

Abstract
We previously demonstrated that baseline synovial overexpression of the interleukin-7 receptor α-chain (IL-7R) is associated with poor response to tumour necrosis factor (TNF) blockade in rheumatoid arthritis (RA). We found that IL-7R gene expression is induced in fibroblast-like synovial cells (FLS) by the addition of TNF-α, IL-1β and combinations of TNF-α+ IL-1β or TNF-α+ IL-17, thereby suggesting that these cytokines play a role in the resistance to TNF blockade in RA. Because FLS and CD4 T cells also produce a soluble form of IL-7R (sIL-7R), resulting from an alternative splicing of the full-length transcript, we wondered whether expression of sIL-7R is similarly regulated by pro-inflammatory cytokines. We also investigated whether sIL-7R is detectable in the serum of RA patients and associated with response to TNF blockade. RA FLS were cultured in the presence of pro-inflammatory cytokines and sIL-7R concentrations were measured in culture supernatants. Similarly, sIL-7R titres were measured in sera obtained from healthy individuals, early untreated RA patients with active disease and disease-modifying anti-rheumatic drug (DMARD)-resistant RA patients prior to initiation of TNF-blockade. Baseline serum sIL-7R titres were correlated with validated clinical measurements of disease activity. We found that exposure of RA FLS to pro-inflammatory cytokines (TNF-α, IL-1β and combinations of TNF-α and IL-1β or TNF-α and IL-17) induces sIL-7R secretion. Activated CD4 T cells also produce sIL-7R. sIL-7R serum levels are higher in RA patients as compared to controls. In DMARD-resistant patients, high sIL-7R serum concentrations are strongly associated with poor response to TNF-blockade. In conclusion, sIL-7R is induced by pro-inflammatory cytokines in RA FLS. sIL-7R could qualify as a new biomarker of response to therapy in RA.
Keywords: rheumatoid arthritis, TNF blockade, biomarker, interleukin-7 receptor α-chain
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease of the synovium that can lead to severe joint damage if insufficiently treated. Central to the pathogenesis of RA is the proliferation of synovial fibroblasts (synovial ‘pannus’) in response to the production of autocrine, but also paracrine molecules produced by infiltrating mononuclear cells. Among these molecules, pro-inflammatory cytokines such as tumour necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6 or IL-17 play an important role, and this observation led to the development of targeted therapies. In this context, TNF blocking agents are used routinely in RA patients who have failed first-line disease-modifying anti-rheumatic drugs (DMARD) therapy.
Several TNF blocking agents are presently available, which display similar effects in terms of efficacy, tolerability and side effects in RA [1–5]. Importantly, about 25% to 30% of RA patients treated with TNF antagonists do not display any significant clinical improvement after initiation of therapy. Thus far, however, there are no validated tools that can predict whether an individual RA patient will respond or not to TNF blockade. Yet the identification of poor responders prior to initiation of therapy would direct the use of other biologics, thereby preventing disease progression in these patients and saving unnecessary costs. Of note, an additional 30–40% of the patients will display feature of secondary resistance to the delivered drug within the first 5 years of therapy. This phenomenon is, at least partially, associated with the development of human anti-chimeric or human anti-human antibodies, and is clearly distinct from primary resistance to TNF blockade.
In earlier experiments, we performed global gene expression studies on synovial biopsies obtained from RA patients prior to the initiation of TNF-blockade [6]. We found that several genes were significantly overexpressed at baseline in non-responders. Strikingly, the expression of these genes is induced in vitro in fibroblast-like synovial cells (FLS) by the addition of TNF-α, IL-1β and combinations of TNF-α and IL-1β or TNF-α and IL-17, thereby suggesting that these cytokines play a role in the mechanisms of primary resistance to TNF blockade in RA.
The interleukin-7 receptor α-chain (IL-7R) is one of the genes overexpressed in the synovial tissue of poor responders to TNF blockade in RA [6]. Fibroblasts are known to produce a soluble form of the molecule (sIL-7R), which results from an alternative splicing of the full-length transcript (excision of exon 6, containing the transmembrane domain) and is able to bind and block IL-7 [7, 8]. We therefore wondered whether sIL-7R expression was also regulated in synovial fibroblasts by the addition of pro-inflammatory cytokines and whether serum sIL-7R determination had any value in predicting primary response to TNF blockade in RA.
Materials and methods
Patients
Sera were collected from healthy individuals (n= 75), patients with early (n= 52) or DMARD-resistant (n= 75) RA and stored at –80°C. All RA patients met the American College of Rheumatology criteria for the diagnosis of RA [9]. Patients with early RA had disease duration of less than 1 year. They were (mean ± S.D.) 45 ± 14 years old. All had active disease at the time of serum sampling [mean disease activity score (DAS)28-C-reactive protein (CRP) score: 5.87 ± 1.28] and none of them was treated, except for non-steroidal anti-inflammatory drugs.
