

Abstract
Substantial genetic evidence suggests that chromosome 11q is involved in regulating initiation and progression of malignant melanomas. Mutations of the MEN1 gene, located in chromosome 11q13, predispose individuals to the multiple endocrine neoplasia type 1 (MEN1) familial syndrome. MEN1 patients develop primary malignant melanoma, suggesting a potential link between MEN1 syndrome and development of melanomas, but the precise molecular mechanism is poorly understood. Here we show that the MEN1 gene suppresses malignant phenotypes of melanoma cells through multiple signalling pathways. Ectopic expression of menin, the product of MEN1 gene, significantly inhibited melanoma cell proliferation and migration in vitro and in vivo. The inhibition was partly achieved through suppressing expression of growth factor pleiotrophin (PTN) and receptor protein tyrosine phosphatase (RPTP) β/ζ, accompanied with the reduced expression of phosphatidylinositol 3-kinase (pI3K) and decreased phosphorylation of focal adhesion kinase (FAK) and extracellular signal regulated kinase (ERK1/2). Interestingly, reduced expression of menin was associated with hypermethylation of the CpG islands of the MEN1 promoter in melanoma cells. Taken together, these findings suggest a previously unappreciated function for menin in suppressing malignant phenotypes of melanomas and unravel a novel mechanism involving in regulating PTN signalling by menin in development and progression of melanomas.
Keywords: melanoma, menin, pleiotrophin, RPTP β/ζ
Introduction
The MEN1 gene is located on chromosome 11q13, which is mutated in patients with an inherited tumour syndrome, multiple endocrine neoplasia type 1 (MEN1) [1]. MEN1 knockout mice develop parathyroid, pancreatic, pituitary and adrenal tumours, mimicking human MEN1 syndrome, indicating that MEN1 as a bona fide tumour suppressor gene in endocrine tumours [2]. Recently, several reports have demonstrated that MEN1 is associated with non-endocrine tumours. For instance, menin has been shown to associate with trxG family proteins in a histone methyltransferase complex including trxG proteins MLL (mixed lineage leukaemia), retinoblastoma binding protein 5, WD repeat domain 5 and ASH2 (absent, small or homeotic) and promote histone 3 Lysine 4 (H3K4) methylation at the promoter of target genes [3, 4], and it is required for maintenance of Hox family gene expression, and initiation of MLL-mediated leukemogenesis and myeloid transformation [3, 5, 6]. Recently, we have found that menin represses PTN transcription through Polycomb gene-mediated trimethylation of H3K27 and development of lung adenocarcinoma [7].
Malignant melanoma is the deadliest form of skin cancer, which is an increasing worldwide health problem because of its highly aggressive and drug-resistant nature [8]. Recent advances in understanding development and maintenance of melanoma provide novel insights into the molecular mechanisms. Kit/stem-cell factor (SCF) signalling and Mitf-dependent transcription is essential for melanoma initiation and development [8]. Disruption of Mitf in melanocytes or melanoma triggered apoptosis that can be blocked by B cell lymphoma 2 (Bcl-2) overexpression [9]. Genome-wide RNA-interference screening has uncovered 17 genes, including insulin like growth factor binding protein (IGFBP7), which have a central role in activated BRAF oncogene (BRAFV600E)-mediated apoptosis of melanocyte [10]. Selective inhibition of B-Raf drives oncogenic RAS-dependent BRAF binding to C-Raf, CRAF activation and mitogen-activated protein kinase kinase (MEK)-extracellular signal regulated kinase (ERK) signalling, revealing another paradigm of BRAF-mediated signalling that promotes tumour progression [11]. These findings indicate a key role of Mitf, BRAF and BCL2 in promoting progression of melanoma, and partly explained the well-known treatment resistance of melanoma. Pleiotrophin (PTN) is a heparin-binding growth factor that is highly expressed in certain solid cancers, including melanoma [12, 13]. Targeted disruption of PTN decreases melanoma tumour growth, metastasis and angiogenesis [14, 15]. PTN-dependent cell growth required both mitogen-activated protein kinase (MAPK) and pI3-kinase activity [16]. In melanoma, both MAPK and phosphatidylinositol 3-kinase (pI3K)-serine/threonine protein kinase (AKT) signalling pathways are constitutively activated through multiple mechanisms, and they exert a crucial regulating role in malignant phenotype of melanoma [17].These advances highlight the importance of understanding signalling pathways in clinical practice and genotyping of tumours prior to administering gene selective drugs, to identify patients who are likely to respond to the treatment with the drugs.
At present, it is unclear whether menin’s function is associated with melanoma. In patients with MEN1 syndrome, various skin tumours of mesenchymal origin, including angiofibromas, collagenomas and lipomas, as well as malignant melanoma had been reported [18, 19]. Nord et al. have found that LOH in 11q13 was detected in six tumours of melanoma, and the deletion including the MEN1 locus in 19 cases of sporadic metastatic melanoma [18]. Previous implications of multiple melanoma tumour suppressors are localized in chromatin 11q, including the MEN1 region [8], raising the possibility of an association between MEN1 and melanoma.
Given these observations, we explored menin’s potential role in suppressing malignant melanoma. Our findings suggest a previously unappreciated function for menin in suppressing malignant phenotypes of melanoma. Menin suppresses proliferation and migration of mouse and human melanoma cells in vitro and in vivo, partly through regulating PTN/RTPT β/ζ signalling. In addition, inactivation of menin was associated with hypermethylation of CpG islands of the MEN1 promoter region in A375 melanoma cells. These data suggest a novel mechanism involving regulation of PTN signalling by menin in controlling malignant phenotypes of melanoma.
Materials and methods
Cell Culture and gene transfection
The non-pigmented human melanoma A375 cells and pigmented mouse melanoma (B16) cell lines were cultured in Dulbecco’s modified Eagle’s medium (HyClone, Logan, UT, USA) supplemented with 10% foetal bovine serum (Hyclone), 100 U/ml penicillin and 1× Penicillin-Streptomycin (100 U/ml– 100 μg/ml) (Invitrogen, Carlsbad CA, USA). Plasmids were introduced into cells by polyethylenimine-mediated transfection [7] or pLNCX2 retrovirus vector (BD, Franklin Lakes, NJ, USA) system according to the protocol. The transfected cells were selected by either G418 or puromycin, and continuously cultured until harvested for analysis.
