

Abstract
E2F-1-deleted mutant, ‘truncated E2F’ (E2Ftr, E2F-1[1–375]), lacking the carboxy-terminal transactivation domain, was shown to be more potent at inducing cancer cell apoptosis than wild-type E2F-1 (wtE2F-1; full-length E2F-1). Mechanisms by which wtE2F-1 and E2Ftr induce apoptosis, however, are not fully elucidated. Our study demonstrates molecular effects of pro-apoptotic BH3-only Bcl-2 family member Harakiri (Hrk) in wtE2F-1- and E2Ftr-induced melanoma cell apoptosis. We found that Hrk mRNA and Harakiri (HRK) protein expression was highly up-regulated in melanoma cells in response to wtE2F-1 and E2Ftr overexpression. HRK up-regulation did not require the E2F-1 transactivation domain. In addition, Hrk gene up-regulation and HRK protein expression did not require p53 in cancer cells. Hrk knockdown by Hrk siRNA was associated with significantly reduced wtE2F-1- and E2Ftr-induced apoptosis. We also found that an upstream factor, ‘downstream regulatory element antagonist modulator’ (DREAM), may be involved in HRK-mediated apoptosis in response to wtE2F-1 and E2Ftr overexpression. DREAM expression levels increased following wtE2F-1 and E2Ftr overexpression. Western blotting detected increased DREAM primarily in dimeric form. The homodimerization of DREAM resulting from wtE2F-1 and E2Ftr overexpression may contribute to the decreased binding activity of DREAM to the 3′-untranslated region of the Hrk gene as shown by electromobility shift assay. Results showed wtE2F-1- and E2Ftr-induced apoptosis is partially mediated by HRK. HRK function is regulated in response to DREAM. Our findings contribute to understanding the mechanisms that regulate wtE2F-1- and E2Ftr-induced apoptosis and provide insights into the further evaluation of how E2Ftr-induced apoptosis may be used for therapeutic gain.
Keywords: E2F-1 (wild-type E2F-1, wtE2F-1); truncated E2F-1 (E2Ftr); apoptosis; Hrk; downstream regulatory element antagonist modulator (DREAM)
Introduction
E2F-1 is the best-characterized member of the E2F family of transcription factors, which comprise nine E2F subunits with the ability to control transcription, cell cycle, apoptosis and senescence [1-4]. The full-length E2F-1 contains a highly conserved DNA binding domain, a hydrophobic heptad repeat domain required for dimerization (dimerization domain), a transactivation domain and other domains required for associations with cyclin A/cdk2 and Rb family members (Fig. 1A) [5-11]. The E2Ftr (1–375) is a C-terminal truncation of E2F-1 that lacks the entire transactivation and pRb-binding domains (Fig. 1A). The full-length E2F-1, but not a truncation mutant (E2Ftr), was able to induce colony formation in NIH-3T3 cells in semi-solid agar [12]. This implies that the transactivation domain of E2F-1 is required for the oncogenic activity of this gene. Studies on the molecular basis of E2F-1-induced apoptosis demonstrated that the transactivation and apoptosis functions of E2F-1 are separable [13-15]. A number of reports indicate that the E2Ftr has stronger pro-apoptotic activity than the full-length protein but without stimulating DNA synthesis [13-15]. Therefore, the clinical development of E2Ftr, which maintains its apoptotic function but no longer possesses oncogenic function, may improve the potential clinical utility of this therapeutic gene. In order to further investigate the death-inducing function of E2Ftr and explore its potential therapeutic activity in vitro and in vivo, we have constructed an E2Ftr adenovirus and tested its in vitro and in vivo anti-tumoural activity [16, 17]. We showed that E2Ftr induced apoptosis in a variety of cancer cell lines with little cytotoxicity to normal cell lines, and that E2Ftr exhibited more than an 80% decrease in tumour size in a mouse melanoma xenograft model [16, 17]. Hence, E2Ftr may be a suitable transgene with significant potential therapeutic activity both in vitro and in vivo.
Fig 1.
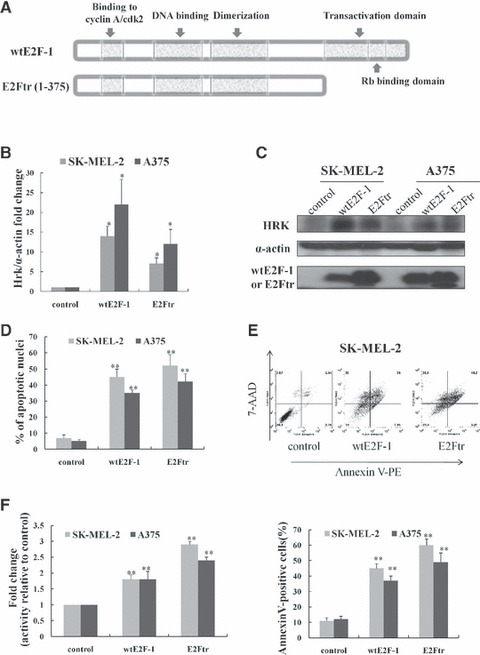
Overexpression of wtE2F-1 and E2Ftr leads to increased Hrk expression and apoptosis in melanoma cells. (A) Schematic representation of wtE2F-1 and E2Ftr domain structure. Two melanoma cell lines (SK-MEL-2 and A375) were infected with Ad-LacZ (control vector), Ad-wtE2F-1 or Ad-E2Ftr at MOI 100. After 24 hrs of infection, real-time RT-PCR (B) and Western blot analysis (C) were performed as described in Materials and methods. (B) RT-PCR results are expressed as Hrk fold increase relative to control vector-infected cells adjusted for α-actin as an internal control. Each experiment is a representation of three independent experiments performed in duplicate (bars: mean ± S.D.; *P < 0.05 compared with control; n = 3). (C) Cell lysates were subjected to Western blot analysis using HRK, wtE2F-1 or E2Ftr and α-actin antibody. (D) After 48 hrs of infection, the percentage of apoptotic cells showing typical apoptotic nuclei by Hoechst 33258 staining was counted under a fluorescence microscope. The percentage of apoptotic cells (annexin V-PE+ cells) were also determined by flow cytometry analysis (E) after 48 hrs of infection, as described in Materials and methods. (F) After 48 hrs of designated infection, the OD405 values of the cell lysates were measured as caspase-9 activity. The results were expressed as the fold change in treated cells over that of the control cells. Values represent mean ± S.D. of three independent experiments (bars: mean ± S.D.; **P < 0.01 compared with control; n = 3).
Until recently, little was known about the pathways by which E2F-1 and E2Ftr exert such apoptotic effects [10, 18, 19]. We, and others, have identified several E2F-1 targets involved in the execution of the apoptotic program [20, 21]. For example, in our previous studies, we found that full-length E2F-1 can induce melanoma cell apoptosis via PUMA (p53 up-regulated modulator of apoptosis) up-regulation and Bcl-2-associated X protein (BAX) translocation, whereas E2Ftr could not activate PUMA promoter activity, suggesting that increased PUMA expression by E2F-1 is dependent on its transactivation domain [21].
