

Abstract
Inflammatory bowel diseases (IBD) result from complex interactions between environmental and genetic factors. Low blood levels of vitamin B12 and folate and genetic variants of related target enzymes are associated with IBD risk, in population studies. To investigate the underlying mechanisms, we evaluated the effects of a methyl-deficient diet (MDD, folate, vitamin B12 and choline) in an experimental model of colitis induced by dextran sodium sulphate (DSS), in rat pups from dams subjected to the MDD during gestation and lactation. Four groups were considered (n= 12–16 per group): C DSS− (control/DSS−), D DSS− (deficient/DSS−), C DSS+ (control/DSS+) and D DSS+ (deficient/DSS+). Changes in apoptosis, oxidant stress and pro-inflammatory pathways were studied within colonic mucosa. In rat pups, the MDD produced a decreased plasma concentration of vitamin B12 and folate and an increased homocysteine (7.8 ± 0.9 versus 22.6 ± 1.2 μmol/l, P < 0.001). The DSS-induced colitis was dramatically more severe in the D DSS+ group compared with each other group, with no change in superoxide dismutase and glutathione peroxidase activity, but decreased expression of caspase-3 and Bax, and increased Bcl-2 levels. The mRNA levels of tumour necrosis factor (TNF)-α and protein levels of p38, cytosolic phospolipase A2 and cyclooxygenase 2 were significantly increased in the D DSS+ pups and were accompanied by a decrease in the protein level of tissue inhibitor of metalloproteinases (TIMP)3, a negative regulator of TNF-α. MDD may cause an overexpression of pro-inflammatory pathways, indicating an aggravating effect of folate and/or vitamin B12 deficiency in experimental IBD. These findings suggest paying attention to vitamin B12 and folate deficits, frequently reported in IBD patients.
Keywords: choline, inflammation, inflammatory bowel diseases, folate, methyl donors, vitamin B12
Introduction
The inflammatory bowel diseases (IBD), encompassing Crohn’s disease (CD) and ulcerative colitis, are chronic inflammatory disorders of the gastrointestinal tract. Current concepts of IBD pathogenesis suggest a complex interplay between genetic, nutritional and environmental factors, and gut microbiota [1, 2]. Over the past decade, we, and others have reported mild to moderate hyperhomocysteinemia in the plasma of patients with IBD [3, 4]. Markedly elevated concentrations of homocysteine were also found in the colonic mucosa of IBD patients [5, 6]. Hyperhomocysteinaemia should only be considered a surrogate marker for methyl donor deficiency. Homocysteine plasma level is mainly influenced by nutritional status in folate, vitamin B12, vitamin B6 and by genetic polymorphisms of key enzymes of its metabolism [4]. Vitamin B12 and methylenetetrahydrofolate reductase 677TT genotype may be the main determinants of hyperhomocysteinemia in CD patients [3].
Methyl donor deficiency (folate and/or vitamin B12 deficiency) leads to increased homocysteine in tissues and might promote mucosal inflammation by activating a wide range of pathways related with inflammation, cellular and oxidative stress and apoptosis [4]. Homocysteine can induce the production of several inflammatory cytokines and chemokines, including the monocyte chemoattractant protein-1 (MCP-1, also termed CCL2) and interleukin (IL)-8 [5, 7]. Homocysteine-induced expression of these pro-inflammatory molecules requires NF-κB (nuclear factor-kappa B) activation, a crucial transcription factor involved in mediating downstream inflammatory process [8]. Elevated homocysteine levels were also shown to enhance superoxide dismutase (SOD) activity, a well-established marker of oxidative stress [9, 10]. We recently showed that the level of SOD was significantly correlated with Crohn’s Disease Activity Index (DAI) in CD patients carrying methionine synthase reductase MTRR AA genotype [3]. Methyl donor deficiency affects the cell organization and function in the gastric mucosa of rat [11]. Apoptosis of T-cells has a key role in the pathophysiology of IBD [12]. A disturbed ratio between pro-apoptotic and anti-apoptotic pathways was shown to mediate apoptosis resistance in patients with CD [13]. By modulating apoptotic pathways [14], methyl donor status might contribute to intestinal homeostasis [4].
Despite the association of nutritional and genetic determinants of homocysteine with IBD, the effects of methyl donor deficiency on intestinal homeostasis remain poorly investigated from a molecular standpoint. Therefore, the aim of our study was to examine the impact of a methyl-deficient diet (MDD; diet deprived of folate, vitamin B12 and choline) on colonic lesions induced by dextran sodium sulphate (DSS) in rats and to identify mediators of inflammation, apoptosis and oxidative stress potentially contributing to colonic inflammation.
Materials and methods
Animals, diet and induction of colitis
Animal experiments were performed in accredited establishments (Inserm U 954) according to governmental guidelines N°86/609/CEE. Wistar rats (Charles River, l’Arbresle, France) were constantly maintained under standard laboratory conditions on a 12 hr light–dark cycle (lights on at 6:00 a.m.) with food and water available ad libitum, as described recently [11]. One month before pregnancy, adult females were fed with either standard food (n= 2) (Maintenance diet M20, Scientific Animal Food and Engineering, Villemoisson-sur-Orge, France) or a diet without vitamins B12, folate and choline (n= 3) (Special Diet Service, Saint-Gratien, France), as previously described [11]. The assigned diet was constantly maintained until the weaning of offspring, i.e. postnatal day 21 and the pups were fed with the same diet as their mother until the killing. Colitis was induced by administration of 5% DSS (molecular weight 36,000–50,000, MP Biomedicals, Strasbourg, France) dissolved in water. After 23 days of age, 14 pups of the control group (n= 26) and 16 pups of the deficient group (n= 29) were treated with DSS in the drinking water for 4 days. Age-matched control rats of the two groups were offered tap water. The pups were killed at 26 days of age (the fourth day of DSS treatment). A total of 55 pups (sex ratio females versus males = 0.96) were divided into four groups: (1) rats fed with standard diet, not treated with DSS and used as controls (noted C DSS−, n= 12); (2) rats fed with deficient diet and treated with DSS (noted D DSS−, n= 13); (3) rats fed with standard diet, treated with DSS and used as controls (C DSS+, n= 14) and (4) rats fed with deficient diet and treated with DSS (D DSS+, n= 16).
Disease activity index
Daily weight, physical condition, stool consistency, water/food consumption and the presence of gross and occult blood in excreta and at the anus were determined. The colitis score was calculated by assigning scores of these parameters resulting in the DAI. DAI was used to evaluate grade and extent of intestinal inflammation based on a previously published grading system [15]. The score ranges from 0 to 4 (total score), which represents the sum of scores for weight loss, stool consistency and rectal bleeding divided by three. DAI has been shown to be well correlated with tissue damage scores and with specific measurements of inflammation, such as myeloperoxidase activity index [16].