DMARD-resistant RA patients were 55 ± 12 year old. Disease duration was 10.8 ± 6.7 years. They were treated at baseline with a median 15 mg methotrexate/week schedule (range 7.5–25 mg/week). A total of 9% were treated with other DMARDs after having failed methotrexate therapy in the past. A total of 13% were treated with another DMARD together with methotrexate. Patients had taken an average three DMARD’s including methotrexate (range 1–7) before starting infliximab therapy. All of them had erosive changes imaged on conventional x-rays of the hands and/or the feet. They all had active disease at the time of serum sampling (mean DAS28-CRP score: 5.69 ± 1.12). After baseline serum collection, DMARD-resistant RA patients were treated with a standard schedule of infliximab: 3 mg/kg at weeks 0, 2, 6 and then every other month in addition to their DMARD therapy. Follow-up DAS28-CRP evaluations were performed between 4 and 6 months after baseline and patients were categorized into non-responders versus responders according to European League Against Rheumatism (EULAR) response criteria [10].
The study was approved by the ethics committee of the Université catholique de Louvain, and informed consent was obtained from all patients.
Isolation and culture of cells
FLS were purified from synovial biopsies from eight additional RA patients as previously described [11]. Briefly, minced synovial fragments were digested in 1 mg/ml hyaluronidase solution (Sigma-Aldrich, St. Louis, MO, USA) for 15 min. at 37°C and 6 mg/ml collagenase type IV (Invitrogen, Paisley, UK) for 2 hrs at 37°C. Next, cells were washed, resuspended in high-glucose Dulbecco’s modified Eagle’s medium (Invitrogen) supplemented with 1% antibiotics-antimycotics (Invitrogen), 1% minimum essential medium sodium pyruvate (Invitrogen) and 10% foetal calf serum (FCS), and seeded at 10,000 cells per square centimetre in six-well plates. When the cells reached confluence, adherent cells were detached using sterile 0.5% trypsin-ethylenediaminetetraacetic acid (EDTA; Invitrogen) and used between passages 3 and 9.
Culture of cells and cytokine stimulation experiments
For the cytokine stimulation experiments, FLS were seeded in 24-well plates at 25,000 per well. Unless stated otherwise, the following cytokine concentrations were used: TNF-α (R&D Systems, Minneapolis, MN, USA) 10 ng/ml, IL-1β (R&D Systems) 10 ng/ml and IL-17 (R&D Systems) 50 ng/ml. After overnight incubation with the indicated cytokines, supernatants were collected for sIL-7R determination and cells were harvested for total RNA extraction. Because of the limited number of cells, all the cytokine stimulation experiments were not performed on the same cells. IL-7R gene expression in response to TNF-α, IL-1β and combinations of both cytokines was studied in FLS from five different patients; IL-7R gene expression in response to IL-17, TNF-α, IL-1β and combinations of TNF-α and IL-17 or IL-1β and IL-17 was studied in FLS from four different patients. sIL-7R gene expression in response to TNF-α, IL-1β and combinations of both cytokines was studied in FLS from four different patients; sIL-7R gene expression in response to IL-17, TNF-α, IL-1β and combinations of TNF-α and IL-17 or IL-1β and IL-17 was studied in FLS from two different patients. For co-culture experiments, CD4 T cells were purified from the synovial fluid using magnetic beads (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany) according to manufacturer’s instructions. FLS (5000/well) and autologous CD4 T cells (50,000/well) were seeded in flat-bottomed 96-well plates in the presence of autologous serum, IL-2 (25 U/ml) and either 100 ng/ml IL-7 (R&D Systems) or 5 μg/ml sIL-7R-Fc fusion protein (R&D Systems), as indicated. After 3 days, proliferative responses were measured in quadruplicates after an overnight pulse with 0.5 μCi 3H-thymidine (GE Healthcare, Amersham, UK).
Peripheral blood mononuclear cells (PBMC) from healthy donors were purified by lymphoprep (Nycomed Pharma, Oslo, Norway) density gradient centrifugation. In some experiments, PBMC were cultured for 24 hrs in the presence of IL-2 and phytohemagglutinin (PHA), and CD4 T cells were purified using magnetic beads (Miltenyi Biotec) as per manufacturer’s instructions. CD8 T cells clones (obtained from Pierre van der Bruggen, Ludwig Institute for Cancer Research, Brussels Branch) were stimulated with their cognate antigen presented by irradiated autologous B-Ebstein-Barr virus (EBV) cells in the presence of. IL-2 (Chiron, Amsterdam, The Netherlands, 50 U/ml) and IL-7 (R&D Systems, 5 ng/ml). After 24 hrs, CD8 T cells clones were purified using magnetic beads (Miltenyi Biotec) as per manufacturer’s protocol. cDNA from B-EBV cell derived from two healthy donors were obtained from Pierre Coulie (Unité de Génétique Cellulaire, Université catholique de Louvain, Brussels, Belgium).