In this study, we sought to clarify the role of HRK, another BH3-only family member, in wild-type (wt) E2F-1- and E2Ftr-induced apoptosis. The product of the Harakiri gene, HRK, is one of the mammalian Bcl-2 homology region 3 (BH3)-only proteins identified during screening for proteins that interact with Bcl-2. HRK physically interacts with the death-repressor proteins BCL-2 and BCL-XL and exhibits death-inducing activity in mammalian cells [22, 23]. Extensive studies have shown that BH3-only proteins, including HRK, can promote permeabilization of the mitochondrial membrane in response to apoptotic stimuli [24]. However, no reports have documented the role of HRK in mediating wtE2F-1- and E2Ftr-induced apoptosis.
Materials and methods
Cell lines and culture reagents
The human melanoma cell lines SK-MEL-2 and A375, the lung cancer cell line H1299 and the osteosarcoma cell line SAOS2 were purchased from American Type Culture Collection (Rockville, MD, USA). Cells were cultured in a 5% CO2 incubator at 37°C and subcultured every 3 to 4 days (about 80% confluent) in α minimal essential medium (α-MEM) for SK-MEL-2 cells and H1299 cells or DMEM for A375 cells or sarcoma osteogenic (SAOS2) cells, supplemented with 10% heat-inactivated foetal bovine serum and penicillin (100 U/ml)/streptomycin (100 μg/ml) solution. All cell culture reagents were obtained from Gibco/BRL (Bethesda, MD, USA).
Adenoviral vectors and infection conditions
Four replication-defective recombinant adenoviral vectors deleted at the viral E1 gene were used in this study. The Ad5CMV-E2F-1 (Ad-wtE2F-1) and Ad5CMV-LacZ (Ad-LacZ) (as a control) vectors contained the transgenes wtE2F-1 and nuclear-localized β-galactosidase, respectively, under control of the cytomegalovirus (CMV) promoter as described [16]. The Ad-E2Ftr vector expresses E2Ftr protein, which retains E2F-1 DNA binding, but lacks its transactivation domain. Our group constructed this based on the widely used Ad-Easy system [16, 17]. The cells were infected at a multiplicity of infection of 100 (MOI 100). The infection procedure has been described in detail previously [16].
siRNA transfection
Control (non-sense) siRNA, Hrk siRNA and siGLO Green Transfection Indicator were obtained from Dharmacon, Inc. (Lafayette, CO, USA). The transfection of siRNA was carried out using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). After adenovirus vector infection, the medium was changed to Opti-Minimum Essential Medium I (MEMI). Transfection was carried out according to the manufacturer’s instructions.
Detection of apoptosis
Hoechst staining was performed by adding Hoechst dye to a final concentration of 10 μM to each well in a 12-well plate. Apoptotic cell death was determined by quantification of apoptotic nuclei (i.e. fragmentation and condensation of nuclei) following Hoechst 33258 staining for 10 min. at 37°C. A total of 400 nuclei were counted for each sample under a fluorescence microscope (Olympus Microsystems, Redwood City, CA, USA).
Quantification of apoptosis was further assessed by annexin V staining according to the manufacturer’s instructions [annexin V-PE (phycoerythrin) apoptosis kit; Pharmingen, San Diego, CA, USA] and as described previously [17]. Apoptotic cells were evaluated by FACScan flow cytometry (Becton Dickinson, Franklin Lakes, NJ, USA) with FlowJo software (Tree Star, Inc., Ashland, OR, USA).
Caspase-9 activity assay
The caspase-9 colorimetric assay kit (R&D Systems, Minneapolis, MN, USA) was used for caspase-9 activity assay, according to the manufacturer’s instructions and as described previously [21]. No cell lysates and no substrates were used as blanks. The results were expressed as the fold increase in treated cells over that of the control cells.
Real-time RT-PCR
Real-time RT-PCR was performed with the SYBR Green PCR Master Mix kit, according to the manufacturer’s protocol (ABI, Foster City, CA, USA). Briefly, at 24 hrs of designated treatment, total RNA was isolated using the RNeasy mini kit, along with RNase Free DNase set to remove any traces of DNA contamination (QIAGEN, Valencia, CA, USA). cDNA was prepared from 500 ng of total RNA with TaqMan reverse transcription reagents (ABI/Roche, Branchburg, NJ, USA). Real-time RT-PCR was performed with 12.5 ng of cDNA as a template for each gene. Reactions were carried out in a 96-well optical reaction plate (ABI) using the ABI Prism 7000 sequence detection system. Primers for the Hrk gene were 5′-CGA TCC ACA CGG AGT ACT TG-3′ and 5′-GGA TGC AGA AGG AGA TCA CTG-3′. α-actin served as an internal control. The primers for α-actin were 5′-GAA GTA GCC GTT TAC AAG CTA AGC A-3′ and 5′-GCC TGG ATT ATC TGG GCT TCT-3′. Data were analysed using the comparative CT method [14] and were presented as mean ± S.D. from three independent experiments performed in duplicate.
Western blot
Cells were treated as indicated. Western blotting was performed as described previously [21]. Human HRK and downstream regulatory element antagonist modulator (DREAM) were detected using the HRK antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and the DREAM antibody (Upstate Biotechnology, Billerica, MA, USA), respectively. wtE2F-1 and E2Ftr were detected by the same E2F-1 antibody from Santa Cruz Biotechnology. wtE2F-1 is about 66 kD and E2Ftr is about 60 kD, which can be differentiated by a 10% SDS-PAGE, but not by a 12% SDS-PAGE. Equal loading of proteins was verified by probing the membrane again with an anti-α-actin primary antibody (1:5000; Sigma-Aldrich, St. Louis, MO, USA).
Immunocytochemistry and confocal microscopy
Cells were seeded on cover slips in 12-well plates and cultured in a 5% CO2 incubator at 37°C. After the designated treatment, cells were incubated with mitotracker Red 580 (Invitrogen) with the final concentration of 300 nM at 37°C for 40 min. The cover slips were rinsed with warm medium twice and fixed with 3.7% formaldehyde at 37°C for 15 min. Primary antibody E2F-1 or HRK (Santa Cruz Biotechnology) or DREAM (Upstate Biotechnology) was applied at 1:100 at room temperature for 45 min. Subsequently, the cells were washed three times for 30 min. and incubated with the secondary antibody [goat antimouse Alexa 488 (green), donkey anti-goat Alexa 488 (green) or donkey anti-goat Alexa 594 (red) at 1:500; Invitrogen] at room temperature for 40 min. Cells were counterstained at room temperature with 200 nM of 4′, 6-diamidino-2-phenylindole (DAPI; shown in blue at left upper panel of all the confocal microscopy images; Santa Cruz Biotechnology) for 2 min. The cells were then washed extensively and mounted with Mowiol (Calbiochem, La Jolla, CA, USA). Scans were performed in a sequential mode to avoid channel crosstalk. Confocal microscope images were obtained on an Olympus Fluoview 500 confocal microscope.
