

Abstract
Mesenchymal stem cells (MSCs) are studied for their potential clinical use in regenerative medicine, tissue engineering and tumour therapy. However, the therapeutic application of MSCs in tumour therapy still remains limited unless the immunosuppressive role of MSCs for tumour growth in vivo is better understood. In this study, we investigated the mechanism of MSCs favouring tumour escape from immunologic surveillance in inflammatory microenvironment. We first compared the promotive capacity of bone marrow-derived MSCs on B16 melanoma cells growth in vivo, pre-incubated or not with the inflammatory cytokines interferon (IFN)-γ and tumour necrosis factor (TNF)-α. We showed that the development of B16 melanoma cells is faster when co-injected with MSCs pre-incubated with IFN-γ and TNF-α compared with control groups. Moreover, tumour incidence increases obviously in allogeneic recipients when B16 melanoma cells were co-injected with MSCs pre-incubated with IFN-γ and TNF-α. We then demonstrated that the immunosuppressive function of MSCs was elicited by IFN-γ and TNF-α. These cytokine combinations provoke the expression of inducible nitric oxide synthase (iNOS) by MSCs. The impulsive effect of MSCs treated with inflammatory cytokines on B16 melanoma cells in vivo can be reversed by inhibitor or short interfering RNA of iNOS. Our results suggest that the MSCs in tumour inflammatory microenvironment may be elicited of immunosuppressive function, which will help tumour to escape from the immunity surveillance.
Keywords: mesenchymal stem cells, proinflammatory cytokines, inducible nitric oxide synthase
Introduction
Mesenchymal stem cells (MSCs) have the ability to differentiate into multiple lineages such as chondrocytes, osteocytes, adipocytes, myocytes and astrocytes, which are a potential source of stem cells for cellular and genetic therapy [1, 2]. MSCs are found in the bone marrow but have also been isolated from other sites in the body such as adipose tissue and uterus [3]. The phenotype of MSCs is identified by the absence of the CD34 and CD45 haematopoietic cell markers and is positive for CD29, CD90 and CD105 [4]. MSCs express the major histocompatibility complex (MHC) class I but do not express MHC class II, B7–1, B7–2, CD40 or CD40L molecules. MSCs can be expanded more than 104-fold in culture without loss of their multilineage differentiation potential. Therefore, MSCs are not only considered as an acceptable source of stem cells in haematopoietic recovery, but also as an available cell in tissue regeneration [2].
MSCs have been shown to be highly immunosuppressive, which is a character unlike embryonic stem cells. MSCs were found to suppress T-cell proliferation and cytokine production, but the mechanisms by which MSCs mediate these immunosuppressive effects have not been fully elucidated [5–8]. Koc et al. found that when 2 × 106 allogeneic MSCs per kg were infused along with allogeneic bone marrow into patients with metachromatic leukodystrophy, or Hurler’s syndrome, there were no evidence of alloreactive T cells and no incidence of Graft-versus-Host Disease (GvHD) [9]. Others have attempted using MSCs to prevent or treat autoimmune diseases, such as experimental autoimmune encephalomyelitis and collagen-induced arthritis [10, 11]. It has been demonstrated that interleukin (IL)-10, transforming growth factor (TGF)-β, nitric oxide, indoleamine 2,3-dioxygenase [12] and prostaglandin E2 [13] have been involved in MSC-mediated immunosuppression, others report that IL-10 and TGF-β are not involved [14]. Meanwhile, the immunomodulatory effect of MSCs is not always achieved; in some cases, no effect is observed [15, 16]. Therefore, the mechanisms of MSCs suppress immune reactions need to be determined for a better utility of these cells. Ren et al. reported that the immunosuppressive function of MSCs is elicited by proinflammatory cytokines and the immunosuppression of MSCs is through the concerted action of chemokines and nitric oxide [17].
Djouad et al. showed that MSCs had displayed side effects related to systemic immunosuppression favouring tumour growth in vivo[18]. On the contrary, MSCs have been shown to be anti-tumorigenic in a mouse model of Kaposi’s sarcoma by inhibiting Akt (a serine-threonine protein kinase known to promote cell survival) activity [19]. MSCs have a tropism for tumours [20] and tumour environment is always accompanied with proinflammatory cytokines. Therefore, it is important to investigate the effect of MSCs favouring tumour growth in inflammatory environment.
The aim of this study was to investigate the mechanism of MSCs favouring tumour escape from immunologic surveillance in inflammatory microenvironment. We first compared the promotive capacity of BM-derived MSCs on B16 melanoma cells growth in vivo, pre-incubated or not with the inflammatory cytokines interferon (IFN)-γ and tumour necrosis factor (TNF)-α. We showed that the development of B16 melanoma cells is faster when co-injected with MSCs pre-incubated IFN-γ and TNF-α compared with control groups. Moreover, the tumour incidence improves obviously in allogeneic recipients when the B16 melanoma cells were co-injected with MSCs pre-incubated with IFN-γ and TNF-α. We then demonstrated that the immunosuppressive function of MSCs was elicited by IFN-γ and TNF-α. These cytokine combinations provoke the expression of inducible nitric oxide synthase (iNOS) by MSCs. The impulsive effect of MSCs treated with inflammatory cytokines on B16 melanoma cells in vivo can partly be reversed by iNOS inhibitor. Our results suggest that the MSCs in tumour inflammatory microenvironment may be elicited of immunosuppressive function, which will help tumour to escape from the immunity surveillance.
