

Abstract
The interaction between depression and stroke is highly complex. Post-stroke depression (PSD) is among the most frequent neuropsychiatric consequences of stroke. Depression also negatively impacts stroke outcome with increased morbidity, mortality and poorer functional recovery. Antidepressants such as the commonly prescribed selective serotonin reuptake inhibitors improve stroke outcome, an effect that may extend far beyond depression, e.g., to motor recovery. The main biological theory of PSD is the amine hypothesis. Conceivably, ischaemic lesions interrupt the projections ascending from midbrain and brainstem, leading to a decreased bioavailability of the biogenic amines – serotonin (5HT), dopamine (DA) and norepinephrine (NE). Acetylcholine would also be involved. So far, preclinical and translational research on PSD is largely lacking. The implementation and characterization of suitable animal models is clearly a major prerequisite for deeper insights into the biological basis of post-stroke mood disturbances. Equally importantly, experimental models may also pave the way for the discovery of novel therapeutic targets. If we cannot prevent stroke, we shall try to limit its long-term consequences. This review therefore presents animal models of PSD and summarizes potential underlying mechanisms including genomic signatures, neurotransmitter and neurotrophin signalling, hippocampal neurogenesis, cellular plasticity in the ischaemic lesion, secondary degenerative changes, activation of the hypothalamo-pituitary-adrenal (HPA) axis and neuroinflammation. As stroke is a disease of the elderly, great clinical benefit may especially accrue from deciphering and targeting basic mechanisms underlying PSD in aged animals.
Keywords: stroke, aging, depression, SSRI, mechanisms
Introduction
The biological underpinnings of PSD
Animal models of PSD
Genome profiling
Neurogenesis
Neurotrophin signalling secondary degenerative brain changes following ischaemia
Role of ageing in the developmen of PSD
Mechanisms underlying antidepressant actions in PSD
Human imaging studies
Functional Imaging
Conclusions
Introduction
Post-stroke depression (PSD) is of high clinical importance. At least one third of stroke survivors display mood symptoms at some time after the insult [1–4]. Depression after stroke generally runs a chronic course and is related to a variety of adverse health outcome including increased disability, morbidity and mortality [5–11]. Interestingly, symptoms of depression may even worsen during the chronic phase after stroke [12]. Depression persists after 20 months in 34% of elderly patients with acute stroke [13] and has been linked to both worse cognitive and physical outcome.
Mental distress associated with physical disability may contribute to the development of PSD. However, the higher prevalence of symptoms of depression in stroke patients compared with orthopaedic patients with a similar degree of disability may be a good argument against purely psychological explanations of PSD [14]. Furthermore, anosognosia does not shield against PSD [15]. Common mood symptoms after stroke include anxiety and feelings of despair as well as anhedonia [16–20].
Antidepressant treatment initiated soon after stroke may prevent the emergence of PSD [21–24]. A number of studies have also reported beneficial effects of antidepressant pharmacotherapy on long-term functional recovery after stroke including activities of daily living as well as cognitive and executive functioning [23, 25–28]. Of note, propitious antidepressant effects may even extend beyond mood symptoms, e.g., to motor recovery, and thus significantly increase the proportion of patients reaching partial or full independence [29]. Moreover, a placebo-controlled clinical trial of antidepressant pharmacotherapy early in the recovery period after stroke demonstrated a significant advantage of active treatment in terms of patients’ survival. Importantly, this benefit was independent of whether or not the patient reported mood symptoms on enrollment [30]. Unfortunately, we do not have animal models for all of the successful treatments, most specifically for the psychotherapy interventions.
The biological underpinnings of PSD
Despite the fact that a high proportion of stroke patients develop mood symptoms, there is as yet little insight into the underlying neurobiological mechanisms. Relevant animal models have only sparsely been investigated. This research gap becomes even more relevant when one considers the growing body of clinical evidence indicating a beneficial effect of antidepressants and especially of selective serotonin reuptake inhibitors (SSRIs) on post-ischaemic outcome.
If we cannot prevent stroke, we shall try to alleviate its long-term consequences. Although PSD may also occur in younger stroke victims, it is primarily a condition that affects the elderly. Therefore, great clinical benefit may specifically accrue from deciphering and targeting basic mechanisms underlying chronic depression in aged animals after stroke. So far, the majority of experimental stroke studies have concentrated heavily on acute stroke outcome, which, after all, represents only a snapshot of a complex sequence of events. This limitation may have majorly contributed to the conspicuous discrepancy between laboratory and clinical findings that has been a recurrent theme in stroke research in recent years (‘translational road block’). In addition, most candidate therapies have so far been developed in young experimental animals, whereas most natural strokes occur in the elderly. Since old age as such is also associated with an enhanced susceptibility to stroke along with a poorer recovery from brain injury, it deserves to be investigated as a key modulatory factor.
Antidepressants may also exert direct actions on the brain independent of, or beyond their effects on depression, such as providing neuroprotection and promoting brain plasticity/neurogenesis. To validate these hypotheses, we will have to translate experimental findings into the clinic and investigate brain correlates of depression in stroke patients. As SSRIs were also found to improve motor outcome by diminishing abnormalities in brain motor areas, studies will have to disentangle their effects on either depression or motor function and on the respective brain correlates.
Animal models of PSD
Depression is a heterogeneous, multifaceted disorder with psychological, behavioural and vegetative components. Translating the complexities of human affective disease (e.g., guilt/suicidality symptoms) or of behavioural interventions, such as psychotherapy into animal models poses a huge challenge to the experimental researcher. Still, various behavioural tests have been proposed to investigate some of the central aspects of human depression in rodents. For example, the forced swim test (FST) [31] and the tail suspension test [32] are widely used as behavioural paradigms that measure behavioural despair in an inescapable stressful situation. These tests have been extensively characterized and have also been pharmacologically validated with drugs possessing antidepressant properties in humans [33]. Another core symptom of depression, namely anhedonia, can be evaluated in rodents by assessing sucrose consumption [34].
In contrast to global cerebral ischaemia, spontaneous post-stroke depressive-like symptoms have not been consistently described after focal brain ischaemia. For example, 1 week after intraluminal transient middle cerebral artery occlusion (MCAo) in the mouse, no depressive-like symptoms were detected in the FST [35–37]. By contrast, another study at a later time point after the ischaemic event (8 weeks) reported an ‘affective’ phenotype with increased anxiety [38]. Craft and DeVries [39] found decreased sucrose consumption 2 weeks after transient focal ischaemia in mice, suggesting a hedonic deficit in MCAo animals. Another publication reports that focal ischaemia induces shuttle-box escape deficits in rats and that this failure is reversed by treatment with a monoamine oxidase inhibitor [40].
