expressing NB cells, which were resistant to parental NK-92. In the case of intrinsically NK-sensitive NB cell lines, we observed markedly increased cell killing activity of retargeted NK-92 cells. Enhanced cell killing was strictly dependent on specific recognition of the target antigen and could be blocked by GD2-specific antibody or anti-idiotypic antibody occupying the CAR’s cell recognition domain. Importantly, strongly enhanced cytotoxicity of the GD2-specific NK cells was also found against primary NB cells and GD2 expressing tumour cells of other origins, demonstrating the potential clinical utility of the retargeted effector cells.
Keywords: neuroblastoma, natural killer cells, chimeric antigen receptor, GD2, adoptive immunotherapy
Introduction
Natural killer (NK) cells are part of the innate immune system and the body’s first line of defence against virally infected and malignant cells. Unlike T cells, which are major histocompatibility complex (MHC) restricted and need to be sensitized and educated about the target, cytotoxicity of NK cells can be activated rapidly and is regulated by a balance of signals from germline-encoded activating and inhibitory cell surface receptors [1, 2]. Over the last decade, significant progress has been made towards realizing the potential of NK cells for cancer immunotherapy [3, 4]. In addition to autologous or donor-derived primary NK cells, also clinically applicable, continuously growing cytotoxic cell lines such as NK-92 are being developed for adoptive cancer immunotherapy. NK-92 cells exhibit functional characteristics of activated NK cells and express typical NK-cell surface receptors, but lack FcγRIII [5]. In preclinical models high intrinsic cytotoxicity of NK-92 against leukaemia, lymphoma, melanoma and other malignancies has been demonstrated [6-8]. This high endogenous cytotoxic potential of NK-92 has been attributed to the absence of most of the inhibitory NK cell receptors, although high numbers of activating lectin-like and immunoglobulin-like receptors are being expressed [9]. General safety and tolerability of NK-92 cell infusion has been established in phase I clinical studies, with some patients experiencing clinical responses [10, 11].
We extended this approach to target cells that display intrinsic resistance to NK cells by genetic modification of NK-92 cells with chimeric antigen receptors (CARs), which recognize protein antigens differentially expressed on the surface of cancer cells [12-14]. Such CAR are transmembrane proteins that consist of an extracellular scFv single-chain Fv antibody as a cell binding domain, genetically linked by a flexible spacer to transmembrane and intracellular parts of signalling molecules such as the CD3-ζ chain [15, 16]. Here we investigated the suitability of this approach to redirect cytolytic activity of NK-92 to cancer cells that express the non-proteinaceous antigen GD2. The disialoganglioside GD2 is a sialic acid-containing glycosphingolipid, which is thought to play a role in the attachment to components of the extracellular matrix [17]. In normal foetal and adult tissues GD2 expression is mainly restricted to the central nervous system, peripheral nerves and skin melanocytes. As a tumour-associated antigen, GD2 is consistently expressed in malignancies of neuroectodermal origin such as neuroblastoma (NB), melanoma, and to a variable degree in other tumours such as bone and soft-tissue sarcoma, small cell lung cancer and brain tumours [18, 19]. Due to its relatively selective expression by tumour cells and its accessibility on the cell surface, GD2 is a suitable antigen for immunotherapy. Murine (3F8 and 14G2a) and chimeric human/mouse (ch14.18) anti-GD2 antibodies have been developed over the past two decades and have been investigated in clinical studies for the treatment of NB and melanoma [18, 19].
Neuroblastoma represents the most common extracranial solid tumour during early childhood. Although survival rates of NB patients with locoregional disease exceed 90%, metastatic NB in patients over 18 months of age is often incurable even with advanced multimodality chemotherapy regimens [20-22]. For more than one decade, high-risk stage IV NB patients have been treated with high-dose chemotherapy followed by autologous stem cell transplantation (auto-SCT), but outcome still remains poor [23]. Clinical trials using chimeric GD2-specific antibody ch14.18 as a single agent [24], or administration of the antibody together with systemic application of GM-CSF and interleukin (IL)-2 [25] were performed. In the latter phase III study treatment of stage IV NB patients with ch14.18 in combination with IL-2 and GM-CSF following auto-SCT resulted in a 20% increase in survival [25]. To enhance the efficacy of antibody therapy, and to minimize systemic toxicities as well as immunogenicity, an immunocytokine (hu14.18-IL2) was developed that targets IL-2 directly to tumour cells by fusing it to the humanized anti-GD2 antibody hu14.18 [26]. Also immunotherapy with highly purified donor NK cells that exhibit cytotoxic activity against NB cells with low MHC class I expression represents a promising strategy to improve clinical outcome of patients with NB stage IV. In an ongoing phase I/II study haploidentical SCT (haplo-SCT) in combination with infusion of IL-2 stimulated allogeneic donor NK cells is being employed for the treatment of high-risk NB patients after unsuccessful auto-SCT [27].
To combine specific recognition of NB cells with the potent cytotoxic activity of NK cells, here we generated clonal NK-92 cells that stably express a GD2-specific CAR which carries a cell binding domain derived from antibody ch14.18. Such NK-92-scFv(ch14.18)-ζ cells displayed markedly enhanced cytotoxicity against established GD2-expressing NB tumour cells, whereas lysis of GD2– targets remained unaffected. Importantly, selective antitumoural activity was also observed against freshly isolated primary NB tumour cells, indicating that this strategy may be suitable for further development as a cell-based immunotherapy for NB.
Materials and methods
Cells and culture conditions
Human SK-N-SH, BE(2)-C, Kelly (ATCC, Manassas, VA, USA) and UKF-NB3 [28] NB cells were maintained in Iscove’s modified Dulbecco’s medium (Biochrom AG, Berlin, Germany), LAN-1 NB cells (kindly provided by Ralph A. Reisfeld, The Scripps Research Institute, La Jolla, CA, USA) in RPMI 1640 (Hyclone Laboratories, Thermo Scientific, Logan, UT, USA). SK-Mel-23 [29] and NW1539 melanoma, and SK-BR-3 breast carcinoma cells (ATCC) were cultured in DMEM (Lonza, Cologne, Germany), each containing standard supplements (10% heat-inactivated foetal bovine serum, 2 mM L-glutamine, 100 units/ml penicillin and 100 μg/ml streptomycin). NK-92 cells (ATCC) and genetically modified NK-92-scFv(ch14.18)-ζ cells were propagated in X-VIVO 10 medium (Lonza) supplemented with 5% heat-inactivated human serum (Red Cross Blood Donor Service Baden-Württemberg – Hessen, Frankfurt, Germany), 100 units/ml IL-2 (Proleukin; Novartis Pharma, Nürnberg, Germany) and 0.6 mg/ml G418 (NK-92-scFv(ch14.18)-ζ).
Primary human NB cells were isolated after surgery from a tumour metastasis of a 4-year-old patient with NB stage IV, and from the bone marrow (BM) of another high-risk patient after relapse. The tumour metastasis from jejunum was minced, treated with papain for digestion and percolated through a 40 μm filter (Cell strainer; BD Biosciences, Heidelberg, Germany) to singularize the cells. Cells were resuspended in DMEM/F-12 medium (Invitrogen, Darmstadt, Germany) containing 100 units/ml penicillin and 100 μg/ml streptomycin, and cultivated until analysis in Iscove’s modified Dulbecco’s medium with the same antibiotics. BM cells of healthy donors and NB patients were obtained after informed consent. Mononuclear cells were applied for analysis after centrifugation with Ficoll Hypaque. Research use of anonymized samples of peripheral blood stem cells (PBSC) and CD34+ cells from healthy donors remaining after allogeneic transplantation was approved by the University Hospital Ethics Committee at the University of Frankfurt, Germany.