Electrophoretic mobility shift assay (EMSA) for DREAM-Hrk binding activity
SK-MEL-2 and A375 cells were seeded at a density of 3.5 × 105 cells in a 6-well plate. After 24 hrs, the cells were transfected as designated. After 24 hrs of infection, nuclear protein and cytosolic protein were extracted using NE-PER nuclear and cytoplasmic extraction reagent (Pierce, Rockford, IL, USA). For EMSA, binding reactions were carried out using 40 fmol of biotin end-labelled double-stranded DRE-Hrk oligonucleotide: DRE-Hrk sense 5′-biotin-GAAACACAGACAGAGGAAGCCCCTCGGGAG-3′ DRE-Hrk antisense 5′-biotin-CTCCCGAGGGGCTTCCTCTGTCTGTGTTTC-3′ (synthesized by Integrated DNA Technologies, Inc., Coralville, IA, USA) with 4 μg of nuclear protein extract. No protein extract was used as negative control. A 200-fold amount of unlabelled DRE-Hrk was used as the specific competitor where indicated. Supershift assay was performed by adding the DREAM antibody to the nuclear extract and incubating at room temperature for 20 min., followed by the steps for a normal gel shift. Assays were loaded onto 5% polyacrylamide gels and electrophoresed at 100 V before being transferred onto a positively charged nylon membrane. Transferred DNAs were crosslinked to the membrane and detected using horseradish peroxidase-conjugated streptavidin (LightShift chemiluminescent EMSA kit; Pierce), according to the manufacturer’s instructions. For loading control, 10 μg of nuclear proteins from each sample were subjected to Western blot analysis for the proliferating cell nuclear antigen (PCNA, 1:800; Santa Cruz Biotechnology).
Data analyses
Experiments presented in the figures are representative of three or more independent experiments. The data are presented as the mean ± S.D. Comparisons between groups were evaluated by a two-tailed Student’s t-test. A P value of <0.05 was considered to be statistically significant.
Results
Overexpression of wtE2F-1 and E2Ftr leads to increased HRK expression and apoptosis in melanoma cells
We have previously reported [21] that wtE2F-1 can induce apoptosis in melanoma cells via PUMA up-regulation, and that the transactivation domain of wtE2F-1 is a requirement for this process. To further investigate whether another BH3-only family member, HRK, is involved in wtE2F-1- and E2Ftr-induced apoptosis, we examined the effect of wtE2F-1 and E2Ftr expression on Hrk mRNA expression and protein expression over time in two melanoma cell lines (SK-MEL-2 and A375). Quantitative real-time PCR analysis showed that there was a 14-fold (SK-MEL-2) and 22-fold (A375) increase of Hrk mRNA levels in melanoma cells at 24 hrs after wtE2F-1 infection compared with the control virus-infected cells. After E2Ftr infection at 24 hrs, there was an increase of 7-fold (SK-MEL-2) and 12-fold (A375 cells) of Hrk mRNA levels as compared with the control virus infection (Fig. 1B).
To determine whether Hrk mRNA up-regulation was associated with a corresponding increase in HRK protein levels, we performed Western blot analysis after wtE2F-1 and E2Ftr expression. The HRK protein levels were also increased at 24 hrs after wtE2F-1 and E2Ftr infection (Fig. 1C). The alterations of the HRK protein level corresponded to changes in the Hrk mRNA level.
Along with the overexpression of HRK, we examined apoptosis by Hoechst staining to observe the typical apoptotic nuclear morphological changes, including chromatin condensation and nuclear fragmentation under the fluorescence microscope. The percentage of apoptotic cells was 45% in SK-MEL-2 cells and 35% in A375 cells, respectively, after 48 hrs of wtE2F-1 infection. After 48 hrs of E2Ftr infection, the percentage of apoptotic cells was 52% in SK-MEL-2 cells and 39% in A375 cells, respectively (Fig. 1D). After 48 hrs of wtE2F-1 and E2Ftr infection, a similar percentage of apoptotic cells was also observed in SK-MEL-2 and A375 cells by using annexin V-PE/7-aminoactinomycin D (7 AAD) flow cytometric analysis (Fig. 1E). Additionally, consistent with apoptosis, caspase-9 activity was induced after 48 hrs of wtE2F-1 and E2Ftr infection in SK-MEL-2 and A375 cells (Fig. 1F). These data demonstrate an association between apoptosis and increased HRK expression following wtE2F-1 and E2Ftr overexpression in melanoma cells. Our data also showed that up-regulation of HRK was independent of the E2F-1 transactivation domain.
Up-regulation of HRK in wtE2F-1- and E2Ftr-induced apoptosis is independent of p53 status
Lack of p53, p53 mutation or aberrant p53 expression is generally associated with aggressive malignancy and chemotherapy resistance in most cancers [25]. As shown above, even though the SK-MEL-2 melanoma cell line has a mutant p53, it still shows up-regulation of HRK and apoptosis after wtE2F and E2Ftr expression. We surmised that this HRK up-regulation by wtE2F and E2Ftr was p53-independent. Therefore, to further explore this, we took advantage of two cell lines: H1299 (p53-null lung cancer cell line) and SAOS2 (osteosarcoma cell line that lacks p53 and the retinoblastoma gene product, pRB). We found that Hrk mRNA (Fig. 2A) and protein levels (Fig. 2B) were elevated after 24 hrs of overexpression of wtE2F and E2Ftr in both cell lines. Concurrent with the increased HRK elevation, we observed significant wtE2F-1- and E2Ftr-induced apoptosis in both cell lines after 48 hrs of wtE2F-1 and E2Ftr infection. This was based on the number of apoptotic cells counted by using Hoechst staining (Fig. 2C) and by flow cytometric analysis (Fig. 2D). These data indicate that enhanced HRK up-regulation, as well as enhanced apoptosis after overexpression of wtE2F and E2Ftr, are not dependent on p53 among some different types of cancer cells.
Fig 2.
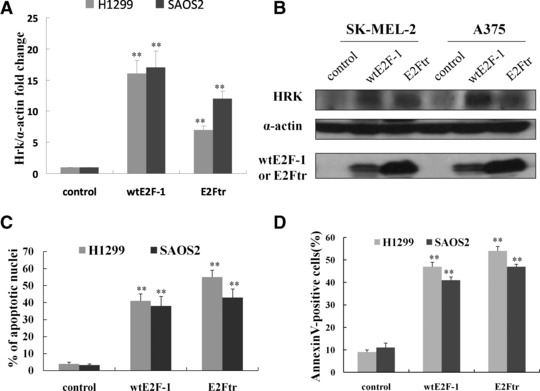
Up-regulation of HRK in wtE2F-1- and E2Ftr-induced apoptosis is independent of p53 status. H1299 and SAOS2 cells, which lack p53, were infected at MOI 100. After 24 hrs of infection, RT-PCR (A) and Western blot analysis (B) were performed as described in Figure 1B and C. After 48 hrs of infection, the percentage of apoptotic cells was counted by Hoechst 33258 staining under a fluorescence microscope (C) and determined by annexin V-PE staining by flow cytometry analysis (D). Values represent mean ± S.D. of three independent experiments (bars: mean ± S.D.; **P < 0.01 compared with control; n = 3).