Materials and methods
Reagents
Recombinant mouse IFN-γ, TNF-α were from Peprotech (La Jolla, CA, USA); 1400W were from Biotime (Haimen, Jiangsu, China); antimouse CD34, CD45, CD90, CD105 and CD29 antibodies were from BioLegend (San Diego, CA, USA).
Cells and animal
MSCs were generated from bone marrow flushed out of tibia and femur of 4–6-week-old mice. Cells were cultured in α-Minimum Essential Medium (MEM) medium supplemented with 10% Fetal Bovine Serum (FBS), 2 mM glutamine, 100 U/ml penicillin and 100 mg/ml streptomycin (all from Invitrogen, Carlsbad, CA, USA). Non-adherent cells were removed after 72 hrs, and adherent cells were maintained with medium replenishment every 3 days. Cells were used at 5th to 20th passage.
Murine B16 melanoma cells were cultured at 37°C, with 5% CO2, in DMEM with 10% fetal bovine serum, supplemented with 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. Cells were subcultured every 3 days when they reached 70–80% confluence.
Male C57BL/6 and Balb/c mice, 6–8 weeks old, were purchased from Shanghai Experimental Animal Center of the Chinese Academy of Sciences, Shanghai, China. Mice in this study were housed in pathogen-free conditions, and all procedures were performed in accordance with the guideline of the Committee on Animals of the Chinese Academy of Sciences.
Differentiation of MSCs
MSCs were induced to differentiate into adipocytes in vitro by supplementation with 60 μM indomethacin, 0.5 mM isobutylmethylxanthin, 10 nM dexamethasone and 10 μg/ml insulin for 14 days. The presence of adipocytes was verified by staining for triglycerides with Oil red O (Sigma-Aldrich, St. Louis, MO, USA), to reveal intracellular lipid accumulation. MSCs were cultured with osteoinductive medium consisting of DMEM supplemented with 10% FBS, β-mercaptoethanol, 100 μM L-ascorbic acids, 10 nM dexamethasone and 10 mM β-glycerophosphate for 14 days. These cells were stained with Von Kossa to identify calcium deposition characteristic of osteoblasts.
B16 melanoma murine tumour model
B16 melanoma cells and MSCs were prepared either as single-cell type suspensions (5 × 106 cells in 100 μl PBS) or a mix of cells (5 × 106 B16 cells and 1 × 106 MSCs in 100 μl PBS). Subcutaneous administration of B16 cells (alone or mixed with MSCs) was performed in the armpit area of C57BL/6 or Balb/c mice. Mice were examined three times a week and tumour growth was evaluated by measuring the length and width of tumour mass. The animals were sacrificed and tumours were recovered at the end of the experiment. Tumour masses were weighed and analysed by histology.
MLR (mixed lymphocyte reaction)
Splenocytes were isolated from mouse spleen by disaggregation into 10 ml RPMI Medium 1640 (Invitrogen). Erythrocytes were lysed with NH4Cl 0.84% and subsequently washed three times in RPMI 1640. Cell count and viability were assessed by trypan blue dye exclusion. In mitogen proliferative assays, responder splenocytes were incubated with 5 μg/ml concanavalin A (ConA; Sigma-Aldrich, St. Louis, MO, USA) then cultured with IL-2 (200 U/ml) alone for 48 hrs. All splenocytes cultures were maintained in RPMI 1640 medium supplemented with 10% heat-inactivated FBS, 2 mM glutamine, 100 U/ml penicillin, 100 mg/ml streptomycin and 50 mM β-ME (complete medium). MSCs were added to the MLR to obtain a 200 μl final volume. After 3 days of incubation, 1 μCi/well (0.037 MBq/well) 3H-thymidine was added overnight and thymidine incorporation was measured using a β-scintillation counter. The data are presented as the percent of the relative proliferative response, corresponding to the mean counts per minute of a responder stimulator pair in the absence of MSCs and was attributed a 100% value.
Real-time PCR
The MSCs were incubated with IFN-γ (20 ng/ml) and TNF-α (20 ng/ml), both or alone, for 12 hrs. The cells were collected to extract the total cellular mRNA with Trizol Reagent (Invitrogen). Expression of mRNA was determined by real-time RT-PCR using SYBR Green Master Mix (Applied Biosystems, Foster City, CA, USA). Total sample RNA was normalized to endogenous β-actin mRNA. Primers sequences for iNOS were: forward: 5′-CAGCTGGGCTGTACAAACCTT-3′; reverse: 5′-CATTGGAAGTGAAGCGTTTCG-3′. Thermocycler conditions included an initial hold at 50°C for 2 min. and then 95°C for 10 min.; this was followed by a two-step PCR program of 95°C for 15 sec. and 60°C for 60 sec. repeated for 40 cycles on an Mx4000 system (Stratagene, La Jolla, CA, USA), on which data were collected and quantitatively analysed. Expression level of mRNA is presented as fold change relative to an untreated control.
