

Abstract
The proline hydroxylase domain-containing enzymes (PHD) act as cellular oxygen sensors and initiate a hypoxic signal cascade to induce a range of cellular responses to hypoxia especially in the aspect of energy and metabolic homeostasis regulation. AMP-activated protein kinase (AMPK) is recognized as a major energetic sensor and regulator of cardiac metabolism. However, the effect of PHD signal on AMPK has never been studied before. A PHD inhibitor (PHI), dimethyloxalylglycine and PHD2-specific RNA interference (RNAi) have been used to activate PHD signalling in neonatal rat cardiomyocytes. Both PHI and PHD2-RNAi activated AMPK pathway in cardiomyocytes effectively. In addition, the increased glucose uptake during normoxia and enhanced myocyte viability during hypoxia induced by PHI pretreatment were abrogated substantially upon AMPK inhibition with an adenoviral vector expressing a dominant negative mutant of AMPK-α1. Furthermore, chelation of intracellular Ca2+ by BAPTA, inhibition of calmodulin-dependent kinase kinase (CaMKK) with STO-609, or RNAi-mediated down-regulation of CaMKK α inhibited PHI-induced AMPK activation significantly. In contrast, down-regulation of LKB1 with adenoviruses expressing the dominant negative form did not affect PHI-induced AMPK activation. We establish for the first time that activation of PHD signal cascade can activate AMPK pathway mainly through a Ca2+/CaMKK-dependent mechanism in cardiomyocytes. Furthermore, activation of AMPK plays an essential role in hypoxic protective responses induced by PHI.
Keywords: proline hydroxylase domain-containing enzymes (PHD), AMP-activated protein kinase (AMPK), neonatal rat cardiomyocyte, dimethyloxalylglycine, hypoxia
Introduction
The heart has developed exquisite sensitivity to oxygen levels due to its high consumption of oxygen. A decline in oxygen tension (hypoxia) triggers a variety of cellular adaptive responses to preserve cell and tissue viability. Recent studies have identified the proline hydroxylase domain-containing enzymes (PHD1, PHD2, PHD3) and asparaginyl hydroxylase domain protein factor-inhibiting HIF (FIH) as cellular oxygen sensors [1, 2]. Under normoxic conditions, hydroxylation of specific proline residues within the oxygen-dependent degradation domain of hypoxia-inducible factor (HIF)-1α by the three PHD isoforms earmarks HIF-1α protein for proteasomal degradation by promoting its interaction with the von Hippel–Lindau (VHL) protein [3], whereas further hydroxylation of an asparagine residue by FIH reduces its transcriptional activity [2]. Thus, hydroxylases repress both the expression and transcriptional activity of HIF-1. During hypoxia, the reduction of proline- and asparagine-hydroxylation leads to HIF-1 stabilization and transactivation. Consequently, HIF-1 activates the expression of over 70 genes that contain hypoxia-response elements in their promoter regions and mediate a myriad of adaptive responses. Moreover, accumulating evidence suggests that PHDs may act as a master regulator in initiating the O2-sensing signal cascade, rather than an ‘obligatory HIF-regulator’. In addition to HIF-1α, PHDs directly hydroxylate other targets to initiate HIF-1α-independent cellular changes [4, 5]. More importantly, activation of the PHD signal pathway via PHD inhibitor (PHI) mediates a variety of cellular responses to hypoxia and preconditioning stimuli [6]. However, the underlying molecular mechanisms remain poorly understood.
Cardiac energy metabolism is essential for the maintenance of mechanical function, and any disturbances of energetics translate directly into cardiac dysfunction. The compensatory effects, which are induced by opening the PHD O2 sensing pathway usually with PHI, are highlighted by its energy and metabolism regulation under varying metabolic stress conditions, such as maintenance of ATP, accumulation of glycogen, increase of glucose uptake and preservation of myocyte viability [7–9].
AMP-activated protein kinase (AMPK) is recognized as the ‘guardian of energy status’ in the heart [10] and a critical regulator of energy homeostasis by coordinating a number of adaptive responses in ATP-depleting metabolic states, such as hypoxia, ischaemia/reperfusion and exercise [11]. The activation of AMPK is also considered as one of the typical cellular responses elicited by low oxygen tension. Previous studies have indicated that AMPK can be rapidly activated in mildly ischaemic heart tissue before any obvious energy deficiency develops [12, 13], implying a direct link between hypoxia signalling and AMPK activation. However, the molecular mechanisms responsible for AMPK activation in hypoxic heart remain incompletely understood.
