

Abstract
Sound data support the concept that in atherosclerosis, inflammation and dyslipidemia intersect each other and that irrespective of the initiator, both participate from the early stages to the ultimate fate of the atheromatous plaque. The two partakers manoeuvre a vicious circle in atheroma formation: dyslipidaemia triggers an inflammatory process and inflammation elicits dyslipidaemia. Independent of the initial cause, the atherosclerotic lesions occur focally, in particular arterial-susceptible sites, by a process that, although continuous, can be arbitrarily divided into a sequence of consecutive stages that lead from fatty streak to the fibro-lipid plaque and ultimately to plaque rupture and thrombosis. In the process, the initial event is a change in endothelial cells (EC) constitutive properties. Then, the molecular alarm signals send by dysfunctional EC are decoded by specific blood immune cells (monocytes, T lymphocytes, neutrophils, mast cells) and by the resident vascular cells, that respond by initiating a robust inflammatory process, in which the cells and the factors they secrete hasten the atheroma development. Direct and indirect crosstalk between the cells housed within the nascent plaque, complemented by the increase in risk factors of atherosclerosis lead to atheroma development and outcome. The initial inflammatory response can be regarded as a defense/protective reaction mechanism, but its further amplification, speeds up atherosclerosis. In this review, we provide an overview on the role of inflammation and dyslipidaemia and their intersection in atherogenesis. The data may add to the foundation of a novel attitude in the diagnosis and treatment of atherosclerosis.
Keywords: inflammation, dyslipidaemia, cardiovascular diseases, atherosclerosis, lipoproteins, endothelial dysfunction
-
Dyslipidaemia and inflammation: partake in atherosclerosis
– Dyslipidaemia induces vascular cell dysfunctions
– Inflammation alone may generate atheroma
-
Irrespective of the initial cause atheroma develops in specific arterial-susceptible areas
– Disturbed blood flow
– Endothelial cell phenotype
-
Consecutive stages delineate atheroma development and fate
– Stage I. Endothelial cell activation/modulation of constitutive functions: the initial event in atheroma formation
– Amendments in EC controlled permeability
– Changes in endothelial cell phenotype
– Stage II. Endothelial cell dysfunction
– Cell adhesion molecules
– Cytokines and chemokines
– Stage III. Recruitment of blood immune cells and commencement of a robust inflammatory reaction
– Monocytes/macrophages
– T lymphocytes
– Dendritic cells (DC)
– Platelets
– Polymorphonuclear neutrophils (PMN)
– Mast cells
– B cells
– Smooth muscle cells
– Stage IV. SMC-key participants to fibrous plaque formation
– Stage V. Resident and immune cells and the factors they secrete generate a calcified fibro-lipid plaque
– Stage VI. The unstable fibro-lipid plaque: rupture and thrombosis
Inflammatory cells and pro-inflammatory molecules are therapeutic targets in atherosclerosis
Inflammation and dyslipidaemia partake to a vicious circle in atheroma formation
Conclusions and Perspectives
Dyslipidaemia and inflammation: partake in atherosclerosis
Atherosclerosis is an inflammatory disorder. Dyslipidemia induces atherosclerosis. Both assertions are supported by strong evidence and, what is more, they reveal that in atherosclerosis inflammation and dyslipidaemia intersect each other. Atherosclerosis is a complex multifactorial, multigenic disease, which may result from either a lipid disorder and/or an inflammatory process, having as ultimate outcome, the atheromatous plaque, a focal lesion located within the intima of small and medium size arteries. The progressive process of atheroma formation is initiated by aggressive risk factors like dyslipidaemia, pro-inflammatory cytokines, hypertension, products of glycoxidation associated with diabetes, and others.
Dyslipidaemia induces vascular cell dysfunctions
Changes in plasma homoeostasis like hypercholesterolaemia, is one of the major risk factors in atherosclerosis. It affects initially the endothelial cells (EC), which upon activation express new adhesion molecules and chemotactic factors that provoke an inflammatory process. The latter involves the recruitment of circulating immune cells that aggravates and accelerates the development of atheroma. The initial optimistic view that correcting dyslipidaemia will eradicate atherosclerosis failed, as extensive evidence revealed that inflammation alone could be an instigator or a key contributor to all stages of this disease, from the initial lesion to the ruptured plaque [1].
Inflammation alone may generate atheroma
About 50% of patients develop atherosclerosis in the absence of systemic hypercholesterolaemia. Putative antigens, heat shock proteins, components of plasma lipoproteins (Lp), and various microbial structures induce an inflammatory process that on its own may generate the atherosclerotic plaque formation [2].
Low-grade systemic inflammation as revealed by the increase in the stable plasma biomarkers (i.e. the C-reactive protein, a circulating pentraxin) was consistently associated with the recurrent risk of cardiovascular events in patients with stable angina and established cardiovascular disease (CVD) [3]. Interestingly, the atherosclerotic lesions occur focally, in particular arterial-susceptible sites.
Irrespective of the initial cause atheroma develops in specific arterial-susceptible areas
Although the entire vascular tree is evenly exposed to systemic risk factors (i.e. hyperlipidaemia, hyperglycaemia, hypertension, chronic infections, or genetic predisposition), atherosclerosis develops preferentially at specific arterial sites, such as branch points, outer wall of bifurcations, inner wall of curvatures, and cardiac valves [4, 5] characterized by variations in shear stress or flow disturbances. The existence of structural- functional variations of EC phenotype along the vascular tree [6] was substantiated elegantly by genomic and proteomic analysis [7].
Disturbed blood flow
Blood pressure-derived tensile stress and in particular, flow generated endothelial shear stress (ESS) shifts the EC functions and structure towards an atherosclerotic phenotype. Low ESS, reduces NO-dependent athero-protection and enhances the uptake and permeability of low–density lipoprotein (LDL). ESS promotes oxidative stress, and inflammation in EC by a process dependent on activation of the transcription factor nuclear factor kappa- B (NF-kB) (reviewed in [8]) and activation of sterol regulatory elements binding proteins (SREBPs) a family of endoplasmic reticulum (ER)–bound transcription factors that up-regulates the expression of genes encoding LDL receptors, cholesterol synthase, and fatty acid synthase [9].
Endothelial cell phenotype
Genomic analysis revealed that in arterial regions with non-disturbed flow and ESS within physiological range, EC express various athero-protective genes and suppress several pro-atherogenic genes, generating stability and quiescence of the area [6, 10, 11]. Conversely, in regions with low and disturbed flow and low ESS, the athero-protective genes are suppressed, whereas the pro-atherogenic genes are up-regulated, thereby promoting atherosclerosis [12, 13]. Moreover, EC gene and protein expressions differ significantly at predictable sites for the development of atherosclerosis compared with athero-resistant locations: differential transcriptional, translational and posttranslational phenotypes including sensitization of inflammatory regulators, coagulation, redox balance, ER stress, unfolded protein response (UPR) and microRNA (miRNA) heterogeneity have been identified [6, 7, 11, 14–16]. Hence, in athero-susceptible sites, the EC are primed or sensitized for atherogenesis, but additional risk factors are required to initiate the disease.
Consecutive stages delineate atheroma development and fate
Atheroma development is a gradual and continuing process; however, the progressive implication of vascular resident and non-resident cells and their secretory products define an arbitrary sequence of consecutive stages that lead from the fatty streak to fibro-lipid plaque, and ultimately to plaque rupture and atherothrombosis (Fig. 1). Inflammation is implicated in all stages of atherosclerotic lesion evolution as described below.
Fig 1.
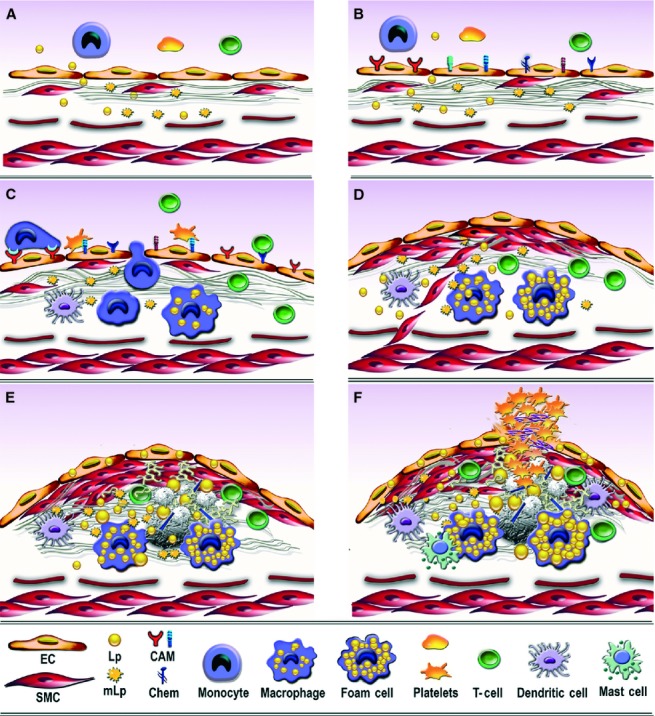
Consecutive arbitrary stages occurring in the development of atherosclerotic lesion in arterial lesion-prone areas. (A) Stage I. Endothelial cell activation/modulation of constitutive functions. The initial stage in atheroma formation in dyslipidaemia consists in endothelial cell (EC) increased transcytosis of plasma lipoproteins (Lp) and their housing in the subendothelium and a switch of the cells to a secretory phenotype responsible for the development of a hyperplasic basal lamina. Within the subendothelium, Lp, especially LDL by interaction with extracellular matrix components, changes its attributes becoming the highly atherogenic, oxidized modified lipoproteins (mLp). (B) Stage II. EC dysfunction. Affected on both sides, luminal by alterations of plasma homoeostasis and abluminal by the accrual of mLp, the EC initiate an inflammatory process manifested by the expression of new or more cell adhesion molecules, cytokines and chemokines (CAM and Chem), an indication of endothelial dysfunction. (C) Stage III. Recruitment of blood immune cells and commencement of a robust inflammatory reaction. Blood monocytes and T cells adhere to activated/dysfunctional EC and undergo directed diapedesis into the intima. Adhered platelets assist leucocytes migration. Within the intima, monocytes become activated macrophages that express scavenger receptors, which function in the uptake of mLp and the formation of foam cells that secrete a variety of proinflammatory mediators. Lymphocytes switch to activated pro-inflammatory (Th 1) and anti-inflammatory (Th 2 and TREG) cells that secrete cytokines and chemokines. The direct or indirect crosstalk between resident and migrated cells within the intima dictates the lesion progression. Activated dendritic cells contribute to T cells recruitment and activation within the plaque. (D) Stage IV. SMC-key participants to fibrous plaque formation. The proliferation of intima-resident SMC and of SMC migrated from the media to the intima leads to the formation of a fibrous cap that is accompanied by increased synthesis of extracellular matrix components. (E) Stage V. Resident and immune cells and the factors they secrete generate a calcified fibro-lipid plaque. SMC-, macrophages-derived foam cells, apoptotic cells-derived lipids and calcification centres form a lipid loaded necrotic core rich in cholesterol crystals. (F) Stage VI. The unstable fibro-lipid plaque: rupture and thrombosis EC apoptosis and erosion, thinning of the fibrous cap, cell apoptosis, macrophages, dendritic and mast cells-secreted pro-inflammatory mediators generate the physical rupture of the plaque. This ends in direct contact between tissue factors and circulating platelets and blood coagulation components triggering the thrombus formation that may partially or totally imped the blood flow leading to either myocardial infarction or stroke.