Construction of chimeric antigen receptors
cDNA of GD2-specific ch14.18 scFv antibody fragments was derived by PCR using plasmids encoding scFv(ch14.18)-Fc fusion proteins as templates (kindly provided by EMD Serono, Billerica, MA, USA) and oligonucleotide primers introducing 5′ SfiI and 3′ NotI restriction sites. The resulting scFv sequences contain the variable domains of heavy (VH) and light chains (VL) of antibody ch14.18 connected by a synthetic (G4S)4 linker either in the orientation VH-linker-VL, designated scFv(ch14.18)HL, or VL-linker-VH, designated scFv(ch14.18)LH. For construction of GD2-specific antigen receptors, each scFv(ch14.18) fragment was assembled stepwise in frame with an immunoglobulin heavy-chain signal peptide sequence 5′ of the scFv, and sequences encoding a Myc-tag, the hinge region of CD8α (amino acids 105–165) and CD3-ζ chain 3′ of the scFv in plasmid pGEM-1 (Promega, Mannheim, Germany). Complete CAR sequences were derived from the resulting pGEM-1-scFv(ch14.18)-ζ constructs as SalI, SmaI fragments, and cloned into the SalI, HpaI restriction sites of a modified pLXSN retroviral vector [30] yielding pL-scFv(ch14.18)HL-ζ-SN and pL-scFv(ch14.18)LH-ζ-SN.
Production of amphotropic retroviral vectors and transduction of NK-92 cells
FLYA-JET packaging cells [31] were transfected with pL-scFv(ch14.18) HL-ζ-SN and pL-scFv(ch14.18)LH-ζ-SN constructs by electroporation using the Easyject Optima electroporation system (Equibio, Ashford, UK) with the following parameters: 20 μg of plasmid DNA per 1 × 106 cells in 0.8 ml of DMEM medium in a 0.4 cm cuvette, and ‘standard’ settings according to the manufacturer’s recommendations. Stable transfectants were selected for 1 week in DMEM growth medium containing 2.4 mg/ml G418. For production of amphotropic retroviral vector, selected packaging cells were grown over night in NK-92 medium. Culture supernatants were passed through a 0.2 μm filter and incubated with NK-92 cells in the presence of 8 μg/ml polybrene for 5 hrs at 37°C. Then NK-92 cells were grown over night in fresh X-VIVO 10 medium, before G418 was added to a final concentration of 0.6 mg/ml for selection of NK-92-scFv(ch14.18)-ζ cells.
Generation of clonal NK-92 cell lines expressing scFv(ch14.18)-ζ
After G418 selection, NK-92-scFv(ch14.18)-ζ cells expressing high levels of CARs were enriched by immunomagnetic cell separation. G418-resistant cells were incubated with Myc-tag-specific monoclonal antibody (mAb) 9E10 (Sigma-Aldrich, Deisenhofen, Germany) (1.5 μg/5 × 105 cells) and selected using goat antimouse IgG MicroBeads and MACS LS+ separation columns (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer’s instructions. Then single-cell clones were derived from sorted cell pools by limiting dilution. During all steps, CAR expression was monitored by flow cytometry. For FACS analysis 5 × 105 NK-92-scFv(ch14.18)-ζ or NK-92 cells were incubated for 30 min. at 4°C with 1.5 μg of mAb 9E10. Cells were washed twice with phosphate-buffered saline and then treated for another 30 min. at 4°C with FITC-labelled goat antimouse IgG secondary antibody (BD Biosciences). Fluorescence of cells was analysed with FACScan or FACSCalibur cytometers (BD Biosciences).
Surface expression of GD2
Expression of GD2 on the surface of established cell lines and primary human cells was determined by flow cytometry using custom PE/Cy5 conjugated 14.G2a anti-GD2 antibody and mouse IgG2a isotype control (BD Biosciences). For quantification of GD2 molecules, cellular antigen expression was measured as antibody binding capacity (ABC) units using the Quantum Simply Cellular kit (Bangs Laboratories, Indianapolis, IN, USA) according to the manufacturer’s recommendations. The ABC values calculated for GD2 after subtraction of the values of the appropriate isotype control were expressed as molecules per cell. Standard curves were set with the maximum at 250,000 ABC units, and the detection threshold was <1000 ABC units. The quantitative analyses were performed on an FC 500 cytometer and data were analysed using the CXP v2.2 software (Beckman Coulter, Krefeld, Germany) as described previously [27].
Cytotoxicity assays
Specific cytotoxic activity of NK cells towards target cells was analysed in europium (Eu3+)-release, FACS-based and 51Cr-release assays. Europium release was measured as described previously [12]. Briefly, 5 × 106 target cells were incubated for 10 min. at 4°C in 800 μl of europium solution containing 50 mM Hepes, pH 7.4, 93 mM NaCl, 5 mM KCl, 2 mM MgCl2, 10 mM Diethylenetriamine-pentaacetic acid (Sigma-Aldrich, Munich, Germany), 2 mM europium(III)-acetate (Sigma-Aldrich) and electroporated at 200 W, 960 μFa, 250 V using a GenePulser (Bio-Rad, Munich, Germany). Then the cells were washed and seeded in triplicates in 96-well tissue culture plates in X-VIVO 10 medium, followed by addition of NK-92 or NK-92-scFv(ch14.18)-ζ cells at different effector to target ratios (E/T) and incubation for 2 hrs at 37°C. To determine maximal lysis, europium-labelled target cells suspended in 100 μl of X-VIVO 10 medium were incubated in the absence of effector cells with 100 μl of lysis buffer (PerkinElmer Wallac, Freiburg, Germany) followed by repeated freezing/thawing. Cells were centrifuged for 5 min. at 500 ×g, 20 μl of culture supernatant were collected and added to 200 μl/well of enhancement solution (PerkinElmer Wallac) in FluoroNunc 96-well plates (Nunc, Wiesbaden, Germany). After incubation on a shaker at 50 rpm for 15 min. at room temperature, fluorescence was determined using an LKB-Wallac 1230 Arcus fluorometer (PerkinElmer Wallac). Specific cytotoxicity was calculated as: % cytotoxicity = (experimental lysis – spontaneous lysis) × 100/(maximal lysis – spontaneous lysis).
For FACS-based cell killing assays, adherent tumour cells were harvested by treatment with Accutase (PAA Laboratories, Pasching, Austria) for 5 to 10 min. at 37°C and singularized. To prevent unwanted cell clumping, the cells were then incubated with 50 μg/ml of DNase I (Roche Applied Science, Mannheim, Germany) in 1× reaction buffer prepared from a 10× stock solution (100 mM Tris-HCl, pH 7.6, 25 mM MgCl2, 5 mM CaCl2) (Roche Applied Science) for 15 min. at 37°C under constant gentle shaking.
The reaction was blocked by addition of 250 mM ethylenediaminetetraacetic acid to a final concentration of 50 mM. Target cells were incubated with NK-92 or NK-92-scFv(ch14.18)-ζ cells for 2 and 4 hrs in X-VIVO 10 medium at different E/T ratios. Lytic activity was measured by single platform 5-colour flow cytometric analysis for effector cells alone, and for co-cultured effector and target cells on an FC 500 cytometer (Beckman Coulter) as described previously [32, 33]. Briefly, calculation of cytotoxicity was based on the loss of living propidium iodide (PI)– target cells (CD9+ CD81+ CD45neg PIneg). Absolute count-dedicated beads (Flow-Count Fluorospheres; Beckman Coulter) were used for absolute cell enumeration. For GD2 blocking experiments, target cells were pre-incubated for 120 min. with 20 μg/ml of parental murine anti-GD2 antibody 14G2a prior to co-culture with NK-92-scFv(ch14.18)-ζ cells.
Cytotoxicity of NK-92 and NK-92-scFv(ch14.18)-ζ towards LAN-1 NB cells was measured by 51Cr release. Target cells were loaded with 51Cr (0.125 mCi/5 × 105 cells in 500 μl) (PerkinElmer, Billerica, MA, USA) for 2 hrs at 37°C, and washed twice with X-VIVO-10 medium containing 5% human AB Serum. Then 5 × 103 target cells were co-cultured in quadruplicates for 6 hrs with NK-92 or NK-92-scFv(ch14.18)-ζ cells in 100 μl of medium at an E/T ratio of 6.3:1. Radioactivity in supernatants was measured using a Wizard 2 gamma counter (PerkinElmer). Maximum 51Cr release was induced by addition of 100 μl of 1% SDS solution to 100 μl of target cell suspension. Specific cytotoxicity was calculated as described above for europium release. For CAR blocking experiments, 10 μg/ml of anti-idiotype antibody 1A7 (kindly provided by M. Bhattacharya-Chatterjee, University of Cincinnati Medical Center, Cincinnati, OH, USA) were added during co-culture of cells. Mouse IgG1 clone X40 (BD Biosciences, San Jose, CA, USA) served as an isotype control.