Colon tissue and blood samples
For subsequent studies, pups were killed at 26 days of age by exposure to excess halothane. Intracardiac blood samples were drawn for the measurement of plasma concentrations of vitamin B12, folate and Hcy. The colon was quickly removed, open longitudinally and gently washed in PBS1X (2.7 mmol/l KCl, 140 mmol/l NaCl, 6.8 mmol/l Na2HPO4•2H2O, 1.5 mmol/l KH2PO4, pH 7.4). Subsequently, the colon tissue were either fixed in formaldehyde, embedded in paraffin or snap-frozen in liquid nitrogen and stored at −80°C until usage.
Determination of methyl donor and oxidative stress status
Levels of vitamin B12 and folate were determined in plasma by radio-dilution isotope assay (simulTRAC-SNB; ICN Pharmaceuticals, Costa Mesa, CA, USA) and homocysteine by the fluorescence polarization immunoassay (FPIA, IMx analyser, Abbott Laboratories, Rungis, France) [11]. The Cu/Zn and Mn SOD activities were assessed using the Ransod kit (Randox, Oceanside, CA, USA), while the glutathione peroxidase (GPX) activity was carried out on a Kone Pro automate (Kone, Evry, France) using the Paglia et al. method [17]. We measured the activity of myeloperoxidase in colon samples, using the method previously described by Xia and Zweier [18].
Quantification of mRNA of tumour necrosis factor (TNF)-α by Q-RT-PCR
Total RNA was purified from nitrogen frozen colon samples of control (n= 5) and MDD (n= 5) pups with the RNeasy Lipid Tissue kit following the recommendation of Qiagen (Courtaboeuf, France), which includes treatment with DNase. To check for possible DNA contamination of the RNA samples, reactions were also performed in the absence of Omniscript RT enzyme (Qiagen). The PCR was performed with the Quantitect SYBR Green PCR kit from Qiagen and the Light Cycler instrument from Roche Diagnostics (Manheim, Germany). Specific amplification of TNF-α mRNA was performed with as primers, forward: 5′ATG GGC TCC CTC TCA TCA GT 3′, reverse: 5′ GCT TGG TGG TTT GCT ACG AC 3′. Quantization was performed with ribosomal protein S29 (RPS29) as internal standard with the following primers: forward, 5′ATG GGT CTA CAG CAG CTC TA 3′; reverse: 5′GCC CGT ATT TAC GGA TCA GA 3′. Real-time PCR was carried out using the DNA binding dye SYBR Green I for the detection of PCR products. Temperature cycling for TNF-α run proceeded such as: 15 min. at 95°C to activate the enzyme, followed by 50 cycles consisting of: 90°C for 10 sec., 58°C for 15 sec. and 72°C for 15 sec. Temperature cycling for RPS29 run proceeded as: 15 min. at 95°C to activate the enzyme, followed by 50 cycles consisting of: 94°C for 10 sec., 55°C for 20 sec. and 72°C for 15 sec. Then melting curves analyses were performed by increasing temperature from 65 to 95°C. Results were expressed as arbitrary units (AU) by calculating the ratio of crossing points of amplification curves of TNF-α mRNA and internal standard, respectively, using the RelQuant software (Roche Diagnostics).
Immunoblot analysis
Soluble extracts (30 μg/lane) were separated by 5–10% SDS-PAGE in reducing conditions and transferred to Immobilon membranes (Millipore Corp., Bedford, MA, USA). Blots were blocked in 5% non-fat milk in TBST (10 mM Tris-HCl, 150 mM NaCl, pH 7.6, containing 0.1% Tween 20) for 1 hr at 37°C and then incubated with primary antibodies overnight at 4°C. Membrane were extensively washed with TBST and then incubated for 1 hr at room temperature with horseradish peroxidase-conjugated secondary antibody. After future washings, blots were developed using enhanced chemiluminescence (ECL; Amersham Biosciences, Amersham, UK). Bands were revealed and quantified by densitometry using the ImageQuant 5.1 program. Primary antibodies for caspase-3, p38 and cytosolic phospolipase A2 (cPLA2) were obtained from Cell Signaling Technology (Beverly, MA, USA); antibodies for cyclooxygenases (COX1 and COX2), and 5-lipooxygenase (5-LOX) were obtained from Cayman (Tallinn, Estonia), TIMP3, Bcl-2, Bax and actin were obtained from Santa Cruz (Santa Cruz, CA, USA), TNF-α convertase [TACE/ADAM (a disintegrin and metallopeptidase domain)17] from Chemicon (Temecula, CA, USA). Appropriate secondary antibodies conjugated to HRP were used for detection with ECL or ECL PLUS reagent (Amersham Biosciences).
Immunohistochemistry and immunofluorescence microscopy
Colonic sample were fixed in 4% (m/v) formaldehyde pH 7.4 for 24 hrs and were routinely processed and embedded in paraffin. Paraffin blocks were used to generate 5-μm-thick haematoxylin- and eosin-stained sections. Experimenters assigned the histological scores in blind condition of sample identity. Colonic epithelial damage was assigned scores as follows: 0 = normal; 1 = hyperproliferation, irregular crypts and goblet cell loss; 2 = mild to moderate crypt loss (10–50%); 3 = severe crypt loss (50–90%); 4 = complete crypt loss, surface epithelium intact; 5 = small- to medium-sized ulcer (<10 crypt widths) and 6 = large ulcer (>10 crypt widths). Infiltration with inflammatory cells was assigned scores separately for mucosa (0 = normal, 1 = mild, 2 = modest, 3 = severe), submucosa (0 = normal, 1 = mild to modest, 2 = severe) and muscle/serosa (0 = normal, 1 = moderate to severe). Scores for epithelial damage and inflammatory cell infiltration resulted in a total scoring range of 0–12, according to Katakura et al. [19].
To identify the expression of cPLA2, cryosections of rat colon were fixed in acetone for immunofluorescence microscopy. After washing in PBS1X, non-specific binding was blocked with 10% bovine serum albumin for 1 hr prior to the addition of the cPLA2 antibody (1:200), and incubated overnight at 4°C in a humidified chamber. After repeated washings in PBS1X, the sections were incubated with a fluorescein isothiocyanate-conjugated antimouse secondary antibody for 1 hr 1/1000, Alexa Fluor 488, Cell Signaling Technology). The sections were washed again in PBS1X and mounted in Vectashield media (Vector Labs, Burlingame, CA, USA).