Stage I. Endothelial cell activation/modulation of constitutive functions: the initial event in atheroma formation
In physiological conditions, vascular EC monitor vessel-wall permeability, maintain a non-thrombogenic interface and a non-adhesive (negatively charged) surface for circulating blood cells by sequestering and suppressing transcription of molecules involved in EC-leucocyte interactions. Subtle changes in the microenvironment, within either the blood or interstitia, trigger initially the EC adaptation through modulation of their constitutive functions [17].
Amendments in EC controlled permeability
In experimental and human atherosclerosis, we and others found that the increase in plasma LDL generates a concentration gradient that induces in arterial lesion-susceptible areas, a prominent augmented transcytosis [4] that in conjunction with the reduced efflux [18] concur to the intimal trapping of LDL within and outside the subendothelial basal lamina (Fig. 1A). The intima-confined LDL interacts with matrix proteins, especially proteoglycans, that participate to their atherogenic conversion into oxidatively modified Lp (mLp) [4, 19, 20] that ultrastructurally appear as heterogeneous vesiculated particles (Fig. 2A) rich in unesterified cholesterol [4, 19, 20]. The atherogenic modifications of LDL may take place either within the plasma, when crossing the EC or within the subendothelial extracellular matrix (ECM); alternatively, it may occur, to different degree, in all these locations [21, 22].
Fig 2.
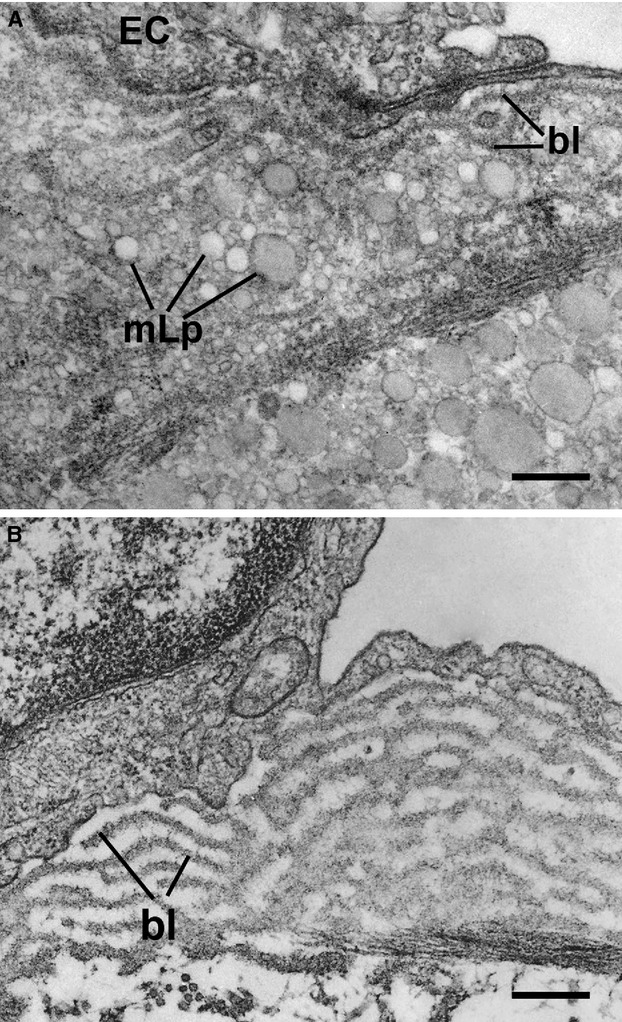
Ultrastructural evidence of the initial endothelial cell (EC) changes occurring in hamster atheroma formation. (A) Accumulation beneath the activated EC of modified lipoproteins (mLp) under and within the basal lamina (bl). Bar: 0.1 μm. The mLp appear as a heterogeneous population of vesiculated, aggregated, or fused particle (arrows). (B) Hyperplasia of EC basal lamina (bl) as a result of the switch of the endothelium to a secretory phenotype; the bl appears in multiple interconnected rows. Bar: 0.1 μm.
Changes in endothelial cell phenotype
Concurrently with increased transcytosis and subendothelial retention of mLp, the EC switch to a secretory phenotype, having as outcome the development of a hyperplasic multilayered basal lamina (Fig. 2B). The extreme proliferation of ECM, hinders the communications through myo-endothelial junctions and via gap junctions between neighbouring smooth muscle cells (SMC) generating an altered response of the vessel wall to external stimuli. The reduction of the EC net negative surface charge, particularly evident in long-term hyperlipidaemia, may account, in part, for the augmented adhesive characteristics of the cells in specific locations [23].
All the above changes are the attributes of an ‘activated’ EC, a generic name that entails a gradual instalment of several modifications that are dependent on the extent and intensity of the insults and prelude further cell dysfunction [17].
Stage II. Endothelial cell dysfunction
Alteration of plasma lipid homoeostasis and the subendothelial accrual of mLp act as warning signals that initiate a defence reaction, manifested by stimuli-generated new EC properties that further trigger a multipart inflammatory process. Thus, in early human and experimental atherosclerosis, the EC plasmalemma expresses new or more cell adhesion molecules (CAM) and synthesize chemokines and cytokines that assist in the recruitment of specific blood inflammatory cells (Fig. 1B).
Cell adhesion molecules
The activated EC express vascular cell adhesion molecule (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), E-selectin, P-selectin, and fractalkine (CX3CL1) that bind to the cognate receptors on leucocytes and platelets triggering their selective recruitment and adherence to the endothelium. Other CAM are implicated in leucocytes transmigration through the EC. Thus, PECAM-1 (CD 31), is expressed at high density on the lateral borders of EC in athero-prone areas, and in the neovessel regions of the human atherosclerotic plaques [24]; their role in the development of atheroma was revealed in knockout mice [25]. Endothelial junctional adhesion molecules (JAMs) are found at high level in atherosclerotic Apoe −/− mice and in atherosclerotic plaques of cardiovascular patients [26]. JAM A and JAM C are involved in the specific recruitment of monocytes and T cells into arteries [27, 28]. Endothelial cell-selective adhesion molecule (ESAM), have been specifically localized to EC tight junctions and in activated platelets [29]. In humans, soluble ESAM, was independently associated with coronary and peripheral atherosclerosis as well as with increased vascular stiffness [30].
Tetraspanins are integral membrane proteins that by interaction with other CAM form microdomains that contribute to the enhancement of receptor signalling, migration and homotypic and heterotypic cell-cell adhesion [31]. In live primary EC, tetraspanin microdomains organize specialized adhesion platforms (distinct from the membrane lipid rafts) in which ICAM-1, VCAM-1, JAM A, PECAM-1, ICAM-2 or CD 44 cluster with tetraspanin functioning in fast kinetics and efficient leucocyte extravasation [31]. Endothelial tetraspanin CD 81 is up-regulated in the initial stage of atherosclerosis and via clustering of ICAM-1 and VCAM-1, enhances substantially monocyte adhesion; the protein was suggested as a possible diagnostic and therapeutic marker of atherogenesis in humans [32].
Cytokines and chemokines
In response to inflammatory stimuli activated EC synthesize a large array of cytokines and chemokines (Fig. 3) that modulate leucocyte recruitment and adhesion [33].
Fig 3.
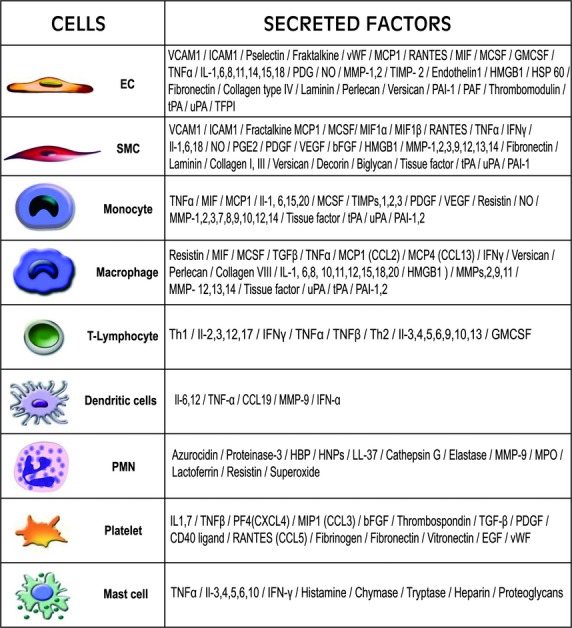
Diagrammatic representation of the implicated vascular resident and recruited cells and the factors they secrete in the course of atheroma development.