Is PHD signal implicated in AMPK activation in heart cells? What are the underlying molecular mechanisms and link between PHD signalling and the AMPK cascade? What is the functional significance of AMPK in cellular protective effects elicited by PHI against hypoxia? In this study, we employed PHI and PHD2-specific RNAi adenovirus vector to address these important questions in neonatal rat cardiomyocytes.
Materials and methods
Ethics statement
All animal procedures were approved by the Institutional Animal Care and Use Committee of the Third Military Medical University and which conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health.
Cell culture and treatment
Ventricles of 1–3-day-old Wistar rats were minced and digested with 0.1% trypsin (Gibco-BRL, New York, NY, USA). The cells were pelleted and suspended in complete DMEM (Gibco-BRL) containing 10% foetal bovine serum (FBS; Gibco-BRL). A single pre-plating step was used to further increase the cardiomyocyte to non-cardiomyocyte ratio due to the propensity of non-cardiomyocytes to attach readily to the bottom of the culture dishes. The remaining unattached viable cells were collected and seeded at a density of 1 χ 106 cells/ml in complete DMEM/F-12 medium plus 0.1 mmol/l 5-bromo-2-deoxyuridine (Sigma-Aldrich, St. Louis, MO, USA) for 48 hrs to prevent low-level nonmyocardial cell proliferation, then replaced with complete DMEM medium and used for experimental treatment as detailed below.
1,2-bis(2-aminophenoxy)ethane-N,N,N’,N’-tetraacetic acid tetrakis (acetoxymethyl ester) (BAPTA-AM) (Sigma-Aldrich) were dissolved in PBS. DMOG (Alexis Biochemicals, San Diego, CA, USA), Ionomycin (Sigma Chemical Co, Deisenhofen, Germany), STO-609 (Tocris, Ellisville, MO, USA) and 5-Aminoimidazole-4-carboxamide ribonucleoside (AICAR) (Calbiochem, Darmstadt, Germany) were dissolved in dimethyl sulfoxide (DMSO). The final concentration of DMSO did not exceed 0.2%, and control cells were treated with the same amount of solvent.
In those experiments in which the effect of a blocker was investigated, cells were pretreated with blocker for 15–30 min., and then treated with PHI or vehicle for three hrs.
Hypoxia treatment was achieved by exposure of isolated cardiomyocytes in a hypoxia chamber filled with 5% CO2, 1% O2 and 94% N2 at 37°C as described previously [14].
Western blot analysis
After treatment, cells were lysed in ice-cold Tris buffer [50 mM Tris–HCl (pH 7.4)] containing 1% Triton, 50 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1 mM sodium orthovanadate, 1 mM NaF, 1 μg/ml aprotinin, 1 μg/ml leupeptin and 1 μg/ml pepstatin. Cell lysates including 20–30 μg protein were subjected directly to Western blot analysis with specific antibodies such as anti-AMPKα-pan, anti-phosphoThr-172 AMPK, anti-phosphoSer-79 ACC, anti-ACC (Cell Signaling Technologies, Beverly, MA, USA), HIF-1α (R&D Systems, Minneapolis, MN, USA), CaMKKα (BD Biosciences-Pharmingen, San Diego, CA, USA). Blots were developed using an enhanced chemiluminescence reagent (Amersham Biosciences, Piscataway, NJ, USA), and scanned and quantified using a calibrated densitometer (Bio-Rad, Hercules, CA, USA).
Measurement of ATP, ADP and AMP in cardiomyocytes by high performance liquid chromatography (HPLC)
The intracellular AMP, ADP and ATP contents were determined in neutralized perchloric acid extracts of cardiomyocytes using HPLC as described previously [15].
AMPK activity assay
AMPK was immunoprecipitated from 100 μg cell lysate protein using a rabbit anti-pan-α antibody (Upstate Biotechnology, Lake Placid, NY, USA) and protein A-Sepharose. AMPK activity was measured using the SAMS peptide (Upstate Biotechnology) phosphorylation assay as reported previously [16]. AMPK activity was calculated and expressed as pmol/min./mg of protein.