Exposure of animals to unpredictable chronic mild stress (CMS) associated with isolated housing after ischaemia is another way to elicit experimental depression. It has been shown that rats subjected to ischaemia show decreased locomotor activity in the open field test and decreased sucrose consumption when exposed to the CMS paradigm for 18 days after surgery. These depressive-like behaviours, associated with a decrease in hippocampal neurogenesis, are again reversed by treatment with an antidepressant, namely SSRI citalopram, corroborating both the face validity and the predictive validity for this PSD model [41]. Nevertheless, future experimental studies should use aged animals that might be even more prone to develop a depressive phenotype post-stroke [42].
Genome profiling
Despite its analytical power, transcriptional profiling has not yet been employed to identify genetic pathways associated with PSD. Most studies reporting the use of gene expression profiling to investigate rodent models of depression focused on stress models instead and did not reach a consensus regarding a specific genomic signature. Promising results have been recently reported from FSL and FRL rats. Flinders rats are a genetic model of depression, derived by selective breeding of Sprague–Dawley rats, which show many key behavioural features of depression in humans. Using these two rat strains small, but statistically significant increases were confirmed in the prefrontal cortex and hippocampus of the FSL rats for genes involved in cholinergic (Chrm2, Chrna7) and in serotonergic (Htr1a, Htr2a) neurotransmission. Much larger gene changes were found for transmembrane protein 176A (TMEM176A) in the cortex of FRL animals compared with FSL rats [43–45]. TMEM176A has been implicated in adaptive immunity and in the induction and maintenance of immune tolerance [46]. In another study, five (Htr2a, Ntrk3, Crhr1, Ntrk2, Crh) of the genes classically related to human major depression were differentially expressed in three animal models of depression: acute treatment with reserpine, olfactory bulbectomy and chronic treatment with corticosterone. In addition, two new genes, complement component 3 and fatty acid-binding protein 7, have recently been described [45].
Patients with either the serotonin transporter protein promoter polymorphism/s or with the serotonin transporter genotype STin2 (variable number tandem repeats) 9/12 or 12/12 were more likely to suffer from PSD than patients with the 1/1 or 1/_l genotype or the STin2 (tandem repeats) 10/10 type. Increased functional activity of the amygdala in response to negative stimuli appears to be a mood-congruent phenomenon, and is likely moderated by the 5-HT transporter gene (SLC6A4) promoter polymorphism (5-HTTLPR).
Neurogenesis
Neurogenesis in the adult brain is an activity-dependent phenomenon. In rodents, neurogenesis has been clearly established in the hippocampal dentate gyrus (DG) as well as in the subventricular zone (SVZ) from which newly born neurons travel through the rostral migratory stream to populate the olfactory bulb. Even to date, relatively little is known about the biological significance of neurogenesis in the adult mammalian brain. Two major hypotheses have driven most of the studies on neurogenesis in the DG, namely that it plays a pivotal role in hippocampus-dependent learning and memory [47] as well as in protecting against and promoting recovery from depression [48]. As far as learning and memory are concerned, the precise role of neurogenesis remains controversial. For example, genetic ablation of cell cycle regulatory protein cyclin D2 that results in virtual absence of newly born neurons in the adult brain does not lead to appreciable learning and memory deficits [49, 50]. Similarly, the involvement of hippocampal neurogenesis in depression and in the efficacy of antidepressive treatments is also not fully established.
Global ischaemia increases neurogenesis in the adult brain, especially in DG [51]. Other more circumscribed experimental stroke models, such as occlusion of the middle cerebral artery were also shown to increase neurogenesis in SVZ [52] as well as in DG [53]. Furthermore, the newly generated cells were shown to migrate from SVZ, but not form SGZ, to the site of the insult, e.g., into the ischaemic penumbra of the stroke-damaged striatum and to develop and mature into the phenotype of the neurons destroyed by ischaemia [54–56]. Also, recovery from stroke was shown to be associated with growth factor-induced neurogenesis in SVZ as well as exercise-induced neurogenesis in DG [57]. Therefore, neurogenesis has been postulated to be an adaptive process involved in brain regeneration and recovery following an ischaemic insult [55]. A schematic representation of interactions between stroke, depression and neurogenesis as a function of age is shown in Fig. 1.
Fig 1.
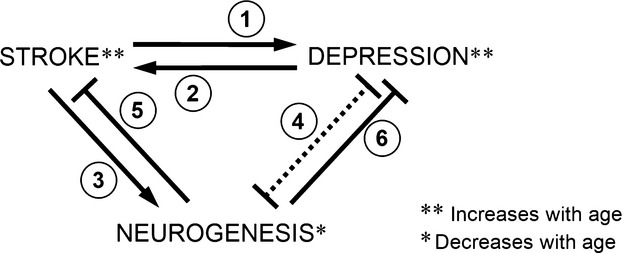
Scheme of interaction between ageing, stroke, neurogenesis and depression. Stroke causes depression (1), depression increases negative effects of stroke (2), stroke induces neurogenesis (3), depression is associated with reduced neurogenesis (4), neurogenesis might help to alleviate the effects of stroke (5), neurogenesis might help to combat depression (6), ageing increases the incidence of stroke and depression [**], neurogenesis decreases with age [*].
Most importantly, very little is known about the relationship between PSD and neurogenesis. Depressive behaviours in ischaemic rats were accompanied by reduced ischaemia-evoked hippocampal neurogenesis and this effect was reversed by citalopram administration. Using pharmacological interventions, the involvement of serotonergic neurotransmission was then further corroborated [58]. However, these findings that originate from a single research laboratory, need further confirmation along with a clear demonstration of functional significance.
Neurotrophin signalling
Neurotrophins are a group of molecules that promote the development and survival of neurons. In the context of mood disorders, most research has focused on the brain-derived neurotrophic factor (BDNF). Antidepressants and the mood stabilizer lithium have both been shown to increase BDNF levels in the brain [59]. Furthermore, BDNF produces antidepressant effects in behavioural models of depression [60] and confers resilience to chronic stress [61]. By contrast, chronic mild stress depresses BDNF protein expression [62]. BDNF is an attractive candidate molecule to explain the complex interaction between depression, antidepressants and post-stroke recovery. First, BDNF protects against ischaemic brain injury [63, 64] and attenuates apoptosis in cultured neurons after glucose deprivation [65]. In addition, BDNF signalling is crucially involved in hippocampal neurogenesis [66]. Furthermore, BDNF in periinfarct cortex improves functional recovery after stroke [67] whereas intraventricular administration of BDNF antisense oligonucleotides impedes re-learning of a motor skill in a focal ischaemia model in rats [68].