Allogeneic implantation of B16
Balb/c mice were used in this study. B16 melanoma cells and MSCs were prepared either as single-cell type suspensions (5 × 106 cells in 100 μl PBS) or a mix of cells (5 × 106 B16 cells and 1 × 106 MSCs in 100 μl PBS). Subcutaneous administration of B16 melanoma cells (alone or mixed with MSCs) was performed in the armpit area of Balb/c. The tumour incidence was examined three times a week to evaluate the situation of tumour growth.
Short interfering RNA (siRNA) synthesis and transient transfection
Three siRNA sequences of iNOS were designed by using Oligoengine software and confirmed by nucleotide Basic Local Alignment Search Tool (BLAST) searches. The three putative candidate sequences and a scrambled sequence with no significant homology were listed in Table 1. Transfections were performed with a Lipofectamine 2000 kit (Invitrogen) according to the manufacturer’s instructions. Cells (1–3 × 106) grew to a confluency of 50–60% in 10 cm Petri dishes were transfected with siRNA sequence or their relative mock sequences, then the cells were observed under fluorescence microscope and harvested 48 hrs after transfection.
Table 1.
Sequence of the oligonucleotides for real-time PCR and siRNA construct-making assays
| Assays | Gene | Sequence (5′→ 3′) | |
|---|---|---|---|
| Real-time PCR | iNOS | F | CAGCTGGGCTGTACAAACCTT |
| R | CATTGGAAGTGAAGCGTTTCG | ||
| β-actin | F | CTCCATCCTGGCCTCGCTGT | |
| R | GCTGTCACCTTCACCGTTCC | ||
| iNOS siRNA | Sequence 1 | Sense | AUUGUACUAUUGUGGACUAdTdT |
| Antisense | UAGUCCACAAUAGUACAAUdAdC | ||
| Sequence 2 | Sense | AGUAUUAUGGCUCCUUUAATT | |
| Antisense | UUAAAGGAGCCAUAAUACUdGdG | ||
| Sequence 3 | Sense | CACAGCAAUAUAGGCUCAUTT | |
| Antisense | AUGAGCCUAUAUUGCUGUGdGdC | ||
| Control | Sense | UUCUCCGAACGUGUCACGUTT | |
| Antisense | ACGUGACACGUUCGGAGAATT | ||
Statistical analysis
Statistical analysis of the data was done by using GraphPad Prism 4. Student’s t-test was used to compare between mean values of two groups. Final values are expressed as mean ± S.E.M. A difference of at least P < 0.05 was considered statistically significant.
Results
MSCs pretreated by proinflammatory cytokines promotes the development of B16 melanoma cells in vivo
MSCs adhered to the plastic surface, presenting a small population of single cells which were spindle-shaped and contained one nucleus after 72 hrs of the primary culture. During 7 to 10 days after the initial plating, the cells looked like long spindle-shaped fibroblastic cells and began to form colonies. MSCs could differentiate into adipocytes and osteoblast-like cells. The surface antigen profile of mouse MSCs was detected by flow cytometry was positive for CD90, CD105 and CD29 and negative for CD34 and CD45 (Fig. 1A, B)
Fig 1.
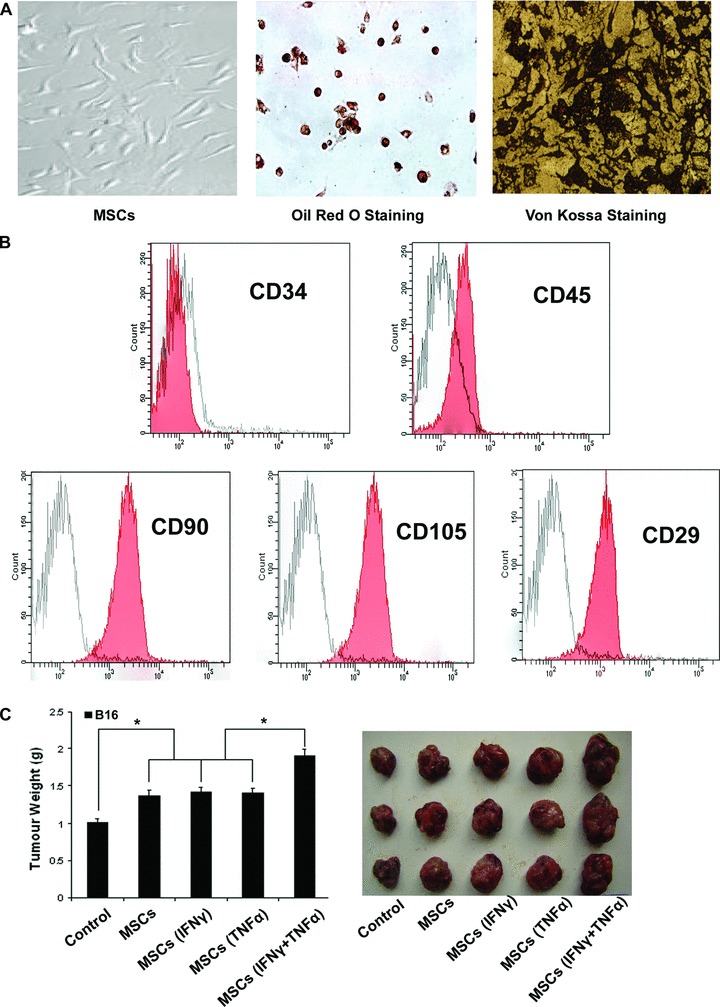
MSCs treated with proinflammatory cytokines favour the growth of tumour. (A) MSCs at passage 8 were grown under different conditions that favour differentiation into either adipocytes (for 14 days), or osteoblasts (for 14 days), as described in ‘Materials and methods’. The presence of triglycerides, characteristic of adipocytes, was revealed by staining with oil red O. Calcium deposition, indicative of osteoblasts, was stained with Von Kossa stain. (B) MSCs derived from C57BL/6 mice were stained with commercially available antibodies to analysis the surface marker by flow cytometry (red). Corresponding antibodies of the same isotype were used as controls (white). (C) C57BL/6 MSCs (1 × 106) were pretreated with proinflammatory cytokines IFN-γ and TNF-α (20 ng/ml each) for 12 hrs and then mixed with B16 melanoma cells (5 × 106) to perform subcutaneous administration in the C57BL/6 mice armpit area. After 14 days of implantation, the animals were sacrificed and tumours were dissected. The weight and volume of B16 tumours were measured. The weight of tumour were measured after been removed from the mice. (*P < 0.05).
To investigate whether BM-derived MSCs could favour the growth of B16 cells in inflammatory microenvironment, the MSCs, which were pre-incubated or not with the inflammatory cytokines IFN-γ and TNF-α, were subcutaneously co-injected with B16 cells in syngeneic C57BL/6 mice. Tumour nodules were firstly formed in the group of MSCs that was pre-incubated with the inflammatory cytokines IFN-γ and TNF-α. We found the growth of tumour that was mixed with MSCs was faster than that of B16 cells alone. Compared with MSCs unpretreated or pretreated with either IFN-γ or TNF-α, the ones pretreated with both inflammatory cytokines displayed the most striking tumour-stimulating effect (Fig. 1C).
The rejection from Balb/c mice on B16 melanoma cells are reduced by the MSCs treated with proinflammatory cytokines
We implanted B16 melanoma cells in Balb/c mice with MSCs, which were pre-incubated or not with the inflammatory cytokines, to test the immunosuppression properties of MSCs that favour the B16 melanoma cells escaping from the rejection of Balb/c mice. While B16 melanoma cells developed into tumours rapidly after implanted subcutaneously in C57BL/6 mice, they were fully rejected in Balb/c mice, indicating no tumour formation at all. Once we implanted B16 melanoma cells mixed with MSCs in Balb/c mice, the incidence of tumour increased. Moreover, the tumour incidence could increase higher if the MSCs were pretreated with proinflammatory cytokines before they were mixed with B16 cells (Fig. 2A). These results indicate that MSCs have the ability to assist the B16 cells escaping from immune surveillance and this capacity is induced by proinflammatory cytokines.
Fig 2.
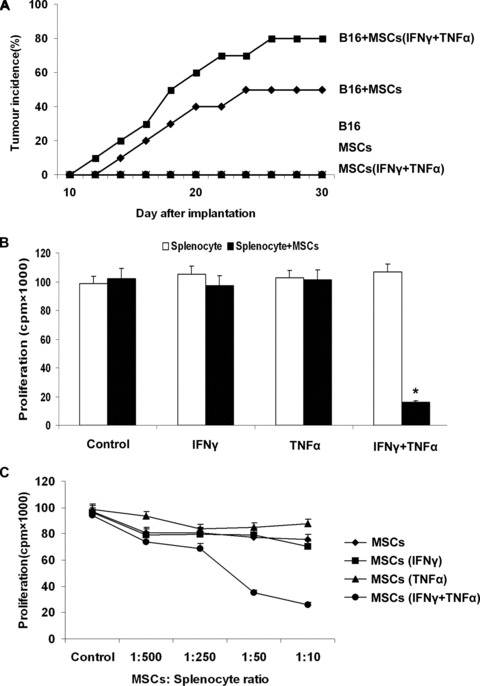
The rejection from Balb/c mice on B16 melanoma cells are reduced by the MSCs treated with proinflammatory cytokines. (A) Balb/c MSCs (1 × 106) were pretreated with inflammatory cytokines IFN-γ and TNF-α (20 ng/ml each) for 12 hrs and then mixed with B16 melanoma cells (5 × 106) to perform subcutaneous administration in the Balb/c mice armpit area. Tumour incidence was observed to evaluate the immunosuppressive function of MSCs that assisted the B16 melanoma cells from escaping the immunological rejection of Balb/c mice. As negative controls, B16 cells or MSCs alone were injected in Balb/c mice. (B) C57BL/6 MSCs were pretreated with inflammatory cytokines IFN-γ and TNF-α (20 ng/ml each) for 12 hrs. Fresh C57BL/6 splenocytes were activated by ConA (5 μg/ml) for 72 hrs and IL-2 (20 ng/ml) was added for proliferation. MSCs were cocultured in a 96-well with splenocytes (1 × 105/well) plate at a 1:10 ratio and cell proliferation was assessed 72 hrs later by 3H-Tdr incorporation. (C) MSCs were mixed with splenocytes (1 × 105/well) at different ratios (1:500, 1:250, 1:50, 1:10), and the proliferation was assessed 72 hrs later (*P < 0.05).