Hrk knockdown by siRNA in wtE2F-1 and E2Ftr-overexpressed cells was associated with significantly reduced apoptosis
We sought to determine whether HRK was functionally involved in wtE2F-1- and E2Ftr-mediated apoptosis. We theorized that if HRK was truly a mediator in E2Ftr-induced melanoma cell apoptosis, there should be a significant reduction in E2Ftr-induced apoptosis after Hrk knockdown. We used Hrk siRNA transfection to confirm our prediction. We first used the siGLO to check the siRNA transfection efficiency in these two cell lines. Both cell lines demonstrated >70% transfection efficiency (data not shown). Transfection of control siRNA in wtE2F- or E2Ftr-infected cells increased Hrk expression in SK-MEL-2 and A375 cell lines after 24 hrs of transfection. Transfection of Hrk siRNA significantly repressed wtE2F-1- and E2Ftr-induced Hrk expression after 24 hrs of transfection as shown by real-time RT-PCR (Fig. 3A). Accordingly, Hrk knockdown by siRNA was associated with significantly reduced wtE2F-1- and E2Ftr-induced apoptosis (Fig. 3B). Forty-eight hours after transfection of control siRNA in wtE2F-1- and E2Ftr-infected cells, the apoptotic nuclei increased to about 50% to 60% in these two cell lines. The apoptotic nuclei were reduced to 30% to 35% after 48 hrs of transfection of Hrk siRNA in wtE2F-1- and E2Ftr-infected melanoma cells (P< 0.05; Fig. 3B). A similar trend was also observed in SK-MEL-2 and A375 cells by annexin V/7-AAD flow cytometric analysis (Fig. 3C). Moreover, in concordance with significantly reduced apoptosis by Hrk siRNA knockdown in wtE2F-1 and E2Ftr overexpressed cells, caspase-9 activity was also repressed (Fig. 3D). These results indicate that wtE2F-1- and E2Ftr-induced melanoma cell apoptosis is mediated, at least in part, by HRK.
Fig 3.
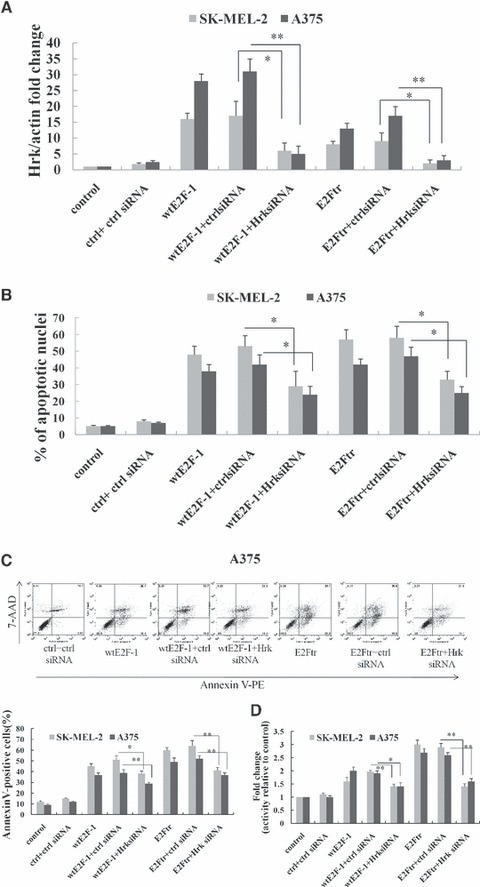
Hrk knockdown by siRNA was associated with significantly reduced wtE2F-1- or E2Ftr-induced apoptosis. SK-MEL-2 and A375 cells were infected with control vector, Ad-wtE2F-1 or Ad-E2Ftr at MOI 100, followed by transfection with control siRNA or Hrk siRNA as indicated. (A) RT-PCR was performed after 24 hrs of transfection as described in Materials and methods. The bar graph is expressed as Hrk fold increase relative to control vector-infected cells adjusted for α-actin as an internal control. Each experiment is a representation of three independent experiments performed in duplicate (Ctrl: control; bars: mean ± S.D.; *P < 0.05; **P < 0.01 compared with either wtE2F-1 or E2Ftr infection plus control siRNA transfection; n = 3). After 48 hrs of transfection, the percentage of cells showing typical apoptotic nuclei was counted by Hoechst 33258 staining under a fluorescence microscope (B) and determined by annexin V-PE staining by flow cytometry analysis (C). (D) After 48 hrs of transfection, caspase-9 activity was determined as in Figure 1F. Values represent mean ± S.D. of three independent experiments. (Ctrl: control; bars: mean ± S.D.; *P < 0.05; **P < 0.01 compared with either wtE2F-1 or E2Ftr infection plus control siRNA transfection; n = 3).
wtE2F-1 and E2Ftr do not directly act to induce apoptosis at mitochondria
Because HRK is a mitochondria protein, we were interested in determining whether wtE2F-1 and E2Ftr would act directly to induce apoptosis at the mitochondria, as p53 does [26, 27]. To determine this, we first stained the cell mitochondria with mitotracker red 580 (left lower panel, Fig. 4A) and then applied the HRK antibody followed by the second antibody with Alexa 488 (green fluorescence, right upper panel, Fig. 4A). Confocal microscope sequential scanning shows red (mitochondria) and green fluorescence (HRK) with an overlaying yellow (right lower panel, Fig. 4A), which represents co-localization of HRK in mitochondria. The subcellular distribution of wtE2F-1 or E2Ftr is shown after staining the mitochondria with mitotracker red 580 (left lower panel, Fig. 4B) and with E2F-1 antibody followed by the second antibody with Alexa 488 (green fluorescence, right upper panel, Fig. 4B). wtE2F-1 and E2Ftr (green fluorescence, right upper panel, Fig. 4B) were primarily distributed at the nucleus and cytosol. Confocal microscope sequential scanning did not show an overlay of yellow colour (right lower panel, Fig. 4B), which suggests that wtE2F-1 and E2Ftr are not located at the mitochondria.
Fig 4.