RT-PCR and real-time qRT-PCR
Regular RT-PCR and quantitative RT-PCR (qRT-PCR) were performed as previously described [7], using an ABI PRISM 7300 detection system (ABI, Foster, CA, USA) with primers listed in Table S1. The RT-PCR reactions were repeated at least for three times.
Western blotting
The Western blot detections were performed as described [7]. Antibodies are listed in Table S2.
Methylation-specific PCR (MSP)
Genomic DNA from cells was prepared using a DNA Extraction Kit (GE Healthcare, Buckinghamshire, UK) according to the manufacturer’s instructions. The genomic DNA was modified and purified using an CpGenome™ DNA Methylation Kit (Chemicon International, Temecula, CA, USA), following the manufacturer’s protocol. MSP was performed with methylation-specific primers, which were designed to recognize bisulphite-induced modifications of unmethylated cytosines. The primer was used to target the CpG islands located in the putative promoter region of MEN1.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed as previously described [7]. Briefly, 1 × 106 cells were treated with 1% formaldehyde, followed by pulsed ultrasonication to shear cellular DNA according to the protocol of the ChIP assay kit (Millipore, Billerica, MA, USA). After overnight incubation with the antibodies, protein G-agarose beads were added. The crosslinks between nuclear proteins and genomic DNA were reversed, and the antibody pulled down DNA was purified by phenol/chloroform extraction. The primer pair sequences and antibodies for ChIP assays were shown in Tables S1 and S2.
Xenograft of melanoma cells and tumour development in mice
Tumours were generated in female C57BL/6J mice (vitalriver, Beijing, China) by subcutaneous injection of B16 cells (5 × 105 cells in 100 μl PBS) into the right dorsum of each mouse. Tumour measurements were converted to tumour volume (V) using the formula (L×W2× 0.52), where L and W are the length and width, respectively, and the growth of tumours was measured once every 2 days using vernier callipers. For the pulmonary metastasis model, the B16 cells (5 × 105 cells in 25 μl PBS) were injected into the foot pad of C57BL/6J mice, which were performed as previously described [20]. The mice were killed around day 40. The number and the size of metastatic foci on the pulmonary surface were macroscopically quantified. Macroscopic lung pictures were acquired with Canon camera and processed with Adobe Photoshop CS Version 8.0. All procedures were performed according to animal welfare and other related ethical regulations approved by the Institutional Animal Care Committee of Medical College at Xiamen University.
5-Aza-2′-deoxycytidine treatment
A375 cells were grown for 7 days in the presence of various concentrations of 5′-aza-dc (0, 3 and 5 μM). Fresh drug was added every 24 hrs. RNA and genomic DNA were separately isolated.
The study of clinical melanoma samples
The study was approved by the Xiamen University Medical Ethics Committee, and written informed consent was obtained from all participants or from patients’ representatives if direct consent could not be obtained. We collected 12 malignant melanoma and 6 pigmented nerves samples from Zhongshan Hospital of Xiamen University. The method of immunohistochemistry was as described previously [7].
Data analysis and statistics
Data were presented as the mean ± S.D. or ±S.E. as indicated for each figure. Statistical comparisons between groups were performed with the Student’s t-test. P < 0.05 was considered statistically significant.
Results
Menin inhibits proliferation and migration of melanoma cells
Loss or mutation of MEN1 acutely promotes pancreatic islet cell proliferation [21, 22]. We have also found that menin suppresses proliferation of lung cancer cells, but the MEN1 point mutations, A242V and L22R, which were identified from inherited MEN1 patients [23], lost or partially lost ability to repress cell proliferation [7]. Melanomas secrete melanin just like endocrine organs secrete their respective hormones. To explore whether menin affects proliferation of pigmented melanoma cells, we stably transfected B16 cells with either a control vector or a menin-expressing construct. The 3-(4,5)-dimethylthiahiazo (-z-y1)-3,5-di- phenytetrazoliumromide (MTT) assay showed that ectopic expression of menin significantly reduced the number of B16 cells on day 4 (P < 0.05) (Fig. 1A and B). Furthermore, B16 cells with Men1 knockdown significantly increased cell proliferation (P < 0.05) (Fig. S1a). To further confirm whether menin affects non-pigment melanoma cell phenotype, we generated menin overexpressing A375 cells, a human non-pigmented melanoma cell line, through transduction with either vector or menin-expressing pLNCX2 retroviruses. The BrdU assay clearly showed that overexpression of menin (Fig. 1C) reduced the proliferation of A375 cells on days 2 and 4 (Fig. 1D, P < 0.05, respectively). Next, another pair of control and menin overexpressing A375 cell line was established via using retrovirus-mediated transduction, and similar results on the role of menin in regulating proliferation of A375 cells were observed by using cell counting assays (Fig. S1b). In malignant melanoma, dysregulation of cell adhesion molecules is associated with tumour progression and metastasis [14]. Menin has been shown to control endocrine cell migration and cell–cell adhesion through interacting with a scaffold protein, IQ motif containing guanosine triphosphatase (GTPase) activating protein 1 [24]. We also found that menin expression was markedly reduced in 23% of certain lung adenocarcinoma, which was correlated with lymph node metastasis [7]. Therefore, we performed a modified transwell chamber assay to evaluate the impact of stably ectopic menin expression on migration of melanoma cells. The results indicated that MEN1 overexpression significantly decreased migration of B16 cells (Fig. 1E, P < 0.05) and A375 cells (Fig. S1c and d). We next used an alternative approach, the scratch wound assay, to compare the motility of mock and menin overexpressing B16 cells. The extent of wound closure achieved by control cells within 48 hrs of wounding was much higher than that menin overexpressed B16 cells (Fig. 1F and G). The dramatic difference in wound healing between these two types of cells reinforces the notion that menin represses migration of melanoma cells. These results reveal a previously unappreciated function for menin in suppressing proliferation and migration of melanoma cells.
Fig 1.