Cytotoxic activity of NK cells upon prolonged co-culture with target cells at low E/T ratios was visualized by microscopy. Microscopic images of cells were taken after 18 hrs of co-culture using an Axiovert 135 microscope (Carl Zeiss, Göttingen, Germany) and a Sony 3CCD camera (Sony, Berlin, Germany).
Microscopic control of the interaction between NK-92 and primary NB cells
Primary NB cells were seeded at low density on 20 mm chamber slides (Nunc, Langenselbold, Germany) and grown for 24 hrs. Then adherent NB cells were co-cultured with gene-modified NK-92-scFv(ch14.18)-ζ or parental NK-92 cells for 4 hrs at an E:T ratio of 5:1. NK-92 cells were identified with CD45-specific antibody followed by FITC-conjugated secondary antibody. CAR expressing NK-92 cells were detected with mAb 9E10 followed by PE-conjugated secondary antibody. All cells were counter-stained with DAPI. Fluorescence microscopy was performed with an Olympus IX71 microscope (Olympus, Hamburg, Germany), and microscopic images were acquired with a charge-coupled device camera.
Statistical analysis
Kruskal–Wallis test with Dunn’s multiple comparison and Wilcoxon matched pairs test were applied to assess statistical significance of differences between groups. P values <0.05 were considered as significant. Data were analysed using GraphPad Prism software (GraphPad Software, San Diego, CA, USA).
Results
Generation of NK cells carrying GD2-specific chimeric antigen receptors
GD2-specific scFv(ch14.18) antibody fragments were derived from constructs encoding scFv(ch14.18)-Fc fusion proteins that carry heavy and light chain variable domains of the chimeric mAb ch14.18 [34, 35]. To address potential differences in the functionality of scFv(ch14.18) molecules that depend on the orientation of the variable domains, we employed scFv fragments where VH and VL of antibody ch14.18 were either assembled in the orientation VH-linker-VL, or VL-linker-VH, with the synthetic (G4S)4 sequence serving as a flexible linker. Chimeric antigen receptors were constructed by inserting the scFv fragments designated scFv(ch14.18)HL and scFv(ch14.18)LH between a sequence encoding an N-terminal immunoglobulin heavy-chain signal peptide, and sequences encoding a Myc-tag, the CD8α hinge region (amino acids 105–165) and the CD3-ζ chain in the retroviral transfer vector pLXSN [30] (Fig. 1A).
Fig 1.
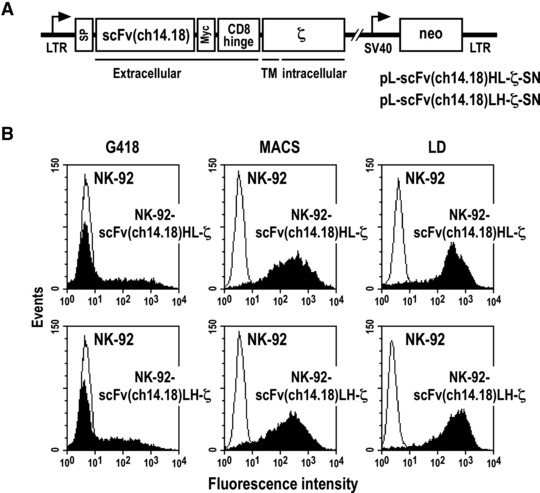
Transduction of NK-92 cells with retroviral vectors encoding chimeric scFv(ch14.18)-ζ antigen receptors. (A) Schematic representation of pL-scFv(ch14.18)-ζ-SN constructs. The Moloney murine leukaemia virus 5′ long terminal repeat (LTR) controls the expression of chimeric scFv(ch14.18)-ζ antigen receptors which consist of an N-terminal immunoglobulin heavy-chain leader peptide (signal peptide), a GD2-specific single-chain antibody scFv(ch14.18) with heavy (VH) and light chain (VL) variable domains in VH-linker-VL (HL) or VL-linker-VH (LH) orientation, a Myc-tag, the hinge region of CD8α and the CD3-ζ chain. The neomycin-resistance gene for G418 selection of transduced cells is driven by the SV40 early promoter. (B) Surface expression of chimeric scFv(ch14.18)-ζ antigen receptors. After G418 selection of transduced cells (G418), NK-92-scFv(ch14.18)HL-ζ and NK-92-scFv(ch14.18)LH-ζ cells expressing homogenous levels of the CARs on their surface were enriched by immunomagnetic separation with mAb 9E10 and goat antimouse IgG-coated magnetic beads (MACS). Single cell clones were derived by limiting dilution (LD). Representative clones are shown. After each selection step, surface expression of scFv(ch14.18)-ζ receptors was determined by flow cytometry using mAb 9E10 and FITC-labelled goat antimouse secondary antibody. NK-92 cells transduced with empty pLXSN served as a control.
Amphotropic retroviral vector particles were produced by stable transfection of FLYA-JET packaging cells [31], and used for transduction of human NK-92 cells. After selection with G418, expression of scFv(ch14.18)HL-ζ and scFv(ch14.18)LH-ζ receptor proteins on the cell surface was analysed by flow cytometry. At this step the majority of cells in the selected cell pools displayed low or undetectable expression of the CARs (Fig. 1B, left panels). To enrich NK-92 cells that express more homogenous receptor levels, cells were sorted with Myc-tag specific mAb 9E10 and immunomagnetic beads (Fig. 1B, middle panels), followed by limiting dilution to obtain single cell clones. This yielded stable NK-92 cell clones consistently expressing high levels of CARs (Fig. 1B, right panels). We did not observe a difference in expression levels between clones carrying scFv(ch14.18)HL-ζ or scFv(ch14.18)LH-ζ (Fig. 1B and data not shown), indicating that the orientation of VH and VL within scFv(ch14.18) had no influence on the overall expression or surface display of the receptors.
Surface expression of GD2 on NB cells
As a prerequisite for the analysis of CAR functionality and activity of retargeted NK-92 cells, first surface expression of GD2 by established NB cell lines and primary NB cells was investigated by flow cytometry using fluorochrome-labelled GD2-specific murine mAb 14.G2a. Control cells were treated with an irrelevant isotype-matched antibody. Established human UKF-NB3, Kelly, BE(2)C and LAN-1 NB cells displayed intermediate to high levels of GD2 on their surface, whereas only a very weak signal was determined with anti-GD2 antibody for SK-N-SH cells (Fig. 2A). Analysis of primary NB cells from the BM of 12 relapsed NB patients revealed markedly enhanced GD2 expression in these samples when compared to established GD2+ NB cell lines (data not shown). To illustrate this pronounced difference exemplarily, BM with NB cells from one of these patients was mixed with established UKF-NB-3 cells, and GD2 expression was determined by flow cytometry (Fig. 2B).
Fig 2.