So far, only few studies have specifically investigated the role of BDNF in the pathogenesis of PSD. In stroke survivors diagnosed with PSD, serum BDNF concentrations were found to be decreased at 3–6 months post-event [69]. Furthermore, low serum levels on day 1 after admission were reported to predict the risk of subsequent PSD [70]. Preliminary evidence also suggests that the BDNF Val66Met polymorphism may modify the association between stroke and depression [71]. As yet, BDNF signalling has not been investigated in animal models of PSD. Important questions that will have to be addressed will be whether antidepressant treatment is able to increase BDNF levels in and around the ischaemic lesion and whether BDNF expression in a particular brain area (e.g., the hippocampus) is especially involved in the pathogenesis of PSD.
Secondary degenerative brain changes following ischaemia
There has been great interest in elucidating the impact of lesion characteristics on PSD. Indeed, among the many theories that have been espoused, one considers PSD as a consequence of specific brain lesions. In support of this theory, differences in the frequency of depression between left and right hemispheric lesions have been reported [72]. Thus, left hemispheric cortical strokes, mainly frontal lesions, have been suggested to be linked with increased risk for depression. However, a systematic review offered no support for the hypothesis that the risk of depression after stroke was affected by the hemispheric location of the brain lesion nor by the position of the lesion along the antero-posterior axis [73]. Thus, a definitive statement regarding lateralization and risk for depression cannot be made at this point.
Robinson and others postulated that dysfunction of (cortico)-striato-pallido-thalamic-cortical projections predisposes to PSD [74]. This loop could even be disrupted indirectly by secondary degeneration when not included in the primary ischaemic lesion. Indeed, focal ischaemia is known to cause neuronal loss in remote brain areas. For instance, after a stroke in the most commonly affected MCA territory, brain regions, such as the ipsilateral thalamus, and substantia nigra will display delayed shrinkage due to synaptic contacts to the primary lesion site. Retrograde degeneration due to cortical lesion as well as anterograde (Wallerian) degeneration due to basal ganglia lesion, and/or extensive vasogenic edema [75] are thought to be responsible for these forms of secondary neurodegeneration. These changes seem to be, at least partly, apoptotic [76]. In addition, such extrafocal neurodegeneration may also affect neurotransmitter synthesis. Furthermore, the ageing brain seems to be even more sensitive to these mechanisms of secondary degeneration [77].
Role of ageing in the development of PSD
It is estimated that ischaemic brain injury is responsible for approximately half of all patients hospitalized for acute neurological disorders. As old age is associated with an enhanced susceptibility to stroke along with a poorer recovery from brain injury, it deserves to be investigated as a key modulatory factor. Depression is also a highly significant problem in the elderly. It is often associated with cognitive impairments including cognitive impairments after stroke [78, 79]. It is therefore unfortunate that most experimental stroke studies are performed in young or middle-aged animals.
Vascular risk factors, such as hypertension, obesity, diabetes, dyslipidaemia, hyperhomocystaeinemia and systemic inflammation increase the probability of silent strokes. Even in the absence of clinically apparent ischaemia, microvascular changes and microstrokes in vulnerable regions may lead to depression (‘vascular depression’; [80]). Several genes, such as protein kinase Cη (PRKCH) gene, angiotensin-converting enzyme, and apolipoprotein (a) [apo(a)] andLp(a), play an important role in this context [81].
Mechanisms underlying antidepressant actions in PSD
The neurotransmitter and cytokine hypotheses represent the two main biological theories of PSD [82]. Robinson and Bloom (1977) [74] postulated that ischaemic lesions may interrupt the biogenic amine-containing axons ascending from the brainstem to the cerebral cortex leading to a decreased availability of biogenic amines in limbic structures of frontal and temporal lobes as well as in basal ganglia. The monoamine theory postulates that depression is associated with low levels of monoamines, particularly 5-HT, NE and dopamine [83]. Along the same lines, a high global receptor density for the monoamine oxidase A (MAO-A), which non-specifically metabolizes these neurotransmitters, has also been reported. The diverse symptoms of depression (mood, cognition and pain) may be attributable to the involvement of different neural systems. Alterations of the dopaminergic mesolimbic reward system may lead to anhedonia [84]. Serotonergic and noradrenergic fibres originate from brainstem nuclei and innervate the limbic system, prefrontal cortex and associated structures involved in the regulation of mood. Furthermore, descending pathways project through the dorsolateral spinal column and are instrumental in the regulation of pain. In addition to monoamines, the cholinergic system, through nicotinic acetylcholine receptors, has recently been suggested to be involved in the aetiology of major depressive disorder. All these pathways may be disrupted by the stroke lesion, which may result in depression [85] (Fig. 2).
Fig 2.

The acetyl-amine hypothesis of post-stroke depression. The serotonergic pathway (green), the acetylcholinergic pathway (red), the dopaminergic pathway (violet) and the noradrenergic pathway (blue). Dotted lines represents a stroke lesion disrupting these pathways.
In vivo and in vitro studies have shown that fluoxetine and paroxetine, two of the most commonly prescribed antidepressants, prevented degeneration of nigrostriatal dopaminergic neurons. This was accompanied by marked repression of microglia activation, neutrophil infiltration and expression of proinflammatory markers. In addition, these neuroprotective effects in Parkinson's models were accompanied by improvement of motor impairment and neurological deficits [86–88]. As stroke in aged rodents is accompanied by a strong inflammatory reaction in the brain [89], we expect that treatment with antidepressants will reduce pro-inflammatory cytokines released by the lesion [90].
Elevated levels of stress hormone cortisol are consistently found in severely depressed patients. Increased cortisol may cause impairment in neuroplasticity and cellular resilience, and down-regulation of the glucocorticoid receptor's sensitivity. Subsequently, hyperactivation of the hypothalamo-pituitary-adrenal axis, in conjunction with amygdala activation, leads to increased sympathetic tone, which promotes the release of cytokines from macrophages, in addition to the release of cytokines from the ischaemic brain. Increases in pro-inflammatory cytokines (IL-1beta, IL-6, and IL-18) and tumour necrosis factor-alpha have been associated with (i) activation of indoleamine 2,3-dioxygenase which metabolizes tryptophan to kynurenin, thus depleting serotonin in paralimbic regions such as the ventral lateral frontal cortex, polar temporal cortex and basal ganglia and precipitating the onset of depression [88, 90]; (ii) loss of sensitivity of insulin and glucocorticoid receptors, respectively, which further perpetuates metabolic and neuroendocrine dysregulation. It may also compromise neurotrophic support and monoaminergic neurotransmission.