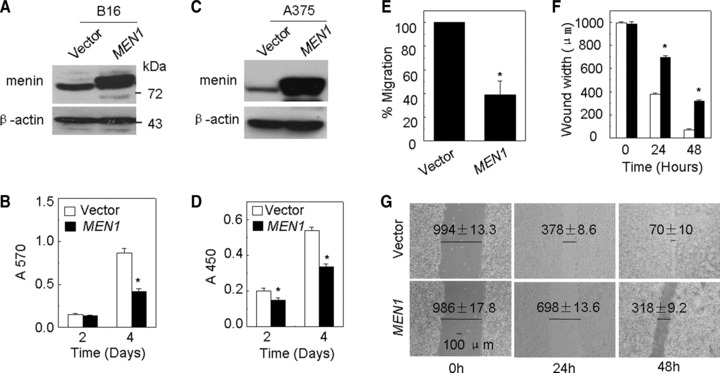
Menin inhibits proliferation and migration of melanoma cells. (A) The efficiency of menin overexpression was detected by Western blot in B16 cells. (B) The proliferation of B16 cells which was stably transfected with either pMX-puro or pMX-menin was estimated by MTT assay. (C) The efficiency of menin overexpression was detected by Western blot in A375 cells. (D) The proliferation of A375 cells, which were stably transfected with either vector or menin, was detected by BrdU assay. (E) Stably transfected B16 cells were added to the upper filter, and cell migration was determined. (F and G) Quantification of the time-dependent effects of menin overexpression on cell motility (wound width). Confluent monolayers of B16 menin overexpression cells were wounded with a pipette tip. Wound closure was monitored by microscopy at the indicated time, *P < 0.05, N= 3.
Menin inhibits melanoma cells partly through repressing PTN signalling
To elucidate how menin represses proliferation and migration of melanoma cells, we turned our attention to the impact of menin on expression of certain signalling pathways. Our previous work has shown that menin suppresses lung cancer cell proliferation and migration partly through epigenetically repressing transcription of growth factor PTN [7]. PTN is a heparin-binding growth factor involved in the differentiation and proliferation of neuronal tissue during embryogenesis, and is highly expressed in certain solid tumours including melanoma and breast carcinoma cells [12, 13]. PTN binds to cell surface receptor RPTP β/ζ and exerts multiple functions including cell proliferation, adhesion and migration [25–27]. Thus, we initially evaluated the impact of menin overexpression on expression of PTN and its receptor RPTP β/ζ in melanoma cells. The results indicate that menin overexpression substantially reduced mRNA levels of PTN and RPTP β/ζ, but not other growth factor vascular endothelial growth factor (VEGF), VEGF-C and basic fibroblast growth factor (bFGF) in B16 cells (Fig. 2A). Menin overexpression also reduced protein levels of PTN and RPTP β/ζ, but not VEGF (Fig. 2B). We further evaluated if PTN/RPTP β/ζ signalling is required for menin-mediated repression of migration of melanoma cells. Two distinct PTN shRNAs and a control Luc shRNA vector were stably transfected into B16 cells, and RT-PCR results showed that shRNA1 substantially reduced PTN expression, but shRNA2 failed to knockdown PTN expression (Fig. 2C). Interestingly, correlated with the levels of PTN knockdown by the shRNAs, shRNA1 significantly decreased cell proliferation (P < 0.05), but control vector and PTN shRNA2, which were unable to reduce PTN expression, did not significantly decrease proliferation of B16 cells (P > 0.05) (Fig. 2D). Notably, PTN knockdown by shRNA1 also reduced migration of B16 cells (Fig. 2E). Furthermore, RPTP β/ζ knockdown effectively reduced intracellular RPTP β/ζ mRNA (Fig. 2F) and protein expression (Fig. 2G), concomitant with reduced migration of B16 cells (Fig. 2H). Together, these data indicate that menin inhibits proliferation and migration of B16 cells at least partly through regulating expression of PTN and RPTP β/ζ.
Fig 2.
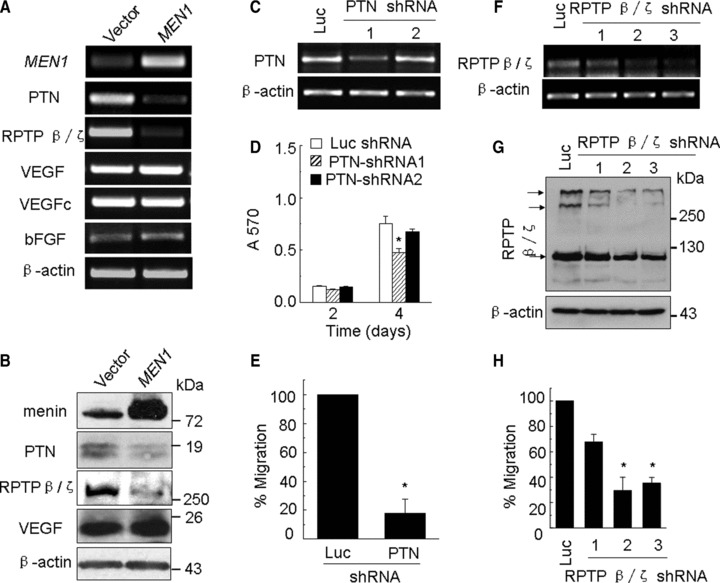
Menin represses proliferation and migration of melanoma cells partly through PTN signalling. (A) Men1, PTN, RPTP β/ζ, VEGF, VEGFc and bFGF mRNA levels were detected by RT-PCR. (B) The efficiency of menin overexpression and the effect of Men1 expression on PTN, RPTP β/ζ and VEGF expression were determined by Western blotting and β-actin was used as loading control. (C) B16 cells were transfected with either vector expressing shRNAs against Luc or one of the two shRNAs against PTN and selected by G418. The efficiency of PTN silencing was determined by RT-PCR. (D) The proliferation of the selected B16 cells was estimated by MTT assay. (E) The selected B16 cells were added to upper filter and cell migration was determined. (F and G) B16 cells were transfected with either vector expressing shRNAs against Luc or one of the there shRNAs against RPTP β/ζ and selected by G418. The efficiency of RPTP β/ζ silencing was determined by RT-PCR and Western blotting. (H) The selected B16 cells were added to upper filter and cell migration was determined.