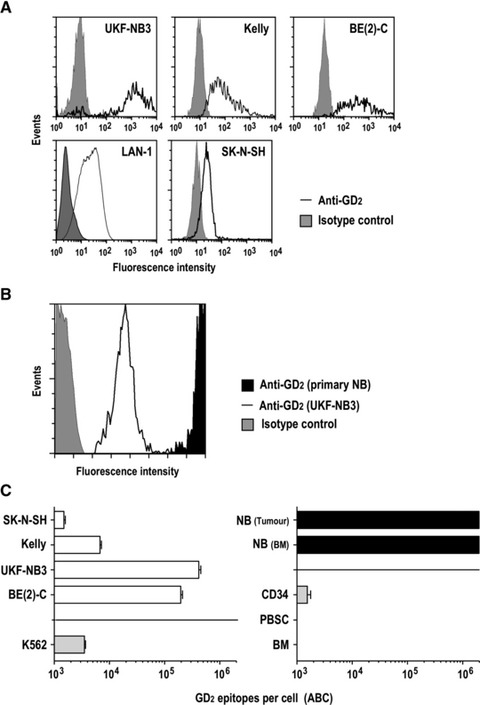
Surface expression of GD2 on established and primary NB cells. (A) Expression of GD2 on established human UKF-NB3, Kelly, BE(2)C, LAN-1 and SK-N-SH NB cells was determined by flow cytometry using PE-Cy5-labelled GD2-specific mAb 14.G2a (open areas). Control cells were treated with isotype control (grey areas). (B) To illustrate high level expression of GD2 on primary cells, BM with NB cells from a relapsed patient was mixed with established UKF-NB3 NB cells before analysis by flow cytometry as described in (A). (C) For quantification of GD2 molecules on the cell surface, antigen expression was measured as ABC molecules/cell as described in the methods section. GD2 expression on established NB cell lines (left panel, open bars), K562 erythroleukaemic cells (left panel, shaded bar), primary cells from an NB tumour metastasis (NB tumour) and BM infiltrating NB cells (NB BM) (right panel, filled bars) as well as haematopoietic cells (selected 34+ cells, CD34; lymphocytes from peripheral blood, PBSC; lymphocytes from bone marrow, BM) from healthy donors (right panel, shaded bars) is indicated. The graphs are shown with a logarithmic scale. Lower and upper detection limits of this assay were 1 × 103 and 2 × 106 ABC molecules/cell, respectively. Data are represented as mean ± S.D.
For quantification of GD2 molecules, cellular antigen expression on the surface of established and primary NB cells as well as haematopoietic cells from healthy donors was determined in comparison to antibody-binding microbeads as a standard (Fig. 2C). We found very high expression of GD2 in the range of 2 to 4 × 105 antibody binding capacity (ABC) molecules/cell for established BE(2)C and UKF-NB3 NB cells, 7 × 103 ABC molecules/cell for Kelly cells and 1.5 × 103 ABC molecules/cell for SK-N-SH (Fig. 2C), which is near the lower detection limit of 1 × 103 epitopes/cell for this assay. Malignant cells of non-NB origin such as K562 (erythroleukaemia) expressed low levels of GD2 (3.5 × 103 ABC molecules/cell). Similar results were obtained for A673 peripheral neuroepithelioma tumour (PNET) and rhabdomyosarcoma cells (data not shown). GD2 levels on primary NB cells isolated from a tumour metastasis and the BM of a relapsed NB patient with stage IV disease were above 2 × 106 ABC molecules/cells, which was the upper detection limit of the assay (Fig. 2C). Haematopoietic cells from healthy donors expressed very low GD2 levels (sorted CD34+ cells), or yielded ABC signals below the lower detection limit of the assay (lymphocytes in PBSC and BM).
Cytotoxic activity of NK-92-scFv(ch14.18)-ζ against NB cells
Next we investigated whether expression of GD2-specific antigen receptors can augment cell killing activity of NK-92 towards established NB cells. Cytotoxicity of clonal NK-92-scFv(ch14.18)HL-ζ and parental NK-92 cells against UKF-NB3, Kelly and SK-N-SH cells during co-culture at different E/T ratios was analysed in FACS-based assays. Highly GD2+ UKF-NB3 cells displayed already pronounced sensitivity to parental NK-92 cells (58% specific lysis at an E/T ratio of 10:1), but were more potently killed by CAR-expressing NK-92-scFv(ch14.18)HL-ζ (79% specific lysis; P < 0.01) (Fig. 3A). Similar results were obtained with NK-92-scFv(ch14.18)LH-ζ cells (data not shown). Kelly cells were relatively resistant to NK-92 (22% specific lysis at an E/T ratio of 5:1), but like UKF-NB-3 highly sensitive to NK-92-scFv(ch14.18)HL-ζ cells (58% specific lysis; P < 0.05). This cytotoxicity was strongly reduced when we blocked GD2 on the surface of Kelly cells with parental murine anti-GD2 antibody 14G2a prior to co-culture with NK-92-scFv(ch14.18)HL-ζ (18% specific lysis at an E/T ratio of 5:1), which demonstrates that the enhanced activity of CAR-expressing NK cells was dependent on the accessibility of GD2 on the tumour cell surface. For SK-N-SH cells which do not overexpress the target molecule, similar lytic activity was found for parental NK-92 and retargeted NK-92-scFv(ch14.18)HL-ζ cells (43% versus 41% specific lysis at an E/T ratio of 5:1; not significant).
Fig 3.
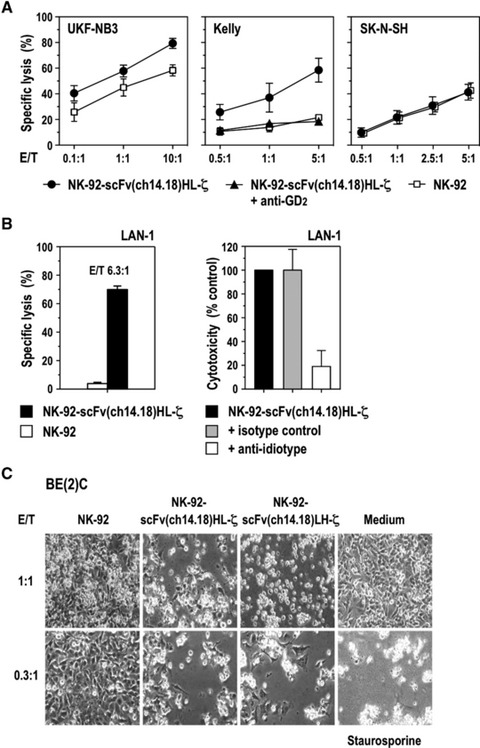
NK-92-scFv(ch14.18)-ζ cells display enhanced cell killing activity towards established GD2-expressing NB cells. (A) Cytotoxic activity of NK-92-scFv(ch14.18)HL-ζ cells towards UKF-NB3, Kelly and SK-N-SH NB cells was determined in FACS-based cytotoxicity assays at different effector to target ratios (E/T). Parental NK-92 cells were included for comparison. To investigate dependence of cell killing on GD2 recognition, GD2 on Kelly NB cells was blocked with parental murine anti-GD2 antibody 14G2a prior to co-culture with NK-92-scFv(ch14.18)HL-ζ as indicated. Data are represented as mean ± S.D. (B) Cytotoxic activity of NK-92-scFv(ch14.18)HL-ζ towards LAN-1 NB cells was determined in 51Cr-release assays at an E/T ratio of 6.3:1. Parental NK-92 cells were included for comparison (left panel). To block interaction of CAR with GD2 on the target cell surface, LAN-1 and NK-92-scFv(ch14.18)HL-ζ cells were co-cultured in the presence of 10 μg/ml of anti-idiotype antibody 1A7 (right panel). Data are represented as mean ± S.D. (C) Cytotoxic activity of NK-92-scFv(ch14.18)HL-ζ and NK-92-scFv(ch14.18)LH-ζ towards BE(2)C NB cells was investigated by microscopical analysis after 18 hrs of co-culture at low E/T ratios of 1:1 and 0.3:1. Parental NK-92 cells were included for comparison. Control cells were incubated in the absence of NK cells, or were treated with 8 μM of the apoptosis-inducing drug staurosporine as indicated. Representative fields are shown.