Hippocampal volume loss is characteristic of elderly or chronically ill samples and may be impacted by the Val66Met BDNF gene variant (see above) and the 5-HTTLPR SLC6A4 polymorphism.
Human imaging studies
Neuroimaging markers of cellular function, functional and structural abnormalities have been found in prefrontal and limbic structures after major depression [91–93]. We have tempted to summarize in a table the main and more consistent effects found in the literature like the reduction in volume of these structures. Neuroimaging of MAO-A binding, 5-HT2A, 5-HTT, 5-HT1A and 5-HT1B receptors binding, levels of dopamine transporters D1 and D2 have been investigated and elevated MAO-A binding with [11C]harmine PET imaging has been found inprefrontal and anterior cingulate cortex [92]. Also increased or decreased basal activity were reported (Table 1). For example, a hyperactivity of the ventromedial prefrontal cortex (was associated with enhanced sensitivity to pain, anxiety, depressive ruminations and tension), whereas a hypoactivity of the dorsolateral prefrontal cortex may produce psychomotor retardation, apathy and deficits in attention and working memory. More precisely, depression was associated with increases in hippocampal activity and decreases in posterior cingulate and prefrontal cortex activity in 17 patients [94]. After 6 weeks of treatment, patients who had responded to treatment showed a reversal of this pattern whereas non-responders continued to show abnormalities. Remission was also associated with decreasing ventral frontal lobe metabolism, ventral anterior cingulate gyrus (AC) and anterior insula activity, and increasing dorsolateral prefrontal cortex metabolism [95]. It has been postulated that ACC or amygdala activity was predictive of clinical response to antidepressant medication [94, 96]. Using diffusion tensor imaging, it was found that fractional anisotropy was significantly lower and apparent diffusion coefficient was significantly higher in white matter tracts that connect to the emotional regulation in depressed patients: prefrontal lobe, frontal lobe and limbic structures.
Table 1.
Main effects of depression on structural and basal activity of brain regions. Summarized from [93, 94]
| Volume | Basal activity | Basal activity on Remission | Prediction | Functional activity | Functional activity on remission | MAO-A binding* | |
|---|---|---|---|---|---|---|---|
| Prefrontal cortex | ↗ | ||||||
| Ventromedial | ↘ | ↗ | ↘ | ||||
| Lateral orbital | ↘ | ↗ | |||||
| Dorsolateral | ↘ | ↗ | + | ||||
| Anterior cingulate cortex (SGPFC) | ? | + | ↘ | ↗ | ↗ | ||
| Posterior cingulate cortex | ↘ | ↗ | |||||
| Nucleus accumbens | ↗ | ↘ | |||||
| Amygdala | ↗beginning ↘end | + | ↗ | ↘ | |||
| Hippocampus | ↘ | ↗ | ↘ | ↗ |
Monoamine oxidase-A (MAO-A) [92].
Functional imaging
Using fMRI paradigms, hypoactivity to fearful faces in the rostral ACC (rACC) and hyperactivity in the limbic regions [97–99, 102, 103] have been found in depressed patients. Normalization of these abnormalities was correlated with remission of depressive symptoms (Table 1). Connectivity fMRI studies have also suggested a decrement in the ‘communication’ between amygdala and ACC regions [103] that could lead to a failure of the ACC to serve its inhibitory role in emotional regulation [104]. Functional connectivity of the resting state yielded several results. Compared with controls, in depressed individuals each of the following three networks (the cognitive control network, the default mode network and the affective network) was shown to have increased connectivity to the same bilateral dorsal medial prefrontal cortex region, an area that was termed the dorsal nexus [98]. This further suggests that reducing increased connectivity of the dorsal nexus presents a potential therapeutic target. Finally, refractory depression was associated with disrupted functional connectivity mainly in thalamo-cortical circuits, whereas non-refractory depression was associated with more distributed decreased connectivity in the limbic-striatal-pallidal-thalamic circuit [105].
Conclusions
Depression is the most frequent non-cognitive neuropsychiatric complication of brain ischaemia, affecting up to 50% of all such patients. PSD is associated with increased morbidity and mortality following stroke. Despite its great clinical relevance, there is as yet little insight into the underlying neurobiological mechanisms. PSD has remained under-researched and, in particular, suitable animal models have not been established in sufficient depth or number. As PSD negatively interferes with functional outcome, it is of crucial importance to better understand its pathophysiology and target PSD by therapeutic interventions. The authors hope that this review will contribute to closing this research gap by spurring further experimental studies in this important field.
Acknowledgments
APW was supported was supported by the European Union's Seventh Framework Programme (FP7/2007–2013) under grant agreement no 229750. LK was supported by the European Union's Seventh Framework Programme (FP7/2007–2013) under grant agreements no 201024 and no 202213 (European Stroke Network/ARISE).