Sections were processed for peroxidase immunostaining using the Dako Laboratories, (Trappes, France) system following the manufacturer’s recommendations. Immunohistochemistry was performed on formalin-fixed, paraffin-embedded tissue sections using the streptavidin-biotin-peroxydase method in a Dakocytomation AutoStainer (Glostrup, Denmark). Sections were first deparaffinized and rehydrated. Antigen retrieval was performed by incubating the slides in Tris-citrate buffer pH 6.0 for 20 min. at 97°C (PT Link, Dakocytomation). Endogenous peroxydase activity was blocked by incubation in 3% hydrogen peroxide for 10 min. Polyclonal rabbit anti-myeloperoxydase (dilution: 1/4000; Dakocytomation) and monoclonal mouse anti-E-Cadherin (dilution: 1/120, clone 4A2C7; Invitrogen, Carlsbad, CA, USA) were incubated on slides for 30 min. at temperature room. Biotinylated secondary antibodies were a polyclonal swine anti-rabbit (Dakocytomation) or polyclonal goat antimouse (Dakocytomation) antibodies.
Sections were incubated with 3,3-diaminobenzidine substrate (Dako Laboratories) for 1 min. before the reaction was stopped in distilled water, and counterstained with haematoxylin. Withdrawal of the primary antibody and replacement with a non-specific antibody were used as negative controls.
Statistical analysis
Data were prospectively collected and analysed with SAS software (SAS Institute, Berkley, CA, USA). Continuous variables were reported as means ± S.D. Raw data were compared by using one-way anova with Fisher’s test. A P-value < 0.05 was considered to indicate statistical significance.
Results
Confirmation of methyl donor deficiency
Levels of vitamin B12, folate and homocysteine were measured in blood samples of rats to evaluate the influence of MDD on these parameters. As expected, at 26 days of age, compared with the standard diet, the deficient diet significantly decreased the plasma concentration of both vitamin B12 (919 ± 54 versus 489 ± 27 pmol/l, respectively, P < 0.01) and folate (99 ± 38 versus 13 ± 2 nmol/l, respectively, P < 0.01) and was accompanied by an increase in the plasma concentration of homocysteine (7.8 ± 0.9 versus 22.6 ± 1.2 μmol/l, respectively, P < 0.01). Of note, treatment of rats with DSS did not influence folate, vitamin B12 and homocysteine plasma levels.
Effects of methyl donor deficiency during DSS-induced colitis
Treatment with DSS induced a colitis characterized by inflammatory cell infiltrations, as previously described [20, 21]. Of note, DSS intake was similar in C DSS+ and D DSS+ rats. The DAI provides a well-characterized scoring system to quantify disease severity that is correlated with histological lesions [21]. Maximum severity of colonic inflammation was reached at 6 days after initiation of DSS treatment. As expected, the DAI was higher in the C DSS+ group than in the C DSS− group (P < 0.05). Importantly, methyl donor deficient diet further aggravated the severity of colitis induced by DSS, as reflected by the dramatically higher DAI in the D DSS+ group compared with the C DSS+ group (P < 0.01, Fig. 1). There was no difference in terms of DAI when comparing the D DSS− group with the C DSS− group. The total score for epithelial damage and inflammatory cell infiltration was estimated to 0.0 ± 0.0, 0.0 ± 0.0, 2.0 ± 0.5 and 5.7 ± 0.6 for the C DSS−, D DSS−, C DSS+ and D DSS+ groups, respectively, showing a dramatic increase of the score in the deficient rats exposed to DSS, compared to the other groups (P < 0.001).
Fig 1.

Effects of MDD on DAI during DSS−-induced colitis in rats. Deficient diet and DSS-treated rats *C DSS+ versus C DSS−, P < 0.05; **D DSS+ versus C DSS+, P < 0.01, anova, Fisher test.
Oxidative stress markers
In animals drinking tap water, levels of both Cu/Zn SOD and Mn SOD activities were significantly higher in the colon of rat pups of the D/DSS− group compared to the C DSS− group, suggesting that methyl donor deficiency promotes oxidative stress under physiological conditions (Table 1). Methyl donor deficiency did not influence SOD activity after the rats were treated with DSS. There was no difference in GPX activity between the four groups of rats (Table 1).
Table 1.
Activity (per mg or g protein) of SOD and GPX in the colon of rats
| C/DSS− | D/DSS− | C/DSS+ | D/DSS+ | |
|---|---|---|---|---|
| Total SOD (UI/mg) | 7.98 ± 1.85 | 13.46 ± 7.01* | 8.11 ± 2.41 | 7.65 ± 1.08 |
| SOD Mn (UI/mg) | 0.76 ± 0.23 | 1.05 ± 0.27* | 0.53 ± 0.12 | 0.74 ± 0.17 |
| SOD Cu/Zn (UI/mg) | 7.22 ± 1.73 | 12.42 ± 6.77* | 7.58 ± 2.3 | 6.91 ± 1.01 |
| GPX (μmol/min/g) | 64.86 ± 6.95 | 84.55 ± 31.77 | 66.97 ± 15.33 | 69.03 ± 14.88 |
Results are expressed as mean ± S.D. *D DSS− versus C DSS−, P < 0.05, anova, Fisher test.
Apoptosis
At the protein level, neither MDD nor DSS treatment alone affected the expression of caspase-3, Bax and Bcl2. When combined, DSS treatment and MDD decreased the expression of both caspase-3 and Bax, two pro-apoptotic factors, while increasing the expression of Bcl2, an anti-apoptotic factor. As shown in Figure 2, there was an increase in the protein level of the 17 kD active subunit of caspase-3 in D DSS− rats when compared with C DSS− animals, but this did not reach statistical significance. The protein level of the 17 kD active subunit of caspase-3 was significantly higher in the colon of rats of the C/DSS+ group compared with the D/DSS+ group. Similarly, the expression level of Bax was lower in the D/DSS+ group than in the C/DSS+ group (Fig. 2), indicating that MDD may further decrease the expression of pro-apoptotic factors during experimental colitis. In the colon of rats fed a MDD and treated with DSS (D DSS+ group), the expression levels of both caspase-3 and Bax were significantly decreased while the expression level of Bcl2 was significantly increased when compared with animals fed a methyl-deficient diet but drinking tap water (D DSS− group), even though the magnitude of the difference was lower for Bcl2 than for caspase-3 and Bax. Collectively, these findings show that during experimental colitis in rats, methyl donor deficiency further stimulates anti-apoptotic pathways.
Fig 2.
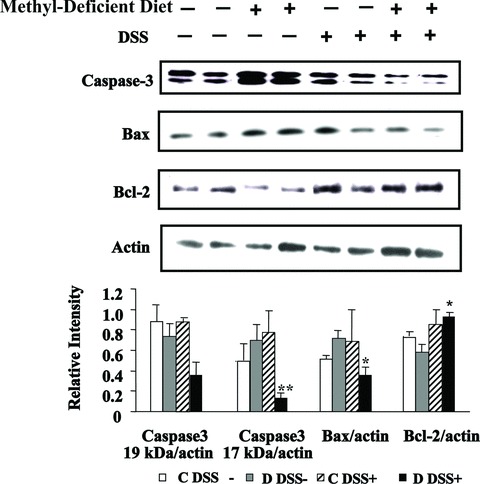
Effects of MDD on apoptosis. Western blot analyses of caspase3, Bax and Bcl2 were performed in rats with DSS-induced colitis. ** Caspase3 of D DSS− compared with D DSS+, P < 0.05; * Bax/actin and Bcl-2/actin ratios of D DSS− compared with D DSS+, P < 0.05.