Cytotoxic activity of NK-92-scFv(ch14.18)HL-ζ towards established GD2+ LAN-1 NB cells was determined in 51Cr release assays at an E/T ratio of 6.3:1. Similar to Kelly cells, LAN-1 displayed resistance to parental NK-92 but were readily killed by NK-92-scFv(ch14.18)HL-ζ cells (4% versus 70% specific lysis, respectively; P < 0.01) (Fig. 3B, left panel). When effector and target cells were co-cultured in the presence of anti-idiotypic antibody 1A7 which blocks the antigen binding site of ch14.18 [36], cytotoxicity of NK-92-scFv(ch14.18)HL-ζ cells was inhibited by 81%, confirming dependence on the functionality of the target recognition domain of the CAR (Fig. 3B, right panel). Cell killing activity of NK cells at low E/T ratios was investigated with BE(2)C cells as targets. The NB cells were incubated for 18 hrs with clonal NK-92-scFv(ch14.18)HL-ζ, NK-92-scFv(ch14.18)LH-ζ or parental NK-92 cells. NK-cell mediated cytotoxicity was analysed by light microscopy (Fig. 3C). After exposure to NK-92-scFv(ch14.18)HL-ζ or NK-92-scFv(ch14.18)LH-ζ cells at an E/T ratio of 1:1 only very few viable BE(2)C cells could be detected, comparable to exposure to the apoptosis-inducing reagent staurosporine. Even at an E/T ratio of 0.3:1 almost all NB cells were eliminated, suggesting that NK-92-scFv(ch14.18)HL-ζ and NK-92-scFv(ch14.18)LH-ζ cells are able to sequentially attack several target cells. In contrast, parental NK-92 cells had no apparent effect on the survival of BE(2)C cells after prolonged incubation. In clinical studies with untargeted NK-92, the cells were irradiated with 10 Gy before infusion to prevent permanent engraftment [10, 11]. Because similar safety measures may be important for clinical use of retargeted NK-92 variants, we tested the effects of irradiation on cytotoxic activity of GD2-specific NK-92-scFv(ch14.18)HL-ζ cells towards established UKF-NB3 cells. We found that irradiated effector cells retained marked cytotoxicity, which was comparable to that of non-irradiated NK-92-scFv(ch14.18)HL-ζ cells (Fig. 4).
Fig 4.
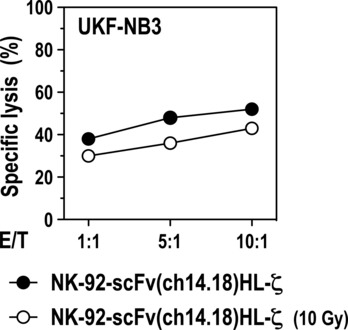
Cytotoxic activity of irradiated NK-92-scFv(ch14.18)HL-ζ cells. Established UKF-NB3 NB tumour cells were incubated at different E/T ratios with untreated NK-92-scFv(ch14.18)HL-ζ, or NK-92-scFv(ch14.18)HL-ζ cells 24 hrs after irradiation with a dose of 10 Gy. Cytotoxic activity was determined after 2.5 hrs of co-incubation in FACS-based cytotoxicity assays.
These results demonstrate that expression of scFv(ch14.18)-ζ receptors on NK-92 triggers markedly enhanced cell killing towards NB cells, which is dependent on specific recognition of GD2 on the target cell surface. Thereby NK-92-scFv(ch14.18)HL-ζ and NK-92-scFv(ch14.18)LH-ζ cells were similarly active, indicating that the orientation of VH and VL in the scFv(ch14.18) antibody fragment does not influence target cell recognition and GD2-specific cytotoxicity.
Enhanced cell killing activity of NK-92-scFv(ch.14.18)-ζ against primary NB cells
To investigate cytotoxic activity of NK-92-scFv(ch14.18)-ζ towards primary tumour cells, human NB cells were freshly isolated from a tumour metastasis of a 4-year-old patient with NB stage IV, singularized and cultivated for several days. Quantitative flow cytometric analysis revealed very high levels of GD2 expression on the surface of these cells (Fig. 2C). The primary NB cells were co-cultured at different E/T ratios with NK-92-scFv(ch14.18)HL-ζ or parental NK-92 cells for 2 hrs before analysis of specific lysis in FACS-based assays. Primary tumour cells displayed intermediate sensitivity to parental NK-92 cells (43% specific lysis at an E/T ratio of 10:1), but were highly sensitive to CAR-expressing NK-92-scFv(ch14.18)HL-ζ cells, resulting in 82% specific lysis at an E/T ratio of 10:1 (P < 0.01) (Fig. 5A). This enhanced cytotoxic activity corresponded well with the high level of GD2 expression on the target cells. In accordance with the results of the cytotoxicity analysis, interaction between gene-modified NK-92 and adherent, primary NB cells was substantially enhanced when compared to untargeted parental NK-92 cells (Fig. 5B).
Fig 5.
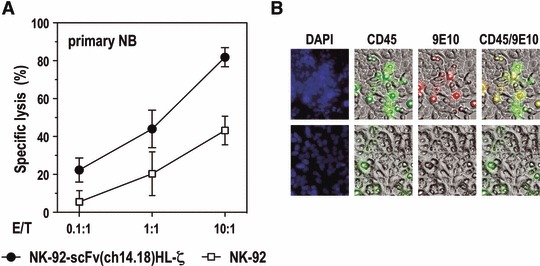
Cytotoxic activity of NK-92-scFv(ch14.18)HL-ζ cells towards primary human NB cells. (A) Cytotoxic activity of NK-92-scFv(ch14.18)HL-ζ cells towards primary NB cells isolated from a tumour metastasis was determined in FACS-based cytotoxicity assays at different E/T ratios as indicated. Parental NK-92 cells were included for comparison. Data are represented as mean ± S.D. (B) Gene-modified NK-92-scFv(ch14.18)HL-ζ cells (upper panels) and parental NK-92 cells (lower panels) were identified in co-cultures with adherent NB tumour cells by staining with a CD45-specific antibody (green). NK-92-scFv(ch14.18)HL-ζ cells were also detected by staining with 9E10 antibody (red) which recognizes the Myc-tag included in the CAR. All cells were counter-stained with DAPI (blue).
Cytotoxicity of NK-92-scFv(ch.14.18)-ζ against melanoma and breast carcinoma cells
To investigate cell killing activity of GD2-specific NK-92 against GD2-expressing tumour cells of origins other than NB, we analysed the sensitivity of established melanoma and breast carcinoma cells for NK-cell mediated lysis. Flow cytometric analysis revealed high levels of GD2 on SK-Mel-23 and NW1539 melanoma cells, whereas SK-BR-3 breast carcinoma cells expressed only moderate GD2 levels (Fig. 6A). In europium release assays, SK-BR-3 cells were resistant to lysis by parental NK-92 cells, confirming previous findings [12]. In contrast, clonal NK-92-scFv(ch14.18)LH-ζ cells were able to kill SK-BR-3 cells achieving specific lysis of 44% at an E/T ratio of 10:1 (P < 0.01) (Fig. 6B). Although SK-Mel-23 and NW1539 melanoma cells displayed moderate sensitivity already to unmodified NK-92 cells, cell killing activity was markedly enhanced upon expression of GD2-specific scFv(ch14.18)-ζ receptors with specific lysis of SK-Mel-23 cells at an E/T ratio of 10:1 of 22% and 92%, respectively (P < 0.001) (Fig. 6B, C). At a low E/T ratio of 1:1 prolonged co-culture of NW1539 cells with NK-92-scFv(ch14.18)HL-ζ or NK-92-scFv(ch14.18)LH-ζ resulted in almost complete elimination of the target cells, comparable to exposure to the apoptosis-inducing reagent staurosporine. Even at an E/T ratio below 1:1 only very few viable melanoma cells remained, suggesting that individual NK-92-scFv(ch14.18)HL-ζ and NK-92-scFv(ch14.18)LH-ζ cells may sequentially attack several NW1539 targets as in the case of BE(2)C NB cells (Fig. 3C).
Fig 6.