Flow cytometry experiments
FLS were harvested and resuspended in a sodium phosphate (1 mM, pH = 7.4) buffer containing 137 nM NaCl, 5 mM KCl, 0.4 mM MgSO4, 0.3 mM MgCl2, 5 mM Glucose, 4 mM NaHCO3, 1 mM EDTA and 3% FCS in the presence of a phycoerythrin (PE)-conjugated IL-7R antibody (Becton Dickinson, Mountain View, CA, USA) or a control isotype (Becton Dickinson). Cells were washed and fixed in paraformaldehyde (0.6%) before being analysed by flow cytometry (Becton Dickinson).
Western blot experiments
FLS and PBMC were washed with ice-cold PBS, and lysed in cold lysis buffer (20 mM Hepes pH7.8, 75 mM KCl, 0.1 mM EDTA, 1 mM sodium-orthovanadate, 2 mM MgCl2, 1 mM dithiothreitol (DTT), 10% glycerol, 0.5% Triton-X 100 and one tablet of Complete Protease Inhibitor (Roche, Vilvoorde, Belgium) per 20 ml). Lysates were subjected to sonication and spun at 13,000 ×g to remove debris. Supernatants were resolved on a polyacrylamide gel and immunoblotted with an monoclonal mouse anti-IL-7R antibody (Sigma-Aldrich) at 1:1,000. Immunoreactive proteins were visualized using a goat antimouse-HRP antibody (Santa Cruz Biotechnology, Heidelberg, Germany) at 1:1,000, with a femto-range-sensitive ECL detection system (Thermo Fisher Scientific, Rockford, IL, USA).
RT-PCR experiments
Total RNA was extracted using the Nucleospin® RNA II extraction kit (Macherey-Nagel, Düren, Germany), including DNase treatment of the samples. cDNA was synthesized using RevertAid Moloney murine leukaemia virus RT (Fermentas, St. Leon-Rot, Germany) and Oligo(dT) primers. IL-7R PCR amplification was carried out using Taq DNA polymerase (Fermentas) and the following primers: Forward: tccctcccttcctcttactctc and Reverse: tctggcagtccaggaaactt. PCR products were analysed by agarose gel electrophoresis. PCR fragments were gel purified and sequenced using the same primers and the BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems, Foster City, CA, USA) before being analysed on a 3130xl Genetic Analyzer (Applied Biosystems). Quantitative RT-PCR was performed on a MyiQ single-colour RT-PCR detection system (Bio-Rad Laboratories, Nazareth Eke, Belgium) using SYBR Green detection mix. For each sample, 5 ng of cDNA was loaded in triplicate with 1x SYBR Green Mix (Applied Biosystems) and the following 10 mM primers: β-actin : Forward: ggcatcgtgatggactccg and Reverse: ctggaaggtggacagcga; IL-7R: Forward: tttctctgtcgctctgttggt and Reverse: gcttgaatgtcatccacccta; sIL-7R: Forward: agccaatgactttgtggtgac and Reverse: tacgataggcttaatcctgag. The IL-7R forward primer is located in exon 6 of the gene, which is absent in the sIL-7R mRNA sequence. The sIL-7R reverse primer spans the junction between exon 5 and exon 7 of the mRNA sequence of the IL-7R gene. PCR amplification of plasmids containing either an IL-7R or a sIL-7R cDNA confirmed the specificity of the primer pairs for each isoform of the molecule. The melting curves obtained after each quantitative polymerase chain reaction (qPCR) amplification confirmed the specificity of the SYBR Green assays. Relative expression of the target genes in the studied samples was obtained using the difference in the comparative threshold (ΔΔCt) method.
sIL-7R ELISA
sIL-7R serum titres were determined by sandwich-ELISA in serum samples and culture supernatants using a goat polyclonal hIL-7R antibody (Sigma-Aldrich) as coating antibody and a mouse monoclonal hIL-7R antibody as detecting antibody (Sigma-Aldrich). Microlon ELISA plates (Greiner Bio One, Wemmel, Belgium) were coated overnight at 4°C with a 100 ml goat polyclonal hIL-7R antibody solution diluted at 0.5 μg/ml in a 50 mM sodium carbonate (stock at 1 M, pH 10) coating buffer. Plates were blocked with milk and then incubated at 37°C with the control and patient’s sera diluted in PBS supplemented with 0.5% bovine serum albumin (BSA). Sera were tested in duplicates. For the standard curve, we used serial dilutions of a commercial IL-7R-Fc fusion protein (R&D Systems) containing the extracellular portion of the receptor bound to an Fc fragment of human IgG. For this reason, the results were expressed in pmol/ml instead of μg/ml. The detection antibody was used at 1 μg/ml in PBS 0.5% BSA. The third antibody was a rat horseradish peroxydase (HRPO)-antimouse IgG monoclonal antibody (LO-MK1, Unit of Experimental Immunology, Université catholique de Louvain, Brussels, Belgium), diluted at 0.5 μg/ml in PBS 0.5% BSA. That antibody does not cross-react with human IgG, and is therefore unable to detect IgG rheumatoid factors bound to the coated anti-IL-7R goat antibody. Each incubation step lasted 2 hrs. Plates were washed five times with PBS Tween (Sigma-Aldrich) 1/1000 between each step. The reactions were revealed with 1-Step Ultra-TMB (Thermo Fisher Scientific) and stopped by the addition of 2M H2SO4. The sensitivity of the ELISA is 20 pmol/ml. Inter-assay coefficient of variation is <8%. Pre-incubation of serum samples or standard curve with increasing concentrations of IL-7 does not affect the results of the ELISA readings.