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Hachinsky V. Post-stroke depression, not to be underestimated. Lancet. 1999;353:1728. doi: 10.1016/S0140-6736(99)00139-7. [DOI] [PubMed] [Google Scholar]
- 2.Robinson RG, Bolduc PL, Price TR. Two-year longitudinal study of poststroke mood disorders: diagnosis and outcome at one and two years. Stroke. 1987;18:837–43. doi: 10.1161/01.str.18.5.837. [DOI] [PubMed] [Google Scholar]
- 3.Whyte EM, Mulsant BH, Vanderbilt J, et al. Depression after stroke: a prospective epidemiological study. J Am Geriatr Soc. 2004;52:774–8. doi: 10.1111/j.1532-5415.2004.52217.x. [DOI] [PubMed] [Google Scholar]
- 4.Robinson RG, Spalletta G. Poststroke depression: a review. Can J Psychiatry. 2010;55:341–9. doi: 10.1177/070674371005500602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morris PL, Robinson RG, Andrzejewski P, et al. Association of depression with 10-year post-stroke mortality. Am J Psychiatry. 1993;150:124–9. doi: 10.1176/ajp.150.1.124. [DOI] [PubMed] [Google Scholar]
- 6.Downhill JE, Jr, Robinson RG. Longitudinal assessment of depression and cognitive impairment following stroke. J Nerv Ment Dis. 1994;182:425–31. doi: 10.1097/00005053-199408000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Paolucci S, Antonucci G, Pratesi L, et al. Poststroke depression and its role in rehabilitation of inpatients. Arch Phys Med Rehabil. 1999;80:985–90. doi: 10.1016/s0003-9993(99)90048-5. [DOI] [PubMed] [Google Scholar]
- 8.Gainotti G, Antonucci G, Marra C, et al. Relation between depression after stroke, antidepressant therapy, and functional recovery. J Neurol Neurosurg Psychiatry. 2001;71:258–61. doi: 10.1136/jnnp.71.2.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Williams LS, Ghose SS, Swindle RW. Depression and other mental health diagnoses increase mortality risk after ischemic stroke. Am J Psychiatry. 2004;161:1090–5. doi: 10.1176/appi.ajp.161.6.1090. [DOI] [PubMed] [Google Scholar]
- 10.Pohjasvaara T, Vataja R, Leppävuori A, et al. Depression is an independent predictor of poor long-term functional outcome post-stroke. Eur J Neurol. 2001;8:315–9. doi: 10.1046/j.1468-1331.2001.00182.x. [DOI] [PubMed] [Google Scholar]
- 11.Chemerinski E, Robinson RG, Kosier JT. Improved recovery in activities of daily living associated with remission of poststroke depression. Stroke. 2001;32:113–7. doi: 10.1161/01.str.32.1.113. [DOI] [PubMed] [Google Scholar]
- 12.Hackett ML, Yapa C, Parag V, et al. Frequency of depression after stroke: a systematic review of observational studies. Stroke. 2005;36:1330–40. doi: 10.1161/01.STR.0000165928.19135.35. [DOI] [PubMed] [Google Scholar]
- 13.Baumann K, Rotter M, Schippan B. Posttraumatic embitterment disorder in comparison to other mental disorders. Psychother Psychosom. 2008;77:50–6. doi: 10.1159/000110060. [DOI] [PubMed] [Google Scholar]
- 14.Folstein MF, Maiberger R, McHugh PR. Mood disorder as a specific complication of stroke. J Neurol Neurosurg Psychiatry. 1977;40:1018–20. doi: 10.1136/jnnp.40.10.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Biran I, Chatterjee A. Depression with anosognosia following a left subcortical stroke. Clin Neurol Neurosurg. 2003;105:99–101. doi: 10.1016/s0303-8467(02)00113-0. [DOI] [PubMed] [Google Scholar]
- 16.Angeleri F, Angeleri VA, Foschi N, et al. The influence of depression, social activity, and family stress on functional outcome after stroke. Stroke. 1993;24:1478–83. doi: 10.1161/01.str.24.10.1478. [DOI] [PubMed] [Google Scholar]
- 17.Castillo CS, Starkstein SE, Fedoroff JP, et al. Generalized anxiety disorder after stroke. J Nerv Ment Dis. 1993;181:100–6. doi: 10.1097/00005053-199302000-00005. [DOI] [PubMed] [Google Scholar]
- 18.Piamarta F, Iurlaro S, Isella V, et al. Unconventional affective symptoms and executive functions after stroke in the elderly. Arch Gerontol Geriatr Suppl. 2004;9:315–23. doi: 10.1016/j.archger.2004.04.042. [DOI] [PubMed] [Google Scholar]
- 19.Barker-Collo SL. Depression and anxiety 3 months post stroke: prevalence and correlates. Arch Clin Neuropsychol. 2007;22:519–31. doi: 10.1016/j.acn.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 20.Carod-Artal FJ, Ferreira Coral L, Trizotto DS, et al. Poststroke depression: prevalence and determinants in Brazilian stroke patients. Cerebrovasc Dis. 2009;28:157–65. doi: 10.1159/000226114. [DOI] [PubMed] [Google Scholar]
- 21.Rasmussen A, Lunde M, Poulsen DL, et al. A double-blind, placebo-controlled study of sertraline in the prevention of depression in stroke patients. Psychosomatics. 2003;44:216–21. doi: 10.1176/appi.psy.44.3.216. [DOI] [PubMed] [Google Scholar]
- 22.Niedermaier N, Bohrer E, Schulte K, et al. Prevention and treatment of poststroke depression with mirtazapine in patients with acute stroke. J Clin Psychiatry. 2004;65:1619–23. doi: 10.4088/jcp.v65n1206. [DOI] [PubMed] [Google Scholar]
- 23.Narushima K, Robinson RG. The effect of early versus late antidepressant treatment on physical impairment associated with poststroke depression: is there a time-related therapeutic window? J Nerv Ment Dis. 2003;191:645–52. doi: 10.1097/01.nmd.0000092197.97693.d2. [DOI] [PubMed] [Google Scholar]
- 24.Robinson RG, Jorge RE, Moser DJ, et al. Escitalopram and problem-solving therapy for prevention of poststroke depression: a randomized controlled trial. JAMA. 2008;299:2391–400. doi: 10.1001/jama.299.20.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Narushima K, Paradiso S, Moser DJ, et al. Effect of antidepressant therapy on executive function after stroke. Br J Psychiatry. 2007;190:260–5. doi: 10.1192/bjp.bp.106.025064. [DOI] [PubMed] [Google Scholar]
- 26.Acler M, Robol E, Fiaschi A, et al. A double blind placebo RCT to investigate the effects of serotonergic modulation on brain excitability and motor recovery in stroke patients. J Neurol. 2009;256:1152–8. doi: 10.1007/s00415-009-5093-7. [DOI] [PubMed] [Google Scholar]
- 27.Jorge RE, Acion L, Moser D, et al. Escitalopram and enhancement of cognitive recovery following stroke. Arch Gen Psychiatry. 2010;67:187–96. doi: 10.1001/archgenpsychiatry.2009.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pariente J, Loubinoux I, Carel C, et al. Fluoxetine modulates motor performance and cerebral activation of patients recovering from stroke. Ann Neurol. 2001;50:718–29. doi: 10.1002/ana.1257. [DOI] [PubMed] [Google Scholar]