Inflammatory mediators and pathways
The mRNA level of TNF-α was determined by semi-quantitative real time RT-PCR, using PR29 (Ribosomal protein 29) as reference gene. The TNF-α mRNA level was significantly higher in colon from methyl-deficient rats compared to the rats fed a standard diet (Fig. 3). In line with the increased expression of TNF-α, Western blot analysis showed a decreased expression of TIMP3, a potent inhibitor of TNF-α in rats subjected to DSS and/or the MDD, compared with control animals. By contrast, expression levels of TACE were broadly similar between the four groups (Fig. 3). Western blot analysis revealed that the expression level of p38 was markedly increased by the MDD in the absence of DSS treatment (Fig. 4). The MDD dramatically increased the p38 protein level in DSS-treated rats, compared to animals receiving DSS alone (C/DSS+ group) (Fig. 4).
Fig 3.
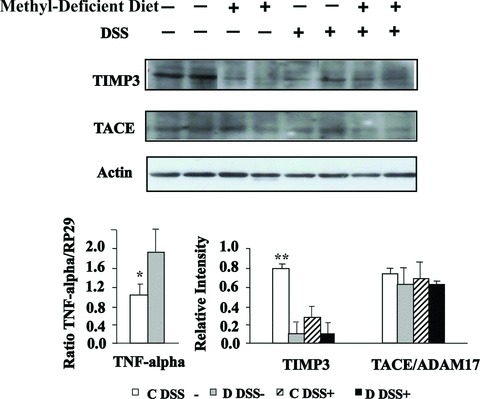
Effects of MDD on expression levels of TNF, TIMP3 and TACE in DSS-induced colitis in rats. Western blot analysis of TIMP3 and TACE expression (top and bottom right) and mRNA level of TNF determined by semi-quantitative real time RT-PCR, using PR29 as reference gene (bottom left). *TNF of C DSS− compared with D DSS−, P < 0.05; **TIMP3 of C DSS− compared with either D DSS−, C DSS+ or D DSS+, P < 0.01.
Fig 4.
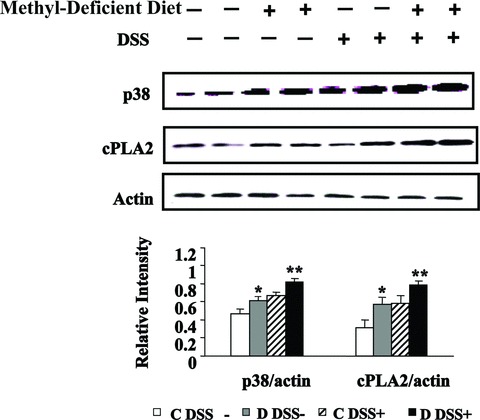
Effects of MDD on expression levels of p38 and cPLA2 during DSS-induced colitis in rats. Western blot analysis of p38 and cPLA2 expression (top) and quantitative analysis (bottom). *p38 and cPLA2 of C DSS− compared with D DSS−, P < 0.05; **p38 and cPLA2 of D DSS+ compared with either C DSS+ or D DSS−, P < 0.01.
Because p38 MAPK may be an upstream regulator of cPLA2 [22], we investigated whether MDD and DSS treatment may affect the expression of cPLA2 at the protein level. Similar to p38, in animals drinking tap water, the cPLA2 protein level was significantly increased in the D DSS− group compared with the C DSS− group. The MDD also increased the expression of cPLA2 in the rats treated with DSS (Fig. 4). Immunohistochemistry confirmed an increased expression of cPLA2 in the colon of rats fed a MDD compared with animals of the control group, in absence of DSS treatment (Fig. 5). Histopathological examination of the mucosa of the deficient animals treated by DSS showed necrotic lesions in the glands, an oedema and an infiltration by mononuclear cells in the sub-mucosa. The vessels were dilated and shared thrombotic lesions (Fig. 5).
Fig 5.
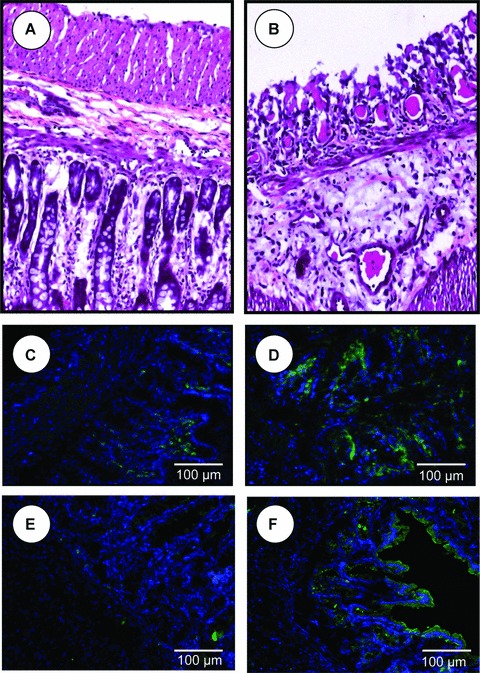
Histopathological examination of the mucosa of the methyl donor deficient rats treated with DSS (B) compared with controls (A). Immunohistochemistry of cPLA2 in colonic mucosa of control (C) and deficient rats (D). Immunohistochemistry of COX2 in colonic mucosa of control (E) and methyl-deficient rats (F). Data shown are representative of triplicate experiments and magnifications are indicated by bars.
The mucosal damage produced by DSS did not allow evaluating the expression of cPLA2 by immunohistochemistry in the DSS-treated animals. Arachidonic acid is released from membrane phospholipids by cPLA2 [23]. Arachidonic acid is then converted into eicosanoids by three enzymes, namely COX, LOX and cytochrome P450 [23, 24]. To further evaluate the consequences of the increase in cPLA2 expression during MDD and experimental colitis, the protein levels of COX-1, COX-2 and 5-LOX were measured in the colon of rats [23]. As showed in Figure 6, the expression level of COX-2 was significantly increased in the colon of rats of the D DSS+ group compared with the C DSS+ group. By contrast, the protein levels of both COX-1 and 5-LOX were broadly similar among the four groups of rats. Immunohistochemistry suggested an increased expression of COX2 in the colon of rats fed a MDD in the absence of DSS treatment (Fig. 5). However, this finding was not confirmed by Western blot analysis (Fig. 6). Taken together, these results showed that the methyl donor deficiency may activate pro-inflammatory cascades, such as the p38/TNF/cPLA2/COX-2 pathway, in the colon of rats under both physiological and pathological conditions.