Statistical analyses
In vitro data were analysed using unpaired Student’s t-tests. Serum data were analysed using Mann-Whitney U-tests. Correlations between DAS scores, CRP values, Delta DAS scores and serum sIL-7R were analysed using Spearman rank tests.
Results
Fibroblast-like synovial cells express two isoforms of the gene encoding the IL-7R α-chain
In our previous experiments, we demonstrated that RA patients have higher synovial IL-7R gene and protein expression as compared to osteoarthritis or lupus patients [12]. Moreover, we found that synovial overexpression of IL-7R in DMARD-resistant RA patients is associated with poor response to TNF blockade [6]. In these earlier experiments, anti IL-7R antibodies stained not only synovial mononuclear cells, but also synovial fibroblasts. Therefore, we wanted to further study the production and regulation of IL-7R by cultured FLS obtained from the joints of RA patients. Western blot and real-time qPCR experiments indicated that FLS express two isoforms of the molecule (Fig. 1A). Sequencing of the amplicons demonstrated that these isoforms correspond to the native IL-7R, and an alternatively spliced variant lacking exon 6 (transmembrane domain), which encodes a soluble form of the IL-7R (sIL-7R) (Fig. 1B).
Fig 1.
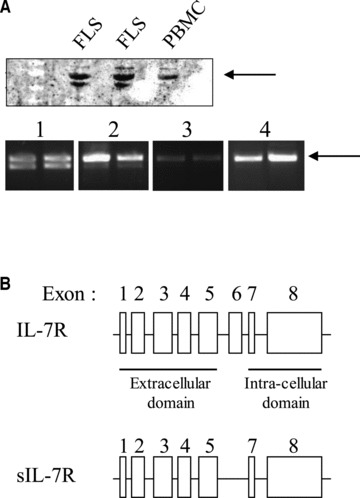
FLS and activated CD4 T cells produce two IL-7R α-chain isoforms. (A) IL-7R Western blots on protein extracts from FLS and PBMC, and IL-7R PCR on cDNA from duplicate TNF-α and IL-1β activated FLS (1), IL-2 and PHA activated CD4 T cells (2), antigen-activated CD8 T cell clones (3) and B-EBV cells (4). Arrows indicate the expected sizes of the membrane-bound IL-7R. (B) Sequencing of purified IL-7R PCR fragments indicates that FLS produce a full-length IL-7R α-chain and a truncated form of the IL-7R α-chain lacking exon 6 encoding the transmembrane domain, thereby resulting in a secreted form of the molecule.
sIL-7R expression by FLS is induced by pro-inflammatory cytokines
Because sIL-7R transcripts result from an alternative splicing of IL-7R mRNAs, we wondered whether expression of both IL-7R isoforms is similarly regulated in FLS by the addition of pro-inflammatory cytokines known to play a role in the pathogenesis of RA. We found that mRNA levels of both the membrane-bound and soluble forms of IL-7R are up-regulated by the addition of TNF-α, IL-1β and combinations of TNF-α and IL-1β or TNF-α and IL-17 (Fig. 2A). Flow cytometry experiments were, however, unable to detect any cell surface expression of the membrane-bound IL-7R on FLS under any of these conditions (Fig. 2B). By contrast, ELISA experiments performed on culture supernatants indicated that sIL-7R secretion is induced in FLS by TNF-α, IL-1β and the combination of both cytokines (Fig. 2C). Interestingly, sIL-7R gene expression is negative in activated CD8 T and B cells but slightly positive in activated CD4 T cells (Fig. 1A). Taken together, these results indicate that sIL-7R is a marker of fibroblast and, to a lesser extent, CD4 T-cell activation.
Fig 2.