The immunosuppressive function of MSCs depended on proinflammatory cytokines treatment
We examined the effect of proinflammatory cytokines on immunosuppressive function of MSCs by co-cultured the MSCs with splenocytes. The splenocytes were activated with ConA followed by expansion with IL-2 for 3 days. We cocultured the activated splenocytes with MSCs that were pre-treated with or not with IFN-γ and TNF-α. The results showed that MSCs could not inhibit the proliferation of splenocytes. And if we pretreated MSCs with proinflammatory cytokines IFN-γ and TNF-α, the inhibitory effect of MSCs on splenocytes proliferation were arised (Fig. 2B). We also cocultured proinflammatory cytokines pretreated MSCs with activated splenocytes at graded ratios and found that the proliferation of splenocytes was blocked by MSCs at ratios as low as 1:50 (MSC to splenocyte) (Fig. 2C). These observations suggest that the MSCs immunosuppressive ability can be induced by proinflammatory cytokines IFN-γ and TNF-α.
MSCs exert immunosuppression requires nitric oxide
High concentrations Nitric oxide is known to inhibit T-cell responses [21]. We examined whether iNOS and nitric oxide expression could increase in MSCs that were exposed to proinflammatory cytokines. MSCs were treated with IFN-γ and TNF-α, and the level of iNOS mRNA was assayed by real-time PCR. As was shown in the histogram and electropherogram, iNOS mRNA was up-regulated significantly in MSCs after being exposed to proinflammatory cytokines (Figure 3A).
Fig 3.
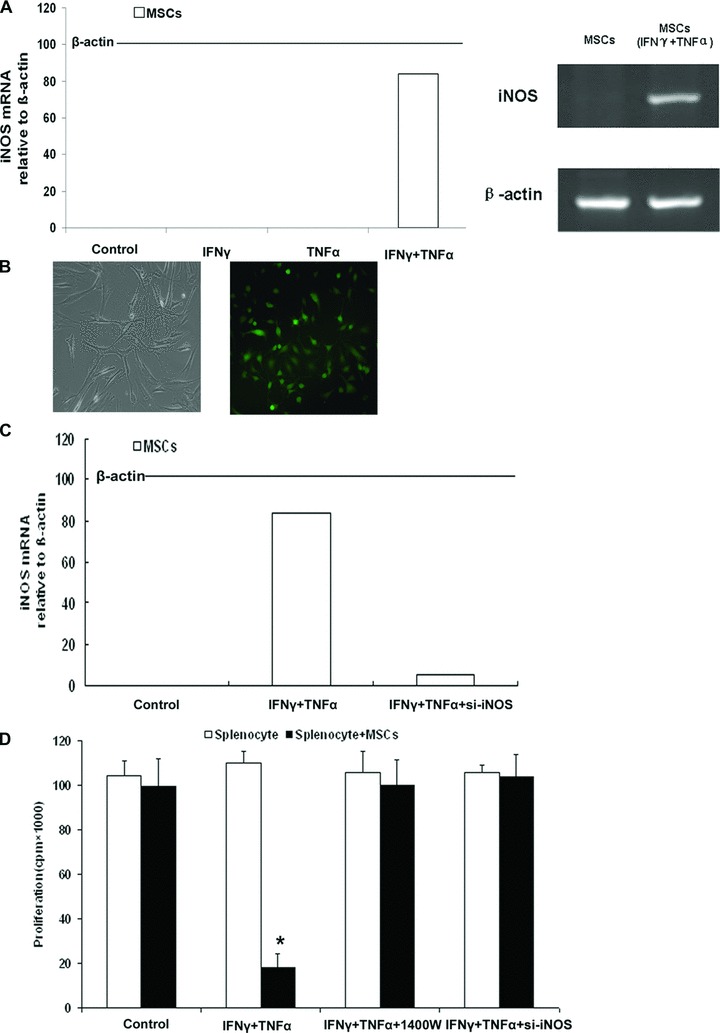
Immunosuppression by MSCs requires nitric oxide. (A) C57BL/6 MSCs were pretreated with proinflammatory cytokines IFN-γ and TNF-α (20 ng/ml each) for 12 hrs. iNOS mRNA in MSCs was assayed by real-time PCR and compared to β-actin mRNA, defined as 100 arbitrary unit. The electropherogram of iNOS cDNA demonstrated the same result as real-time PCR. (B) Transfections of iNOS siRNA into MSCs were performed with a Lipofectamine, and FAM was observed under fluorescence microscope (right, ×200). (C) Real-time-PCR result showed that the expression of iNOS was markedly decreased in MSCssi-iNOS stimulated by both IFN-γ and TNF-α, the inhibitory efficiency was more than 90% compared with the MSCsvector stimulated by both IFN-γ and TNF-α. (D) MSCs from C57BL/6 mice were cocultured with fresh C57BL/6 splenocytes plus Con A and IL-2, with or without the iNOS inhibitor – 1400W (100 μM) and siRNA of iNOS. Cell proliferation was assayed by 3H-Tdr incorporation after 72 hrs.