Construction of recombinant adenoviruses and infection
An adenoviral vector expressing a dominant negative mutant of AMPK-α1 (DN-AMPK) was constructed by mutation of Thr172 to Ala172 as described previously [17]. An adenoviruse expressing a dominant negative form of LKB1 was constructed by single amino acid substitution of Alanine for Asparagine 194 of LKB1 (kinase inactive, D194A mutant) as previously described [18]. Ad-GFP, a replication-defective adenoviral vector expressing green fluorescence protein (GFP), was used as control. Both constructs were made by Genechem Co., Ltd, Shanghai, China. Cardiomyocytes were infected with Ad-GFP, or DN-AMPK or DN-LKB1 at a multiplicity of infection (MOI) of 30, 20 and 40, respectively. After 24–26 hrs, the medium was renewed and the cells were either harvested or cultured for subsequent experiments.
For PHD2 siRNA knockdown in cardiomyocytes, short hairpin RNA was designed according to the complementary DNA sequence of the EGLN1 (GenBank NM_178334). The PHD2 small hairpin RNA (shRNA) expression vector was built using pGenesil-1/U6 (Genesil Biotechnology, Wuhan, China) as parental plasmid and nucleotide positions 554 to 572 of PHD2 (5′-CTGGTCAGCCAGAAGAGTG-3′) as the target. A negative control sequence was also designed which had no homology with rat, mouse or human. All of the constructs used in this study were verified by DNA sequencing. The shRNA adenoviral vector was linearized with PacI and transfected into 293A cells using Lipofectamine following the protocols. Approximately 12 days after transfection, the adenovirus-containing 293A cells were harvested and lysed to prepare a crude viral stock. The resultant viral stock was amplified, and the viral concentration was determined. Two days after plating, cardiomyocytes were infected with the virus at MOI 20.
Lentiviral siRNA vector for CaMKKα and transduction
Small hairpin RNA of rat CaMKKα lentivirus gene transfer vector encoding GFP was constructed by Genechem Co., Ltd, Shanghai, China. The targeting sequence of the shRNA was 5’-GAAACTGGACCACGTGAAT-3’ (GenBank Accession No.NM_031662), and confirmed by sequencing. The recombinant lentivirus of shRNA targeting CaMKKα (CaMKKα-RNAi-Lentivirus) and the negative control lentivirus (5′-TTCTCCGAACGTGTCACGT-3′-lentivirus) which contain a scrambled, non-targeting sequence were prepared and titred to 109 TU (transfection unit)/ml. The cardiomyocytes were plated for 2 days, then incubated with the lentiviral vectors with the addition of polybrene (10 μg/ml) and enhanced transfect solution at various MOI for 3, 4 and 5 days, respectively. At day 4 after transduction, cells were either harvested or treated with PHI and then harvested.
RNA isolation and real-time PCR
Total RNA was extracted from the cardiomyocytes using the TRIZOL (Invitrogen, Carlsbad, CA, USA). RNA was reverse transcribed using the Ambion reverse transcription Retroscript kit, Real-time PCR was performed using the SYBR Green Real-time PCR Master Mix (TOYOBAO, Japan), DNA templates were amplified by real-time PCR on the Applied Biosystems SDS 7500 instrument (ABI, Foster City, CA, USA) using the Sybr green method [19]. GAPDH and β-actin were used as internal control. The primer sequences were: GAPDH 5′-TTCAACGGCACAGTCAAGG-3′ and 5′-CTCAGCACCAGCATCACC-3′. β-actin 5′-GTT GAC ATC CGT AAA GAC C-3′ and 5′-GAC TCA TCG TAC TCC TGC T-3′. CaMKKα5′-GGAAAGACCAGCGGGAAG-3′ and 5′-CAGCCTCACCACACCATAG-3′. PHD2 5′-CCCTGGTGGACATCTT-3′ and 5′-TCTCCGCTCTGAACAA-3′. Experimental data were analysed and the relative levels of gene expression were calculated by 7500 System SDS software.
2-(3H)deoxyglucose uptake assay
After treatments, the cells were incubated with 2-(3H)deoxyglucose (1μCi/ml) (Beijing Atom Hightech, Beijing, China) at room temperature for 15 min. Then reactions were terminated by washing cells with ice-cold phloretin buffer, and cells were lysed directly on plates with 0.05 M NaOH. Extracts were counted via γ-scintillation (Beckman LS3801 version-3.0-D). Parallel non-radiolabelled cultures treated as above were harvested for protein determinations. Protein concentration was determined using the Bradford reagent (Sigma-Aldrich).