The chemokines (chemotactic cytokines) MCP-1 (CCL2) (monocytes chemotactic protein-1), RANTES(CCL5) (regulated on activation of normal T cells expressed and secreted), interleukin-8/Il-8 (CXCL8), fractalkine (CX3CL1), expressed by EC and their cognate leucocyte receptors (CCR2, CCR1, CCR5, CXCR2 and CX3CR1 respectively) promote leucocyte adhesion in early atherosclerosis [reviewed in [34]. Interestingly, CX3CL1 has a dual role acting as both a cell adhesion molecule and a chemokine, mediating direct capture, firm adhesion and transmigration of leucocytes [35]. EC express CX3CL1 and its receptor, CX3CR1 [36], both being increased in the human atherosclerotic plaques. The expression of CX3CL1 is up-regulated by pro-inflammatory mediators. Recently we reported that in human EC, the expression of CX3CL1 is induced by resistin, a cytokine assumed to be primarily involved in inflammation in humans [37]. As resistin is present and considered a marker of human atherosclerosis [38] and up-regulates CX3CL1 [37] it is safe to consider that resistin and fractalkine constitute molecular links between chronic inflammation and atherosclerosis. Recently, CX3CL1 was proposed as an early endothelial marker of atherosclerosis by a global gene expression profile study of human arterial EC [13]. This suggests that the CX3CL1/CX3CR1 axis is a promising therapeutic target in atherosclerosis [39]. The EC associated chemokine, IP10/CXCL10 predominates together with CX3CL1 in early human atherosclerotic lesions. Nevertheless, CX3CL1 is nominated as a more suitable early marker of activated EC being anchored in the cell plasmalemma, whereas IP 10 is an endothelial glycocalix–bound cytokine [13].
Lately, it was shown that chemokines are engaged in heterophilic interactions (interactome) forming functional complexes as is the case of platelets CCL5 and CXCL4 heteromerization that function in atherosclerotic lesion progression [40]. The concept of heterophilic chemokine interactome could explain the functional diversity and plasticity of chemokines and may assist the development of new therapeutic strategies for selective targeting [41].
Stage III. Recruitment of blood immune cells and commencement of a robust inflammatory reaction
Molecular alarm signals sent by dysfunctional EC are decoded by specific immune cells (monocytes, T lymphocytes, neutrophils, mast cells) and by the resident vascular cells (SMC, dendritic cells), that respond by initiating the inflammatory process, which has a major contribution to the escalation of the atheromatous plaque formation and fate (Fig. 1C).
Monocytes/macrophages
Monocytes subsets: Human peripheral blood monocytes are heterogeneous in terms of size, granularity, nuclear morphology and phenotype [42]. On the basis of the expression of CD14 and CD16 receptors, three subsets of monocytes were defined in humans: the ‘classical’ monocytes (CD14++CD16−) which represent ∼90% of all monocytes in normal conditions, the intermediate monocytes (CD14++CD16+) and the non-classical monocytes (CD14+CD16++) [43]. These subsets differ in many respects, including the expression of adhesion molecules and chemokine receptors [44]. The CD14++CD16+ monocytes express CCR2, CD62L and CD64 whereas the CD14+CD16++ cells lack CCR2 and have higher level of major histocompatibility complex II and CD32. Although both subsets express CX3CR1, the expression is higher in CD14+CD16++ monocytes. The role of each subset in human atherosclerosis is debated. There are indications that patients with coronary artery disease have significantly higher number of CD14+CD16++ monocytes than healthy controls and their counts correlate negatively with the concentration of HDL and positively with the level of atherogenic lipids. There is a consensus that this subset is associated with the production of high levels of TNF-alpha [45]. However, other studies indicate that the inflammatory genes and surface markers are down-regulated in monocytes of patients with coronary artery disease [46]. The exact role of CD14+CD16+ and CD14+CD16++ monocytes in human atherosclerosis remains to be established.
Recruitment of monocytes subsets in atherosclerotic plaque: The pathway for leucocyte recruitment to the growing plaque is from either the vascular lumen or trafficking through the vasa vasorum and neovessels. The latter are a common occurrence in the developing atheroma, appearing as immature, leaky microvessels, expressing leucocyte adhesion molecules and thus supporting leucocyte infiltration and determining plaque progression. Monocyte recruitment is a highly regulated process involving CAM, chemokines and their cognate receptors [reviewed in [47].
Monocyte diapedesis: Both EC and monocytes proactively contribute to diapedesis. Novel structures, termed ‘transmigratory cups’ or docking structures were described to function both in strengthening adhesion and in facilitating/guiding diapedesis of leucocytes [48, 49]. Moreover, tetraspanins are largely implicated in forming/stabilizing lateral protein-protein associations, and recent in vitro studies indicate that CD81 enhance monocyte adhesiveness in a transmigratory cup-dependent manner [32]. Within the plaque, activated monocytes secrete pro-inflammatory mediators (Fig. 3) and may differentiate into macrophages.
Macrophage subsets in atherosclerosis: Plaque macrophages are also heterogeneous expressing a continuum of pro- and anti-atherogenic programs (differentiation/activation) in response to environmental signals [50, 51]. The up-regulation of scavenger receptors in plaque-activated macrophages are operational in the uptake of mLp, which leads to their transformation into cholesterol loaded macrophage-derived foam cells. Accumulation of the latter within the plaque is a characteristic of fatty-streak type lesion, which ultimately may evolve to advanced fibro-lipid plaque. In a nomenclature similar to type 1 T helper (Th1) and type 2–helper (Th2) cells polarization, macrophages are defined as M1 (classically activated) and M2 (alternatively activated). The IFNγ-producing TH1 cells dominate during atherogenesis, and atherosclerotic lesions are characterized by macrophages with a ‘classic’ M1 phenotype producing inflammatory cytokines. The latter coexist with the ‘alternative’ M2 phenotype in human atheroma [51].
Macrophages in atheroma are a major source of inflammatory mediators (Fig. 3).The actual characterization of plaque-associated macrophages remains fragmentary, and ‘omics’ approaches will be required to define their heterogeneity and polarization.
T lymphocytes
T cells are recruited to the arterial vessel wall by endothelial VCAM-1 that binds to their VLA-4 receptor [52]. The cells migrate in response to the local chemokines, including RANTES, and the chemokines trio CXCL9, (or MIG), CXCL10 (or IP-10) and CXCL11 (or ITAC) which bind to a common T cell receptor, CXCR3 [53]. Within the plaque, upon interaction with antigen-presenting cells (macrophages, dendritic cells), T cells assume different programs of activation, becoming TH1 and TH2 cells [50, 51]. The TH1 cells (the arm of the adaptative immunity) secrete a large array of inflammatory cytokines (Fig. 3). These mediators induce polarization towards ‘classical’ activated macrophages (M1), which in turn produce pro-inflammatory cytokines, MMPs and tissue factor that are major factors in plaque instability. Locally expressed cytokines (IL-12, IL-18) induce a TH1-dominated response [53].
The TH2 cytokines promote alternatively activated macrophages (M2) and produce anti-inflammatory cytokines (Fig. 3) [54].
The regulatory -T cells or Treg, identified by the presence of Foxp3 antigen are present in the atherosclerotic plaques of mice and in lower number in humans. They produce the anti-inflammatory cytokines, Il-10 and TGF-β and counteract inflammation in experimental atherosclerosis [55].
In general, TH1- cytokines promote the development and progression of the disease, whereas TH2 and Treg cytokines exert anti-atherogenic activities [56, 57].
Dendritic cells (DC)
Dendritic cells are residents of healthy arteries. In the inflamed vessel wall, the recruitment of DC from the plasma is mediated by endothelial CAM and by platelets covering the lesion; the chemokines CCL2, CCL5 and fractalkine are also instrumental in the process [58, 59]. Two types of DC have been identified in the plaque: the classical myeloid DC (mDC) which mainly recognize bacterial signatures and plasmacytoid DC (pDC) which sense viral fragments and have the potential to produce large amounts of type I interferon. The latter up-regulates the expression of cytotoxic molecules, which contribute to apoptosis of plaque-resident cells [59]. An additional source of DC is the circulating monocytes that under inflammatory conditions and the effect of the granulocyte/macrophage colony-stimulating factor are transformed into DC. In the plaque, DC secrete inflammatory mediators (Fig. 3), recognize danger signals, and with advancement of atherosclerosis, activate and play a role in plaque rupture [reviewed in [59].
Platelets
Besides their role in late stages of atherosclerosis, it is now recognized that platelets play a part in early stages of atheroma formation. In the initial stage, the interaction of platelets with leucocytes and EC triggers autocrine and paracrine activation processes and the consequent recruitment of leucocyte into the vascular wall. The platelet glycoprotein Ibα (GPIbα) and P-selectin glycoprotein ligand (PSGL-1) receptors bind to the endothelial P-selectin initiating platelet rolling, whereas the subsequent firm adhesion is mediated through αIIbβ3 integrin and P-selectin. The platelet release inflammatory mediators (Fig. 3) that stimulate EC and provide an inflammatory milieu, which supports further pro-atherogenic alterations of the endothelium [60].