Menin represses tumour growth and metastasis of melanoma cells in vivo
To determine whether menin affects growth of melanoma cell-derived tumours in animal model, we stably transfected B16 cells with either control or menin-expressing construct, and the resulting cells were subcutaneously transplanted into C57BL/6J mice (n= 8 per group). Ectopic expression of menin was confirmed by Western blotting (Fig. 3A). The size of the solid tumour was measured after various periods of time following transplantation. Ectopic menin expression in B16 cells significantly reduced the size of B16 cell-derived solid tumour in C57BL/6J mice after transplantation (Fig. 3B, P < 0.05). To determine if menin affects the growth of the established tumours in C57BL/6J mice partly through PTN, the PTN knockdown B16 cells were generated and subcutaneously transplanted into C57BL/6J mice (n= 8 per group). The efficiency of PTN silencing was determined by Western blotting (Fig. 3C). As expected, reduction in PTN expression also significantly suppressed the growth of B16 cell-derived solid tumours on indicated days (Fig. 3D, P < 0.05). These results suggest that menin represses, but PTN promotes, growth of B16 solid tumour in mice, highlighting a crucial role of menin and PTN in controlling growth of melanoma in vivo. In the syngeneic murine metastasis models, we also found that either menin overexpression (Fig. 3E and F) or PTN knockdown (Fig. 3G and H) significantly repressed the number of macroscopic pulmonary metastatic foci. Together, these data show that menin suppresses growth and pulmonary metastasis of solid melanomas partly through repressing PTN signalling in vivo.
Fig 3.
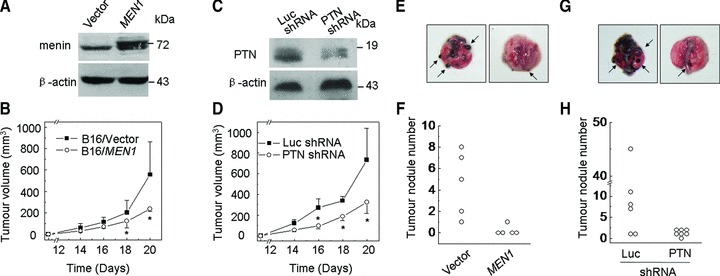
Menin represses tumour growth and metastasis of melanoma cells in vivo. (A) The efficiency of menin overexpression was determined by Western blotting. (B) Menin overexpressing B16 cells were injected subcutaneously into nude mice and tumour formation was examined day 14 after transplantation. N= 8, *P < 0.05. (C) The efficiency of PTN silencing was determined by Western blotting. (D) The PTN-shRNA expression B16 cells were injected subcutaneously into nude mice, and tumour formation was examined day 14 after transplantation, N= 8, *P < 0.05. (E and F) The number of macroscopic pulmonary metastases from each mouse treated with menin overexpressing B16 cells, N= 5. (G and H) The number of macroscopic pulmonary metastases from each mouse treated with PTN-shRNA B16 cells, N= 6 or 7.
pI3K and ERK1/2 were crucial for menin-mediated regulation of melanoma cells
To further elucidate cell signalling underlying menin/PTN regulated cell proliferation and migration, we tested the impact of menin on pI3K and ERK1/2, which is essential for regulating phenotype of melanoma [17]. The results showed that ectopic expression of menin reduced expression of pI3K as well as phosphorylation (Thr202/Tyr204) of ERK1/2 in A375 cells (Fig. 4A). FAK (focal adhesion kinase) is a protein tyrosine kinase that is recruited at an early stage to focal adhesions and mediates many of the downstream responses, including activation of the MAPK and pI3K p85-subunit in epithelial tumour cells and fibroblasts [28, 29]. To further dissect the potential relationship between menin, FAK, ERK1/2 and pI3K, the stable menin-expressing A375 cells were analysed. Our results showed that menin overexpression did not affect the total amount (Fig. 4A) and cell localization (data not shown) of FAK, but reduced the level of its Tyr 397-phosphorylated form (Figs 4A and S2a). Next, serum-starved A375 cells were stimulated by addition of rhPTN and allowed to progress for various periods of time prior to analysis. The results indicated that pERK1/2 was rapidly increased after exposure to rhPTN at 15–60 min. (Fig. 4B). It showed that menin regulated activation of ERK1/2 partly through repressing PTN. These results suggest that FAK signalling may link menin/PTN to cell proliferation and migration partly through regulating pI3K and ERK1/2 pathways. To further confirm this observation, we determined whether pI3K and ERK1/2 signalling were necessary for the menin/PTN regulating phenotypes of melanoma cells. To this end, A375 cells were treated with either LY294002 or U0126, which are specific inhibitors for pI3K and MEK1/2, respectively. As expected, both LY294002 and U0126 decreased proliferation of A375 cells in a dose-dependent manner (Fig. 4C and D). Migration of A375 cells treated with either LY294002 or U0126 was also reduced (Fig. 4E and F). β-catenin acts as a key factor in E-cadherin-mediated cell–cell adhesion [30]. We further determined if menin/PTN regulated cell migration was dependent on β-catenin signalling. However, menin did not effectively suppress expression and phosphorylation (Tyr 142) of β-catenin (Fig. S2b). Cell morphology and migration were regulated by members of the Rho family of small GTPases, including Rho, Rac1 and Cdc42 [31]. Hence, we further examined if menin controls cell migration partly through Rho family signalling. Ectopic menin expression did not alter the amount of either activated forms (GTP bound) or the total amount of Rho, Rac1 and Cdc42 in A375 cells (Fig. S2b).
Fig 4.
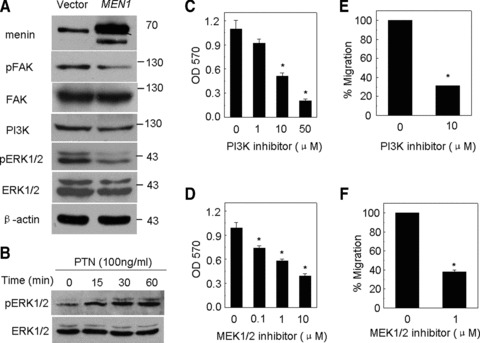
pI3K and ERK1/2 were crucial for menin-mediated regulation of melanoma cells. (A) Menin, pFAK, pI3K and pERK1/2 protein level were detected by Western blot. (B) Serum-starved A375 cells were treated with 100 ng/ml rhPTN and harvested at various time-points. The activation of ERK1/2 was detected by Western blotting. (C) A375 cell lines were treated using LY294002, a pI3K inhibitor 48 hrs, and cell proliferation was measured by MTT. (D) A375 cell lines were treated with U0126 at 0.1, 1 and 10 μM, a MEK1/2 inhibitor 48 hrs and cell proliferation was measured by MTT. (E) A375 cells treated with LY294002 were added to upper filter and cell migration was determined. (F) A375 cells treated with U0126 were added to upper filter and cell migration was determined.