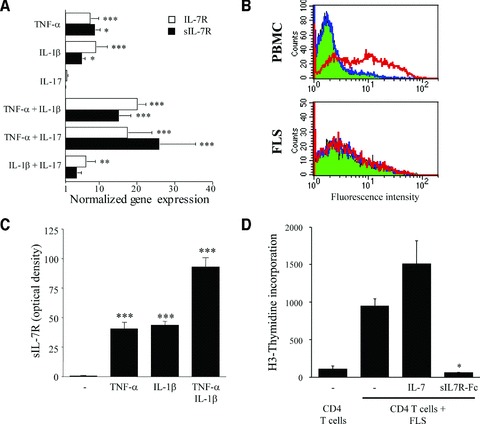
Pro-inflammatory cytokines stimulate sIL-7R production by FLS. (A) IL-7R (open bars) and sIL-7R (closed bars) real-time qPCR studies carried out on FLS stimulated with the indicated cytokines. Results are expressed as mean fold changes in IL-7R and sIL-7R gene expression (±S.E.M.) over unstimulated FLS, obtained from two to five different experiments each. (B) Flow cytometric evaluation of IL-7R expression by PBMC and FLS. Cells were incubated with a PE-conjugated IL-7R antibody (red) or a PE-conjugated isotype control (blue). Autofluorescence of the cells is depicted in green. Graphs are representative of three different experiments. Similar results were obtained using FLS stimulated with pro-inflammatory cytokines, alone or in combination. (C) sIL-7R measurements were performed by sandwich-ELISA in supernatants of FLS cultures stimulated with the indicated cytokines. Results are expressed as mean optical density (O.D.) units ×1000 (after subtraction of the baseline O.D.) ±S.E.M. obtained from three different experiments. (D) Effect of IL-7 and sIL-7R-Fc fusion protein on proliferation of synovial CD4 T cells cultured in the presence of autologous FLS, autologous serum and IL-2. Results are expressed as mean cpm (±S.E.M.) obtained from two different experiments. *P < 0.05; **P < 0.005; ***P < 0.0005 versus unstimulated cells.
IL-7 blockade inhibits FLS-induced CD4 T-cell proliferation
Because sIL-7R is known to display IL-7 binding and blocking properties, we investigated whether it could interfere with synovial CD4 T-cell proliferation. RA FLS are known to express major histocompatibility complex (MHC) class II molecules and stimulate the proliferation of autologous synovial CD4 T cells. We found that the addition of IL-7 stimulates the proliferation of synovial CD4 T cells cultured in the presence of autologous FLS. By contrast, addition of a sIL-7R-Fc fusion protein blocks the proliferation of these cells (Fig. 2D). These results suggest that sIL-7R production by activated synovial fibroblasts could play a role in a negative feedback loop resulting in a decreased proliferation of synovial CD4 T cells.
sIL-7R serum levels are higher in RA patients compared to controls
Next, we wondered whether sIL-7R could be detected in sera of healthy individuals and whether it was increased in patients with RA. sIL-7R is readily detectable in the sera of healthy individuals (n= 75) (median: 618 pmol/ml). In untreated early RA patients (n= 52) and in treated RA patients with active disease despite DMARD therapy (n= 75), sIL-7R serum levels are significantly higher (median: 921 and 1080 pmol/ml, respectively) as compared to controls (Fig. 3). We defined serum sIL-7R upper normal concentration as the mean serum sIL-7R concentration in the controls + 2 standard deviations. A total of 1.3% of the controls have an abnormal serum sIL-7R titre, versus 17.3% of the early RA patients and 24% of the DMARD-resistant RA patients. In both early and DMARD-resistant RA patients, baseline sIL-7R serum levels did not correlate with serum CRP values or with DAS28-CRP scores (data not shown).
Fig 3.
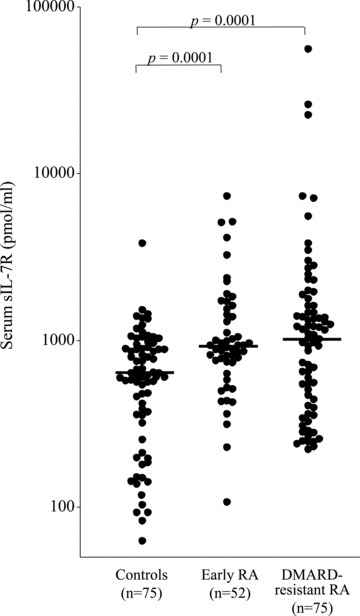
Serum sIL-7R titres are significantly higher in early and DMARD-resistant RA patients. sIL-7R titres measured by sandwich ELISA in duplicate serum samples from healthy individuals, early RA and DMARD-resistant RA patients. The horizontal bar depicts the median value in each group.