The crosstalk between platelets and leucocytes occurs by the interaction of activated platelets P-selectin and the leucocyte PSGL-1 that triggers rapid leucocytes β2 integrin activation and induces the gene expression and protein synthesis (delayed response) that generates the inflammatory phenotype of leucocytes [61]. The delayed response requires the concerted actions of outside-in signalling and of signals transduced by chemokine or cytokine receptors. For example, P-selectin and RANTES act in concert to induce nuclear translocation of NF-kB, gene expression and synthesis of MCP-1 and IL-8 in monocytes [60]. The platelet chemokines released onto the EC surface may serve as signals that are decoded by circulating monocytes, thus contributing to their recruitment at sites prone to atherosclerosis.
Activated platelets are a source of circulating microparticles (Mp), small vesicles formed by membrane budding that are present in patient's blood in different clinical conditions [62]. Mp bind to EC, leucocytes and vascular ECM and activate various signal transduction pathways; interestingly, platelets-derived Mp transfer the GPIIbIIIa receptor to other cells (i.e. PMN, EC, SMC) and influence their behaviour [63]. However, their role in human atherosclerosis is yet to be defined. Platelet Mp represent the most abundant population of all circulating microparticles (70–90%). Other sources are the EC derived Mp believed to function as circulating signalling modules or as cargos to communicate protective activation signals to vascular cells in atherosclerosis. Collected data indicate that Mp may be considered as components of a communication network for the local and systemic intercellular exchange of biological information [reviewed in [64].
Polymorphonuclear neutrophils (PMN)
These cells were considered of marginal relevance to atherosclerosis. However, recent data obtained in humans and animal models indicate a close relationship between the number of circulating activated PMN, the coronary artery disease and their presence in the culprit lesions suggesting that PMN have a role in early atherogenesis and during destabilization of advanced plaque [65]. It is assumed that activated PMN release superoxide and pro-inflammatory mediators at the blood – endothelial interface (Fig. 3) that affect the EC properties, and promote or amplify the recruitment of other inflammatory cells. Recent studies indicate that PMN launch monocyte adhesion and mobilization to the site of inflammation [66]. Moreover, within the plaque, by the molecules they secrete, PMN may contribute to its vulnerability [65]. More research is necessary to uncover the mechanisms by which PMN contribute to atherogenesis. Thus far, the data imply a causative role to the inflammatory state associated with atherogenesis and indicate that the control of neutrophilic inflammation may help the treatment of atherosclerosis [reviewed in [65].
Mast cells
Within the atherosclerotic lesions, mast cells secrete mediators, such as histamine, tryptase and chymase, and large amounts of inflammation activators including cytokines, chemokines that act on the neighbouring cells, on mLp and on the ECM (Fig. 3). In turn, Immunoglobulin G immune complexes containing mLp present within the human atherosclerotic lesions, activate mast cells and induce the secretion of numerous pro-inflammatory cytokines (TNF-α, IL-8 and MCP-1) and the release of histamine and tryptase [67]. In addition, mast cells release large amounts of granule remnants, which non-specifically bind to LDL that is further phagocytosed by macrophages thus contributing to foam cell formation [68].
B cells
These cells are not commonly found in lesions, but are present in the adjacent adventitia of established lesions [69]. Recent evidence on experimental animals revealed that B cells direct the immune response during the development of the atheroma and that their immunoglobulin products may perform protective functions throughout the plaque progression [70].
Smooth muscle cells
Although the majority of SMC in the human vessel wall are located within the medial layer, a significant number is present within the intima, in areas, known as ‘intimal thickenings’ that consist almost exclusively of SMC and the proteoglycans they produce. Intimal SMC differ significantly from medial SMC by their unique atherogenic properties that make them a fertile ground for the initiation of the plaques [71]. In contrast to human medial SMC, which predominantly express proteins involved in the contractile function, intimal SMC express lower levels of these proteins, have a higher proliferative index and a greater synthetic capacity for ECM proteins, proteases and cytokines. In response to atherogenic stimuli, SMC switch from the ‘contractile’ to the ‘synthetic’ phenotype. The latter migrate and proliferate more readily than contractile SMC, synthesize 25–46 times more collagen and express a higher number of receptors implicated in lipid uptake and formation of SMC-derived foam cell [reviewed in [71]. Plaque SMC synthesize abnormal ECM proteins, express new adhesion molecules for monocytes and lymphocytes and produce various inflammatory mediators (Fig. 3) [72]. It was recently reported that oxidized LDL induces transdifferentiation of vascular SMC towards an osteoblastic-like phenotype by a mechanism mediated by the nuclear factor of activated T cells (NFAT) signalling pathway. This process may represent a key feature in atheroma calcification and a possible target to prevent vascular calcification associated with oxidative stress [73].
SMC-monocyte/macrophages interaction: Within the human atheroma, SMC are in direct contact with macrophages [74] via the surface expressed ICAM-1, VCAM-1, CX3CL1 that endorse their ability to retain monocyte/macrophages in the lesion. Moreover, soluble factors, synergistically enhanced by IL-6 and MCP-1 production, may augment the pro-inflammatory status and acceleration of early atherogenesis [75].
Stage IV. SMC-key participants to fibrous plaque formation
The crosstalk between the resident and the recruited inflammatory cells either directly (Fig. 4) or via molecular messages govern the plaque development, including the clonal accumulation of SMC within the intima [17]. SMC migrate also from the vessel's media into the intima, through the fragmented, partially degraded internal elastic lamina forming the fibrous cap (Fig. 1D). MMPs in particular MMP-2 and -9 promote SMC migration and fibrous cap formation and thickening thus contributing to plaque stability. Other sources of intimal SMC, are the circulating bone marrow cells and the vascular progenitor cells present in the adventitia of all arteries [76]. The migrated SMC switch to a secretory phenotype is characterized by increased synthesis of ECM components. As atherosclerosis progresses, the atherogenic cytokines stimulate SMC to augment the production of proteoglycans and fibronectin; the ECM content influences the cellularity of the lesion. When SMC are bound to healthy fibrillar collagen or laminin, they quickly become arrested in G1 phase. In contrast, when SMC are bound to fibronectin and proteoglycans (as in the atheroma), cdk2 inhibitors, such as p27kip1, are down-regulated, thus promoting SMC proliferation that in turn produce more proteoglycans than quiescent cells, amplifying the effect [71]. Like for EC, the conversion of SMC to a secretory phenotype may represent an adaptation/modulation of these cells to protect themselves from the vicious microenvironment [17].
Fig 4.
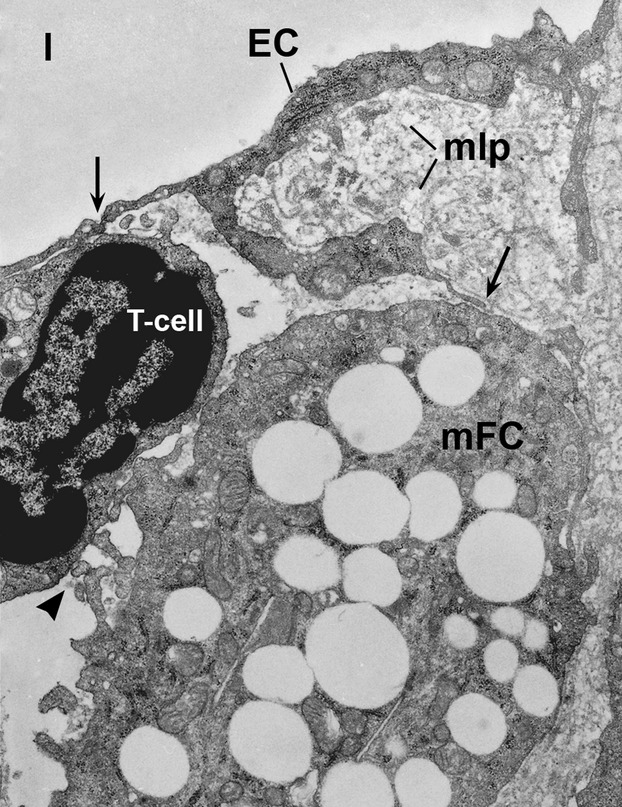
Ultrastructural evidence of the cross-talk between an endothelial cell (EC), a T cell and a monocytes-derived foam cell (mFC) located in an area of subendothelial accumulation and retention of modified lipoproteins (mLp) in a hypercholesterolaemic hamster valve. Note the direct contact between the EC and the T cell and via pseudopodes between EC and mFC (arrows) and between mFC and T cell (arrow head). l, vascular lumen. ×14,000.
Stage V. Resident and immune cells and the factors they secrete generate a calcified fibro-lipid plaque
Advanced atherosclerotic plaque contains macrophages, SMC-, and macrophage-derived foam cells, extracellular lipid droplets and calcification cores, which may develop into large calcification centres occupying a sizeable sector of the arteries (Fig. 1E). The free cholesterol accumulated in the plaque is a potent inducer of foam cells and SMC and T cells apoptosis [77]. The content of apoptotic cells, including the lipids, contributes to the formation of the lipid-rich necrotic core encapsulated by fibrous tissue, which is a defining feature of calcified atherosclerotic fibro-lipid plaque [78]. The latter, which is endowed with a robust SMC- rich fibrous cap, is considered a stable plaque. Thinning of the fibrous cap concomitantly with its infiltration by macrophages and T lymphocytes, cell apoptosis and the accumulation of large cholesterol crystals generate the unstable (vulnerable) plaque prone to rupture and the consequent thrombosis [79].
Stage VI. The unstable fibro-lipid plaque: rupture and thrombosis
The cap thinning, excess of inflammatory cytokines and proteases (inducing ECM digestion), decreased collagen synthesis, and the accumulation of cell debris within the necrotic core are part of the mechanisms that account for plaque rupture (Fig. 1F). The neovessel density is increased in both unstable and ruptured plaque and participate intraplaque haemorrhage and proteolytic degradation of angiogenic factors [80].