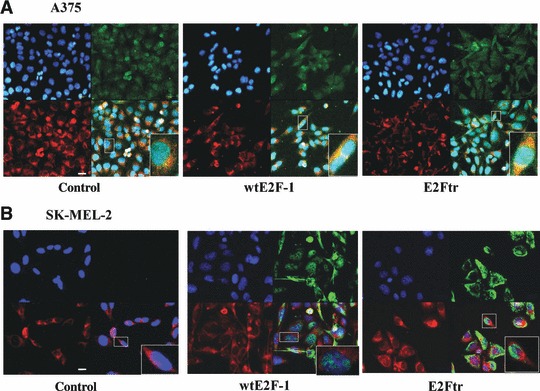
wtE2F-1 and E2Ftr are not directly acting to induce apoptosis at mitochondria. After 24 hrs of infection, A375 cells (A) and SK-MEL-2 cells (B) were stained with mitotracker red 580 and HRK (A) or E2F-1 (B) and then counterstained with DAPI (blue) (upper left panel) as described in Materials and methods (magnification, ×600). (A) An overlay (lower right panel) of the red (mitochondria, lower left panel) and the green (HRK, upper right panel) is provided in yellow to show co-localization of HRK in mitochondria. (B) wtE2F-1 or E2Ftr is primarily distributed in the nuclei and cytoplasma as shown in green (right upper panel). An overlay (lower right panel) of the red (mitochondria, lower left panel) and the green (wtE2F-1 or E2Ftr, right upper panel) is not shown in yellow, which suggests wtE2F-1 or E2Ftr is not located in mitochondria. Insets represent higher magnifications of the boxed areas (bar = 20μm).
Our Western blot analysis of the subcellular fraction of wtE2F-1 and E2Ftr confirmed the results from the confocal microscopy (unpublished data, Hongying Hao). These results demonstrate that wtE2F-1 and E2Ftr do not localize to the mitochondria to induce apoptosis.
wtE2F-1- and E2Ftr-induced melanoma cell apoptosis is mediated by Hrk that is dependent on DREAM function
Previous studies [28, 29] have shown that there is a silencer sequence in the 3′-untranslated region of the Hrk gene (DRE-Hrk) binding to the transcriptional repressor DREAM, and that Hrk is transcriptionally silenced via DREAM. To delineate how DREAM is involved in the regulation of Hrk in E2Ftr-induced apoptosis, we performed an EMSA using DREAM-hrk. The nuclear extracts were prepared and used to analyze the binding of DREAM to a biotin end-labelled probe that contained the DREAM binding site (DRE-Hrk). After wtE2F-1 and E2Ftr overexpression, the binding activity of DREAM to the Hrk gene was reduced (Fig. 5A), which correlates with the up-regulation of Hrk and induction of apoptosis (Fig. 5B). The supershift assay (Fig. 5C) showed the specific binding band shifted due to the formation of a larger complex after the addition of anti-DREAM antibody. Overexpression of wtE2F-1 or E2Ftr inhibited binding of the transcriptional inhibitor DREAM to the DRE-Hrk sequence, which may explain increased Hrk gene expression in response to wtE2F-1 and E2Ftr. As a whole, these results implicate HRK as a mediator of wtE2F-1- and E2Ftr-induced apoptosis, and that this response is modulated by DREAM.
Fig 5.
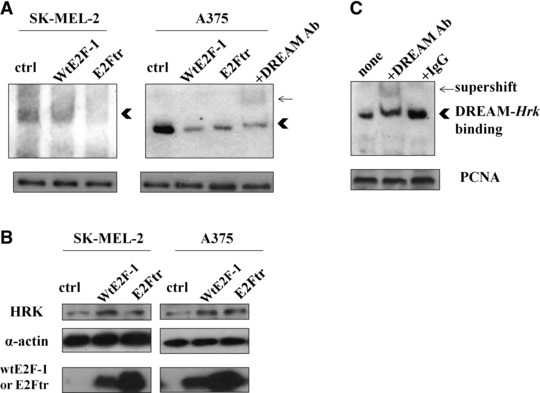
wtE2F-1 and E2Ftr expression resulted in reduced binding of DREAM to the Hrk gene and was correlated with increased HRK up-regulation and apoptosis. SK-MEL-2 and A375 cells were infected as indicated. After 24 hrs of infection, nuclear and cytoplasma protein were extracted. (A) Nuclear proteins (4 μg) were subjected to EMSA using biotin-labelled DRE-Hrk. PCNA Western blot analysis was used as loading control of nuclear proteins from each sample (Ctrl: control virus infected cell nuclear protein lysate; arrow: supershifted band; arrowhead: specific DREAM-Hrk binding). (B) 50 μg of cytoplasma proteins were subjected to Western blot analysis using HRK, wtE2F-1 or E2Ftr, and α-actin antibody. (C) Supershift assay using 10 μg of control virus-infected cell nuclear protein lysate is shown. The specific binding band of DREAM-Hrk (arrowhead) was shifted upward by the addition of anti-DREAM antibody (arrow), but not by control IgG.
Co-localization of DREAM with wtE2F-1 or E2Ftr and homodimerization of DREAM after wtE2F-1 and E2Ftr overexpression
To better understand how wtE2F-1 and E2Ftr interfere with the binding of DREAM to DRE-Hrk sequence, we used confocal microscopy to detect the cellular distribution of DREAM after wtE2F-1 and E2Ftr infection. DREAM (shown in red, left lower panel, Fig. 6A) was primarily located in the cell nuclei in the control vector (Ad-LacZ) infected cells. After wtE2F-1 and E2Ftr infection (shown in green, right upper panel, Fig. 6A), an overlay image of DREAM and wtE2F-1 or E2Ftr is yellow (right lower panel), suggesting that DREAM co-localized with wtE2F-1 or E2Ftr in the nuclei. This result demonstrated that the co-localization of DREAM with wtE2F-1 or E2Ftr is specific for these proteins and not simply a result of increased protein due to infection, because the Ad-LacZ was used as a control vector. We then performed RT-PCR to examine DREAM mRNA expression. The DREAM level increased after overexpression of wtE2F-1 and E2Ftr (Fig. 6B). This increased DREAM level was mostly in the dimeric form, as is shown in the Western blot analysis of the whole cell lysates (Fig. 6C). We isolated cytosolic and nuclear fraction of the cells to perform Western blot analysis and detected the monomeric and dimeric forms of DREAM, as previously characterized in another cell system [30]. We found that the DREAM monomer cannot be detected in the cytoplasm, but is present in the nuclei. The DREAM dimeric form was more obvious in the nuclei than in that of the dimeric form in the cytoplasm (Fig. 6D and E). The DREAM dimeric form was significantly present in wtE2F-1-treated cells and was more evident in E2Ftr-treated cells. These data suggest that the homodimerization of DREAM may cause the decreased binding activity of DREAM to the DRE-hrk sequence, thus allowing up-regulation of HRK and subsequent apoptosis.
Fig 6.