To investigate the role of nitric oxide, its production was shut down using siRNA to inhibit the expression of iNOS and a selective inhibitor of iNOS activity, 1400W. All the three candidate sequences could effectively inhibit iNOS expression in MSCs stimulated by IFN-γ and TNF-α (data not shown). As shown in Figure 3B, the transfection efficiency of iNOS siRNA labelled with FAM (commonly used fluorophores, which is attached at the 5′ end of Taqman probe) was more than 70% through observed under fluorescence microscope. Real-time PCR was performed to assess the expression of iNOS in MSCs to further confirm the transfection efficiency of iNOS siRNA. Compared with the MSCsvector stimulated by both IFN-γ and TNF-α, the expression of iNOS was markedly decreased in MSCssi-iNOS stimulated by both IFN-γ and TNF-α, which showed a more than 90% inhibitory efficiency (Fig. 3C).
In mixed cocultures of splenocytes and MSCs pre-stimulated by IFN-γ and TNF-α, splenocytes proliferation was restored to normal level by si-iNOS and 1400W (Fig. 3D). These results strongly suggest that nitric oxide produced by proinflammatory cytokine-induced MSCs mediates the suppression of splenocytes.
The growth of B16 melanoma favoured by MSCs in vivo can be inhibited by iNOS inhibitor and siRNA of iNOS
As was showed above that the growth of B16 cells was enhanced by MSCs that had been pretreated with inflammatory cytokines IFN-γ and TNF-α both compared with control groups. The enhancement might be correlated with MSCs immunosuppressive function that could help B16 melanoma cells to escape from the local immunosurveillance. The immunosuppressive capability of MSCs was dependent upon the expression of iNOS which was induced by inflammatory cytokines. Therefore, we demonstrated that the stimulating effect of MSCs on allograft tumour growth could be inhibited by iNOS inhibitor – 1400W and iNOS siRNA (Fig. 4A). The result of tumour volume was same as the weight (data not shown). And the tumour incidence of B16 melanoma cells that had been co-injected with MSCs, which were pretreated with inflammatory cytokines, in Balb/c mice was reduced significantly after the use of 1400W and siRNA of iNOS (Fig. 4B). These data support that immunosuppression mediated by MSCs via nitric oxide play an important role in favouring the escape of B16 melanoma cells from immune surveillance.
Fig 4.
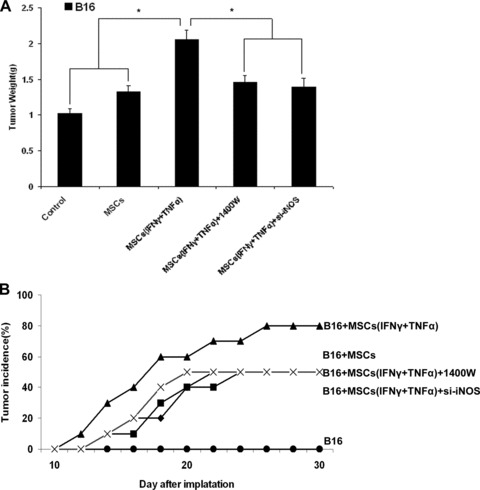
The growth of tumour favoured by MSCs in vivo can be inhibited by iNOS inhibitor. (A) C57BL/6 MSCs (1 × 106) were pretreated with inflammatory cytokines IFN-γ and TNF-α (20 ng/ml each) for 12 hrs and then mixed with B16 cells (5 × 106) to perform subcutaneous administration in the C57BL/6 mice armpit area. On days 2, 5, 8 and 11 after implantation, recipients were injected i.p. with iNOS inhibitor – 1400W. After 14 days of implantation, the animals were sacrificed and tumours were dissected. The weight and volume of B16 tumours were measured. Transfections of iNOS siRNA into C57BL/6 MSCs were performed with a Lipofectamine. Then the MSCssi-iNOS stimulated by both IFN-γ and TNF-α for 12 hrs before been mix with B16 melanoma cells (5 × 106) to perform subcutaneous administration in the C57BL/6 mice armpit area. After 14 days of implantation, the animals were killed and tumours were dissected. The weight and volume of B16 tumours were measured. (B) Balb/c MSCs (1 × 106) were pretreated with inflammatory cytokines IFN-γ and TNF-α (20 ng/ml each) for 12 hrs and then mixed with B16 melanoma cells (5 × 106) to perform subcutaneous administration in the Balb/c mice armpit area. On days 2, 5, 8 and 11 after implantation, recipients were injected i.p. with iNOS inhibitor – 1400W. Tumour incidence was observed. Transfections of iNOS siRNA into Balb/c MSCs were performed with a Lipofectamine. Then the MSCssi-iNOS stimulated by both IFN-γ and TNF-α for 12 hrs before been mixed with B16 melanoma cells (5 × 106) to perform subcutaneous administration in the Balb/c mice armpit area. Tumour incidence was observed.