Next, we determined whether the level of expression of menin in melanoma cell lines is correlated with cell sensitivity to the cytotoxic effects of cisplatin and dacarbazine, the two most commonly used drugs for treating malignant melanoma. The time-course results indicate that menin was gradually increased after exposure to cisplatin at 0–24 hrs (Fig. S3a). Meanwhile, the dose–response result also indicates that menin was increased after exposure to indicated concentrations of cisplatin at 16 hrs (Fig. S3b). However, there was no significant correlation between menin levels and sensitivity of melanoma cell lines to dacarbazine (Fig. S3c and d). Because it is well known that menin can induce cell apoptosis [32], we determined whether menin could serve as a means to enhance killing of malignant melanoma cells. Overexpression of menin indeed increased cisplatin induced apoptosis of A375 cells (Fig. S3e). Further studies indicated that menin repressed phosphorylation (S139) of γ-H2AX, a marker of DNA damage repair, and cell cycle regulators, such as cyclin B1 and B2 (Fig. S3f). These results raise a possibility that menin also regulates apoptosis of melanoma cells, and this process may be associated with controlling DNA damage response and cell cycle progression. The precise mechanism for menin regulated apoptosis remains to be investigated. Together, our results suggest that menin inhibits ERK1/2 phosphorylation partly through PTN expression, and FAK, pI3K and ERK1/2 signalling might be involved in menin-mediated repression of phenotype of melanoma cells.
DNA Methylation of the MEN1 promoter correlates with menin inactivation in A375 cells
Since we observed a crucial role for menin in repressing phenotype of melanoma cells, we wondered if the menin protein level is altered in patients’ primary melanoma. We examined 12 malignant melanoma samples and 6 pigmented nevus. These tumours were from male and female patients with ages ranging from 28 to 88 (Table S3). Sections from paraffin-embedded samples were stained with affinity-purified anti-menin antibody for immunohistochemistry (IHC) staining, and the specificity of the anti-menin antibody was verified in menin-null and menin-expressing cells [7]. Menin was easily detected in the nucleus of the normal six pigmented nevus cells (Fig. 5A). However in melanoma tumours, staining for menin was slightly weaker (three cases) or undetectable (nine cases), as compared to that in the pigmented nevus cells (Fig. 5B, C and Table S3). To determine the cause for inactivation of menin in A375 cells, we designed the primers to determine if MEN1 was mutated (Fig. S4). Unexpectedly, DNA sequencing data did not reveal any mutation in the sequence of MEN1.
Fig 5.
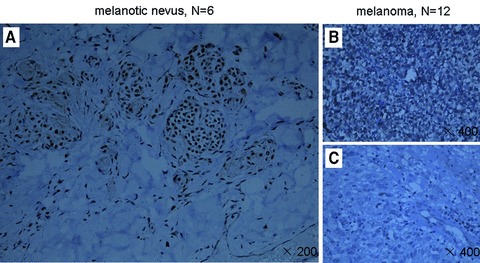
Menin expression is reduced in certain primary melanoma cells. Sections from paraffin-embedded samples were stained with affinity-purified anti-menin antibody for immunohistochemistry staining. (A) Menin was easily detectable in the nucleus of the pigmented nerves (×200). (B and C) In melanoma, staining for menin was slightly weaker (three cases) or undetectable (nine cases), as compared to that in the pigmented nevus cells (×200).
Transcriptional silencing of tumour suppressor genes, associated with DNA hypermethylation of CpG islands [33]. Hence we considered if reduced menin expression is related to epigenetic regulation. In MSP analysis, we designed methylation-specific primers and unmethylation-specific primers which were targeted to CpG sites (Fig. 6A), and examined the methylation status of the MEN1 promoter in A375 cells using real-time qPCR. In order to clarify the functional association between MEN1 promoter methylation, 5′-aza-dc, an agent reducing DNA methylation, was used to treat A375 cells. The quantitative methylation-specific PCR (qMSP) results showed that the level of DNA hypermethylation at the MEN1 promoter was reduced by treatment with 5′-aza-dc in A375 cells (Fig. 6B). After 7 days treatment with 5′-aza-dc at 3 μM or 5 μM, the increased MEN1 mRNA re-expression was detected by real-time qRT-PCR (Fig. 6C).
Fig 6.
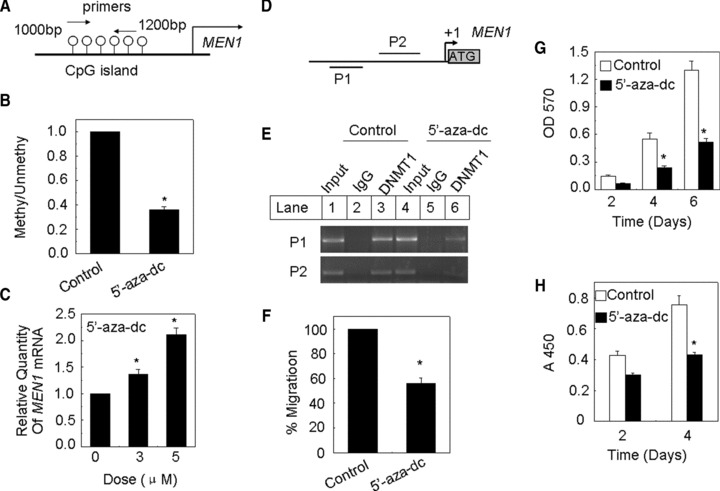
Methylation of the menin promoter correlates with menin expression in A375 cell. (A) Primers for unmethylated and methylated DNA of corresponding CpG islands were used. (B) qMSP assay of MEN1 gene in A375 cells. (C) A375 Cells were treated with 5′-aza-dc at 3 or 5 μM for 7 days with medium changed each day, and MEN1 mRNA level was determined by real-time qPCR. (D and E) ChIP assay to demonstrate the association of DNMT with the MEN1 genes. (F) A375 cells treated with 5′-aza-dc at 5 μM for 7 days were added to the upper filter, and cell migration was determined. (G and H) The proliferation of A375 cells treated with 5′-aza-dc at 5 μM for 7 days was estimated by MTT assay and BrdU cell proliferation assay, respectively.