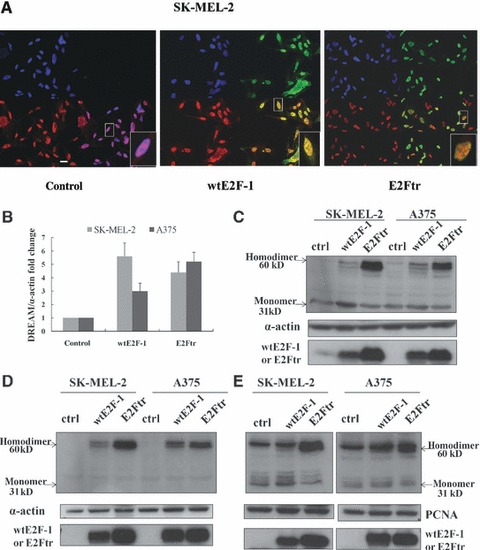
Co-localization of DREAM with wtE2F-1 or E2Ftr in the nuclei and homodimerization of DREAM after wtE2F-1 and E2Ftr overexpression. (A) After 24 hrs of infection in SK-MEL-2 cells, cells were incubated with DREAM antibody followed by Alexa-594 second antibody (shown in red, lower left panel) and E2F-1 antibody followed by Alexa-488 second antibody (shown in green, upper right panel) and then counterstained with DAPI (blue, upper left panel). An overlay (lower right panel) of yellow shows the co-localization of wtE2F-1 or E2Ftr with DREAM in the nuclei (magnification, ×600). Insets represent higher magnifications of the boxed areas (bar = 20 μm). (B) After 24 hrs of infection in SK-MEL-2 and A375 cells, RT-PCR of the DREAM gene was performed as described in Materials and methods. The bar graph is expressed as fold increase relative to control vector-infected cells adjusted for α-actin as an internal control. Each experiment is a representation of three independent experiments performed in duplicate (bars: mean ± S.D.). After 24 hrs of infection in SK-MEL-2 and A375 cells, 60 μg of total proteins from the whole cell lysates (C), cytoplasma (D) and nuclear (E) protein were extracted and subjected to Western blot analysis by using DREAM antibody. α-Actin was used for loading control of the whole cell lysates and cytoplasma fraction. PCNA was used for the loading control of the nuclear fraction (Ctrl: control; arrows: DREAM monomer or homodimer).
Discussion
The biochemical activation of classical apoptosis is mediated by two central pathways: the extrinsic (or death receptor) pathway and the intrinsic (or mitochondrial) pathway. The intrinsic pathway originates from the mitochondrial release of cytochrome c that leads to activation of caspase-9 and subsequent activation of the caspase cascade [24, 31, 32]. The Bcl-2 family proteins play a critical role in the signalling and execution of death signals in the mitochondrial pathway. They are usually grouped into three distinct categories: the anti-death proteins (BCL-2, BCL-XL, Mcl-1 and BCL-W); the pro-death proteins [BAX, Bcl-2 antagonist/killer (BAK) and Bcl-2-related ovarian killer (BOK)] and the larger group of BH3-only proteins [BH3-interacting domain death agonist (BID), Bcl-2 antagonist of cell death (BAD), Bcl-2-like protein 11 (BIM), PUMA, NOXA and HRK] [33].
Several studies have shown that the BH3-only protein HRK is an important mediator of apoptosis. Ma et al. [34] reported that HRK plays a critical role in potassium deprivation-induced apoptosis in cerebellar granule neurons. Kalinec et al. [35] also showed that gentamicin-induced apoptosis of auditory cells is mediated by the extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase pathway through up-regulation of HRK, whereas L-carnitine can prevent gentamicin-induced HRK up-regulation and apoptosis. HRK inactivation is associated with a low apoptotic index in secondary glioblastomas [36]. Previous studies [20, 21] have shown that full-length E2F-1 can up-regulate HRK through a direct transcriptional mechanism. In the present study, we showed that E2Ftr, lacking the transactivation domain, could still induce Hrk up-regulation and cause melanoma cell apoptosis. Our findings show that there may be an indirect transcriptional mechanism, DREAM-HRK, involved in E2Ftr-induced apoptosis, as discussed later. We observed that up-regulation of Hrk by wtE2F-1 was more prominent than that by E2Ftr (Fig. 1B). One possible explanation is that wtE2F-1 could induce apoptosis by both the direct transcriptional pathway and indirect transcriptional pathway, whereas E2Ftr could only induce apoptosis through the indirect transcriptional pathway (Fig. 7).
Fig 7.
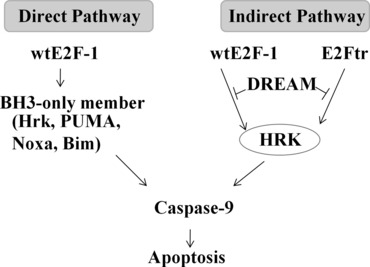
Schematic representation of the differences between the mechanism of wtE2F-1- and E2Ftr-induced apoptosis.
Previous studies have shown that there is a silencer sequence located in the 3′ untranslated region of the Hrk gene [28, 37-39]. This sequence binds to a calcium-binding protein, DREAM, which functions as a transcriptional repressor [28–30]. Loss of the DREAM-DNA binding complex was associated with increased levels of HRK and apoptosis [30]. In our DREAM-Hrk EMSA, we found that overexpression of wtE2F-1 and E2Ftr inhibited binding of DREAM to the DRE-Hrk sequence. This was correlated with increased HRK expression and apoptosis. Further analysis of DREAM expression showed that the DREAM level increased after wtE2F-1 and E2Ftr infection. Most of the increases occurred with DREAM in dimeric form after wtE2F-1 and E2Ftr overexpression, according to Western blot analyses. The homodimerization of DREAM in the cytoplasm and nucleus may explain the decreased binding activity of DREAM-Hrk, the correspondingly increased HRK expression and the eventual cellular apoptosis. In our study, we observed that DREAM occurred primarily in the monomeric and dimeric forms. In DREAM-overexpressed HEK293 cells, the monomeric, dimeric and tetrameric forms were all observed [30]. This might be due to discrepancies in different cell systems. Thus, DREAM appears to be an upstream regulator of Hrk in both wtE2F-1- and E2Ftr-induced melanoma cell apoptosis. This may be a common DREAM-HRK pathway shared by wtE2F-1- and E2Ftr-induced apoptosis.
DREAM, also known as calsenilin and potassium-channel interacting protein-3, is a multi-functional protein. It has roles in the cytoplasm as a potassium channel interacting protein and a presenilin interacting protein. DREAM has been identified as a pro-apoptotic protein. DREAM expression could induce the morphological and biochemical features of apoptosis, such as cell shrinkage, DNA laddering and caspase activation. DREAM-induced apoptosis was suppressed by caspase inhibitor and by Bcl-xL and was potentiated by increasing cytosolic Ca2+ [40, 41]. This pro-apoptotic function of DREAM is mostly observed in its monomeric form in neuron cells. DREAM can also affect cellular calcium homeostasis. We present here the evidence of DREAM as a transcriptional repressor in wtE2F-1 and E2Ftr-mediated apoptosis. Undoubtedly, it is worthy of further investigation on other functions of DREAM in E2F-1-mediated melanoma cell apoptosis.
We observed that even though up-regulation of Hrk by wtE2F-1 was more prominent than that by E2Ftr (Fig. 1B), E2Ftr exhibited stronger apoptotic effects than that of wtE2F-1 (Fig. 1D and E). Western blotting (Figs 1C, 2B, 5B and 6C) showed that the E2Ftr band was significantly stronger than the wtE2F-1 band. This is in accordance with our previous observation that E2Ftr protein was more stable and accumulated to higher levels in cells compared with wtE2F-1 [17]. Higher E2Ftr protein accumulated in melanoma cells may lead to stronger apoptotic effects.