Discussion
MSCs play an important role in treating various degenerative diseases and immune disorders. They have great potential for correcting aberrant immune reactions. Previous studies showed that these cells could be expanded and induced ex vivo, terminally differentiate into osteoblasts, chondrocytes, adipocytes, myotubes, neural cells and haematopoietic supporting stroma [1, 2, 22]. And the immunosuppression demonstrated by MSCs has been reported in several studies [5–8]. However, in certain circumstances the property of immunosuppression may display negative effects such as the promotion of tumour growth. Ren et al. have shown that the immunosuppressive function of MSCs is elicited by proinflammatory cytokines and the immunosuppression of MSCs is through the concerted action of chemokines and nitric oxide [17]. MSCs have a tropism for tumours [20] and the incidence and development of tumour is always accompanied with proinflammatory cytokines. Therefore it is important to investigate the effect of MSCs favouring tumour growth in inflammatory environment. To investigate this hypothesis, we assessed the role of MSCs in inflammatory environment in the development of tumour by B16 melanoma cells implanted in allogeneic mice.
Our results demonstrate that tumour growth is faster when B16 melanoma cells were co-injected with MSCs pre-incubated with IFN-γ and TNF-α compared with control groups. Moreover, the tumour incidence improves obviously in allogeneic recipients when the B16 melanoma cells were co-injected with MSCs pre-incubated with IFN-γ and TNF-α. We then demonstrated that the immunosuppressive function of MSCs was elicited by IFN-γ and TNF-α. These cytokine combinations provoke the expression of iNOS by MSCs. The impulsive effect of MSCs treated with inflammatory cytokines on B16 melanoma cells in vivo can partly be reversed by iNOS inhibitor. Our results suggest that the MSCs in tumour inflammatory microenvironment may be elicited of immunosuppressive function, which will help the tumour to escape from the immunity surveillance.
Recent studies have shown that MSCs displayed an ability to favour tumour growth under certain circumstances. Djouad et al. showed that MSCs had displayed side effects related to systemic immunosuppression favouring tumour growth in vivo[18]. Hall et al. have also shown that stromal cells overexpressing VCAM-1 enhanced survival of leukemic cells in a PI-3 kinase dependent manner, compared with stromal cells expressing only endogenous vascular cell adhesion molecule (VCAM)-1 [23]. On the contrary, MSCs have been reported to be anti-tumorigenic in a mouse model of Kaposi’s sarcoma by inhibiting Akt activity [19]. Our study implies that inflammatory cytokines may be the key factors that regulate the effect of MSCs on tumour growth. The presence of an active immune reaction in tumour environment can lead the MSCs that localized in tumour to acquire immunosuppressive function. On the other hand, the absence of active inflammation in tumour may not only abstract MSCs to localize but also induce them to be immunosuppressive.
Although MSCs have acquired many interests for their potential use in clinical therapy, there are still lots of unsolved problems, which limit the application of MSCs. The study of MSCs clinical application for tissue engineering and regenerative medicine needs to pay attention to the local microenvironment. Our results suggest that the MSCs in tumour inflammatory microenvironment may be elicited of immunosuppressive function, which will help the tumour to escape from the immunity surveillance. Therefore, the use of MSCs in clinical treatment, especially in cancer therapy, should be extremely cautious.
Acknowledgments
This project was supported by the Special Funds for National key Sci-Tech Special Project of China (Grant NO: 2008ZX10002-019, 2008ZX10002-025). National Natural Science Foundation of China (Grant NO: 30870974, 30801347, 30700981, 30901722, 81000970, 81030041). Shanghai Science and Technology Committee (Grant NO: 08XD14003, 10411963100, 10ZR1439900, 10ZR1439600). Key Basic Research Project of China (Grant NO: 2010CB945600, 2011CB966200). Science Fund for Creative Research Groups, NSFC, China, (Grant NO: 30921006) and Stem Cell and Medicine Postgraduate Innovation Experiment Center, The Second Military Medical University (Grant NO: SCOP101913, SCOP102004, SCOP101812).