All cells that contribute to atheroma formation are, to a different degree, implicated in plaque rupture. EC covering the fibrous cap become exceedingly thin, loaded with lipid droplets (EC derived-foam cells) and susceptible to apoptosis and necrosis. Ultimately, erosion of EC leads to matrix exposure to blood components (Fig. 1F).
Macrophage present either within the large lipid core embedded within an extensively degraded ECM or within the hypo-cellular fibrous cap secrete several classes of neutral proteases, such as serine proteases, cathepsins, and MMPs tissue factor, urokinase Plasminogen Activator (uPA), tissue plasminogen activator (tPA), and plasminogen activator inhibitor 1,2 (PAI-1,2) [81–83] (Fig. 3).
The MMP system acting alone or in concert with the fibrinolitic system (plasminogen/plasmin) degrades most ECM components, thus contributing to neovascularization and in the later stages to rupture of the plaque or aneurysm [84, 85].
The MMPs degrade collagen and most other ECM components and modify soluble, and cell surface proteins, including cytokines and chemokines [81, 82]. Elevated levels of MMP1-2, -3, -8, -9, -11, -12, -13, -14 and -16 are found in macrophage-rich regions of human atherosclerotic plaques and MMP1, -3 and -8 co-localize with cleaved collagen. Moreover, the activity of MMP1, -8, -12, -13, and -16 is greater in inflamed, lipid-rich atheromas compared with fibrous plaques whereas MMP-2 has the opposite association [81]. Excessive MMPs cause matrix destruction or increase inflammation associated with plaque rupture either by direct destruction of ECM or indirectly by promoting macrophages and SMC death [86]. Necrosis of the vulnerable plaque is due to a combination of macrophage death and defective phagocyte clearance of apoptotic cells. Dead or dying cells within the lesion release inflammatory cytokines and proteases that together with ECM digestion and accelerated SMC apoptosis cause the thinning of the fibrous cap and the plaque rupture [reviewed in [86].
Plasmin, a serine protease is generated from plasminogen, by tPA and uPA, both activators being overexpressed in the atherosclerotic lesions [87]. It exerts its atherogenic properties through ECM proteolysis-mediated cell apoptosis, chemoattraction of inflammatory cells and modification of Lp [reviewed in [83].
The fibrinolytic and MMP systems cooperate also in thrombus dissolution. Plasminogen/plasmin system activates several MMPs that participate in the dissolution of fibrin deposits. Recent data state that MMP-10 degrades various ECM components and activate other MMP enhancing fibrinolysis via a thrombin-activatable fibrinolysis inhibitor –mediated mechanism. Thus, MMP-10 is proposed as a new profibrinolytic agent and a therapeutic target in arterial thrombosis [88].
Vascular SMC are also implicated in fibrous cap thinning due to their decreased capacity to synthesize collagen and increased apoptosis [89]. Interferon γ secreted by activated T cells may be responsible for the markedly decreased ability of human SMC to express interstitial collagen genes [53]. Uncovering the mechanisms underlying MMP up-regulation during macrophage differentiation and reducing SMC apoptosis could be employed in devising new therapies to prevent plaque instability.
Mast cells, localized especially in the rupture–prone regions of the plaque, secrete proteases (Fig. 3) that assist in the destabilization of the atherosclerotic plaque [90].
Platelets have a major role in the thromboembolitic complications of the vulnerable plaque. When the plaque rupture platelets adhere to the exposed ECM rich in pro-inflammatory factors (i.e. tissue factor), become activated, aggregate and form a thrombus on the surface of the disrupted lesion that is often in continuity with the underlying necrotic core rich in macrophages [91] (Fig. 1F). Thrombotic vascular occlusion (∼80% of sudden coronary deaths) is associated with ischaemic events, such as acute coronary syndrome or cerebral stroke. In some cases, the matrix heals by a concerted biological process involving accumulation of SMC and ECM proteins (i.e. proteoglycans and collagen), neovascularisation and luminal surface re-endothelialization [92].
Inflammatory cells and pro-inflammatory molecules as therapeutic targets in atherosclerosis
Based on the current knowledge on the key role of inflammation in atherosclerosis, the existing therapy, which is to lower the lipids level and modulate the risk factors, could profit from the association with an anti-inflammatory therapy. The new treatments using anti-cytokine drugs (TNF-α and IL-1 receptor antagonists) yielded no appreciable results, yet ongoing clinical studies are in progress [93, 94]. Currently, the best-characterized anti-inflammatory drugs in primary and secondary prevention of CVD are the statins, which beyond the lipid –lowering property have an anti-inflammatory effect. However, ∼70% of clinical events cannot be prevented with available drug therapy, including statins [95].
In ‘treating’ pathologic inflammation, the knowledge of the cellular and molecular mechanisms involved hold promise to unravel new targets for imaging and therapeutics [96]. Thus far, the processes that could serve as targets are the EC dysfunction (markers: CAM), accumulation and activation of cells (monocytes/macrophages), inflammatory mediators (i.e. fractalkine, chemokine receptors), metabolic activities of cells (i.e. glucose transport), apoptosis within atheroma (i.e. phosphatidyl serine), plaque procoagulant activity (tissue factor, factor XIII), proteolytic enzymes (cathepsin K, MMPs), ROS and angiogenic markers (integrin αvβ3) [96]. Additional relevant biomarkers that reflect the stage of the disease are of paramount importance and need to be found. The bone marrow derived endothelial and smooth muscle progenitor cells recruited to the lesions raise the possibility that a cell-based approach could be used to treat atherosclerosis; however, the data are still controversial [97, 98].
Lately, all CAM and their functional complexes formed during leucocyte recruitment elicited great interest as diagnostic tools (markers of EC dysfunction), for molecular imaging and targeted therapy. The new therapies exploit the increased expression of inflammatory molecules on the EC surface for site-directed release of drugs and imaging agents. Antagonists to VCAM-1, ICAM-1, and inhibitors of P-selectin and CD 44 molecules are in preclinical phase of testing [99]. We reported that ‘intelligent’ immunoliposomes recognizing VCAM-1 could be an appropriate target for specific drug delivery to activated human EC [100].
Pharmacological modulation of monocytes/macrophages regulation in atherosclerosis is another strategy for prevention and treatment of associated inflammation Angiotensin converting enzyme inhibitors, beta-blockers, aspirin, clopidogrel, cytokines or antibodies to cytokines, PPARs activators, statins and Liver X receptor (LXR) ligands reduce monocyte adhesion to inflamed tissue and the pro-inflammatory properties of macrophages and of other immune cells. siRNA is a potential new therapeutic class to target inflammatory genes and signalling pathways in macrophages and promising nanotechnologies for targeting plaque macrophages are in development [reviewed in [50].
Effective atherosclerosis treatment would require macrophage manipulation as a function of the stage of the disease process. Plaque stabilization is associated with a diminution in plaque macrophages. A possible avenue to induce plaque regression is to stimulate macrophage migration through lymphatics to limph nodes. Moreover, amelioration of atherosclerotic plaque milieu may determine the transformation of macrophages from an immobile to a mobile state, by expression of dendritic cell markers, such as chemokine (C-C motif) receptor 7 (CCR7), an essential requirement for dendritic cell migration [101]. As macrophages have both pro-atherogenic and anti-atherogenic effects, their total depletion is not a sound therapeutic option. We have reported that in experimental atherosclerosis systemic depletion of monocytes (with clodronate-encapsulated liposomes that selectively induce monocyte apoptosis) is a two-edged sword: it has a beneficial effect by decreasing the expression of IL-1β, MMP-2 and MMP-9 activities and an adverse effect by expanding the cardiac valve lesions that exhibit a significant increase in lipid and collagen content [102].
Strategies targeting other immune cells in atherogenesis have emerged. Drugs are designed to target T cells surface molecules or to selectively deplete activated effectors T cells while promoting Treg cells expansion in vivo [95]. Immunization is a promising therapy to combat atherosclerosis by enhancing protective antibody titres, altering the balance of pro- and anti-inflammatory T cell subtypes and expanding Treg cells [95]. Modulation of dendritic cell migratory properties using an agonist of CCR7 receptor is already in a preclinical study [99].
In SMC, N-cadherin mediated cell–cell adhesion functions as a survival signal and is also effective as a cell–matrix contact. Recently, a soluble form of N-cadherin (SNC) was demonstrated to be a successful pro-survival factor for cultured SMC via a mechanism involving FGF-R, PI-3 kinase, and Akt signalling. Attenuating SMC apoptosis, SNC may suppress atheroma instability suggesting its therapeutic potential to retard plaque rupture [103].
In addition, drugs targeting inflammatory components are designed to recoup the altered processes in the plaque. These are the antagonists to chemokine receptor 2 (CCR2), inhibitors of p38 mitogen–activated protein kinase (MAPK) and the antioxidant, anti-inflammatory agent AGI-1076-which down regulates VCAM-1 and may reduce macrophage accumulation/activity in the plaque [99]. To counteract the harmful effect of MMPs in plaque rupture, inhibitors of MMPs and cathepsins are in preclinical tests [99].
Preventive strategies to reduce atherosclerosis as well as therapeutic interventions for plaque regression are urgently needed. The new imaging technologies using nanocontrast agents targeting plaque constituents may lead to an early diagnostic. In addition, nanoscale drug delivery devices can be tailored for site-specific therapeutic interventions. Thus far, the research leaded to preclinical validation of nanoscale devices that target cellular and molecular components of the atherosclerotic plaque [104].
The increased knowledge on the patient- particular atherosclerotic plaque components will lead to a customized patient-specific clinical management and the design of nanoscale devices to guide a spatial and temporal release of drugs to ‘visible’ molecular cues thus designing a specific plaque-‘tailored’ therapy.