Fig 6.
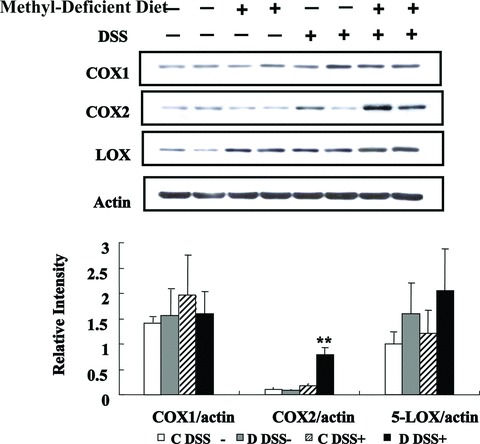
Effects of MDD on expression levels of COX1, COX2 and LOX during DSS-induced colitis in rats. Western blot analysis of COX1, COX2 and LOX expression (top) and quantitative analysis (bottom). ** COX2 of D DSS+ compared with either C DSS+, D DSS− or C DSS−, P < 0.001.
Integrity of the intestinal barrier
The methyl donor deficiency did not influence the mucosal thickness of the colon (0.26 ± 0.02, 0.22 ± 0.01 mm in C DSS− and D DSS− groups, respectively), while the DSS produced a significant reduction in the two DSS+ groups (0.17 ± 0.01 and 0.16 ± 0.02 mm in C DSS+ and D DSS+, respectively), compared with the other groups (P < 0.01). The absence of influence of the methyl donor deficiency on mucosal integrity was also demonstrated by the maintained expression of β-catenin and the absence of MPO in immunohistochemical examination of colon specimens from D DSS− rats, compared with C DSS− rats (Fig. 7). In agreement with these findings, the MPO activity was similar in colon samples of C DSS− and D DSS− rats (2.08 ± 0.55 and 1.67 ± 0.23 μmol/min./g, respectively).
Fig 7.
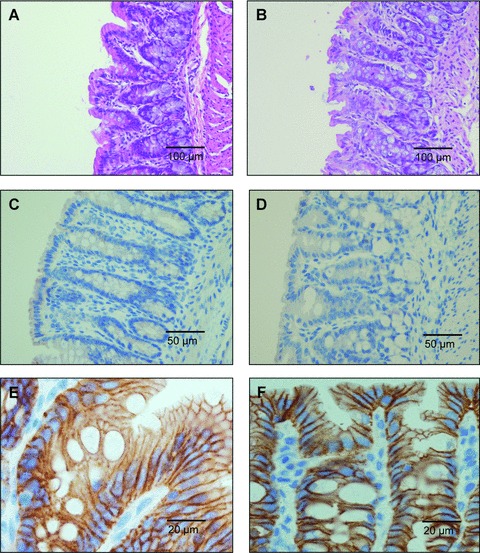
Histopathological examination of the haematoxylin- and eosin-stained colon specimens from methyl donor deficient rats (B) compared with controls (A). Immunohistochemistry of myeloperoxidase in colonic mucosa specimens from control (C) and methyl donor deficient rats (D). Immunohistochemistry of E-Cadherin in colonic mucosa of control (E) and methyl donor deficient rats (F). Data shown are representative of triplicate experiments and magnifications are indicated by bars.
Discussion
We recently demonstrated in rats that methyl donor deficient diet during gestation and lactation induced gastric lesions at weaning characterized by surface layer erosion related with loss of cell polarity, anarchic cell migration, abnormal progenitor differentiation and apoptosis [25]. Herein, we found that this MDD was not capable of increasing the DAI, in the absence of any additional exposure. We were willing to investigate whether the pro-inflammatory pathways and the surface layer erosion specifically activated by the methyl donor deficiency aggravate the mucosal injury produced by DSS, because the latter is a widely used non soluble agent that disrupts the colonic epithelium.
A decrease in antioxidant capacity associated with an increased oxidative stress has been reported in CD patients [26]. In our model, the methyl deficiency diet led to a significant increase in SOD in the absence of DSS administration. This result is in line with the study by Moat et al. showing enhanced activity of SOD in the plasma of patients with hyperhomocysteinemia [9]. Consistently, we recently found a significant association between clinical activity and the SOD activity in CD patients carrying the MTRR 66AA genotype [3]. The effect of homocysteine on GPX activity remains controversial [9, 27]. This may be partly explained by differences between the models studied (human beings, animals, or cell culture). In our study, a MDD did not affect GPX activity, suggesting that SOD rather than GPX may mediate oxidative stress in the colon of rats and contribute to pathological intestinal inflammation.
An increased Bcl2 : Bax ratio resulting in resistance of T cells against apoptosis has been reported in the lamina propria of CD patients [12, 13]. We found that DSS-induced colitis enhanced the expression of the anti-apoptotic factor Bcl2 while decreasing the level of pro-apoptotic markers (capsase-3 and Bax) in rats fed the MDD. Homocysteine treatment was shown to induce apoptosis of different cell types in vitro[14, 28], whereas the expression level of pro-apoptotic markers was increased in the liver from cystathionine β synthase-deficient mice, brain from methyl-deficient rats or in rats restricted in folate and vitamin E [28–31]. The contrasted results between these and our study may be explained by differences in the experimental models and designs and by differences in the cystathionine β synthase expression in tissues. In our rat model, cystathionine β synthase is expressed in the intestine and can degrade homocysteine. In contrast, cystathionine β synthase is not expressed in the brain and in tissues of cbs−/− mice. Consequently, the lack of degradation of homocysteine could have produced more dramatic effects in these studies than in our model.
TNF-α is a key actor of the acute phase of IBD. We failed to detect TNF-α by Western blotting, but we demonstrated an increased expression of TNF gene at the transcriptional level in the deficient rats. TIMP3 is a crucial negative regulator of TNF-α in both tissue homeostasis and tissue response to injury [32–34]. Western blotting of membrane-bound TNF and soluble TNF is increased in the livers of TIMP3−/− mice compared with those of wild-type mice, while no difference is observed in the expression of TNF mRNA [32]. In our experimental model, the MDD produced a dramatic decreased of TIMP3 and no change in TACE (ADAM17). A recent study showed a differential expression of ADAM17 and TIMP3 in acute inflamed intestinal epithelia [33]. These and our data therefore suggest investigating whether TIMP3 exerts regulating effects on the TNF-α pathway by dual mechanisms, the one dependent and the other independent from TACE [34]. Activation of p38 MAPK has been linked to IBD pathogenesis [35]. In line with previous reports showing that homocysteine can activate p38 [5], which in turn increases TNF levels [36], both p38 and TNF expression levels were significantly increased in the colon of rats fed a MDD. Pro-inflammatory stimuli, such as TNF, are well-established inducers of anti-apoptotic pathways [12]. Accordingly, the expression of Bax, an anti-apoptotic factor, is down-regulated in inflamed colonic mucosa of IBD patients [37]. Vitamin B12 deficiency is associated with increased TNF levels and TNF-related pathways in cell models, in human beings and in rats [38–40]. Therefore, during colitis, concomitant methyl donor deficiency may further activate anti-apoptotic factors while decreasing pro-apoptotic factors. Both TNF and homocysteine are known to stimulate NF-κB [8], an anti-apoptotic mediator. Furthermore, overexpression of MnSOD and CuZnSOD is capable of preventing apoptosis [41]. Taken as a whole, these results may explain, at least in part, the mechanisms whereby MDD aggravates experimental colitis in rats.