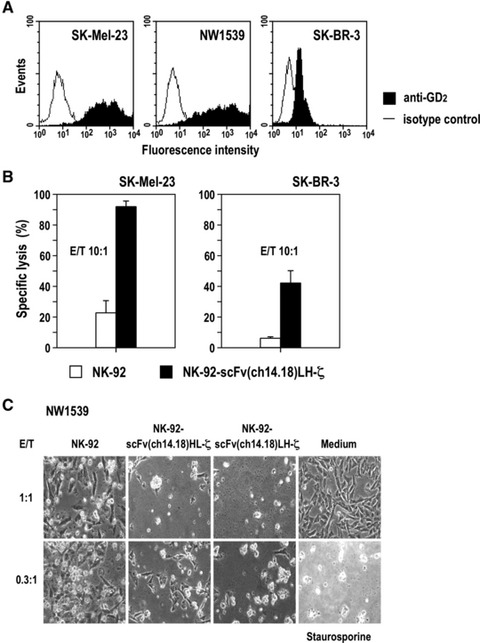
Cell killing activity of NK-92-scFv(ch14.18)-ζ cells towards GD2-expressing melanoma and breast carcinoma cells. (A) Surface expression of GD2 on SK-Mel-23 and NW1539 melanoma cells and SK-BR-3 breast carcinoma cells was determined by flow cytometry using PE-Cy5-labelled GD2-specific mAb 14.G2a (filled areas). Control cells were treated with isotype control (open areas). (B) Cytotoxic activity of NK-92-scFv(ch14.18)LH-ζ towards SK-Mel-23 and SK-BR-3 cells was determined in europium release assays at an E/T ratio of 10:1. Parental NK-92 cells were included for comparison. Data are represented as mean ± S.D. (C) Cytotoxic activity of NK-92-scFv(ch14.18)HL-ζ and NK-92-scFv(ch14.18)LH-ζ towards NW1539 melanoma cells was investigated by microscopical analysis after 18 hrs of co-culture at low E/T ratios of 1:1 and 0.3:1. Parental NK-92 cells were included for comparison. Control cells were incubated in the absence of NK cells, or were treated with 8 μM of the apoptosis-inducing drug staurosporine as indicated. Representative fields are shown.
Discussion
High-risk stage IV NB is characterized by disseminated metastasis, and continues to be a therapeutic challenge in paediatric oncology. Despite intense multimodal treatment, prognosis remains poor with long-term survival in only around 30% to 40% of patients [20-22], justifying efforts to develop alternative, more efficient treatment strategies. In this study, we have redirected continuously growing human NK cells to GD2 expressing NB cells using CARs that harbour a GD2-specific scFv fragment derived from the ch14.18 antibody. This was linked to the CD3-ζ chain to trigger cytolytic activity upon target cell recognition. Gene-modified NK cell lines were derived by transduction with retroviral vectors followed by selection of single cell clones, which displayed high and stable CAR expression over several months in continuous culture. NK-92 cells carrying CAR that harboured scFv(ch14.18) antibody fragments either in VH-linker-VL or VL-linker-VH orientation both displayed high cytotoxic activity towards GD2 expressing NB and melanoma cells. A moderate cell killing activity was achieved against breast cancer cells with more limited GD2 expression.
Immunotherapeutic approaches that target GD2 have first focused on monoclonal antibodies. To date murine antibodies 3F8 and 14G2a, the chimeric human/mouse antibody ch14.18, and the humanized immunocytokine hu14.18-IL2 have been used in clinical trials for the treatment of NB to enhance antibody-dependent cell-mediated cytotoxicity (ADCC) against GD2+ tumour cells [18, 25, 26]. Yet there is no consensus about the benefit of these strategies [37]. Promising responses were seen in a recent phase III study combining ch14.18 with GM-CSF and IL-2 [25], and a recent phase II study demonstrating antitumoural activity of the immunocytokine hu14.18-IL2 in patients with relapsed or refractory NB [26]. Other reports described no advantage over conventional therapy [38]. Yu et al. attributed the difference in outcome to the addition of IL-2 and GM-CSF, which augments ch14.18-mediated ADCC in vivo [25]. Combination of mAbs with different treatment modalities may be required to improve survival of NB patients, which includes utilization of cellular effector mechanisms [39]. Safety and feasibility of anti-GD2 antibody 3F8 together with haploidentical NK cells for the treatment of high-risk disease is currently being investigated in a phase I study (NCT00877110; clinicaltrials.gov). The lack or very low expression of MHC class I molecules on NB cells make them an ideal target for NK cells. Ex vivo stimulation of the effector cells with IL-2 resulted in increased expression of natural cytotoxicity receptors and NKG2D, and enhanced cytotoxicity towards NB cells [27]. Nevertheless, expression of soluble NKG2D ligands such as MHC class I related protein A by NB cells can have a marked inhibitory effect on NK cells [40, 41]. Furthermore, ADCC inducing activity of anti-GD2 antibodies may be affected by Fc receptor polymorphisms, as it has been described for other therapeutic antibodies [42, 43].
Our results show that expression of a GD2-specific CAR in NK cells directly couples antibody-mediated recognition of GD2 with the execution of cytotoxicity. This provides the effector cells with built-in ADCC-like activity, which can bypass limitations such as insufficient FcγRIII activation by antibodies and overcome the tumour cells’ endogenous resistance mechanisms as demonstrated for Kelly and LAN-1 cells. These NB cells were largely resistant to parental NK-92 cells, but were lysed by NK-92-scFv(ch14.18)-ζ with high efficiency. In the case of intrinsically NK-sensitive NB cell lines, we observed markedly increased cell killing activity of NK-92-scFv(ch14.18)-ζ cells. This enhanced activity was strictly dependent on specific recognition of the GD2 target antigen by the NK cells. Blocking of GD2 on the target cell surface with GD2-specific antibody or occupation of the antigen binding site of the scFv(ch14.18) domain of the CAR with an anti-idiotypic antibody abrogated cytotoxic activity of NK-92-scFv(ch14.18)-ζ towards GD2-expressing targets. In addition to GD2-dependent cell killing, retargeted NK-92-scFv(ch14.18)-ζ cells retained endogenous natural cytotoxicity of NK-92, demonstrated by similar activity of NK-92 and the GD2-specific variant against NK-sensitive SK-N-SH NB cells which express only very low GD2 levels. This may be of advantage for the treatment of tumours that consist of cells with heterogeneous GD2 expression. Importantly, strongly enhanced cytotoxicity of NK-92-scFv(ch14.18)-ζ cells was also found in the case of primary NB cells, indicating that the retargeted effector cells may be of clinical utility. GD2 has previously been established as a relevant cancer antigen for NB, and only minimal intra- or intertumoural heterogeneity of GD2 expression has been described [44, 45]. Moreover, persistent GD2 antigen expression after treatment with GD2-specific antibody was demonstrated. Only in 1 out of 62 NB patients the tumour lost GD2 expression after therapy, but in this case underwent phenotypic transformation into a pheochromocytoma-like tumour [45].
High level GD2 expression is restricted to NB, melanoma and other tumours of neuroectodermal origin [18, 19, 39]. We did not observe measurable GD2 expression on lymphocytes from BM and peripheral blood, and only very low GD2 expression on sorted CD34+ progenitor cells. Hence, haematological toxicities induced by treatment with GD2-specific NK cells appear unlikely. Nevertheless, limited antigen expression in other normal tissues can result in unwanted side effects of GD2-targeted therapies. Reported toxicities of treatment with anti-GD2 antibodies with or without GM-CSF included fever, nausea/vomiting, urticara, hypotension, capillary leak syndrome, ocular symptoms, neuropathic pain and neurotoxicity [25, 26, 38, 46, 47]. In general, these adverse effects were considerable but manageable, and did not result in discontinuation of the treatment. Recent data from animal models suggest that such toxicities are to a large part due to complement-dependent cytotoxicity (CDC) induced by such antibodies, and may be reduced by limiting CDC [48]. Introduction of a point mutation in GD2-specific ch14.18 antibody which interferes with CDC while retaining ADCC activity, resulted in markedly reduced allodynia in rats when compared to the parental molecule [48]. In the case of retargeted NK-92-scFv(ch14.18)-ζ cells, the antibody fragments within the GD2-specific CAR are restricted to the variable domains of antibody heavy and light chains, and do not contain the Fc portion that would be required to elicit CDC. Hence, CAR-triggered cytotoxicity of the NK cells is limited to ADCC-like activity, which will likely circumvent CDC-dependent toxicities.