Baseline sIL-7R serum levels predict response to therapy in RA
Synovial overexpression of IL-7R is associated with poor response to TNF blockade in DMARD-resistant RA patients [6]. We therefore wondered whether baseline serum sIL-7R levels would have the same informative value about response to TNF blocking therapy in RA. Strikingly, in patients displaying active disease despite DMARD therapy and subsequently treated with TNF blockade, elevated baseline sIL-7R serum levels strongly predicted poor response to anti-TNF therapy (Fig. 4A, B and C). A total of 55.5% of non-responders to infliximab therapy had an abnormal sIL-7R value at baseline, versus 15.3% of the responders. Conversely, normal baseline sIL-7R serum levels were associated with adequate response to therapy, with a sensitivity of 93% and a specificity of 56%. In case of low sIL-7R serum titres, patients have a 87% probability of being responders (predictive positive value); by contrast, in case of elevated sIL-7R serum levels, patients have a 71% probability of being non-responders (predictive negative value).
Fig 4.
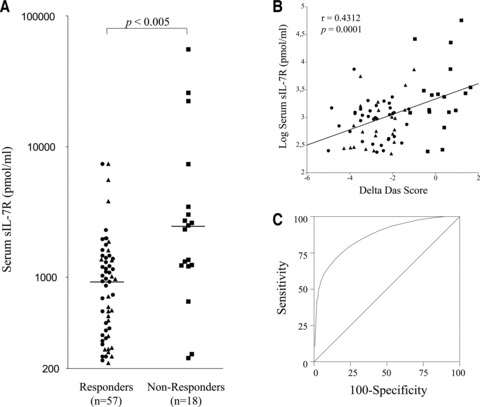
sIL-7R serum concentrations predict response to TNF blockade in RA. (A) sIL-7R titres measured by sandwich ELISA in duplicate baseline serum samples obtained in DMARD-resistant RA patients treated with 3 mg/kg infliximab. Patients were categorized in good-responders (circles), moderate- (triangles) and non-responders (squares) according to EULAR response criteria. The horizontal bar depicts the median value in each group. (B) Linear correlation between baseline sIL-7R serum levels and DAS-Score differences (follow-up minus baseline DAS28-CRP). (C) Receiving operating characteristic curve evaluating the value of baseline sIL-7R in predicting response to therapy. The curve was plotted by calculating sensitivity and specificity of the test at several cut-off values.
Discussion
In a first part of our work, we showed that soluble IL-7R α-chain (sIL-7R) gene expression is induced in FLS by the addition of pro-inflammatory cytokines (TNF-α, IL-1β and combinations of TNF-α and IL-1β or TNF-α and IL-17). Next, we investigated whether sIL-7R was detectable in the sera of healthy individuals and RA patients. We found that sIL-7R serum titres are significantly higher in early and in DMARD-resistant RA patients as compared to controls. We also found that high sIL-7R serum levels are strongly associated with poor response to TNF blockade in DMARD-resistant RA patients.
In a previous work, we performed global gene expression studies in synovial biopsies obtained from RA patients before initiation of anti-TNF therapy. We found, and confirmed by immunohistochemistry, that specific groups of genes were overexpressed in synovial tissue of non-responders at baseline. Interestingly, we demonstrated that these genes are induced in FLS by TNF-α, IL-1β and by combinations of TNF-α, IL-1β and IL-17. These observations indicate that the gene signatures in poor responders reflect synovial cell exposure to larger amounts and/or combinations of pro-inflammatory cytokines, potentially explaining the association with poor response to TNF-blockade alone [6].
Among the genes overexpressed in poor responders, the IL-7R α-chain came out as an interesting biomarker candidate. In an earlier study, we had described this gene to be the most differentially expressed in synovial biopsies from RA patients compared to patients with osteoarthritis or SLE, followed by other genes involved in T- and B-cell activation [12]. The functional IL-7 receptor is mainly expressed on Th1 and Th17 cells (by contrast to regulatory T cells) and is made of two chains: the IL-7R α-chain and the IL-2R γ-chain. Binding of IL-7 to its receptor induces T-cell proliferation and activation and this effect is more pronounced in Th17 cells. By contrast, suppressor of cytokine signaling (SOCS)1 induction by interferon-γ in Th1 cells has an inhibitory effect on IL-7R signalling and IL-7 induced cell proliferation [13]. The role of the IL-7/IL-7R axis in the pathogenesis of RA was recently highlighted by the work of van Roon et al., who demonstrated that IL-7 is present in RA synovial fluid and stimulates proliferation of RA CD4 T cells and macrophages co-cultures [14]. Taken together, these data indicate that the IL-7/IL-7R axis could be a new target for therapy in RA [15, 16].
Here, we studied the expression of the IL-7R α-chain in synovial cells from RA patients, and found that cultured FLS produce the secreted form of the molecule. Although PCR and Western blot experiments indicate that FLS also display a positive signal for the full length IL-7R α-chain, we could not detect it by flow cytometry at the cell surface of resting or stimulated cells, and neither could we find any proliferative effect or induction of cytokines by FLS in response to IL-7 (data not shown). The soluble IL-7R has been described in fibroblast cell lines as the result of alternative splicing of the gene, leading to a transcript lacking exon 6, which encodes the transmembrane domain [7, 8]. We also show that addition of pro-inflammatory cytokines, such as TNF-α and IL-1β, or combinations of TNF-α and IL-1β or TNF-α and IL-17, stimulates the expression and secretion of sIL-7R by FLS. Other than FLS, we also detected expression of the sIL-7R gene in activated CD4 T cells, although to a lesser extent.