Inflammation and dyslipidaemia partake to a vicious circle in atheroma formation
At present, inflammation is acknowledged as the cholesterol's partner and key culprit in CVD. Cholesterol would not be nearly as dangerous without the inflammatory process. Excess LDL transcytosed into the artery wall triggers an inflammatory response, which in turn speeds up the accumulation of cholesterol that consecutively produces more inflammation; a vicious circle is created that eventually accelerates the atheroma formation. Chronic inflammatory diseases also lead to dyslipidaemia and the generation of atheroma [105]. Studies showed that C-reactive protein (as marker of inflammation) may be more effective than cholesterol in gauging the risk of heart attack and other cardiovascular events [106].
Still many questions are to be answered to comprehend the basic biology of inflammation in atherosclerosis as for example, why and how the arteries respond to insults, such as high cholesterol or inflammatory mediators, which are the causes of the vascular inflammation, the triggers of plaque development and rupture leading to atherothrombosis or the key processes and pathways involved in plaque regression.
Conclusions and perspectives
The progress in atherosclerosis research incriminates two partakers that power a vicious circle in atheroma formation: dyslipidemia, which triggers as a defence reaction a vascular inflammatory process and inflammation that alone may elicit dyslipidemia. Thus, inflammation may be either a cause or a result of vascular disorders leading to atherosclerosis.
The new data on inflammation biology and its implications in atherosclerosis advocate for a change in diagnosis, monitoring and treatment of patients with CVDs. However, caution should be exercised when translating the data from experimental models to humans. There is a stringent need for more human specific biomarkers of inflammation, additional clinical end-point trials, genome-wide association studies and refined imaging targets (molecular imaging) to be employed as tools in the by-directional translation of data between bench and bedside.
Acknowledgments
We are indebted to our prominent mentor, Professor Nicolae Simionescu, and our great collaborators for their contribution to the data presented here. We thank Mrs. Marilena Daju for the excellent graphic design and image processing.
The support of the Romanian Academy and Romanian Ministry of Education, Research, Youth and Sport, UEFISCDI research grants 1307 (PNII-IDEI), contract no. 989/2009 and ERANET contract no. 4_001, project acronym NANODIATER under the frame of EuroNanoMed 2011–2013 are gratefully acknowledged.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Libby P, Ridker PM, Hansson GK. Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol. 2009;54:2129–38. doi: 10.1016/j.jacc.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Millonig G, Malcom GT, Wick G. Early inflammatory-immunological lesions in juvenile atherosclerosis from the pathobiological determinants of atherosclerosis in youth (PDAY)-study. Atherosclerosis. 2002;160:441–8. doi: 10.1016/s0021-9150(01)00596-2. [DOI] [PubMed] [Google Scholar]
- 3.Tsimikas S, Willerson JT, Ridker PM. C-reactive protein and other emerging blood biomarkers to optimize risk stratification of vulnerable patients. J Am Coll Cardiol. 2006;47:C19–31. doi: 10.1016/j.jacc.2005.10.066. [DOI] [PubMed] [Google Scholar]
- 4.Simionescu N, Vasile E, Lupu F, et al. Prelesional events in atherogenesis: accumulation of extracellular cholesterol rich liposomes in the arterial intima and cardiac valves of hyperlipidemic rabbits. Am J Pathol. 1986;123:85–101. [PMC free article] [PubMed] [Google Scholar]
- 5.VanderLaan PA, Reardon CA, Getz GS. Site-specificity of atherosclerosis: site-selective responses to atherosclerotic modulators. Arterioscler Thromb Vasc Biol. 2004;24:12–22. doi: 10.1161/01.ATV.0000105054.43931.f0. [DOI] [PubMed] [Google Scholar]
- 6.Davies PF, Civelek M, Fang Y, et al. Endothelial heterogeneity associated with regioneal athero-susceptibility and adaptation to disturbed flow in vivo. Semin Thromb Haemost. 2010;36:265–75. doi: 10.1055/s-0030-1253449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davies PF. Endothelial transcriptome profiles in vivo in complex arterial flow fields. Ann Biomed Eng. 2008;36:563–70. doi: 10.1007/s10439-007-9400-0. [DOI] [PubMed] [Google Scholar]
- 8.Chatzizisis YS, Coskun AU, Jonas M, et al. Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling: molecular, cellular, and vascular behavior. J Am Coll Cardiol. 2007;49:2379–93. doi: 10.1016/j.jacc.2007.02.059. [DOI] [PubMed] [Google Scholar]
- 9.Liu Y, Chen BP, Lu M, et al. Shear stress activation of SREBP1 in endothelial cells is mediated by integrins. Arterioscler Thromb Vasc Biol. 2002;22:76–81. doi: 10.1161/hq0102.101822. [DOI] [PubMed] [Google Scholar]
- 10.Passerini AG, Polacek DC, Shi C, et al. Coexisting proinflammatory and antioxidative endothelial transcription profiles in a disturbed flow region of the adult porcine aorta. Proc Natl Acad Sci USA. 2004;101:2482–7. doi: 10.1073/pnas.0305938101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hajra L, Evans AI, Chen M, et al. The NF-kB signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci USA. 2000;97:9052–7. doi: 10.1073/pnas.97.16.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gimbrone MA, Jr, Topper JN, Nagel T, et al. Endothelial dysfunction, hemodynamic forces, and atherogenesis. Ann NY Acad Sci. 2000;902:203–9. doi: 10.1111/j.1749-6632.2000.tb06318.x. [DOI] [PubMed] [Google Scholar]
- 13.Volger OL, Fledderus JO, Kisters N, et al. Distinctive expression of chemokines and transforming growth factor–beta signaling in human arterial endothelium during atherosclerosis. Am J Pathol. 2007;171:326–37. doi: 10.2353/ajpath.2007.061196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dai G, Vaughn S, Zhang Y, et al. Biomechanical forces in atherosclerosis-resistant vascular regions regulate endothelial redox balance via phosphoinisitol 3-kinase/Akt-dependent activation of Nrf2. Circ Res. 2007;101:723–33. doi: 10.1161/CIRCRESAHA.107.152942. [DOI] [PubMed] [Google Scholar]
- 15.Civelek M, Manduchi E, Rilley RJ, et al. Chronic endoplasmic reticulum stress activates unfolded protein response in arterial endothelium in regions of susceptibility to atherosclerosis. Circ Res. 2009;105:453–61. doi: 10.1161/CIRCRESAHA.109.203711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bartel DP. MicroRNA: target recognition and regulatory functions. Cell. 2009;136:215–33. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simionescu M. Implications of early structural-functional changes in the endothelium for vascular disease. Arterioscler Thromb Vasc Biol. 2007;27:266–74. doi: 10.1161/01.ATV.0000253884.13901.e4. [DOI] [PubMed] [Google Scholar]
- 18.Nordestgaard BG, Hjelms E, Stender S, et al. Different efflux pathways for high and low density lipoproteins from porcine aortic intima. Arteriosclerosis. 1990;10:477–85. doi: 10.1161/01.atv.10.3.477. [DOI] [PubMed] [Google Scholar]
- 19.Kruth HS. Localization of unesterified cholesterol in human atherosclerotic lesions. Demonstration of filipin positive, oil-Red-0-negative particles. Am J Pathol. 1984;114:201–8. [PMC free article] [PubMed] [Google Scholar]
- 20.Tirziu D, Dobrian A, Tasca C, et al. Intimal thickenings of human aortas contain modified reassembled lipoproteins. Atherosclerosis. 1995;112:101–14. doi: 10.1016/0021-9150(94)05405-8. [DOI] [PubMed] [Google Scholar]
- 21.Steinberg D. The LDL modification hypothesis of atherogenesis: an update. J Lipid Res. 2009;50:S376–81. doi: 10.1194/jlr.R800087-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishigaki Y, Oka Y, Katagiri H. Circulating oxidized LDL: a biomarker and a pathogenic factor. Curr Opin Lipidol. 2009;20:363–9. doi: 10.1097/MOL.0b013e32832fa58d. [DOI] [PubMed] [Google Scholar]
- 23.Ghinea N, Leabu M, Hasu M, et al. Prelesional events in atherogenesis. Changes induced by hypercholesterolemia in the cell surface chemistry of arterial endothelium and blood monocytes, in rabbit. J Submicrosc Cytol. 1987;19:209–27. [PubMed] [Google Scholar]
- 24.Li C, Mollahan P, Baguneid MS, et al. A comparative study of neovascularization in atherosclerotic plaque using CD31, CD105 and transforming growth factor (TGF)–ß1. Pathobiology. 2006;73:192–7. doi: 10.1159/000096020. [DOI] [PubMed] [Google Scholar]
- 25.Stevens HY, Melchior B, Bell KS, et al. PECAM-1 is a critical mediator of atherosclerosis. Dis Model Mech. 2008;1:175–81. doi: 10.1242/dmm.000547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ostermann G, Fraemohs L, Baltus T, et al. Involvement of JAM-A in mononuclear cell recruitment in inflamed or atherosclerotic endothelium: inhibition by soluble JAM-A. Arterioscler Thromb Vasc Biol. 2005;25:729–35. doi: 10.1161/01.ATV.0000157154.14474.3b. [DOI] [PubMed] [Google Scholar]