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Pittenger MF, Mackay AM, Beck SC, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–7. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 2.Barry FP, Murphy JM. Mesenchymal stem cells: clinical applications and biological characterization. Int J Biochem Cell Biol. 2004;36:568–84. doi: 10.1016/j.biocel.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 3.da Silva Meirelles L, Chagastelles PC, et al. Mesenchymal stem cells reside in virtually all post-natal organs and tissues. J Cell Sci. 2006;119:2204–13. doi: 10.1242/jcs.02932. [DOI] [PubMed] [Google Scholar]
- 4.Noel D, Djouad F, Jorgense C. Regenerative medicine through mesenchymal stem cells for bone and cartilage repair. Curr Opin Investig Drugs. 2002;3:1000–4. [PubMed] [Google Scholar]
- 5.Sato K, Ozaki K, Oh I, et al. Nitric oxide plays a critical role in suppression of T-cell proliferation by mesenchymal stem cells. Blood. 2007;109:228–34. doi: 10.1182/blood-2006-02-002246. [DOI] [PubMed] [Google Scholar]
- 6.Rasmusson I, Ringden O, Sundberg B, et al. Mesenchymal stem cells inhibit lymphocyte proliferation by mitogens and alloantigens by different mechanisms. Exp Cell Res. 2005;305:33–41. doi: 10.1016/j.yexcr.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 7.Krampera M, Glennie S, Dyson J, et al. Bone marrow mesenchymal stem cells inhibit the response of naive and memory antigen-specific T cells to their cognate peptide. Blood. 2003;101:3722–9. doi: 10.1182/blood-2002-07-2104. [DOI] [PubMed] [Google Scholar]
- 8.Di Nicola M, Carlo-Stella C, Magni M, et al. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood. 2002;99:3838–43. doi: 10.1182/blood.v99.10.3838. [DOI] [PubMed] [Google Scholar]
- 9.Koc ON, Day J, Nieder M, et al. Allogeneic mesenchymal stem cell infusion for treatment of metachromatic leukodystrophy (MLD) and Hurler syndrome (MPS-IH) Bone Marrow Transplant. 2002;30:215–22. doi: 10.1038/sj.bmt.1703650. [DOI] [PubMed] [Google Scholar]
- 10.Djouad F, Fritz V, Apparailly F, et al. Reversal of the immunosuppressive properties of mesenchymal stem cells by tumor necrosis factor alpha in collagen-induced arthritis. Arthritis Rheum. 2005;52:1595–603. doi: 10.1002/art.21012. [DOI] [PubMed] [Google Scholar]
- 11.Zappia E, Casazza S, Pedemonte E, et al. Mesenchymal stem cells ameliorate experimental autoimmune encephalomyelitis inducing T-cell anergy. Blood. 2005;106:1755–61. doi: 10.1182/blood-2005-04-1496. [DOI] [PubMed] [Google Scholar]
- 12.Meisel R, Zibert A, Laryea M, et al. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood. 2004;103:4619–21. doi: 10.1182/blood-2003-11-3909. [DOI] [PubMed] [Google Scholar]
- 13.Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood. 2005;105:1815–22. doi: 10.1182/blood-2004-04-1559. [DOI] [PubMed] [Google Scholar]
- 14.Xu Y, Flies AS, Flies DB, et al. Selective targeting of the LIGHT-HVEM costimulatory system for the treatment of graft-versus-host disease. Blood. 2007;109:4097–104. doi: 10.1182/blood-2006-09-047332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nauta AJ, Westerhuis G, Kruisselbrink AB, et al. Donor-derived mesenchymal stem cells are immunogenic in an allogeneic host and stimulate donor graft rejection in a nonmyeloablative setting. Blood. 2006;108:2114–20. doi: 10.1182/blood-2005-11-011650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sudres M, Norol F, Trenado A, et al. Bone marrow mesenchymal stem cells suppress lymphocyte proliferation in vitro but fail to prevent graft-versus-host disease in mice. J Immunol. 2006;176:7761–7. doi: 10.4049/jimmunol.176.12.7761. [DOI] [PubMed] [Google Scholar]
- 17.Ren G, Zhang L, Zhao X, et al. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell. 2008;2:141–50. doi: 10.1016/j.stem.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 18.Djouad F, Plence P, Bony C, et al. Immunosuppressive effect of mesenchymal stem cells favors tumor growth in allogeneic animals. Blood. 2003;102:3837–44. doi: 10.1182/blood-2003-04-1193. [DOI] [PubMed] [Google Scholar]
- 19.Khakoo AY, Pati S, Anderson SA, et al. Human mesenchymal stem cells exert potent antitumorigenic effects in a model of Kaposi’s sarcoma. J Exp Med. 2006;203:1235–47. doi: 10.1084/jem.20051921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakamizo A, Marini F, Amano T, et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005;65:3307–18. doi: 10.1158/0008-5472.CAN-04-1874. [DOI] [PubMed] [Google Scholar]
- 21.Niedbala W, Cai B, Liew FY. Role of nitric oxide in the regulation of T cell functions. Ann Rheum Dis. 2006;65:iii37–40. doi: 10.1136/ard.2006.058446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu W, Zhang X, Qian H, et al. Mesenchymal stem cells from adult human bone marrow differentiate into a cardiomyocyte phenotype in vitro. Exp Biol Med. 2004;229:623–31. doi: 10.1177/153537020422900706. [DOI] [PubMed] [Google Scholar]
- 23.Hall BM, Fortney JE, Taylor L, et al. Stromal cells expressing elevated VCAM-1 enhance survival of B lineage tumor cells. Cancer Lett. 2004;207:229–39. doi: 10.1016/j.canlet.2003.10.033. [DOI] [PubMed] [Google Scholar]