Eicosanoids are produced de novo from membranes following cell activation by physiological and pathological stimuli, such as pro-inflammatory cytokines [24]. When tissues are exposed to these stimuli, arachidonic acid is released from membrane phospholipids by phospholipases, cPLA2 being the key enzyme in eicosanoid production [24]. Arachidonic acid is then converted into eicosanoids by three enzymes, namely COX, LOX and cytochrome P450 [24]. The activity of cPLA2 was markedly increased in the colon of rats treated with DSS [42]. The concentration of cPLA2 was also increased in colonic mucosa of patients with active IBD [43]. Accordingly, cPLA2 inhibition had an anti-inflammatory effect during experimental colitis in rats [43]. In the absence of colitis, we found that methyl donor deficiency was associated with an increase in cPLA2 levels in the rat colon. Similar to apoptosis, the increase in p38 levels associated with MDD may contribute to high cPLA2 in these animals, as suggested by previous reports [21–23]. Therefore, a MDD may further enhance colonic lesions induced by DSS treatment in rats.
COX and LOX are two enzymes responsible for the synthesis of eicosanoids, using arachidonic acid as substrate [24]. Two COX isoforms have been identified, COX-1, the constitutive form and COX-2, the inducible form [24]. The expression of 5-LOX and COX-1 was unchanged in the colon of IBD patients [44]. Consistently, DSS treatment and/or methyl donor deficiency had no impact on 5-LOX and COX-1 levels in our animal model. COX-2 is undetectable in normal ileum or colon of IBD patients and, but it is induced in apical epithelial cells of inflamed foci in IBD patients [45]. Hendel et al. found a significant correlation between endoscopic activity and COX-2 mRNA levels [46]. COX-2 expression level was dramatically increased in the colon of methyl-deficient rats treated with DSS. This elevation of COX-2 concentration in the rat colon may occur through direct and indirect mechanisms. High TNF levels within the colonic mucosa of rats may directly enhance COX-2 expression [47]. In addition, pro-inflammatory stimuli, such as p38 and TNF, may increase the release of arachidonic acid from membrane phospholipids, which in turns serves as substrate for LOX, COX-1 and COX-2 [24]. Overall, combined elevation of cPLA2 and COX-2 may increase the synthesis of prostaglandins and thromboxanes and contribute to maintenance of mucosal inflammation in the colon of IBD patients. Interestingly, platelet incubation with homocysteine significantly increased thromboxane levels [48], further supporting a role for methyl donor deficiency in the development and/or maintenance of colonic inflammation in rats. From a clinical point of view, beside their direct role in the severity of the disease, inflammation pathways are key players in the development of IBD-associated colon cancer [49]. The decreased expression of pro-apoptotic markers in colon mucosa of the deficient animals has also to be considered in regards to the controversy that exists between the folate status and the risk for colon cancer [50–52]. The mucosal integrity and the transepithelial migration of neutrophils are interacting parameters to be considered in the mechanisms beyond the relations between methyl donor deficiency, inflammation and the severity of the mucosal injury produced by DSS. The apical junctional complex is the main component of the intestinal barrier; it consists of tight and adherens junctions that are compromised in IBD, with disappearance of key proteins such as occluding and E-cadherin [53] and infiltration of neutrophils [54]. These mechanisms had a limited influence, if any, in our model, since we observed a maintained thickness and integrity of the colon mucosa in the DSS− methyl-deficient rats, with no disappearance of E-cadherin expression and no transepithelial migration of neutrophils.
In conclusion, a methyl deficiency diet aggravate experimental colitis in rats by promoting oxidative stress, decreasing cell apoptosis and by activating inter-related pro-inflammatory mechanisms, including the TNF pathway, p38, cPLA2 and COX-2. However, the validity of these findings needs to be investigated by evaluating the clinical implications. In IBD patients, homocysteine levels were positively correlated with activity, number of flares and duration of IBD [52]. Therefore, screening for folate and vitamin B12 deficiencies aimed at restoring a normal methyl donor status might improve the management of IBD patients, possibly modifying the clinical course of the disease. Further studies are now required to assess the benefit/risk ratio for treating the patients with B12 and/or folate deficit in human IBD.