Similar to our results, selective cytotoxicity of GD2-specific primary NK cells and cytotoxic T lymphocytes (CTLs) was found in experimental models [49, 50]. Clinical application of GD2-targeted CTLs appeared safe, and was associated with tumour regression or necrosis in half of the NB patients treated [51]. In the latter study, Epstein-Barr virus-specific CTLs were employed for genetic modification with a GD2-specific CAR to minimize the risk of autoimmunity caused by reactivity of the endogenous T-cell receptors of the retargeted cells with normal tissue antigens. In studies evaluating untargeted NK-92 cells, the effector cells were irradiated as a safety measure prior to infusion into cancer patients. This prevented permanent engraftment, although cells remained viable and retained cytotoxicity for several days [10, 11]. This was also the case for irradiated retargeted NK-92 cells (this study and [12]).
The use of CAR-expressing primary cells for adoptive immunotherapy requires for each individual patient the isolation, expansion and genetic modification of the relevant T- or NK-cell populations. Clinically applicable cytotoxic cell lines such as NK-92 could complement these approaches, especially in cases where autologous effector cells cannot be employed and suitable donors are not available. Methodology for GMP-compliant large scale production of unmodified NK-92 cells is well established [10, 11], and could be readily applied for continuous expansion of retargeted derivatives such as GD2-specific NK-92-scFv(ch14.18)-ζ cells. NK-92 cells express HLA class I molecules. Nevertheless, in the clinical trials conducted so far with unmodified, parental NK-92 cells, only few patients developed anti-HLA class I antibodies upon repeated treatment with the allogeneic effector cells [10, 11]. This was most likely due to the impaired immune status of the patients following chemo- and radiation therapy. Hence, repeated intravenous NK-92 therapy appears feasible when performed under continuous crossmatch testing with the patients’ serum before infusion. Our results demonstrate that clonal NK-92-scFv(ch14.18)-ζ cells selectively and reliably eliminate established and primary NB cells and GD2 expressing tumour cells of other origins. Utilization of NK-92-scFv(ch14.18)-ζ cells could bypass the need for separate genetic modification of effector cells for each individual patient, justifying further development of this approach.
Acknowledgments
This work was supported in part by a grant from Deutsche Krebshilfe to W.S.W., R.E., T.T. and H.N.L. (research grant 102386/10–2244). We thank Merck Serono, Merck KGaA for providing scFv(ch14.18) constructs, Dr. Jindrich Cinatl (University Hospital Frankfurt) for UKF-NB3 and Dr. Ralph A. Reisfeld (The Scripps Research Institute) for LAN-1 NB cells, Dr. Elke Jäger (Krankenhaus Nordwest, Frankfurt) for SK-Mel-23 and NW1539 melanoma cells, Dr. M. Bhattacharya-Chatterjee (University of Cincinnati Medical Center, Cincinnati) for antibody 1A7, and Annemarie Schimpf (Georg-Speyer-Haus), Rabiä El Kalaaeoui, Stephanie Erben and Regine Müller (all Pediatric Hematology and Oncology, University Hospital Frankfurt) for excellent technical assistance.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Smyth MJ, Cretney E, Kelly JM, et al. Activation of NK cell cytotoxicity. Mol Immunol. 2005;42:501–10. doi: 10.1016/j.molimm.2004.07.034. [DOI] [PubMed] [Google Scholar]
- 2.Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. 2008;9:495–502. doi: 10.1038/ni1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smyth MJ, Hayakawa Y, Takeda K, et al. New aspects of natural-killer-cell surveillance and therapy of cancer. Nat Rev Cancer. 2002;2:850–61. doi: 10.1038/nrc928. [DOI] [PubMed] [Google Scholar]
- 4.Terme M, Ullrich E, Delahaye NF, et al. Natural killer cell-directed therapies: moving from unexpected results to successful strategies. Nat Immunol. 2008;9:486–94. doi: 10.1038/ni1580. [DOI] [PubMed] [Google Scholar]
- 5.Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia. 1994;8:652–8. [PubMed] [Google Scholar]
- 6.Klingemann HG, Wong E, Maki G. A cytotoxic NK-cell line (NK-92) for ex vivo purging of leukemia from blood. Biol Blood Marrow Transplant. 1996;2:68–75. [PubMed] [Google Scholar]
- 7.Yan Y, Steinherz P, Klingemann HG, et al. Antileukemia activity of a natural killer cell line against human leukemias. Clin Cancer Res. 1998;4:2859–68. [PubMed] [Google Scholar]
- 8.Tam YK, Miyagawa B, Ho VC, et al. Immunotherapy of malignant melanoma in a SCID mouse model using the highly cytotoxic natural killer cell line NK-92. J Hematother. 1999;8:281–90. doi: 10.1089/106161299320316. [DOI] [PubMed] [Google Scholar]
- 9.Maki G, Klingemann HG, Martinson JA, et al. Factors regulating the cytotoxic activity of the human natural killer cell line, NK-92. J Hematother Stem Cell Res. 2001;10:369–83. doi: 10.1089/152581601750288975. [DOI] [PubMed] [Google Scholar]
- 10.Tonn T, Becker S, Esser R, et al. Cellular immunotherapy of malignancies using the clonal natural killer cell line NK-92. J Hematother Stem Cell Res. 2001;10:535–44. doi: 10.1089/15258160152509145. [DOI] [PubMed] [Google Scholar]
- 11.Arai S, Meagher R, Swearingen M, et al. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy. 2008;10:625–32. doi: 10.1080/14653240802301872. [DOI] [PubMed] [Google Scholar]
- 12.Uherek C, Tonn T, Uherek B, et al. Retargeting of natural killer-cell cytolytic activity to ErbB2-expressing cancer cells results in efficient and selective tumour cell destruction. Blood. 2002;100:1265–73. [PubMed] [Google Scholar]
- 13.Müller T, Uherek C, Maki G, et al. Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. Cancer Immunol Immunother. 2008;57:411–23. doi: 10.1007/s00262-007-0383-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tavri S, Jha P, Meier R, et al. Optical imaging of cellular immunotherapy against prostate cancer. Mol Imaging. 2009;8:15–26. [PubMed] [Google Scholar]
- 15.Wels W, Biburger M, Müller T, et al. Recombinant immunotoxins and retargeted killer cells: employing engineered antibody fragments for tumour-specific targeting of cytotoxic effectors. Cancer Immunol Immunother. 2004;53:217–26. doi: 10.1007/s00262-003-0482-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kershaw MH, Teng MW, Smyth MJ, et al. Supernatural T cells: genetic modification of T cells for cancer therapy. Nat Rev Immunol. 2005;5:928–40. doi: 10.1038/nri1729. [DOI] [PubMed] [Google Scholar]
- 17.Cheresh DA, Pierschbacher MD, Herzig MA, et al. Disialogangliosides GD2 and GD3 are involved in the attachment of human melanoma and neuroblastoma cells to extracellular matrix proteins. J Cell Biol. 1986;102:688–96. doi: 10.1083/jcb.102.3.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Modak S, Cheung NK. Disialoganglioside directed immunotherapy of neuroblastoma. Cancer Invest. 2007;25:67–77. doi: 10.1080/07357900601130763. [DOI] [PubMed] [Google Scholar]
- 19.Navid F, Santana VM, Barfield RC. Anti-GD2 antibody therapy for GD2-expressing tumours. Curr Cancer Drug Targets. 2010;10:200–9. doi: 10.2174/156800910791054167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pearson AD, Pinkerton CR, Lewis IJ, et al. High-dose rapid and standard induction chemotherapy for patients aged over 1 year with stage 4 neuroblastoma: a randomised trial. Lancet Oncol. 2008;9:247–56. doi: 10.1016/S1470-2045(08)70069-X. [DOI] [PubMed] [Google Scholar]