Furthermore, we also determined if DNA methytransferase 1 (DNMT1) binds to the MEN1 promoter using ChIP assay. We designed two primers used for ChIP assays at Men1 promoter loci (Fig. 6D). In A375 cells, an interaction between DNMT1 and the promoter of MEN1 could be detected (Fig. 6E, lane 3). Following exposure to 5′-aza-dc, the interaction between the DNMT1 and the promoter of MEN1 was reduced (Fig. 6E, lane 6). To explore whether treatment with 5′-aza-dc affects proliferation and migration of melanoma cells, we treated A375 cells with 5 μM 5′-aza-dc for 7 days. The transwell assay showed that treatment with 5′-aza-dc significantly reduced the number of migrated A375 cells on days 4 and 6 (P < 0.05, respectively) (Fig. 6F).
In addition, MTT assay confirmed that treatment with 5′-aza-dc reduced the number of A375 cells (Fig. 6G). A similar result was obtained using the BrdU incorporation assay (Fig. 6H). Exposure of A375 cells to 5′-aza-dc effectively demethylated the CpG regions within the MEN1 promoter, leading to MEN1 gene expression and suppressed malignant phenotypes of melanoma, including proliferation and migration. Together, these data indicate that MEN1 silencing was associated with promoter CpG region hypermethylation in melanoma, and suggest a key role for menin in repressing melanomas.
Discussion
MEN1 knockout mice develop parathyroid, pancreatic, pituitary and adrenal tumours [2]. Menin interacted with MLL and promoted the development of leukaemia through binding to the locus of Hox family genes and highlight the level of H3K4me3 [3–6]. Recently, we have found that menin inhibits lung cancer cell proliferation and migration via epigenetic repression of PTN signalling [7]. Various skin tumours of mesenchymal origin, including angiofibromas, collagenomas and lipomas, as well as malignant melanoma, were detected in MEN1 syndrome patients [18, 19]. However, until recently, little has been known about the precise role and regulatory mechanism of menin in melanoma.
In present study, we have shown that menin inhibits proliferation, migration and metastasis of melanoma cells partly through repressing PTN and its receptor, RPTP β/ζ expression. Further investigation revealed that menin regulates cell phenotype of melanoma via PTN/RPTP β/ζ, in conjunction with FAK, pI3K and ERK1/2 signalling. Our previous results show that menin not only inhibits expression of PTN and RPTP β/ζ, but also represses the activation (phosphorylation) of FAK, pI3K and ERK1/2 in lung cancer cells [34]. Therefore, the similar mechanism underlying menin-mediated tumour suppression may exist in lung cancer cells and melanoma cells. However how menin regulates FAK, pI3K and ERK1/2 signalling through PTN and RPTP β/ζ remains unclear. PTN binds to its receptor, RPTP β/ζ and increases tyrosine phosphorylation of many downstream genes including β-catenin, ALK and integrin β3[35–37]. In the present melanoma model, we did not find that menin affects the expression and phosphorylation of β-catenin. Cell morphology and migration were regulated by members of the Rho family of small GTPases [31]. Our results indicate that extopic expression of menin did not alter the amount of either activated forms (GTP bound) or the total amount of Rho, Rac1 and Cdc42 in A375 cells. FAK interacts with integrin β3 and promotes cell migration and invasion [34]. It has been reported that integrin–FAK interaction may serve as a downstream effector of PTN [37], thus PTN may increase activation of FAK by binding to RPTP β/ζ and increasing tyrosine phosphorylation of integrin β3. Then activated FAK promotes pI3K-ERK1/2 signalling. Collectively, menin may inhibit FAK, pI3K and ERK1/2 signalling through repressing the ability of menin to repress phosphorylation of the crucial signalling proteins downstream of PTN. During the progression of cutaneous melanomas, matrix metalloproteinases (MMPs) facilitate the tumour cells to traverse the basement membrane and invade the dermis. Melanoma cell lines with low expression of MMP19 exhibited increased adhesion to various substrates and lower migration in comparison with the cell line with higher expression of MMP19 [38]. Dimethylfumarate inhibits tumour cell invasion and metastasis by suppressing the expression and activities of MMPs in melanoma cells [39]. Whether the menin affects melanoma motility via one of the multiple MMPs will be determined. In addition, menin also promotes the cisplatin-induced apoptosis of A375 cells, and represses expression of phosphor-γ-H2AX, a DNA damage repair marker. However, how menin may regulate apoptosis of melanoma cell and DNA damage response remains to be further determined.
The function of menin in regulating tumorigenesis is opposite between endocrine tumour and leukaemia. What decides the role of menin in different tissues and cell lines? In the present study, we chose the A375 and B16 cell lines, a non-pigmented and pigmented cell lines, respectively, to demonstrate that the impact of menin on non-pigmented and pigmented melanoma cell lines. Our results reveal that menin has similar effects on both pigmented and non-pigmented melanoma cells. These results indicate that menin has broad-spectrum suppressing effect on melanoma. Melanomas secrete melanin just as endocrine organs secrete their respective hormones. A possible link between melanoma and endocrine is that both of them are secretary tissues. Nonetheless, the precise mechanism remains to be determined. Menin expression was significantly reduced in primary melanoma cells from clinical samples. What is the cause for decreased expression of menin in melanoma cells? Our DNA sequencing data did not reveal any mutation in the sequence of MEN1. Treatment of A375 cell with the demethylating agent 5′-aza-dc reactivated menin expression and then repressed proliferation and migration of A375 cells. Based on these results, DNA methylation appears to play a major role in silencing menin expression in A375 cells. And another possibility is that menin has different epigenetic modifications in different tissues and the modifications may determine the various status of menin expression.
Collectively, our findings unravel a previously unrecognized function of menin in controlling melanoma cell proliferation, migration, metastasis and apoptosis. Menin inhibits FAK, pI3K and ERK1/2 signalling through repressing PTN and its receptor, RPTP β/ζ. And this mechanism is similar to what is in lung cancer cells. These findings may suggest that the similar function and regulatory mechanism for menin may exist among lung cancer, melanoma and endocrine tumours, and provide a new insight into further understanding the function of menin in a broader spectrum of tumours.
Acknowledgments
This work is supported by National Natural Science Foundation of China (grant numbers 30701003 and 81071926 to G.H.J.), National Natural Science Foundation of Xiamen (grant number 3502Z20104001 to G.H.J.) and fundamental research funds for the central universities (grant number 2010121106 to G.H.J.). We appreciate the valuable comments from other members of our laboratories.