Cell viability assay
Cardiomyocytes were plated at a density of 2.0 χ 105 cells/well in 96-well plates, after treatment the cell viability was examined using cell counting kit-8 (Dojindo Laboratory, Kumamoto, Japan) following manufacturer’s protocol. The relative number of surviving cells was determined in duplicates with the value of untreated cells as 100%.
Statistical analysis
Data were expressed as the mean ± S.D. Statistical analyses were performed using SPSS 11.0 (SPSS, Chicago, IL, USA) and statistical significance evaluated by ANOVA followed by post-hoc tests. P ≤ 0.05 was considered significant.
Results
PHIs activate AMPK in cardiomyocytes
We first attempted to evaluate the effect of DMOG, the widely used specific PHI, on AMPK activity in neonatal rat cardiomyocytes. Figure 1 shows that, under normoxic conditions DMOG caused a time- and dose-dependent increase in AMPK phosphorylation at Thr172, a crucial site for enzyme activity. This increase in Thr172 phosphorylation occurred simultaneously with the increased phosphorylation of acetyl-CoA carboxylase (ACC) at Ser79 (Fig. 1A and B), which is a putative AMPK substrate; while the total amount of ACC or AMPKα subunit was essentially the same. Similar results were obtained when AMPK activity was measured by 32P incorporation into the AMPK-specific SAMS peptide (Fig. 1C and D). AMPK activation induced by PHI was detectable at 0.5–1 hr, peaking at 6 hrs and persisted for at least 9 hrs. The HIF-1α induction and AMPK activation by DMOG occurred at a similar time point (Fig. 1A and B), thus revealing that PHI could effectively open the PHD-oxygen sensing pathway in cardiomyocytes, and therefore activate the AMPK pathway. Furthermore, there was no obvious alteration in PHD1, 2, 3 protein contents in cardiomyocytes treated with DMOG (Fig. 1E and F).
Fig 1.
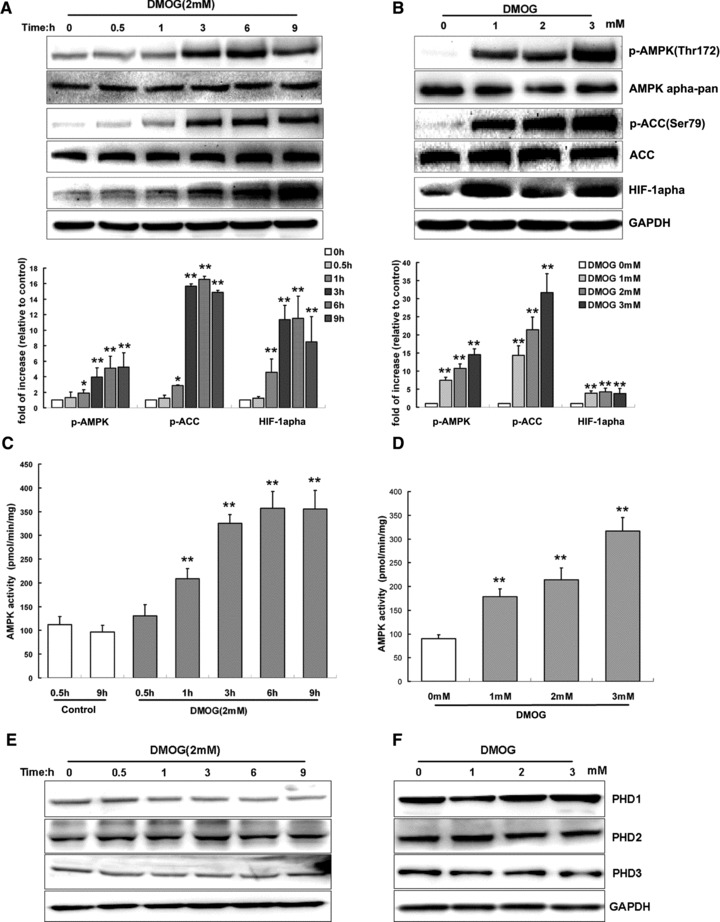
PHIs activate AMPK activity in cardiomyocytes in a time- and dose-dependent manner. Cardiomyocytes were treated with DMOG (2 mM) for indicated time periods (A, C, E) or different dose of DMOG for 3 hrs (B, D, F). For A and B, representative immunoblots together with densitometric analysis of four experiments are shown. Target protein phosphorylation has been normalized to the expression of the total protein. For C and D, AMPK activity was measured and shown as pmol/min./mg of protein. For E and F, representative immunoblots of PHD1, 2, 3 under same DMOG-treated conditions were shown. Each bar represents the mean ± S.D. for three independent assays in duplicate. **P < 0.01 versus control; *P < 0.05 versus control.