- 21.Zage PE, Kletzel M, Murray K, et al. Outcomes of the POG 9340/9341/9342 trials for children with high-risk neuroblastoma: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2008;51:747–53. doi: 10.1002/pbc.21713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matthay KK, Reynolds CP, Seeger RC, et al. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a Children’s Oncology Group study. J Clin Oncol. 2009;27:1007–13. doi: 10.1200/JCO.2007.13.8925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berthold F, Boos J, Burdach S, et al. Myeloablative megatherapy with autologous stem-cell rescue versus oral maintenance chemotherapy as consolidation treatment in patients with high-risk neuroblastoma: a randomised controlled trial. Lancet Oncol. 2005;6:649–58. doi: 10.1016/S1470-2045(05)70291-6. [DOI] [PubMed] [Google Scholar]
- 24.Handgretinger R, Anderson K, Lang P, et al. A phase I study of human/mouse chimeric antiganglioside GD2 antibody ch14.18 in patients with neuroblastoma. Eur J Cancer. 1995;31A:261–7. doi: 10.1016/0959-8049(94)00413-y. [DOI] [PubMed] [Google Scholar]
- 25.Yu AL, Gilman AL, Ozkaynak MF, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010;363:1324–34. doi: 10.1056/NEJMoa0911123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shusterman S, London WB, Gillies SD, et al. Antitumour activity of Hu14.18-IL2 in patients with relapsed/refractory neuroblastoma: a Children’s Oncology Group (COG) phase II study. J Clin Oncol. 2010;28:4969–75. doi: 10.1200/JCO.2009.27.8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huenecke S, Zimmermann SY, Kloess S, et al. IL-2-driven regulation of NK cell receptors with regard to the distribution of CD16+ and CD16- subpopulations and in vivo influence after haploidentical NK cell infusion. J Immunother. 2010;33:200–10. doi: 10.1097/CJI.0b013e3181bb46f7. [DOI] [PubMed] [Google Scholar]
- 28.Cinatl J, Cinatl J, Mainke M, et al. In vitro differentiation of human neuroblastoma cells induced by sodium phenylacetate. Cancer Lett. 1993;70:15–24. doi: 10.1016/0304-3835(93)90069-l. [DOI] [PubMed] [Google Scholar]
- 29.Houghton AN, Real FX, Davis LJ, et al. Phenotypic heterogeneity of melanoma. Relation to the differentiation program of melanoma cells. J Exp Med. 1987;165:812–29. doi: 10.1084/jem.165.3.812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Altenschmidt U, Kahl R, Moritz D, et al. Cytolysis of tumour cells expressing the Neu/erbB-2, erbB-3, and erbB-4 receptors by genetically targeted naive T lymphocytes. Clin Cancer Res. 1996;2:1001–8. [PubMed] [Google Scholar]
- 31.Gerstmayer B, Groner B, Wels W, et al. Stable expression of the ecotropic retrovirus receptor in amphotropic packaging cells facilitates the transfer of recombinant vectors and enhances the yield of retroviral particles. J Virol Methods. 1999;81:71–5. doi: 10.1016/s0166-0934(99)00053-1. [DOI] [PubMed] [Google Scholar]
- 32.Zimmermann SY, Esser R, Rohrbach E, et al. A novel four-colour flow cytometric assay to determine natural killer cell or T-cell-mediated cellular cytotoxicity against leukaemic cells in peripheral or bone marrow specimens containing greater than 20% of normal cells. J Immunol Methods. 2005;296:63–76. doi: 10.1016/j.jim.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 33.Kloss S, Bochennek K, Huenecke S, et al. A novel five-colour flow cytometric assay to determine NK cell cytotoxicity against neuroblastoma and other adherent tumour cells. J Immunol Methods. 2007;325:140–7. doi: 10.1016/j.jim.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 34.Gillies SD, Lo KM, Wesolowski J. High-level expression of chimeric antibodies using adapted cDNA variable region cassettes. J Immunol Methods. 1989;125:191–202. doi: 10.1016/0022-1759(89)90093-8. [DOI] [PubMed] [Google Scholar]
- 35.Mueller BM, Romerdahl CA, Gillies SD, et al. Enhancement of antibody-dependent cytotoxicity with a chimeric anti-GD2 antibody. J Immunol. 1990;144:1382–6. [PubMed] [Google Scholar]
- 36.Sen G, Chakraborty M, Foon KA, et al. Preclinical evaluation in nonhuman primates of murine monoclonal anti-idiotype antibody that mimics the disialoganglioside GD2. Clin Cancer Res. 1997;3:1969–76. [PubMed] [Google Scholar]
- 37.Gray JC, Kohler JA. Immunotherapy for neuroblastoma: turning promise into reality. Pediatr Blood Cancer. 2009;53:931–40. doi: 10.1002/pbc.22153. [DOI] [PubMed] [Google Scholar]
- 38.Simon T, Hero B, Faldum A, et al. Consolidation treatment with chimeric anti-GD2-antibody ch14.18 in children older than 1 year with metastatic neuroblastoma. J Clin Oncol. 2004;22:3549–57. doi: 10.1200/JCO.2004.08.143. [DOI] [PubMed] [Google Scholar]
- 39.Modak S, Cheung NK. Neuroblastoma: therapeutic strategies for a clinical enigma. Cancer Treat Rev. 2010;36:307–17. doi: 10.1016/j.ctrv.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 40.Raffaghello L, Prigione I, Airoldi I, et al. Downregulation and/or release of NKG2D ligands as immune evasion strategy of human neuroblastoma. Neoplasia. 2004;6:558–68. doi: 10.1593/neo.04316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kloess S, Huenecke S, Piechulek D, et al. IL-2-activated haploidentical NK cells restore NKG2D-mediated NK-cell cytotoxicity in neuroblastoma patients by scavenging of plasma MICA. Eur J Immunol. 2010;40:3255–67. doi: 10.1002/eji.201040568. [DOI] [PubMed] [Google Scholar]
- 42.Cartron G, Dacheux L, Salles G, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99:754–8. doi: 10.1182/blood.v99.3.754. [DOI] [PubMed] [Google Scholar]
- 43.Musolino A, Naldi N, Bortesi B, et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol. 2008;26:1789–96. doi: 10.1200/JCO.2007.14.8957. [DOI] [PubMed] [Google Scholar]
- 44.Schulz G, Cheresh DA, Varki NM, et al. Detection of ganglioside GD2 in tumour tissues and sera of neuroblastoma patients. Cancer Res. 1984;44:5914–20. [PubMed] [Google Scholar]
- 45.Kramer K, Gerald WL, Kushner BH, et al. Disialoganglioside G(D2) loss following monoclonal antibody therapy is rare in neuroblastoma. Clin Cancer Res. 1998;4:2135–9. [PubMed] [Google Scholar]
- 46.Ozkaynak MF, Sondel PM, Krailo MD, et al. Phase I study of chimeric human/ murine anti-ganglioside G(D2) monoclonal antibody (ch14.18) with granulocyte-macrophage colony-stimulating factor in children with neuroblastoma immediately after hematopoietic stem-cell transplantation: a Children’s Cancer Group Study. J Clin Oncol. 2000;18:4077–85. doi: 10.1200/JCO.2000.18.24.4077. [DOI] [PubMed] [Google Scholar]
- 47.Kremens B, Hero B, Esser J, et al. Ocular symptoms in children treated with human-mouse chimeric anti-GD2 mAb ch14.18 for neuroblastoma. Cancer Immunol Immunother. 2002;51:107–10. doi: 10.1007/s00262-001-0259-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sorkin LS, Otto M, Baldwin WM, 3rd, et al. Anti-GD(2) with an FC point mutation reduces complement fixation and decreases antibody-induced allodynia. Pain. 2010;149:135–42. doi: 10.1016/j.pain.2010.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Altvater B, Landmeier S, Pscherer S, et al. 2B4 (CD244) signaling by recombinant antigen-specific chimeric receptors costimulates natural killer cell activation to leukemia and neuroblastoma cells. Clin Cancer Res. 2009;15:4857–66. doi: 10.1158/1078-0432.CCR-08-2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rossig C, Bollard CM, Nuchtern JG, et al. Targeting of G(D2)-positive tumour cells by human T lymphocytes engineered to express chimeric T-cell receptor genes. Int J Cancer. 2001;94:228–36. doi: 10.1002/ijc.1457. [DOI] [PubMed] [Google Scholar]
- 51.Pule MA, Savoldo B, Myers GD, et al. Virus-specific T cells engineered to coexpress tumour-specific receptors: persistence and antitumour activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–70. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]