Acknowledgments
Institutional grants were obtained from the region Lorraine and from Inserm, France.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–34. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 2.Peyrin-Biroulet L, Desreumaux P, Sandborn WJ, et al. Crohn’s disease: beyond antagonists of tumour necrosis factor. Lancet. 2008;372:67–81. doi: 10.1016/S0140-6736(08)60995-2. [DOI] [PubMed] [Google Scholar]
- 3.Peyrin-Biroulet L, Gueant-Rodriguez RM, Chen M, et al. Association of MTRR 66A>G polymorphism with superoxide dismutase and disease activity in patients with Crohn’s disease. Am J Gastroenterol. 2008;103:399–406. doi: 10.1111/j.1572-0241.2007.01573.x. [DOI] [PubMed] [Google Scholar]
- 4.Peyrin-Biroulet L, Rodriguez-Gueant RM, Chamaillard M, et al. Vascular and cellular stress in inflammatory bowel disease: revisiting the role of homocysteine. Am J Gastroenterol. 2007;102:1108–15. doi: 10.1111/j.1572-0241.2007.01170.x. [DOI] [PubMed] [Google Scholar]
- 5.Danese S, Sgambato A, Papa A, et al. Homocysteine triggers mucosal microvascular activation in inflammatory bowel disease. Am J Gastroenterol. 2005;100:886–95. doi: 10.1111/j.1572-0241.2005.41469.x. [DOI] [PubMed] [Google Scholar]
- 6.Morgenstern I, Raijmakers MT, Peters WH, et al. Homocysteine, cysteine, and glutathione in human colonic mucosa: elevated levels of homocysteine in patients with inflammatory bowel disease. Dig Dis Sci. 2003;48:2083–90. doi: 10.1023/a:1026338812708. [DOI] [PubMed] [Google Scholar]
- 7.Poddar R, Sivasubramanian N, DiBello PM, et al. Homocysteine induces expression and secretion of monocyte chemoattractant protein-1 and interleukin-8 in human aortic endothelial cells: implications for vascular disease. Circulation. 2001;103:2717–23. doi: 10.1161/01.cir.103.22.2717. [DOI] [PubMed] [Google Scholar]
- 8.Collins T, Cybulsky MI. NF-kappaB: pivotal mediator or innocent bystander in atherogenesis? J Clin Invest. 2001;107:255–64. doi: 10.1172/JCI10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moat SJ, Bonham JR, Cragg RA, et al. Elevated plasma homocysteine elicits an increase in antioxidant enzyme activity. Free Radic Res. 2000;32:171–9. doi: 10.1080/10715760000300171. [DOI] [PubMed] [Google Scholar]
- 10.Wang XL, Duarte N, Cai H, et al. Relationship between total plasma homocysteine, polymorphisms of homocysteine metabolism related enzymes, risk factors and coronary artery disease in the Australian hospital-based population. Atherosclerosis. 1999;146:133–40. doi: 10.1016/s0021-9150(99)00111-2. [DOI] [PubMed] [Google Scholar]
- 11.Nishio E, Watanabe Y. Homocysteine as a modulator of platelet-derived growth factor action in vascular smooth muscle cells: a possible role for hydrogen peroxide. Br J Pharmacol. 1997;122:269–74. doi: 10.1038/sj.bjp.0701391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mudter J, Neurath MF. Apoptosis of T cells and the control of inflammatory bowel disease: therapeutic implications. Gut. 2007;56:293–303. doi: 10.1136/gut.2005.090464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ina K, Itoh J, Fukushima K, et al. Resistance of Crohn’s disease T cells to multiple apoptotic signals is associated with a Bcl-2/Bax mucosal imbalance. J Immunol. 1999;163:1081–90. [PubMed] [Google Scholar]
- 14.Zhang C, Cai Y, Adachi MT, et al. Homocysteine induces programmed cell death in human vascular endothelial cells through activation of the unfolded protein response. J Biol Chem. 2001;276:35867–74. doi: 10.1074/jbc.M100747200. [DOI] [PubMed] [Google Scholar]
- 15.Stevceva L, Pavli P, Husband A, et al. Dextran sulphate sodium-induced colitis is ameliorated in interleukin 4 deficient mice. Genes Immun. 2001;2:309–16. doi: 10.1038/sj.gene.6363782. [DOI] [PubMed] [Google Scholar]
- 16.Cooper HS, Murthy S, Kido K, et al. Dysplasia and cancer in the dextran sulfate sodium mouse colitis model. Relevance to colitis-associated neoplasia in the human: a study of histopathology, B-catenin and p53 expression and the role of inflammation. Carcinogenesis. 2000;21:757–68. doi: 10.1093/carcin/21.4.757. [DOI] [PubMed] [Google Scholar]
- 17.Paglia DE, Valentine WN. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J Lab Clin Med. 1967;70:158–69. [PubMed] [Google Scholar]
- 18.Xia Y, Zweier JL. Measurement of myeloperoxidase in leukocyte-containing tissues. Anal Biochem. 1997;245:93–6. doi: 10.1006/abio.1996.9940. [DOI] [PubMed] [Google Scholar]
- 19.Katakura K, Lee J, Rachmilewitz D, et al. Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. J Clin Invest. 2005;115:695–702. doi: 10.1172/JCI22996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okayasu I, Hatakeyama S, Yamada M, et al. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98:694–702. doi: 10.1016/0016-5085(90)90290-h. [DOI] [PubMed] [Google Scholar]
- 21.Melgar S, Karlsson A, Michaelsson E. Acute colitis induced by dextran sulfate sodium progresses to chronicity in C57BL/6 but not in BALB/c mice: correlation between symptoms and inflammation. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1328–38. doi: 10.1152/ajpgi.00467.2004. [DOI] [PubMed] [Google Scholar]
- 22.Murthy SN, Cooper HS, Shim H, et al. Treatment of dextran sulfate sodium-induced murine colitis by intracolonic cyclosporin. Dig Dis Sci. 1993;38:1722–34. doi: 10.1007/BF01303184. [DOI] [PubMed] [Google Scholar]
- 23.Borsch-Haubold AG, Ghomashchi F, Pasquet S, et al. Phosphorylation of cytosolic phospholipase A2 in platelets is mediated by multiple stress-activated protein kinase pathways. Eur J Biochem. 1999;265:195–203. doi: 10.1046/j.1432-1327.1999.00722.x. [DOI] [PubMed] [Google Scholar]
- 24.Harizi H, Corcuff JB, Gualde N. Arachidonic-acid-derived eicosanoids: roles in biology and immunopathology. Trends Mol Med. 2008;14:461–9. doi: 10.1016/j.molmed.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 25.Bossenmeyer-Pourié C, Blaise S, Pourié G, et al. Methyl donor deficiency affects fetal programming of gastric ghrelin cell organization and function in the rat. Am J Pathol. 2010;176:270–7. doi: 10.2353/ajpath.2010.090153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maor I, Rainis T, Lanir A, et al. Oxidative stress, inflammation and neutrophil superoxide release in patients with Crohn’s disease: distinction between active and non-active disease. Dig Dis Sci. 2008;53:2208–14. doi: 10.1007/s10620-007-0141-6. [DOI] [PubMed] [Google Scholar]
- 27.Handy DE, Zhang Y, Loscalzo J. Homocysteine down-regulates cellular glutathione peroxidase (GPx1) by decreasing translation. J Biol Chem. 2005;280:15518–25. doi: 10.1074/jbc.M501452200. [DOI] [PubMed] [Google Scholar]