Depletion of PHD2 leads to AMPK activation in cardiomyocytes
PHD2 is considered as a main cellular oxygen sensor [20, 21]. To investigate the effects of PHD oxygen-sensing pathway on AMPK, we employed RNA-interference technique to knock down PHD2 in cardiomyocytes. Real-time PCR revealed that adenoviral mediated PHD2-specific shRNA, but not a scrambled sequence, significantly diminished PHD2 at mRNA level (Fig. 2A) and led to decrease of PHD2 protein level and HIF-1α accumulation under normoxic condition (Fig. 2B). PHD2 siRNA did not affect the expression of other genes, such as PHD1, PHD2 or ß-actin (Fig. 2A and B), suggesting that PHD2 shRNA specifically inhibited PHD2 transcription. Consistent with the decrease of PHD2, Ad-PHD2-shRNA induced significant increase in phosphorylation of AMPK and hence ACC in normoxic cardiomyocytes (Fig. 2C). Collectively, these results confirmed that opening the PHD pathway leads to activation of the AMPK pathway.
Fig 2.
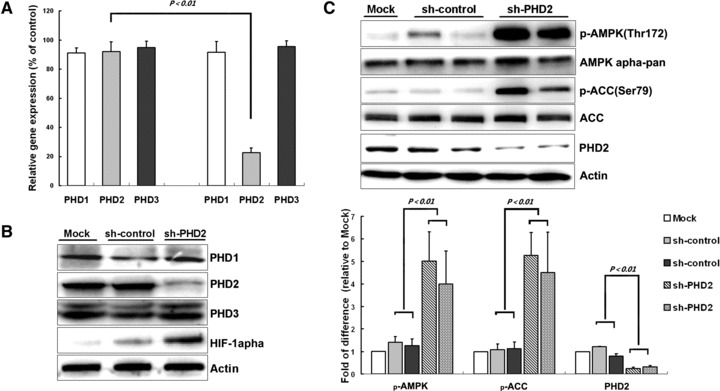
PHD2 knockdown leads to AMPK activation in cardiomyocyte. Cardiomyocytes were infected with adenoviruses for scrambled shRNA or PHD2 shRNA for 26 hrs, and then were collected for molecular analysis. (A) Real-time PCR for PHD1, 2, 3 (n = 3 independent experiments) shows fold change of mRNA. Values represent the mean ± S.D. (B and C) Representative immunoblots together with densitometric analysis (mean ± S.D.) of four independent experiments. AMPK and ACC phosphorylation has been normalized to the expression of total protein, respectively.
Opening of the PHD pathway increases glucose uptake via AMPK activation
Glucose uptake is an important protective manoeuvre against ischaemic/hypoxic injury especially in the heart [22]. To evaluate whether AMPK plays a role in mediating PHI-induced glucose uptake in cardiomyocytes, we first generated a dominant-negative mutant of AMPK-α1 (AMPK-DN) by mutation of Thr172 to alanin. Cardiomyocytes were infected with DN-AMPK vector or Ad-GFP vector as negative control. Western blot analysis confirmed the expression of DN-AMPK (Fig. 3A) and its inhibitory effect on endogenous AMPK activity, as demonstrated by reduced DMOG-mediated phosphorylation of AMPKα subunit (Thr172) and ACC Ser79 (Fig. 3B). To examine the contribution of AMPK to PHI-induced glucose uptake in the cardiomyocytes, myocytes were infected with DN-AMPK or Ad-GFP for 24 hrs, and then treated with DMOG or vehicle (DMSO) for 8 hrs before glucose uptake assay. The results showed that DMOG treatment facilitate 2-(3H)deoxyglucose uptake whereas these effects have been greatly abolished by DN-AMPK (Fig. 3C). These data indicate the critical role of AMPK in this process.
Fig 3.