- 27.Zernecke A, Liehn EA, Fraemohs L, et al. Importance of junctional adhesion molecule-A for neointimal lesion formation and infiltration in atherosclerosis-prone mice. Arterioscler Thromb Vasc Biol. 2006;26:e10–3. doi: 10.1161/01.ATV.0000197852.24529.4f. [DOI] [PubMed] [Google Scholar]
- 28.Keiper T, Al-Fakhri N, Chavakis E, et al. The role of junctional adhesion molecule–C (JAM-C) in oxidized LDL-mediated leukocyte recruitment. FASEB J. 2005;19:2078–80. doi: 10.1096/fj.05-4196fje. [DOI] [PubMed] [Google Scholar]
- 29.Hirata K, Ishida T, Penta K, et al. Cloning of an immunoglobulin family adhesion molecule selectively expressed by endothelial cells. J Biol Chem. 2001;276:16223–31. doi: 10.1074/jbc.M100630200. [DOI] [PubMed] [Google Scholar]
- 30.Rohatgi A, Owens AW, Khera A, et al. Differential associations between soluble cellular adhesion molecules and atherosclerosis in the Dallas Heart Study: a distinct role for soluble endothelial cell-selective adhesion molecule. Arterioscler Thromb Vasc Biol. 2009;29:1684–90. doi: 10.1161/ATVBAHA.109.190553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barreiro O, Yáñez-Mó M, Sala-Valdés M, et al. Endothelial tetraspanin microdomains regulate leukocyte form adhesion during extravasation. Blood. 2005;105:2852–61. doi: 10.1182/blood-2004-09-3606. [DOI] [PubMed] [Google Scholar]
- 32.Rohlena J, Volger OL, van Buul JD, et al. Endothelial CD81 is a marker of early human atherosclerotic plaques and facilitates monocyte adhesion. Cardiovasc Res. 2009;81:187–96. doi: 10.1093/cvr/cvn256. [DOI] [PubMed] [Google Scholar]
- 33.Sprague AH, Khalil RA. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol. 2009;78:539–52. doi: 10.1016/j.bcp.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weber C, Schober A, Zernecke A. Chemokines: key regulators of mononuclear cell recruitment in atherosclerotic vascular disease. Arterioscler Thromb Vasc Biol. 2004;24:1997–2008. doi: 10.1161/01.ATV.0000142812.03840.6f. [DOI] [PubMed] [Google Scholar]
- 35.Umehara H, Bloom ET, Okazaki T, et al. Fractalkine in Vascular Biology. Arterioscler Thromb Vasc Biol. 2004;24:34–40. doi: 10.1161/01.ATV.0000095360.62479.1F. [DOI] [PubMed] [Google Scholar]
- 36.Yang XP, Mattagajasingh S, Su S, et al. Fractalkine upregulates intercellular adhesion molecule-1 in endothelial cells through CX3CR1 and the Jak Stat 5 pathway. Circ Res. 2007;101:101–8. doi: 10.1161/CIRCRESAHA.107.160812. [DOI] [PubMed] [Google Scholar]
- 37.Manduteanu I, Dragomir E, Calin M, et al. Resistin up-regulates fractalkine expression in human endothelial cells, lack of additive effect with TNF-alpha. Biochem Biophys Res Commun. 2009;381:95–101. doi: 10.1016/j.bbrc.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 38.Burnett MS, Lee CW, Kinnaird TD, et al. The potential role of resistin in atherogenesis. Atherosclerosis. 2005;182:239–48. doi: 10.1016/j.atherosclerosis.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 39.Koenen RR, Weber C. Therapeutic targeting of chemokine interactions in atherosclerosis. Nat Rev Drug Discov. 2010;9:141–53. doi: 10.1038/nrd3048. [DOI] [PubMed] [Google Scholar]
- 40.Koenen RR, von Hundelshausen P, Nesmelova IV, et al. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat Med. 2009;15:97–103. doi: 10.1038/nm.1898. [DOI] [PubMed] [Google Scholar]
- 41.Weber C, Koenen RR. Fine-tunning leukocyte response: towards a chemokine interactome. Trends Immunol. 2006;27:268–73. doi: 10.1016/j.it.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 42.Gordon S, Taylor PR. Monocyte and Macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–64. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 43.Ziegler-Heitbrock L, Ancuta P, Crowe S, et al. Nomenclature of monocytes and dendritic cells in blood. Blood. 2010;116:e74–80. doi: 10.1182/blood-2010-02-258558. [DOI] [PubMed] [Google Scholar]
- 44.Weber C, Belge KU, von Hundelshausen P, et al. Differential chemokine receptor expression and function in human monocytes subpopulations. J Leukoc Biol. 2000;67:699–704. doi: 10.1002/jlb.67.5.699. [DOI] [PubMed] [Google Scholar]
- 45.Schlitt A, Heine GH, Blankenberg S, et al. CD14+ CD16+ monocytes in coronary artery disease and their relationship to serum TNF-alpha levels. Thromb Haemost. 2004;92:419–24. doi: 10.1160/TH04-02-0095. [DOI] [PubMed] [Google Scholar]
- 46.Schirmer SH, Fledderus JO, van der Laan AM, et al. Suppression of inflammatory signaling in monocytes from patients with coronary artery disease. J Mol Cell Cardiol. 2009;46:177–85. doi: 10.1016/j.yjmcc.2008.10.029. [DOI] [PubMed] [Google Scholar]
- 47.Woollard KJ, Geissmann F. Monocytes in atherosclerosis: subsets and functions. Nat Rev Cardiol. 2010;7:77–86. doi: 10.1038/nrcardio.2009.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carman CV, Springer TA. A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J Cell Biol. 2004;167:377–88. doi: 10.1083/jcb.200404129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kamei M, Carman CV. New observations on the trafficking and diapedesis of monocytes. Curr Opin Hematol. 2010;17:43–52. doi: 10.1097/MOH.0b013e3283333949. [DOI] [PubMed] [Google Scholar]
- 50.Wilson HM. Macrophages heterogeneity in atherosclerosis – implications for therapy. J Cell Mol Med. 2010;14:2055–65. doi: 10.1111/j.1582-4934.2010.01121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mantovani A, Garlanda C, Locati M. Macrophage diversity and polarization in atherosclerosis: a question of balance. Arterioscler Thromb Vasc Biol. 2009;29:1419–23. doi: 10.1161/ATVBAHA.108.180497. [DOI] [PubMed] [Google Scholar]
- 52.Rosenthal-Allieri MA, Ticchioni M, Breittmayer JP, et al. Influence of b1 integrin intracytoplasmic domains in the regulation of VLA-4-mediated adhesion of human T cells to VCAM-1 under flow conditions. J Immunol. 2005;175:1214–23. doi: 10.4049/jimmunol.175.2.1214. [DOI] [PubMed] [Google Scholar]
- 53.Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6:508–19. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- 54.Rocha VZ, Libby P. Obesity, inflammation, and atherosclerosis. Nat Rev Cardiol. 2009;6:399–409. doi: 10.1038/nrcardio.2009.55. [DOI] [PubMed] [Google Scholar]
- 55.de Boer OJ, van der Meer JJ, Teeling P, et al. Low numbers of FOXP3 positive regulatory T cells are present in all developmental stages of human atherosclerotic lesions. PLoS One. 2007;2:e779. doi: 10.1371/journal.pone.0000779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12:204–12. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- 57.Ait-Oufella H, Taleb S, Mallat Z, et al. Recent advances on the role of cytokines in atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:969–79. doi: 10.1161/ATVBAHA.110.207415. [DOI] [PubMed] [Google Scholar]
- 58.Langer HF, Daub K, Braun G, et al. Platelets recruit human DC via Mac-1/JAM-C interaction and modulate this function in vitro. Arterioscler Thromb Vasc Biol. 2007;27:1463–70. doi: 10.1161/ATVBAHA.107.141515. [DOI] [PubMed] [Google Scholar]
- 59.Niessner A, Weyand CM. Dendritic cells in atherosclerotic disease. Clin Immunol. 2010;134:25–32. doi: 10.1016/j.clim.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gawaz M, Langer H, May AE. Platelets in inflammation and atherogenesis. J Clin Invest. 2005;115:3378–84. doi: 10.1172/JCI27196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Totani L, Evangelista V. Platelet leukocyte interactions in cardiovascular disease and beyond. Arterioscler Thromb Vasc Biol. 2010;30:2357–61. doi: 10.1161/ATVBAHA.110.207480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Van der Zee PM, Biro E, Ko Y, et al. P-selectin- and CD63-exposing platelet microparticles reflect platelet activation in peripheral arterial disease and myocardial infarction. Clin Chem. 2006;52:657–64. doi: 10.1373/clinchem.2005.057414. [DOI] [PubMed] [Google Scholar]
- 63.Li X, Cong H. Platelet-derived microparticles and the potential of glycoprotein IIb/IIIa antagonists in treating acute coronary syndrome. Tex Heart Inst J. 2009;36:134–9. [PMC free article] [PubMed] [Google Scholar]
- 64.Mause SF, Weber C. Microparticles protagonists of a novel communication network for intercellular information exchange. Circ Res. 2010;107:1047–57. doi: 10.1161/CIRCRESAHA.110.226456. [DOI] [PubMed] [Google Scholar]
- 65.Baetta R, Corsini A. Role of polymorphonuclear neutrophils in atherosclerosis: current state and future perspectives. Atherosclerosis. 2010;210:1–13. doi: 10.1016/j.atherosclerosis.2009.10.028. [DOI] [PubMed] [Google Scholar]
- 66.Soehnlein O, Zernecke A, Weber C. Neutrophils launch monocyte extravasation by release of granule proteins. Thromb Haemost. 2009;102:198–205. doi: 10.1160/TH08-11-0720. [DOI] [PubMed] [Google Scholar]
- 67.Metzler B, Xu Q. The role of mast cells in atherosclerosis. Int Arch Allergy Immunol. 1997;114:10–4. doi: 10.1159/000237636. [DOI] [PubMed] [Google Scholar]
- 68.Kovanen PT. Mast cells in atherogenesis: actions and reactions. Curr Atheroscler Rep. 2009;11:214–9. doi: 10.1007/s11883-009-0033-7. [DOI] [PubMed] [Google Scholar]