Our results showed that up-regulation of Hrk by wtE2F-1 occur independently of its transactivation domain. This also supports the idea that the transactivation and apoptosis functions of E2F-1 are separable. A recent study showed that a region of only 75 amino acids within the DNA-binding domain of E2F-1 is responsible for the apoptotic function of the E2Ftr [12]. However, the mechanism of this 75 amino acid region in E2F-1 to induce apoptosis is still unclear. Identifying the factors that might be involved in the regulation of wtE2F-1- and E2Ftr-induced apoptosis is important not only to better understand the E2F family members, but also to clarify targets of E2Ftr for a possible cancer therapeutic strategy. Identification of pathways shared by wtE2F-1, E2Ftr or even smaller amino acid regions in the E2F family that can induce apoptosis may potentially represent therapeutic targets for molecular cancer therapy.
In addition to the DREAM-HRK pathway involved in wtE2F-1- and E2Ftr-induced apoptosis, we were interested in determining whether there might be other pathways involved in this process. Some transcription factors, such as p53, can act in both the cytosol and mitochondria where they can interact with various members of the Bcl-2 family to promote apoptosis through transcription-independent mechanisms [26, 27]. We attempted to determine whether this would be another possible mechanism for wtE2F-1- and E2Ftr-induced apoptosis. Confocal microscopy showed that wtE2F-1 and E2Ftr did distribute in the cytosol and nuclei. However, neither wtE2F-1 nor E2Ftr translocated to the mitochondria. This makes it unlikely that wtE2F-1 or E2Ftr act directly at the mitochondria to induce apoptosis. However, we did observe that wtE2F-1 and E2Ftr co-localized with DREAM in the nuclei. Future investigation is required to clarify the interaction of wtE2F-1 or E2Ftr with DREAM or DREAM homodimer.
p53, often referred to as the guardian of the genome, is mutated in up to 60% of many human malignancies. Such p53 mutations are particularly common in skin cancers. These p53 disruptions may impact on melanoma at all stages and relate directly to a patient’s prognosis [42]. Experimental cancer therapeutics that can target p53-dependent and p53-independent pathways would be given preferable choice. Our results showed that wtE2F-1 and E2Ftr induced HRK up-regulation, and that subsequent apoptosis is independent of p53 status in melanoma, lung cancer and osteosarcoma cells. This confirms that wtE2F and E2Ftr can induce apoptosis in a p53-dependent manner as well as in a p53-independent manner [43]. The data further suggest that wtE2F-1 and E2Ftr may have potential use in therapeutic gene strategies in a wide variety of tumours.
In the present study, we also found that even though Hrk mRNA expression has been successfully repressed by Hrk siRNA, down-regulation of Hrk can only partially repress wtE2F-1 and E2Ftr-induced apoptosis. Eventually, all of the cells underwent apoptosis after wtE2F-1 and E2Ftr overexpression (data not shown). These results suggest that there are other pathways or factors that might be involved in the apoptotic process, such as the death receptor pathway, or autophagy, among others.
Acknowledgments
We are grateful to Dr. Chi Li for helpful discussion and critical reading of the manuscript and Mrs. Margaret Abby for her expert manuscript editing. We thank Mr. Andrew Marsh of the University of Louisville’s Conn Center for Renewable Energy Research for additional technical editing services.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.DeGregori J, Leone G, Miron A, et al. Distinct roles for E2F proteins in cell growth control and apoptosis. Proc Natl Acad Sci USA. 1997;94:7245–50. doi: 10.1073/pnas.94.14.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Phillips AC, Vousden KH. E2F-1 induced apoptosis. Apoptosis. 2001;6:173–82. doi: 10.1023/a:1011332625740. [DOI] [PubMed] [Google Scholar]
- 3.Muller H, Bracken AP, Vernell R, et al. E2Fs regulate the expression of genes involved in differentiation, development, proliferation and apoptosis. Genes Dev. 2001;15:267–85. doi: 10.1101/gad.864201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crosby ME, Almasan A. Opposing roles of E2Fs in cell proliferation and death. Cancer Biol Ther. 2004;3:1208–11. doi: 10.4161/cbt.3.12.1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bandara LR, Buck VM, Zamanian M, et al. Functional synergy between DP-1 and E2F-1 in the cell cycle-regulating transcription factor DRTF1/E2F. EMBO J. 1993;12:4317–24. doi: 10.1002/j.1460-2075.1993.tb06116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Helin K, Wu CL, Fattaey AR, et al. Heterodimerization of the transcription factors E2F-1 and DP-1 leads to cooperative trans-activation. Genes Dev. 1993;7:1850–61. doi: 10.1101/gad.7.10.1850. [DOI] [PubMed] [Google Scholar]
- 7.DeGregori J, Kowalik T, Nevins JR. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol Cell Biol. 1995;15:4215–24. doi: 10.1128/mcb.15.8.4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vigo E, Muller H, Prosperini E, et al. CDC25A phosphatase is a target of E2F and is required for efficient E2F-induced S phase. Mol Cell Biol. 1999;19:6379–95. doi: 10.1128/mcb.19.9.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vorburger SA, Hetrakul N, Xia W, et al. Gene therapy with E2F-1 up-regulates the protein kinase PKR and inhibits growth of leiomyosarcoma in vivo. Mol Cancer Ther. 2005;4:1710–6. doi: 10.1158/1535-7163.MCT-05-0036. [DOI] [PubMed] [Google Scholar]
- 10.Elliott MJ, Dong YB, Yang H, et al. E2F-1up-regulates c-Myc and p14 (ARF) and induces apoptosis in colon cancer cells. Clin Cancer Res. 2001;7:3590–7. [PubMed] [Google Scholar]
- 11.DeGregori J, Johnson DG. Distinct and overlapping roles for E2F family members in transcription, proliferation and apoptosis. Curr Mol Med. 2006;6:739–48. doi: 10.2174/1566524010606070739. [DOI] [PubMed] [Google Scholar]
- 12.Bell LA, O’Prey J, Ryan KM. DNA-binding independent cell death from a minimal proapoptotic region of E2F-1. Oncogene. 2006;25:5656–63. doi: 10.1038/sj.onc.1209580. [DOI] [PubMed] [Google Scholar]
- 13.Hsieh JK, Fredersdorf S, Kouzarides T, et al. E2F-1-induced apoptosis requires DNA binding but not transactivation and is inhibited by the retinoblastoma protein through direct interaction. Genes Dev. 1997;11:1840–52. doi: 10.1101/gad.11.14.1840. [DOI] [PubMed] [Google Scholar]