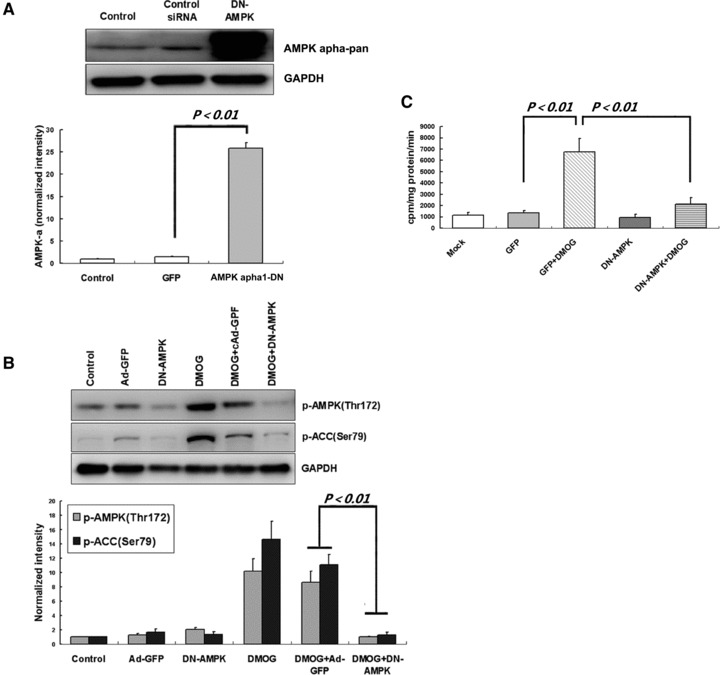
DMOG stimulates glucose uptake via AMPK activation. (A) Cardiomyocytes were exposed to 30 MOI of Ad-GFP or 20 MOI of DN-AMPK adenovirus for 24 hrs, hence, an equal transfection efficacy above 95% was obtained; cells were then harvested for Western blot analysis. (B) After adenovirus infection, cardiomyocytes were then exposed to 2 mM DMOG for three hrs before immunoblot assay. Representative immunoblots together with densitometric analysis (mean ± S.D.) of four independent experiments are shown. AMPK and ACC phosphorylation has been normalized to GAPDH expression. (C) After infection, cells were further incubated with DMOG (2 mM) or vehicle (DMSO) for 8 hrs. Then, 2-[3H]deoxy-D-glucose was added to culture media for 15 min., and glucose uptake was measured 3H accumulation rate which normalized to protein content. Each column represents means ± S.D. for three independent assays in duplicate.
Effects of AMPK inhibition on hypoxic cell viability in cardiomyocytes exposed to PHI
Previous studies have shown that PHI treatment induces tolerance against metabolic inhibition and hypoxic/ischaemic stress. To test whether AMPK is involved in PHI-induced hypoxic tolerance, we infected cardiomyocytes with DN-AMPK vector or Ad-GFP vector as negative control, and then treated them with DMOG before hypoxia exposure. The results showed that pretreatment with DMOG significantly increased cellular resistance to hypoxia as reflected by increased viability of cardiomyocytes infected with Ad-GFP (Fig. 4A). However, DMOG-induced cellular resistance to hypoxia was abrogated when the endogenous AMPK activity was inhibited by DN-AMPK (Fig. 4B). These results indicate that AMPK pathway plays an essential role in cellular protective effects induced by PHI against hypoxic stress.
Fig 4.
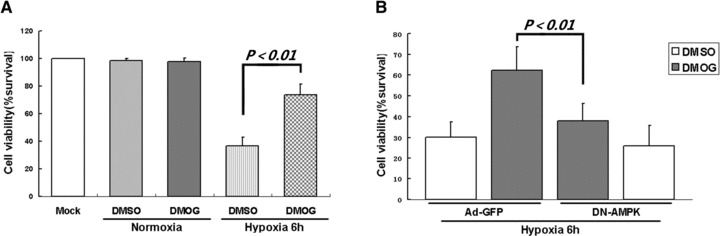
AMPK activation is critical for PHI-induced hypoxic protection in cardiomyocytes. (A) Cardiomyocytes were incubated with DMOG (2 mM) or vehicle (DMSO) for 12 hrs, then subjected to hypoxic condition for six hrs. (B) Cardiomyocytes were infected with Ad-GFP or DN-AMPK constructs for 24 hrs, then exposed to DMOG (2 mM) or vehicle (DMSO) for 12 hrs, and subjected to hypoxia for 6 hrs. Cell viability was determined by CCK-8 assay. Data represent mean ± S.D. of three independent experiments in duplicate, and the cell survival ratio was expressed as a percentage of the control.