- 28.Mangiagalli A, Samuele A, Armentero MT, et al. Effects of homocysteine on apoptosis-related proteins and anti-oxidant systems in isolated human lymphocytes. Eur J Biochem. 2004;271:1671–6. doi: 10.1111/j.1432-1033.2004.04070.x. [DOI] [PubMed] [Google Scholar]
- 29.Robert K, Nehme J, Bourdon E, et al. Cystathionine beta synthase deficiency promotes oxidative stress, fibrosis, and steatosis in mice liver. Gastroenterology. 2005;128:1405–15. doi: 10.1053/j.gastro.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 30.Blaise S, Alberto JM, Nedelec E, et al. Mild neonatal hypoxia exacerbates the effects of vitamin-deficient diet on homocysteine metabolism in rats. Pediatr Res. 2005;57:777–82. doi: 10.1203/01.PDR.0000161406.19231.98. [DOI] [PubMed] [Google Scholar]
- 31.Vijayalakshhmi B, Sesikeran B, Udaykumar P, et al. Effects of vitamin restriction and supplementation on rat intestinal epithelial cell apoptosis. Free Radic Biol Med. 2005;38:1614–24. doi: 10.1016/j.freeradbiomed.2005.02.029. [DOI] [PubMed] [Google Scholar]
- 32.Mohammed FF, Smookler DS, Taylor SE, et al. Abnormal TNF activity in Timp3−/− mice leads to chronic hepatic inflammation and failure of liver regeneration. Nat Genet. 2004;36:969–77. doi: 10.1038/ng1413. [DOI] [PubMed] [Google Scholar]
- 33.Cesaro A, Abakar-Mahamat A, Brest P, et al. Differential expression and regulation of ADAM17 and TIMP3 in acute inflamed intestinal epithelia. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1332–43. doi: 10.1152/ajpgi.90641.2008. [DOI] [PubMed] [Google Scholar]
- 34.Black RA. TIMP3 checks inflammation. Nat Genet. 2004;36:934–5. doi: 10.1038/ng0904-934. [DOI] [PubMed] [Google Scholar]
- 35.Waetzig GH, Seegert D, Rosenstiel P, et al. p38 mitogen-activated protein kinase is activated and linked to TNF-alpha signaling in inflammatory bowel disease. J Immunol. 2002;168:5342–51. doi: 10.4049/jimmunol.168.10.5342. [DOI] [PubMed] [Google Scholar]
- 36.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–69. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 37.Iimura M, Nakamura T, Shinozaki S, et al. Bax is downregulated in inflamed colonic mucosa of ulcerative colitis. Gut. 2000;47:228–35. doi: 10.1136/gut.47.2.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peracchi M, Bamonti Catena F, et al. Human cobalamin deficiency: alterations in serum tumour necrosis factor-alpha and epidermal growth factor. Eur J Haematol. 2001;67:123–7. doi: 10.1034/j.1600-0609.2001.t01-1-00507.x. [DOI] [PubMed] [Google Scholar]
- 39.Hsu-Battaglia SF, Akchiche N, Noel N, et al. Vitamin B12 deficiency reduces proliferation and promotes differentiation of neuroblastoma cells and upregulates PP2A, proNGF and TACE. Proc Natl Acad Sci USA. 2009;106:21930–5. doi: 10.1073/pnas.0811794106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scalabrino G. The multi-faceted basis of vitamin B12 (cobalamin) neurotrophism in adult central nervous system: lessons learned from its deficiency. Prog Neurobiol. 2009;88:203–20. doi: 10.1016/j.pneurobio.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 41.Zemlyak I, Brooke SM, Singh MH, et al. Effects of overexpression of antioxidants on the release of cytochrome c and apoptosis-inducing factor in the model of ischemia. Neurosci Lett. 2009;453:182–5. doi: 10.1016/j.neulet.2009.02.020. [DOI] [PubMed] [Google Scholar]
- 42.Tomita Y, Jyoyama H, Kobayashi M, et al. Role of group IIA phospholipase A2 in rat colitis induced by dextran sulfate sodium. Eur J Pharmacol. 2003;472:147–58. doi: 10.1016/s0014-2999(03)01859-4. [DOI] [PubMed] [Google Scholar]
- 43.Minami T, Shinomura Y, Miyagawa J, et al. Immunohistochemical localization of group II phospholipase A2 in colonic mucosa of patients with inflammatory bowel disease. Am J Gastroenterol. 1997;92:289–92. [PubMed] [Google Scholar]
- 44.Krimsky M, Yedgar S, Aptekar L, et al. Amelioration of TNBS-induced colon inflammation in rats by phospholipase A2 inhibitor. Am J Physiol Gastrointest Liver Physiol. 2003;285:G586–92. doi: 10.1152/ajpgi.00463.2002. [DOI] [PubMed] [Google Scholar]
- 45.Singer II, Kawka DW, Schloemann S, et al. Cyclooxygenase 2 is induced in colonic epithelial cells in inflammatory bowel disease. Gastroenterology. 1998;115:297–306. doi: 10.1016/s0016-5085(98)70196-9. [DOI] [PubMed] [Google Scholar]
- 46.Hendel J, Nielsen OH. Expression of cyclooxygenase-2 mRNA in active inflammatory bowel disease. Am J Gastroenterol. 1997;92:1170–3. [PubMed] [Google Scholar]
- 47.Maier JA, Hla T, Maciag T. Cyclooxygenase is an immediate-early gene induced by interleukin-1 in human endothelial cells. J Biol Chem. 1990;265:10805–8. [PubMed] [Google Scholar]
- 48.Leoncini G, Bruzzese D, Signorello MG. Activation of p38 MAPKinase/cPLA2 pathway in homocysteine-treated platelets. J Thromb Haemost. 2006;4:209–16. doi: 10.1111/j.1538-7836.2005.01708.x. [DOI] [PubMed] [Google Scholar]
- 49.Fantini MC, Pallone F. Cytokines: from gut inflammation to colorectal cancer. Curr Drug Targets. 2008;9:375–80. doi: 10.2174/138945008784221206. [DOI] [PubMed] [Google Scholar]
- 50.Van Guelpen B, Hultdin J, Johansson I, et al. Low folate levels may protect against colorectal cancer. Gut. 2006;55:1461–6. doi: 10.1136/gut.2005.085480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jang H, Mason JB, Choi SW. Genetic and epigenetic interactions between folate and aging in carcinogenesis. J Nutr. 2005;135:2967S–71S. doi: 10.1093/jn/135.12.2967S. [DOI] [PubMed] [Google Scholar]
- 52.Crott JW, Liu Z, Keyes MK, et al. Moderate folate depletion modulates the expression of selected genes involved in cell cycle, intracellular signaling and folate uptake in human colonic epithelial cell lines. J Nutr Biochem. 2008;19:328–35. doi: 10.1016/j.jnutbio.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Drzewoski J, Gasiorowska A, Malecka-Panas E, et al. Plasma total homocysteine in the active stage of ulcerative colitis. J Gastroenterol Hepatol. 2006;21:739–43. doi: 10.1111/j.1440-1746.2006.04255.x. [DOI] [PubMed] [Google Scholar]
- 53.Bruewer M, Samarin S, Nusrat A. Inflammatory bowel disease and the apical junctional complex. Ann N Y Acad Sci. 2006;1072:242–52. doi: 10.1196/annals.1326.017. [DOI] [PubMed] [Google Scholar]
- 54.Kucharzik T, Walsh SV, Chen J, et al. Neutrophil transmigration in inflammatory bowel disease is associated with differential expression of epithelial intercellular junction proteins. Am J Pathol. 2001;159:2001–9. doi: 10.1016/S0002-9440(10)63051-9. [DOI] [PMC free article] [PubMed] [Google Scholar]