Conflict of interest
The authors confirm that there are no conflicts of interest.
Supporting Information
Table S1 Primer sequences and target sequences of shRNA
Table S2 Antibody and reagent
Table S3 Summarize of IHC results from certain primary melanoma samples
Fig. S1 (a) The proliferation of B16 withMEN1 knockdown. (b) The proliferation of A375 cellsstably transfected with either empty vector or menin. (c)Migrated to lower side of the filter A375 cells were stained with0.1% crystal violet. (d) Stably transfected A375 cells wereadded to the upper filter, and cell migration was determined,*P < 0.05, N = 3.
Fig. S2 (a) IF detection of menin (green), pFAK(green), DAPI (blue) and merge in the A375 cells. (b)PAK1-PBD agarose and Rhotekin RBD agarose were used to isolateGTP-Cdc42, GTP-Rac1 and GTP-RhoA from whole cell lysates frommenin-overexpressing A375 cells. The Cdc42-GTP, Rac1-GTP andRhoA-GTP were detected using Western blotting and normalized by thetotal input protein. The pβ-catenin protein level was detectedby Western blot in menin-overexpressing A375 cells.
Fig. S3 (a, c) Melanoma cells were treatedwith 1 μg/ml cisplatin or 250 μg/ml dacarbazine and harvestedat various time-points. And the menin expression was determinedwith Western blotting. (b, d) Melanoma cells weretreated with the indicated concentrations of cisplatin ordacarbazine, and the menin expression was detected by Westernblotting. (e) A375 cells were treated for 24 hrs withvarious doses of Cisplatin and then analysed for apoptosisvia Annexin V-PI staining. (f) menin, γ-H2A.X,cyclinB1 and cyclinB2 protein level were detected by Westernblot.
Fig. S4 Primers for determining whether menin mutated were used.
References
- 1.Chandrasekharappa SC, Guru SC, Manickam P, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276:404–7. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- 2.Crabtree JS, Scacheri PC, Ward JM, et al. A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc Natl Acad Sci USA. 2001;98:1118–23. doi: 10.1073/pnas.98.3.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yokoyama A, Somervaille TC, Smith KS, et al. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell. 2005;123:207–18. doi: 10.1016/j.cell.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 4.Hughes CM, Rozenblatt-Rosen O, Milne TA, et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell. 2004;13:587–97. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- 5.Jin S, Zhao H, Yi Y, et al. c-Myb binds MLL through menin in human leukemia cells and is an important driver of MLL-associated leukemogenesis. J Clin Invest. 2010;120:593–606. doi: 10.1172/JCI38030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thiel AT, Blessington P, Zou T, et al. MLL-AF9-induced leukemogenesis requires coexpression of the wild-type Mll allele. Cancer Cell. 2010;17:148–59. doi: 10.1016/j.ccr.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao SB, Feng ZJ, Xu B, et al. Suppression of lung adenocarcinoma through menin and polycomb gene-mediated repression of growth factor pleiotrophin. Oncogene. 2009;28:4095–104. doi: 10.1038/onc.2009.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445:851–7. doi: 10.1038/nature05661. [DOI] [PubMed] [Google Scholar]
- 9.McGill GG, Horstmann M, Widlund HR, et al. Bcl2 regulation by the melanocyte master regulator Mitf modulates lineage survival and melanoma cell viability. Cell. 2002;109:707–18. doi: 10.1016/s0092-8674(02)00762-6. [DOI] [PubMed] [Google Scholar]
- 10.Wajapeyee N, Serra RW, Zhu X, et al. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132:363–74. doi: 10.1016/j.cell.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heidorn SJ, Milagre C, Whittaker S, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–21. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perez-Pinera P, Chang Y, Deuel TF. Pleiotrophin, a multifunctional tumor promoter through induction of tumor angiogenesis, remodeling of the tumor microenvironment, and activation of stromal fibroblasts. Cell Cycle. 2007;6:2877–83. doi: 10.4161/cc.6.23.5090. [DOI] [PubMed] [Google Scholar]
- 13.Seykora JT, Jih D, Elenitsas R, et al. Gene expression profiling of melanocytic lesions. Am J Dermatopathol. 2003;25:6–11. doi: 10.1097/00000372-200302000-00002. [DOI] [PubMed] [Google Scholar]
- 14.Czubayko F, Schulte AM, Berchem GJ, et al. Melanoma angiogenesis and metastasis modulated by ribozyme targeting of the secreted growth factor pleiotrophin. Proc Natl Acad Sci USA. 1996;93:14753–8. doi: 10.1073/pnas.93.25.14753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Czubayko F, Riegel AT, Wellstein A. Ribozyme-targeting elucidates a direct role of pleiotrophin in tumor growth. J Biol Chem. 1994;269:21358–63. [PubMed] [Google Scholar]
- 16.Bowden ET, Stoica GE, Wellstein A. Anti-apoptotic signaling of pleiotrophin through its receptor, anaplastic lymphoma kinase. J Biol Chem. 2002;277:35862–8. doi: 10.1074/jbc.M203963200. [DOI] [PubMed] [Google Scholar]
- 17.Smalley KS. Understanding melanoma signaling networks as the basis for molecular targeted therapy. J Invest Dermatol. 2010;130:28–37. doi: 10.1038/jid.2009.177. [DOI] [PubMed] [Google Scholar]
- 18.Nord B, Platz A, Smoczynski K, et al. Malignant melanoma in patients with multiple endocrine neoplasia type 1 and involvement of the MEN1 gene in sporadic melanoma. Int J Cancer. 2000;87:463–7. doi: 10.1002/1097-0215(20000815)87:4<463::aid-ijc1>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 19.Baldauf C, Vortmeyer AO, Koch CA, et al. Combination of multiple skin malignancies with multiple endocrine neoplasia type 1: coincidental or pathogenetically related. Dermatology. 2009;219:365–7. doi: 10.1159/000193058. [DOI] [PubMed] [Google Scholar]