PHI has no effect on AMP:ATP ratio in cardiomyocytes
To investigate whether PHI activate AMPK by increasing the AMP:ATP ratio in the cardiomyocytes, we measured intracellular AMP and ATP concentrations in vehicle- or DMOG-treated cardiomyocytes using HPLC. We found that DMOG (3 mM) had not induced significant difference in the intracellular AMP:ATP ratio compared to control (Table 1). These results suggest that PHI activate AMPK by mechanisms other than changing intracellular AMP:ATP ratio.
Table 1.
PHIs do not increase the intracellular AMP:ATP ratio in cardiomyocytes
| ATP | ADP | AMP | AMP/ATP | |
|---|---|---|---|---|
| 1-hr incubation (nmol/mg protein) | ||||
| 3 mM DMOG | 16.8 ± 2.8 | 2.9 ± 0.8 | 0.34 ± 0.06 | 0.02 ± 0.002 |
| Control | 18.1 ± 3 | 2.7 ± 0.3 | 0.33 ± 0.03 | 0.019 ± 0.005 |
| 3-hr incubation (nmol/mg protein) | ||||
| 3 mM DMOG | 17.7 ± 3.2 | 2.7 ± 0.5 | 0.34 ± 0.06 | 0.019 ± 0.003 |
| Control | 17.7 ± 2.2 | 2.6 ± 0.2 | 0.33 ± 0.02 | 0.021 ± 0.004 |
| 6-hr incubation (nmol/mg protein) | ||||
| 3 mM DMOG | 18.3 ± 2.9 | 2.9 ± 0.2 | 0.35 ± 0.05 | 0.019 ± 0.003 |
| Control | 17.4 ± 3.2 | 2.8 ± 0.6 | 0.38 ± 0.08 | 0.022 ± 0.003 |
Cardiomyocytes were treated with vehicle, 3 mM DMOG for the indicated time. The intracellular AMP and ATP concentrations were measured by HPLC and the AMP:ATP ratio was calculated. Values represent mean ± S.D. for at least three independent assays in duplicate.
LKB1 is not required for PHI-induced AMPK activation in cardiomyocytes
LKB1 has been proved to be the main upstream kinase of AMPK in the heart. To test whether LKB1 kinase is involved in PHI-induced AMPK activation, a dominant-negative mutant of LKB1 (DN-LKB1, D194A mutant) was expressed in cardiomyocytes by adenovirus (Fig. 5A). Expression of DN-LKB1 significantly inhibited the AICAR-induced phosphorylation of AMPK and ACC but did not alter the activation of AMPK pathway induced by DMOG treatment, as the phosphorylation of AMPK and ACC had not been inhibited by DN-LKB1 (Fig. 5B). These results strongly suggest that LKB1 is not the main kinase involved in PHI induced AMPK activation.
Fig 5.
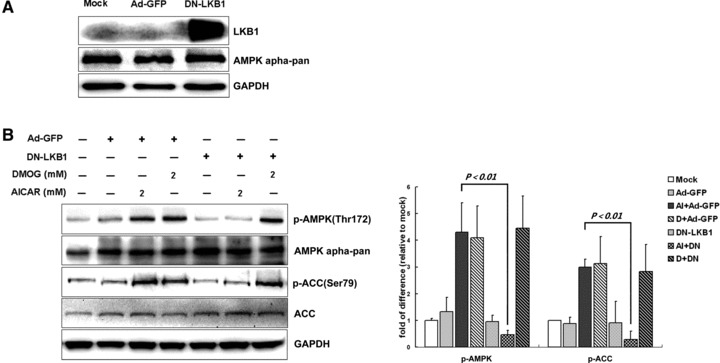
LKB1 is not required for PHI-induced AMPK activation in cardiomyocytes. (A) Cardiomyocytes were exposed for 24 hrs to 30 MOI of Ad-GFP or 40 MOI of DN-LKB1 adenovirus, after that, cells had been harvested for Western blot analysis. (B) After adenovirus infection with Ad-GFP or DN-LKB1 for 24 hrs, cardiomyocytes were then further exposed to 2 mM DMOG or vehicle (DMSO) for 3 hrs before immunoblot assay. Representative immunoblots together with densitometric analysis (mean ± S.D.) of four independent experiments are shown. AMPK and ACC phosphorylation has been normalized to the total protein expression, respectively.