- 29.Chollet F, Tardy J, Albucher JF, et al. Fluoxetine for motor recovery after acute ischaemic stroke (FLAME): a randomised placebo-controlled trial. Lancet Neurol. 2011;10:123–30. doi: 10.1016/S1474-4422(10)70314-8. [DOI] [PubMed] [Google Scholar]
- 30.Jorge RE, Robinson RG, Arndt S, et al. Mortality and poststroke depression: a placebo-controlled trial of antidepressants. Am J Psychiatry. 2003;160:1823–9. doi: 10.1176/appi.ajp.160.10.1823. [DOI] [PubMed] [Google Scholar]
- 31.Porsolt RD, Bertin A, Jalfre M. Behavioral despair in mice: a primary screening test for antidepressants. Arch Int Pharmacodyn Ther. 1977;229:327–36. [PubMed] [Google Scholar]
- 32.Steru L, Chermat R, Thierry B, et al. The tail suspension test: a new method for screening antidepressants in mice. Psychopharmacology. 1985;85:367–70. doi: 10.1007/BF00428203. [DOI] [PubMed] [Google Scholar]
- 33.Cryan JF, Mombereau C, Vassout A. The tail suspension test as a model for assessing antidepressant activity: review of pharmacological and genetic studies in mice. Neurosci Biobehav Rev. 2005;29:571–625. doi: 10.1016/j.neubiorev.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 34.Willner P, Muscat R, Papp M. Chronic mild stress-induced anhedonia: a realistic animal model of depression. Neurosci Biobehav Rev. 1992;16:525–34. doi: 10.1016/s0149-7634(05)80194-0. [DOI] [PubMed] [Google Scholar]
- 35.Yan B, He J, Xu H, et al. Quetiapine attenuates the depressive and anxiolytic-like behavioural changes induced by global cerebral ischemia in mice. Behav Brain Res. 2007;182:36–41. doi: 10.1016/j.bbr.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 36.Gaur V, Kumar A. Behavioral, biochemical and cellular correlates in the protective effect of sertraline against transient global ischemia induced behavioral despair: possible involvement of nitric oxide-cyclic guanosine monophosphate study pathway. Brain Res Bull. 2010;82:57–64. doi: 10.1016/j.brainresbull.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 37.Deplanque D, Venna VR, Bordet R. Brain ischemia changes the long term response to antidepressant drugs in mice. Behav Brain Res. 2011;219:367–72. doi: 10.1016/j.bbr.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 38.Winter B, Juckel G, Viktorov I, et al. Anxious and hyperactive phenotype following brief ischemic episodes in mice. Biol Psychiatry. 2005;57:1166–75. doi: 10.1016/j.biopsych.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 39.Craft TK, DeVries AC. Role of IL-1 in poststroke depressive-like behavior in mice. Biol Psychiatry. 2006;60:812–8. doi: 10.1016/j.biopsych.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 40.Kato M, Iwata H, Okamoto M, et al. Focal cerebral ischemia-induced escape deficit in rats is ameliorated by a reversible inhibitor of monoamine oxidase-a: implications for a novel animal model of post-stroke depression. Biol Pharm Bull. 2000;23:406–10. doi: 10.1248/bpb.23.406. [DOI] [PubMed] [Google Scholar]
- 41.Wang SH, Zhang ZJ, Guo YJ, et al. Hippocampal neurogenesis and behavioural studies on adult ischemic rat response to chronic mild stress. Behav Brain Res. 2008;189:9–16. doi: 10.1016/j.bbr.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 42.Huston JP, Schulz D, Topic B. Toward an animal model of extinction-induced despair: focus on aging and physiological indices. J Neural Transm. 2009;116:1029–36. doi: 10.1007/s00702-009-0210-4. [DOI] [PubMed] [Google Scholar]
- 43.Blaveri E, Kelly F, Mallei A, et al. Expression profiling of a genetic animal model of depression reveals novel molecular pathways underlying depressive-like behaviours. PLoS One. 2010;5:e12596. doi: 10.1371/journal.pone.0012596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eriksson TM, Delagrange P, Spedding M, et al. Emotional memory impairments in a genetic rat model of depression: involvement of 5-HT/MEK/Arc signaling in restoration. Mol Psychiatry. 2012;17:173–84. doi: 10.1038/mp.2010.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Uriguen L, Arteta D, Diez-Alarcia R, et al. Gene expression patterns in brain cortex of three different animal models of depression. Genes, Brain, and Behav. 2008;7:649–58. doi: 10.1111/j.1601-183X.2008.00402.x. [DOI] [PubMed] [Google Scholar]
- 46.Condamine T, Le Texier L, Howie D, et al. Tmem176B and Tmem176A are associated with the immature state of dendritic cells. J Leuk Biol. 2010;88:507–15. doi: 10.1189/jlb.1109738. [DOI] [PubMed] [Google Scholar]
- 47.Deng W, Aimone JB, Gage FH. New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat Rev. 2010;11:339–50. doi: 10.1038/nrn2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee S, Jeong J, Kwak Y, et al. Depression research: where are we now? Mol Brain. 2010;3:8. doi: 10.1186/1756-6606-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kowalczyk A, Filipkowski RK, Rylski M, et al. The critical role of cyclin D2 in adult neurogenesis. J Cell Biol. 2004;167:209–13. doi: 10.1083/jcb.200404181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jaholkowski P, Kiryk A, Jedynak P, et al. New hippocampal neurons are not obligatory for memory formation; cyclin D2 knockout mice with no adult brain neurogenesis show learning. Learn Mem. 2009;16:439–51. doi: 10.1101/lm.1459709. [DOI] [PubMed] [Google Scholar]
- 51.Liu J, Solway K, Messing RO, et al. Increased neurogenesis in the dentate gyrus after transient global ischemia in gerbils. J Neurosci. 1998;18:7768–78. doi: 10.1523/JNEUROSCI.18-19-07768.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang RL, Zhang ZG, Zhang L, et al. Proliferation and differentiation of progenitor cells in the cortex and the subventricular zone in the adult rat after focal cerebral ischemia. Neuroscience. 2001;105:33–41. doi: 10.1016/s0306-4522(01)00117-8. [DOI] [PubMed] [Google Scholar]
- 53.Jin K, Minami M, Lan JQ, et al. Neurogenesis in dentate subgranular zone and rostral subventricular zone after focal cerebral ischemia in the rat. Proc Natl Acad Sci USA. 2001;98:4710–5. doi: 10.1073/pnas.081011098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arvidsson A, Collin T, Kirik D, et al. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med. 2002;8:963–70. doi: 10.1038/nm747. [DOI] [PubMed] [Google Scholar]