- 14.Holmberg C, Helin K, Sehested M, et al. E2F-1-induced p53-independent apoptosis in transgenic mice. Oncogene. 1998;17:143–55. doi: 10.1038/sj.onc.1201915. [DOI] [PubMed] [Google Scholar]
- 15.Phillips AC, Bates S, Ryan KM, et al. Induction of DNA synthesis and apoptosis are separable functions of E2F-1. Genes Dev. 1997;11:1853–63. doi: 10.1101/gad.11.14.1853. [DOI] [PubMed] [Google Scholar]
- 16.Gomez-Gutierrez JG, Rao XM, Garcia-Garcia A, et al. Developing adenoviral vectors encoding therapeutic genes toxic to host cells: comparing binary and single-inducible vectors expressing truncated E2F-1. Virology. 2010;397:337–45. doi: 10.1016/j.virol.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gomez-Gutierrez JG, Garcia-Garcia A, Hao H, et al. Adenovirus-mediated expression of truncated E2F-number>1 suppresses tumour growth in vitro and in vivo. Cancer. 2010;116:4420–32. doi: 10.1002/cncr.25322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ginsberg D. E2F1 pathways to apoptosis. FEBS Lett. 2002;529:122–5. doi: 10.1016/s0014-5793(02)03270-2. [DOI] [PubMed] [Google Scholar]
- 19.Dong YB, DuncaN B, Souza V, et al. E2F1 cancer gene therapy. Gene Ther Mol Biol. 2004;8:147–55. [Google Scholar]
- 20.Hershko T, Ginsberg D. Up-regulation of Bcl-2 homology 3 (BH3)-only proteins by E2F1 mediates apoptosis. J Biol Chem. 2004;279:8627–34. doi: 10.1074/jbc.M312866200. [DOI] [PubMed] [Google Scholar]
- 21.Hao H, Dong Y, Bowling MT, et al. E2F-1 induces melanoma cell apoptosis via PUMA up-regulation and Bax translocation. BMC Cancer. 2007;7:24. doi: 10.1186/1471-2407-7-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Imaizumi K, Tsuda M, Imai Y, et al. Molecular cloning of a novel polypeptide, DP5, induced during programmed neuronal death. J Biol Chem. 1997;272:18842–8. doi: 10.1074/jbc.272.30.18842. [DOI] [PubMed] [Google Scholar]
- 23.Inohara N, Ding L, Chen S, et al. Harakiri, a novel regulator of cell death, encodes a protein that activates apoptosis and interacts selectively with survival-promoting proteins Bcl-2 and Bcl-XL. EMBO J. 1997;16:1686–94. doi: 10.1093/emboj/16.7.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Willis SN, Adams JM. Life in the balance: how BH3-only proteins induce apoptosis. Curr Opin Cell Biol. 2005;17:617–25. doi: 10.1016/j.ceb.2005.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Polager S, Ginsberg D. p53 and E2F: partners in life and death. Nat Rev Cancer. 2009;9:738–48. doi: 10.1038/nrc2718. [DOI] [PubMed] [Google Scholar]
- 26.Speidel D. Transcription-independent p53 apoptosis: an alternative route to death. Trends Cell Biol. 2010;20:14–24. doi: 10.1016/j.tcb.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 27.Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 2009;458:1127–30. doi: 10.1038/nature07986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanz C, Horita M, Fernandez-Luna JL. Fas signaling and blockade of Bcr-Abl kinase induce apoptotic Hrk protein via DREAM inhibition in human leukemia cells. Haematologica. 2002;87:903–7. [PubMed] [Google Scholar]
- 29.Sanz C, Mellstrom B, Link WA, et al. Interleukin 3-dependent activation of DREAM is involved in transcriptional silencing of the apoptotic hrk gene in hematopoietic progenitor cells. EMBO J. 2001;20:2286–92. doi: 10.1093/emboj/20.9.2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carrion AM, Link WA, Ledo F, et al. DREAM is a calcium-regulated transcriptional repressor. Nature. 1999;398:80–4. doi: 10.1038/18044. [DOI] [PubMed] [Google Scholar]
- 31.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–19. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 32.Donovan M, Cotter TG. Control of mitochondrial integrity by Bcl-2 family members and caspase-independent cell death. Biochim Biophys Acta. 2004;1644:133–47. doi: 10.1016/j.bbamcr.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 33.Galonek HL, Hardwick JM. Upgrading the BCL-2 network. Nat Cell Biol. 2006;8:1317–9. doi: 10.1038/ncb1206-1317. [DOI] [PubMed] [Google Scholar]
- 34.Ma C, Ying C, Yuan Z, et al. dp5/HRK is a c-Jun target gene and required for apoptosis induced by potassium deprivation in cerebellar granule neurons. J Biol Chem. 2007;282:30901–9. doi: 10.1074/jbc.M608694200. [DOI] [PubMed] [Google Scholar]
- 35.Kalinec GM, Fernandez-Zapica ME, Urrutia R, et al. Pivotal role of Harakiri in the induction and prevention of gentamicin-induced hearing loss. Proc Natl Acad Sci USA. 2005;102:16019–24. doi: 10.1073/pnas.0508053102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakamura M, Ishida E, Shimada K, et al. Frequent HRK inactivation associated with low apoptotic index in secondary glioblastomas. Acta Neuropathol. 2005;110:402–10. doi: 10.1007/s00401-005-1065-x. [DOI] [PubMed] [Google Scholar]
- 37.Sanz C, Benito A, Inohara N, et al. Specific and rapid induction of the proapoptotic protein Hrk after growth factor withdrawal in hematopoietic progenitor cells. Blood. 2000;95:2742–7. [PubMed] [Google Scholar]
- 38.Fernandez-Luna JL. Regulation of pro-apoptotic BH3-only proteins and its contribution to cancer progression and chemoresistance. Cell Signal. 2008;20:1921–6. doi: 10.1016/j.cellsig.2008.04.015. [DOI] [PubMed] [Google Scholar]
- 39.Harris CA, Johnson Jr EM. BH3-only Bcl-2 family members are coordinately regulated by the JNK pathway and require Bax to induce apoptosis in neurons. J Biol Chem. 2001;276:37754–60. doi: 10.1074/jbc.M104073200. [DOI] [PubMed] [Google Scholar]
- 40.Jo DG, Kim MJ, Choi YH, et al. Pro-apoptotic function of calsenilin/ DREAM/KChIP3. FASEB J. 2001;15:589–91. doi: 10.1096/fj.00-0541fje. [DOI] [PubMed] [Google Scholar]
- 41.Jo DG, Lee JY, Hong YM, et al. Induction of pro-apoptotic calsenilin/DREAM/KChIP3 in Alzheimer’s disease and cultured neurons after amyloid-β exposure. J Neurochem. 2004;88:604–11. doi: 10.1111/j.1471-4159.2004.02159.x. [DOI] [PubMed] [Google Scholar]
- 42.Box NF, Terzian T. The role of p53 in pigmentation, tanning and melanoma. Pigment Cell Melanoma Res. 2008;21:525–33. doi: 10.1111/j.1755-148X.2008.00495.x. [DOI] [PubMed] [Google Scholar]
- 43.Ternovoi W, Curiel DT, Smith BF, et al. Adenovirus-mediated p53 tumour suppressor gene therapy of osteosarcoma. Lab Invest. 2006;86:748–66. doi: 10.1038/labinvest.3700444. [DOI] [PubMed] [Google Scholar]