It has been demonstrated in the past that sIL-7R is able to bind and inhibit IL-7. We wondered whether such a mechanism could also be present in the context of RA and demonstrated that, while IL-7 stimulates synovial CD4 T-cell proliferation in response to FLS, IL-7 inhibition by a sIL-7R-Fc fusion protein induces the opposite effect. Taken together, these results indicate that sIL-7R is induced by pro-inflammatory cytokines in FLS and could play a role in a negative feedback loop through the inhibition of T-cell proliferation and activation. Synovial inflammation is the result of a balance between pro-inflammatory (IL-17, TNF-α, IL-1β, IL-6, . . .) and anti-inflammatory (IL-10, IL-1ra, soluble TNF-receptors, . . .) stimuli. Our results support the hypothesis that sIL-7R is an additional natural inhibitor of synovial cell activation in RA.
Next, we studied whether sIL-7R was detectable in the serum of RA patients and controls. We also wanted to investigate whether serum titres correlate with RA disease severity and progression. We found that sIL-7R is readily detectable in the sera of healthy individuals, an observation that confirms recent findings [17, 18]. In our study, serum levels of the molecule are significantly higher in patients with early RA and in DMARD-resistant RA patients as compared to controls. It is impossible to track the origin of circulating sIL-7R molecules in the serum of RA patients; however, it is tempting to consider that at least part of it could result from synovial or CD4 T cell exposure to pro-inflammatory cytokines.
Strikingly, high baseline sIL-7R serum levels in DMARD-resistant RA patients are significantly associated with poor response to Infliximab. Conversely, lower serum levels of the molecule are strongly associated with adequate response to therapy. If we suggest that high serum sIL-7R titres result from fibroblast and/or CD4 T cell exposure to high levels of TNF-α and/or IL-1β and/or IL-17, then the association with resistance to infliximab monotherapy is straightforward. In this context, however, it is possible that serum sIL-7R is also a marker of poor response to other therapeutic agents, and functions as a marker of disease severity rather than a specific marker of response to TNF blockade. Additional data are needed to clarify this issue.
In any case, with a predictive positive value of 87% and a predictive negative value of 71% for the prediction of good and moderate response to infliximab therapy in our group of patients, serum sIL-7R is a promising clinical biomarker that could aid individual therapeutic decisions in RA. Using such a biomarker in clinical practice would have an important impact in terms of patients’ outcome and drug expenses. In the absence of selection, 25% of the patients described in this work did not respond to infliximab, a proportion in accordance with published data. If we had selected patients based on baseline sIL-7R serum levels, the proportion of non-responders would have dropped to 13%, while only 5% would have been switched to an alternative therapy although they would have responded to TNF blockade. Such differences are highly significant from a clinical point of view, which is why we strongly advocate further validation of serum sIL-7R as a new clinical biomarker in RA.
Acknowledgments
The authors thank Mrs. Anne-Lise Maudoux (Department of Rheumatology, Université catholique de Louvain) for excellent technical assistance, Dr. Catherine Fillée (Department of Clinical Biology, Cliniques Universitaires Saint-Luc) for providing control serum samples and Dr. Nisha Limaye (Department of Human Genetics, Université catholique de Louvain), for critical reading of the manuscript.
This work was supported by grants from the Fondation Saint-Luc (Cliniques universitaires Saint-Luc), Région Wallonne (Biowin), the Fonds de la Recherche Scientifique et Médicale (Belgium), the Fonds Spécial de Recherche (Communauté française de Belgique) and by an unrestricted grant from Abbott (Belgium).
A patent application was submitted by the Université catholique de Louvain and by BRL, VB, FAH and BVDE.
Conflict of interest
The authors have no conflict of interest to disclose.