- 55.Jin K, Sun Y, Xie L, et al. Directed migration of neuronal precursors into the ischemic cerebral cortex and striatum. Mol Cell Neurosci. 2003;24:171–89. doi: 10.1016/s1044-7431(03)00159-3. [DOI] [PubMed] [Google Scholar]
- 56.Yamashita T, Ninomiya M, Hernandez Acosta P, et al. Subventricular zone-derived neuroblasts migrate and differentiate into mature neurons in the post-stroke adult striatum. J Neurosci. 2006;26:6627–36. doi: 10.1523/JNEUROSCI.0149-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Luo CX, Jiang J, Zhou QG, et al. Voluntary exercise-induced neurogenesis in the postischemic dentate gyrus is associated with spatial memory recovery from stroke. J Neurosci Res. 2007;85:1637–46. doi: 10.1002/jnr.21317. [DOI] [PubMed] [Google Scholar]
- 58.Wang SH, Zhang ZJ, Guo YJ, et al. Involvement of serotonin neurotransmission in hippocampal neurogenesis and behavioral responses in a rat model of post-stroke depression. Pharmacol Biochem Behav. 2010;95:129–37. doi: 10.1016/j.pbb.2009.12.017. [DOI] [PubMed] [Google Scholar]
- 59.Yasuda S, Liang MH, Marinova Z, et al. The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol Psychiatry. 2009;14:51–9. doi: 10.1038/sj.mp.4002099. [DOI] [PubMed] [Google Scholar]
- 60.Shirayama Y, Chen AC, Nakagawa S, et al. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci. 2002;22:3251–61. doi: 10.1523/JNEUROSCI.22-08-03251.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Taliaz D, Loya A, Gersner R, et al. Resilience to chronic stress is mediated by hippocampal brain-derived neurotrophic factor. J Neurosci. 2011;31:4475–83. doi: 10.1523/JNEUROSCI.5725-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gersner R, Toth E, Isserles M, et al. Site-specific antidepressant effects of repeated subconvulsive electrical stimulation: potential role of brain-derived neurotrophic factor. Biol Psychiatry. 2010;67:125–32. doi: 10.1016/j.biopsych.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 63.Endres M, Fan G, Hirt L. Stroke damage in mice after knocking the neutrophin-4 gene into the brain-derived neurotrophic factor locus. J Cereb Blood Flow Metab. 2003;23:150–3. doi: 10.1097/01.WCB.0000043949.67811.C6. [DOI] [PubMed] [Google Scholar]
- 64.Jiang Y, Wei N, Lu T, et al. Intranasal brain-derived neurotrophic factor protects brain from ischemic insult via modulating local inflammation in rats. Neuroscience. 2011;172:398–405. doi: 10.1016/j.neuroscience.2010.10.054. [DOI] [PubMed] [Google Scholar]
- 65.Tong L, Perez-Polo R. Brain-derived neurotrophic factor (BDNF) protects cultured rat cerebellar granule neurons against glucose deprivation-induced apoptosis. J Neural Transm. 1998;105:905–14. doi: 10.1007/s007020050101. [DOI] [PubMed] [Google Scholar]
- 66.Chan JP, Cordeira J, Calderon GA, et al. Depletion of central BDNF in mice impedes terminal differentiation of new granule neurons in the adult hippocampus. Mol Cell Neurosci. 2008;39:372–83. doi: 10.1016/j.mcn.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Clarkson AN, Overman JJ, Zhong S, et al. AMPA receptor-induced local brain-derived neurotrophic factor signaling mediates motor recovery after stroke. J Neurosci. 2011;31:3766–75. doi: 10.1523/JNEUROSCI.5780-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ploughman M, Windle V, MacLellan CL, et al. Brain-derived neurotrophic factor contributes to recovery of skilled reaching after focal ischemia in rats. Stroke. 2009;40:1490–5. doi: 10.1161/STROKEAHA.108.531806. [DOI] [PubMed] [Google Scholar]
- 69.Zhou Z, Lu T, Xu G, et al. Decreased serum brain-derived neurotrophic factor (BDNF) is associated with post-stroke depression but not with BDNF gene Val66Met polymorphism. Clin Chem Lab Med. 2011;49:185–9. doi: 10.1515/CCLM.2011.039. [DOI] [PubMed] [Google Scholar]
- 70.Yang L, Zhang Z, Sun D, et al. Low serum BDNF may indicate the development of PSD in patients with acute ischemic stroke. Int J Ger Psychiatry. 2011;26:495–502. doi: 10.1002/gps.2552. [DOI] [PubMed] [Google Scholar]
- 71.Kim JM, Stewart R, Kim SW, et al. BDNF genotype potentially modifying the association between incident stroke and depression. Neurobiol Aging. 2008;29:789–92. doi: 10.1016/j.neurobiolaging.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 72.Sinyor D, Amato P, Kaloupek DG, et al. Post-stroke depression: relationships to functional impairment, coping strategies, and rehabilitation outcome. Stroke. 1986;17:1102–7. doi: 10.1161/01.str.17.6.1102. [DOI] [PubMed] [Google Scholar]
- 73.Carson AJ, MacHale S, Allen K, et al. Depression after stroke and lesion location: a systematic review. Lancet. 2000;356:122–6. doi: 10.1016/S0140-6736(00)02448-X. [DOI] [PubMed] [Google Scholar]
- 74.Robinson RG, Bloom FE. Pharmacological treatment following experimental cerebral infarction: Implications for understanding psychological symptoms of human stroke. Biol Psychiatry. 1977;12:669–80. [PubMed] [Google Scholar]
- 75.Dihné M, Grommes C, Lutzenburg M, et al. Different mechanisms of secondary neuronal damage in thalamic nuclei after focal cerebral ischemia in rats. Stroke. 2002;33:3006–11. doi: 10.1161/01.str.0000039406.64644.cb. [DOI] [PubMed] [Google Scholar]
- 76.De Bilbao F, Guarin E, Nef P, et al. Cell death is prevented in thalamic fields but not in injured neocortical areas after permanent focal ischaemia in mice overexpressing the anti-apoptotic protein Bcl-2. Eur J Neurosci. 2000;12:921–34. doi: 10.1046/j.1460-9568.2000.00984.x. [DOI] [PubMed] [Google Scholar]
- 77.Yan T, Feng Y, Zhai Q. Axon degeneration: mechanisms and implications of a distinct program from cell death. Neurochem Int. 2010;56:529–34. doi: 10.1016/j.neuint.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 78.Snaphaan L, van der Werf S, Kanselaar K, et al. Post-stroke depressive symptoms are associated with post-stroke characteristics. Cerebr Dis (Basel, Switzerland) 2009;28:551–7. doi: 10.1159/000247598. [DOI] [PubMed] [Google Scholar]
- 79.Tang WK, Chen YK, Lu JY, et al. Cerebral microbleeds and symptom severity of post-stroke depression: A magnetic resonance imaging study. J Affect Disord. 2010;129:354–8. doi: 10.1016/j.jad.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 80.Alexopoulos GS, Meyers BS, Young RC, et al. ‘Vascular depression’ hypothesis. Arch Gen Psychiatry. 1997;54:915–22. doi: 10.1001/archpsyc.1997.01830220033006. [DOI] [PubMed] [Google Scholar]