- 20.Mah-Becherel MC, Ceraline J, Deplanque G, et al. Anti-angiogenic effects of the thienopyridine SR 25989 in vitro and in vivo in a murine pulmonary metastasis model. Br J Cancer. 2002;86:803–10. doi: 10.1038/sj.bjc.6600142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schnepp RW, Chen YX, Wang H, et al. Mutation of tumor suppressor gene Men1 acutely enhances proliferation of pancreatic islet cells. Cancer Res. 2006;66:5707–15. doi: 10.1158/0008-5472.CAN-05-4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karnik SK, Chen H, McLean GW, et al. Menin controls growth of pancreatic beta-cells in pregnant mice and promotes gestational diabetes mellitus. Science. 2007;318:806–9. doi: 10.1126/science.1146812. [DOI] [PubMed] [Google Scholar]
- 23.Pannett AA, Thakker RV. Multiple endocrine neoplasia type 1. Endocr Relat Cancer. 1999;6:449–73. doi: 10.1677/erc.0.0060449. [DOI] [PubMed] [Google Scholar]
- 24.Yan J, Yang Y, Zhang H, et al. Menin interacts with IQGAP1 to enhance intercellular adhesion of beta-cells. Oncogene. 2009;28:973–82. doi: 10.1038/onc.2008.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duces A, Karaky R, Martel-Renoir D, et al. 16-kDa fragment of pleiotrophin acts on endothelial and breast tumor cells and inhibits tumor development. Mol Cancer Ther. 2008;7:2817–27. doi: 10.1158/1535-7163.MCT-08-0301. [DOI] [PubMed] [Google Scholar]
- 26.Pariser H, Ezquerra L, Herradon G, et al. Fyn is a downstream target of the pleiotrophin/receptor protein tyrosine phosphatase beta/zeta-signaling pathway: regulation of tyrosine phosphorylation of Fyn by pleiotrophin. Biochem Biophys Res Commun. 2005;332:664–9. doi: 10.1016/j.bbrc.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 27.Lu KV, Jong KA, Kim GY, et al. Differential induction of glioblastoma migration and growth by two forms of pleiotrophin. J Biol Chem. 2005;280:26953–64. doi: 10.1074/jbc.M502614200. [DOI] [PubMed] [Google Scholar]
- 28.Baillat G, Siret C, Delamarre E, et al. Early adhesion induces interaction of FAK and Fyn in lipid domains and activates raft-dependent Akt signaling in SW480 colon cancer cells. Biochim Biophys Acta. 2008;1783:2323–31. doi: 10.1016/j.bbamcr.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 29.Schlaepfer DD, Hanks SK, Hunter T, et al. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature. 1994;372:786–91. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- 30.Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 2004;303:1483–7. doi: 10.1126/science.1094291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–14. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 32.Hussein N, Casse H, Fontaniere S, et al. Reconstituted expression of menin in Men1-deficient mouse Leydig tumour cells induces cell cycle arrest and apoptosis. Eur J Cancer. 2007;43:402–14. doi: 10.1016/j.ejca.2006.08.038. [DOI] [PubMed] [Google Scholar]
- 33.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–59. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 34.Feng ZJ, Gao SB, Wu Y, et al. Lung cancer cell migration is regulated via repressing growth factor PTN/RPTP beta/zeta signaling by menin. Oncogene. 2010;29:5416–26. doi: 10.1038/onc.2010.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meng K, Rodriguez-Pena A, Dimitrov T, et al. Pleiotrophin signals increased tyrosine phosphorylation of beta beta-catenin through inactivation of the intrinsic catalytic activity of the receptor-type protein tyrosine phosphatase beta/zeta. Proc Natl Acad Sci USA. 2000;97:2603–8. doi: 10.1073/pnas.020487997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perez-Pinera P, Chang Y, Astudillo A, et al. Anaplastic lymphoma kinase is expressed in different subtypes of human breast cancer. Biochem Biophys Res Commun. 2007;358:399–403. doi: 10.1016/j.bbrc.2007.04.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mikelis C, Sfaelou E, Koutsioumpa M, et al. Integrin alpha(v)beta(3) is a pleiotrophin receptor required for pleiotrophin-induced endothelial cell migration through receptor protein tyrosine phosphatase beta/zeta. FASEB J. 2009;23:1459–69. doi: 10.1096/fj.08-117564. [DOI] [PubMed] [Google Scholar]
- 38.Muller M, Beck IM, Gadesmann J, et al. MMP19 is upregulated during melanoma progression and increases invasion of melanoma cells. Mod Pathol. 2010;23:511–21. doi: 10.1038/modpathol.2009.183. [DOI] [PubMed] [Google Scholar]
- 39.Yamazoe Y, Tsubaki M, Matsuoka H, et al. Dimethylfumarate inhibits tumor cell invasion and metastasis by suppressing the expression and activities of matrix metalloproteinases in melanoma cells. Cell Biol Int. 2009;33:1087–94. doi: 10.1016/j.cellbi.2009.06.027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Primer sequences and target sequences of shRNA
Table S2 Antibody and reagent
Table S3 Summarize of IHC results from certain primary melanoma samples
Fig. S1 (a) The proliferation of B16 withMEN1 knockdown. (b) The proliferation of A375 cellsstably transfected with either empty vector or menin. (c)Migrated to lower side of the filter A375 cells were stained with0.1% crystal violet. (d) Stably transfected A375 cells wereadded to the upper filter, and cell migration was determined,*P < 0.05, N = 3.
Fig. S2 (a) IF detection of menin (green), pFAK(green), DAPI (blue) and merge in the A375 cells. (b)PAK1-PBD agarose and Rhotekin RBD agarose were used to isolateGTP-Cdc42, GTP-Rac1 and GTP-RhoA from whole cell lysates frommenin-overexpressing A375 cells. The Cdc42-GTP, Rac1-GTP andRhoA-GTP were detected using Western blotting and normalized by thetotal input protein. The pβ-catenin protein level was detectedby Western blot in menin-overexpressing A375 cells.
Fig. S3 (a, c) Melanoma cells were treatedwith 1 μg/ml cisplatin or 250 μg/ml dacarbazine and harvestedat various time-points. And the menin expression was determinedwith Western blotting. (b, d) Melanoma cells weretreated with the indicated concentrations of cisplatin ordacarbazine, and the menin expression was detected by Westernblotting. (e) A375 cells were treated for 24 hrs withvarious doses of Cisplatin and then analysed for apoptosisvia Annexin V-PI staining. (f) menin, γ-H2A.X,cyclinB1 and cyclinB2 protein level were detected by Westernblot.
Fig. S4 Primers for determining whether menin mutated were used.