References
- 1.Weinblatt ME, Keystone EC, Furst DE, et al. Adalimumab, a fully human anti-tumor necrosis factor alpha monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: the ARMADA trial. Arthritis Rheum. 2000;48:35–45. doi: 10.1002/art.10697. [DOI] [PubMed] [Google Scholar]
- 2.Burmester GR, Mariette X, Montecucco C, et al. Adalimumab alone and in combination with disease-modifying antirheumatic drugs for the treatment of rheumatoid arthritis in clinical practice: the Research in Active Rheumatoid Arthritis (ReAct) trial. Ann Rheum Dis. 2007;66:732–9. doi: 10.1136/ard.2006.066761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weinblatt ME, Kremer JM, Bankhurst AD, et al. A trial of etanercept, a recombinant tumor necrosis factor receptor:Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. N Engl J Med. 1999;340:253–9. doi: 10.1056/NEJM199901283400401. [DOI] [PubMed] [Google Scholar]
- 4.Elliott MJ, Maini RN, Feldmann M, et al. Randomised double-blind comparison of chimeric monoclonal antibody to tumour necrosis factor alpha (cA2) versus placebo in rheumatoid arthritis. Lancet. 1994;344:1105–10. doi: 10.1016/s0140-6736(94)90628-9. [DOI] [PubMed] [Google Scholar]
- 5.Emery P, Fleischmann RM, Moreland LW, et al. Golimumab, a human anti-tumor necrosis factor alpha monoclonal antibody, injected subcutaneously every four weeks in methotrexate-naive patients with active rheumatoid arthritis: twenty-four-week results of a phase III, multicenter, randomized, double-blind, placebo-controlled study of golimumab before methotrexate as first-line therapy for early-onset rheumatoid arthritis. Arthritis Rheum. 2009;60:2272–83. doi: 10.1002/art.24638. [DOI] [PubMed] [Google Scholar]
- 6.Badot V, Galant C, Nzeusseu Toukap A, et al. Gene expression profiling in the synovium identifies a predictive signature of absence of response to adalimumab therapy in rheumatoid arthritis. Arthritis Res Ther. 2009;11:R57. doi: 10.1186/ar2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goodwin RG, Friend D, Ziegler SF, et al. Cloning of the human and murine interleukin-7 receptors: demonstration of a soluble form and homology to a new receptor superfamily. Cell. 1990;60:941–51. doi: 10.1016/0092-8674(90)90342-c. [DOI] [PubMed] [Google Scholar]
- 8.Pleiman CM, Gimpel SD, Park LS, et al. Organization of the murine and human interleukin-7 receptor genes: two mRNAs generated by differential splicing and presence of a type I-interferon-inducible promoter. Mol Cell Biol. 1991;11:3052–9. doi: 10.1128/mcb.11.6.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arnett FC, Edworthy SM, Bloch DA, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–24. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 10.van Gestel AM, Prevoo ML, van ‘t Hof MA, et al. Development and validation of the European League Against Rheumatism response criteria for rheumatoid arthritis. Comparison with the preliminary American College of Rheumatology and the World Health Organization/International League Against Rheumatism Criteria. Arthritis Rheum. 1996;39:34–40. doi: 10.1002/art.1780390105. [DOI] [PubMed] [Google Scholar]
- 11.De Bari C, Dell’Accio F, Tylzanowski P, et al. Multipotent mesenchymal stem cells from adult human synovial membrane. Arthritis Rheum. 2001;44:1928–42. doi: 10.1002/1529-0131(200108)44:8<1928::AID-ART331>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 12.Nzeusseu Toukap A, Galant C, Theate I, et al. Identification of distinct gene expression profiles in the synovium of patients with systemic lupus erythematosus. Arthritis Rheum. 2007;56:1579–88. doi: 10.1002/art.22578. [DOI] [PubMed] [Google Scholar]
- 13.Liu X, Leung S, Wang C, et al. Crucial role of interleukin-7 in T helper type 17 survival and expansion in autoimmune disease. Nature Med. 2010;16:191–7. doi: 10.1038/nm.2077. [DOI] [PubMed] [Google Scholar]
- 14.van Roon JA, Verweij MC, Wijk MW, et al. Increased intraarticular interleukin-7 in rheumatoid arthritis patients stimulates cell contact-dependent activation of CD4(+) T cells and macrophages. Arthritis Rheum. 2005;52:1700–10. doi: 10.1002/art.21045. [DOI] [PubMed] [Google Scholar]
- 15.van Roon JA, Hartgring SA, Wenting-van Wijk M, et al. Persistence of interleukin 7 activity and levels on tumour necrosis factor alpha blockade in patients with rheumatoid arthritis. Ann Rheum Dis. 2007;66:664–9. doi: 10.1136/ard.2006.062547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hartgring SA, van Roon JA, Wenting-van Wijk M, et al. Elevated expression of interleukin-7 receptor in inflamed joints mediates interleukin-7-induced immune activation in rheumatoid arthritis. Arthritis Rheum. 2009;60:2595–605. doi: 10.1002/art.24754. [DOI] [PubMed] [Google Scholar]
- 17.Faucher S, Crawley AM, Decker W, et al. Development of a quantitative bead capture assay for soluble IL-7 receptor alpha in human plasma. PLoS ONE. 2009;4:e6690. doi: 10.1371/journal.pone.0006690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rose T, Lambotte O, Pallier C, et al. Identification and biochemical characterization of human plasma soluble IL-7R: lower concentrations in HIV-1-infected patients. J Immunol. 2009;182:7389–97. doi: 10.4049/jimmunol.0900190. [DOI] [PubMed] [Google Scholar]