- 81.Lim JS, Kwon HM. Risk of “silent stroke” in patients older than 60 years: risk assessment and clinical perspectives. Clin Interv Aging. 2010;5:239–51. doi: 10.2147/cia.s7382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Santos M, Kovari E, Gold G, et al. The neuroanatomical model of post-stroke depression: towards a change of focus? J Neurol Sci. 2009;283:158–62. doi: 10.1016/j.jns.2009.02.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature. 2008;455:894–902. doi: 10.1038/nature07455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nestler EJ, Carlezon WA., Jr The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. 2006;59:1151–9. doi: 10.1016/j.biopsych.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 85.Mineur YS, Picciotto MR. Nicotine receptors and depression: revisiting and revising the cholinergic hypothesis. Trends in Pharmacol Sci. 2010;31:580–6. doi: 10.1016/j.tips.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chung YC, Kim SR, Jin BK. Paroxetine prevents loss of nigrostriatal dopaminergic neurons by inhibiting brain inflammation and oxidative stress in an experimental model of Parkinson's disease. J Immunol. 2010;185:1230–7. doi: 10.4049/jimmunol.1000208. [DOI] [PubMed] [Google Scholar]
- 87.Chung YC, Kim SR, Park JY, et al. Fluoxetine prevents MPTP-induced loss of dopaminergic neurons by inhibiting microglial activation. Neuropharmacology. 2011;60:963–74. doi: 10.1016/j.neuropharm.2011.01.043. [DOI] [PubMed] [Google Scholar]
- 88.Lim CM, Kim SW, Park JY, et al. Fluoxetine affords robust neuroprotection in the postischemic brain via its anti-inflammatory effect. J Neurosci Res. 2009;87:1037–45. doi: 10.1002/jnr.21899. [DOI] [PubMed] [Google Scholar]
- 89.Badan I, Buchhold B, Popa-Wagner A. Accelerated glial reactivity to stroke in aged rats correlates with reduced functional activity. J Cereb Blood Flow Metab. 2003;23:845–54. doi: 10.1097/01.WCB.0000071883.63724.A7. [DOI] [PubMed] [Google Scholar]
- 90.Spalletta G, Bossu P, Ciaramella A, et al. The etiology of poststroke depression: a review of the literature and a new hypothesis involving inflammatory cytokines. Mol Psychiatry. 2006;11:984–91. doi: 10.1038/sj.mp.4001879. [DOI] [PubMed] [Google Scholar]
- 91.Meyer JH. Neuroimaging markers of cellular function in major depressive disorder: implications for therapeutics, personalized medicine, and prevention. Clin Pharmacol Ther. 2012;91:201–14. doi: 10.1038/clpt.2011.285. [DOI] [PubMed] [Google Scholar]
- 92.Lorenzetti V, Allen NB, Fornito A, et al. Structural brain abnormalities in major depressive disorder: a selective review of recent MRI studies. J Affect Disord. 2009;117:1–17. doi: 10.1016/j.jad.2008.11.021. [DOI] [PubMed] [Google Scholar]
- 93.Eker C, Gonul AS. Volumetric MRI studies of the hippocampus in major depressive disorder: meanings of inconsistency and directions for future research. World J Biol Psychiatry. 2010;11:19–35. doi: 10.1080/15622970902737998. [DOI] [PubMed] [Google Scholar]
- 94.Mayberg HS, Brannan SK, Tekell JL, et al. Regional metabolic effects of fluoxetine in major depression: serial changes and relationship to clinical response. Biol Psychiatry. 2000;48:830–43. doi: 10.1016/s0006-3223(00)01036-2. [DOI] [PubMed] [Google Scholar]
- 95.Brody AL, Saxena S, Mandelkern MA, et al. Brain metabolic changes associated with symptom factor improvement in major depressive disorder. Biol Psychiatry. 2001;50:171–8. doi: 10.1016/s0006-3223(01)01117-9. [DOI] [PubMed] [Google Scholar]
- 96.DeRubeis RJ, Siegle GJ, Hollon SD. Cognitive therapy versus medication for depression: treatment outcomes and neural mechanisms. Nature Rev. 2008;9:788–96. doi: 10.1038/nrn2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Whalen PJ, Johnstone T, Somerville LH. A functional magnetic resonance imaging predictor of treatment response to venlafaxine in generalized anxiety disorder. Biol Psychiatry. 2008;63:858–63. doi: 10.1016/j.biopsych.2007.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sheline YI, Barch DM, Donnelly JM, et al. Increased amygdala response to masked emotional faces in depressed subjects resolves with antidepressant treatment: an fMRI study. Biol Psychiatry. 2001;50:651–8. doi: 10.1016/s0006-3223(01)01263-x. [DOI] [PubMed] [Google Scholar]
- 99.Chen CH, Ridler K, Suckling J, et al. Brain imaging correlates of depressive symptom severity and predictors of symptom improvement after antidepressant treatment. Biol Psychiatry. 2007;62:407–14. doi: 10.1016/j.biopsych.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 100.Wu F, Tang Y, Xu K, et al. Whiter matter abnormalities in medication-naive subjects with a single short-duration episode of major depressive disorder. Psychiatry Res. 2011;191:80–3. doi: 10.1016/j.pscychresns.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Blood AJ, Iosifescu DV, Makris N, et al. Microstructural abnormalities in subcortical reward circuitry of subjects with major depressive disorder. PLoS One. 2010;5:e13945. doi: 10.1371/journal.pone.0013945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cullen KR, Klimes-Dougan B, Muetzel R, et al. Altered white matter microstructure in adolescents with major depression: a preliminary study. J Am Acad Child Adolesc Psychiatry. 2010;49:173–83. doi: 10.1097/00004583-201002000-00011. e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Anand A, Li Y, Wang Y, et al. Activity and connectivity of brain mood regulating circuit in depression: a functional magnetic resonance study. Biol Psychiatry. 2005;57:1079–88. doi: 10.1016/j.biopsych.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 104.Whittle S, Allen NB, Lubman DI, et al. The neurobiological basis of temperament: towards a better understanding of psychopathology. Neurosci Biobehav Rev. 2006;30:511–25. doi: 10.1016/j.neubiorev.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 105.Lui S, Wu Q, Qiu L, et al. Resting-state functional connectivity in treatment-resistant depression. Am J Psychiatry. 2011;168:642–8. doi: 10.1176/appi.ajp.2010.10101419. [DOI] [PubMed] [Google Scholar]