- 69.Caligiuri G, Nicoletti A, Poirier B, et al. Protective immunity against atherosclerosis carried by B cells of hypercholesterolemic mice. J Clin Invest. 2002;109:745–53. doi: 10.1172/JCI07272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Binder CJ. IL-5 links adaptive and natural immunity specific for epitopes of oxidized LDL and protects from atherosclerosis. J Clin Invest. 2004;114:427–37. doi: 10.1172/JCI20479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Doran AC, Meller N, McNamara CA. The role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:812–9. doi: 10.1161/ATVBAHA.107.159327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Raines EW, Ferri N. Thematic review series: the immune system and atherogenesis. Cytokines affecting endothelial and smooth muscle cells in vascular disease. J Lipid Res. 2005;46:1081–92. doi: 10.1194/jlr.R500004-JLR200. [DOI] [PubMed] [Google Scholar]
- 73.Goettsch C, Rauner M, Hamann C, et al. Nuclear factor of activated Tcells mediates oxidized LDL-induced calcification of vascular smooth muscle cells. Diabetologia. 2011;54:2690–701. doi: 10.1007/s00125-011-2219-0. [DOI] [PubMed] [Google Scholar]
- 74.Barlic J, Zhang Y, Murphy PM. Atherogenic lipids induce adhesion of human coronary artery smooth muscle cells to macrophages by up-regulating chemokine CX3CL1 on smooth muscle cells in a TNF α-NF-kB-dependent manner. J Biol Chem. 2007;282:19167–76. doi: 10.1074/jbc.M701642200. [DOI] [PubMed] [Google Scholar]
- 75.Chen L, Frister A, Wang S, et al. Interaction of vascular smooth muscle cells and monocytes by soluble factors synergistically enhances IL-6 and MCP-1 production. Am J Physiol Heart Circ Physiol. 2009;296:H987–96. doi: 10.1152/ajpheart.01158.2008. [DOI] [PubMed] [Google Scholar]
- 76.Han CI, Campbell GR, Campbell JH. Circulating bone marrow cells can contribute to neointimal formation. J Vasc Res. 2001;38:113–9. doi: 10.1159/000051038. [DOI] [PubMed] [Google Scholar]
- 77.Tiwari RL, Singh V, Barthwal MK. Macrophages: an elusive yet emerging therapeutic target of atherosclerosis. Med Res Rev. 2008;28:483–544. doi: 10.1002/med.20118. [DOI] [PubMed] [Google Scholar]
- 78.Tabas I, Tall A, Accili D. The impact of macrophage insulin resistance on advanced atherosclerotic plaque progression. Circ Res. 2010;106:58–67. doi: 10.1161/CIRCRESAHA.109.208488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Virmani R, Kolodgie FD, Burke AP, et al. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20:1262–75. doi: 10.1161/01.atv.20.5.1262. [DOI] [PubMed] [Google Scholar]
- 80.Le Dall J, Ho-Tin-Noé B, Louedec L, et al. Immaturity of microvessels in haemorrhagic plaques is associated with proteolytic degradation of angiogenic factors. Cardiovasc Res. 2010;85:184–93. doi: 10.1093/cvr/cvp253. [DOI] [PubMed] [Google Scholar]
- 81.Newby AC. Metalloproteinase expression in monocytes and macrophages and its relationship to atherosclerotic plaque instability. Arterioscler Thromb Vasc Biol. 2008;28:2108–14. doi: 10.1161/ATVBAHA.108.173898. [DOI] [PubMed] [Google Scholar]
- 82.Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69:562–73. doi: 10.1016/j.cardiores.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 83.Leclercq A, Houard X, Loyau S, et al. Topology of protease activities reflects atherothrombotic plaque complexity. Atherosclerosis. 2007;191:1–10. doi: 10.1016/j.atherosclerosis.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 84.Lijnen HR. Metalloproteins in development and progression of vascular disease. Pathophysiol Haemost Thromb. 2003;33:275–81. doi: 10.1159/000083814. –2004. [DOI] [PubMed] [Google Scholar]
- 85.Carmeliet P, Moons L, Lijnen R, et al. Urokinase-generated plasmin activates matrix metalloproteinases during aneurysm formation. Nat Genet. 1997;17:439–44. doi: 10.1038/ng1297-439. [DOI] [PubMed] [Google Scholar]
- 86.Newby AC. Matrix metalloproteinases regulate migration, proliferation and death of vascular smooth muscle cells by degrading matrix and non-matrix substrates. Cardiovasc Res. 2006;69:614–24. doi: 10.1016/j.cardiores.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 87.Lupu F, Heim DA, Bachmann F, et al. Plasminogen activator expression in human atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 1995;15:1444–55. doi: 10.1161/01.atv.15.9.1444. [DOI] [PubMed] [Google Scholar]
- 88.Orbe J, Barrenetxe J, Rodriguez JA, et al. Matrix metalloproteinase-10 effectively reduces infarct size in experimental stroke by enhancing fibrinolysis via a thrombin-activatable fibrinolysis inhibitor-mediated mechanism. Circulation. 2011;124:2909–19. doi: 10.1161/CIRCULATIONAHA.111.047100. [DOI] [PubMed] [Google Scholar]
- 89.Dhume A, Soundararajan K, Hunter W, et al. Comparison of vascular smooth muscle cell apoptosis and fibrous cap morphology in symptomatic and asymptomatic carotid artery disease. Ann Vasc Surg. 2003;17:1–8. doi: 10.1007/s10016-001-0331-1. [DOI] [PubMed] [Google Scholar]
- 90.Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis. Annu Rev Immunol. 2009;27:165–97. doi: 10.1146/annurev.immunol.021908.132620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rudd JFH, Narula J, Strauss HW, et al. Imaging atherosclerotic plaque inflammation by fluorodeoxyglucose with positron emission tomography. Ready for prime time? J Am Coll Cardiol. 2010;55:2527–35. doi: 10.1016/j.jacc.2009.12.061. [DOI] [PubMed] [Google Scholar]
- 92.Kolodgie FD, Nakazawa G, Sangiorgi G, et al. Pathology of atherosclerosis and stenting. Neuroimaging Clin N Am. 2007;17:285–310. doi: 10.1016/j.nic.2007.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mann DL, McMurray JJ, Packer M, et al. Targeted anticytokine therapy in patients with chronic heart failure: results of the Randomized Etanercept Worldwide Evaluation (RENEWAL. Circulation. 2004;109:1594–602. doi: 10.1161/01.CIR.0000124490.27666.B2. [DOI] [PubMed] [Google Scholar]
- 94.Crossman DC, Morton AC, Gunn JP, et al. Investigation of the effects of Interleukin-1 receptor antagonist (IL-1ra) on markers of inflammation in non-ST elevation acute coronary syndromes (The MRC-ILA-Heart Study. Trials. 2008;9:8. doi: 10.1186/1745-6215-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Klingenberg R, Hansson GK. Treating inflammation in atherosclerotic cardiovascular disease: emerging therapies. Eur Heart J. 2009;30:2838–44. doi: 10.1093/eurheartj/ehp477. [DOI] [PubMed] [Google Scholar]
- 96.Libby P, DiCarli M, Weissleder R. The vascular biology of atherosclerosis and imaging targets. J Nucl Med. 2010;51:33S–7S. doi: 10.2967/jnumed.109.069633. [DOI] [PubMed] [Google Scholar]
- 97.Bentzon JF, Weile C, Sondergaard CS, et al. Smooth muscle cells in atherosclerosis originate from the local vessel wall and not circulating progenitor cells in ApoE knockout mice. Arterioscler Thromb Vasc Biol. 2006;26:2696–702. doi: 10.1161/01.ATV.0000247243.48542.9d. [DOI] [PubMed] [Google Scholar]
- 98.Sata M, Saiura A, Kunisato A, et al. Hematopoietic stem cells differentiate into vascular cells that participate in the pathogenesis of atherosclerosis. Nat Med. 2002;8:403–9. doi: 10.1038/nm0402-403. [DOI] [PubMed] [Google Scholar]
- 99.Rader DJ, Daugherty A. Translating molecular discoveries into new therapies for atherosclerosis. Nature. 2008;451:904–13. doi: 10.1038/nature06796. [DOI] [PubMed] [Google Scholar]
- 100.Voinea M, Manduteanu I, Dragomir E, et al. Immunoliposomes directed toward VCAM-1 interact specifically with activated endothelial cells-a potential tool for specific drug delivery. Pharm Res. 2005;22:1906–17. doi: 10.1007/s11095-005-7247-3. [DOI] [PubMed] [Google Scholar]
- 101.Francis AA, Pierce GN. An integrated approach for the mechanisms responsible for atherosclerotic plaque regression. Exp Clin Cardiol. 2011;16:77–86. [PMC free article] [PubMed] [Google Scholar]
- 102.Calin MV, Manduteanu I, Dragomir E, et al. Effect of depletion of monocytes/macrophages on early aortic valve lesion in experimental hyperlipidemia. Cell Tissue Res. 2009;336:237–48. doi: 10.1007/s00441-009-0765-2. [DOI] [PubMed] [Google Scholar]
- 103.Lyon CA, Johnson JL, Williams H, et al. Soluble N-cadherin overexpression reduces features of atherosclerotic plaque. Arterioscler Thromb Vasc Biol. 2009;29:195–201. doi: 10.1161/ATVBAHA.108.178087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jayagopal A, Linton MF, Fazio S, et al. Insights into atherosclerosis using nanotechnology. Curr Atheroscler Rep. 2010;12:209–15. doi: 10.1007/s11883-010-0106-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Toms TE, Panoulas VF, Kitas GD. Dyslipidaemia in rheumatological autoimmune diseases. Open Cardiovasc Med J. 2011;5:64–75. doi: 10.2174/1874192401105010064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Libby P. Molecular and cellular mechanisms of the thrombotic complications of atherosclerosis. J Lipid Res. 2009;50:S352–7. doi: 10.1194/jlr.R800099-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]