CaMKKα mediates AMPK activation upon PHI stimulation in cardiomyocytes
Next we explored another mechanism that may account for PHI-induced AMK activation. Since AMPK activation is Ca2+-dependent in some contexts [23, 24], we first treated cardiomyocytes with the Ca2+ ionophore ionomycin (1 μM) and indeed found AMPK phosphorylation (Fig. 6A), suggesting that Ca2+ may be an upstream signal for AMPK activation in cardiomyocytes. Next, we found that chelation of intracellular Ca2+ by BAPTA led to significant inhibition of AMPK phosphorylation at Thr172 stimulated by DMOG (Fig. 6B). Correspondingly, ACC phosphorylation at Ser79 was reduced, which presented inhibition of AMPK activity. Then, we used STO-609, a relatively selective inhibitor of CaMKKα and CaMKKß [25] and found that it caused significant reduction of DMOG-induced AMPK and ACC phosphorylation (Fig. 6C). Although CaMKK β is the most functionally relevant AMPK kinase [24], we could not detect CaMKKβ in cardiomyocytes in this study. It has also been demonstrated that both CaMKK-α and -β function as AMPKK [26], so we examined whether CaMKKα acts as the AMPK kinase in PHI-stimulated cardiomyocytes. To this end, we knocked down CaMKKα in cardiomyocytes by shRNA and found that AMPK and ACC phosphorylation induced by DMOG was significantly reduced while AMPK and ACC phosphorylation induced by AICAR, an activator of AMPK mimicking the action of AMP, were not significantly changed (Fig. 6D), suggesting that CaMKKα was the major AMPK kinase under these conditions. The selective down-regulation of CaMKKα by RNAi was confirmed at both protein and mRNA levels (Fig. 6E and F).
Fig 6.
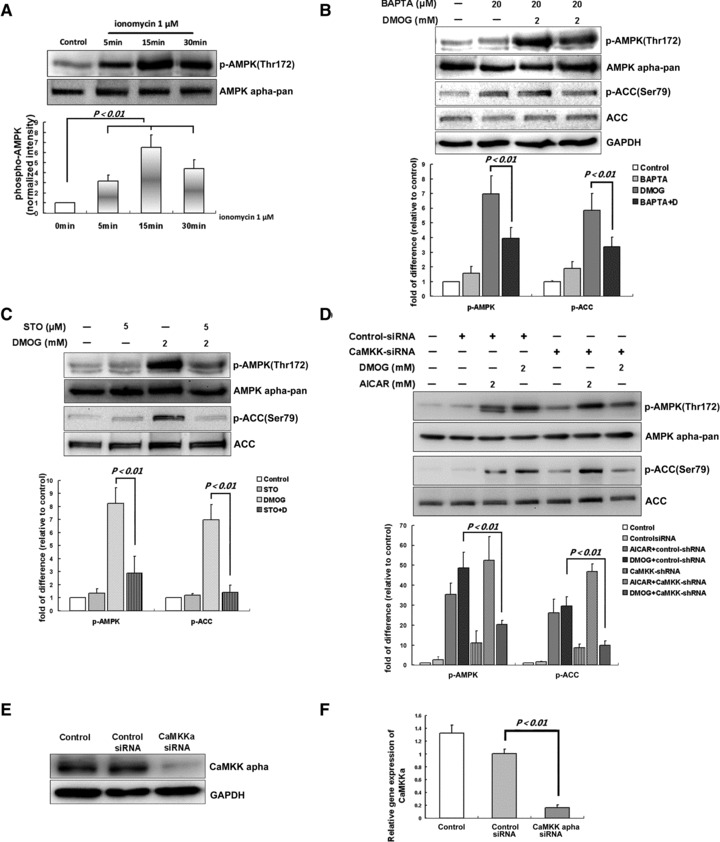
CaMKKα is the major AMPK kinase upon PHI stimulation in cardiomyocytes. (A) Cardiomyocytes were stimulated with ionomycin (1 μM) for the indicated times. (B) Cardiomyocytes were pretreated with BAPTA-AM (20 μM, 30 min.) and subsequently stimulated with DMOG (2 mM) for three hrs. (C) Cardiomyocytes were preincubated with STO-609 (5 μM) for 30 min. before DMOG stimulation for three hrs; cells were lysed and subjected to immunoblotting. (D) Cardiomyocytes were incubated with transfection reagent only [polybrene (10 μg/ml), control] or with lentiviral shRNA vector targeted to rat CaMKKα or lentiviral control-shRNA (1 μg/30-mm dish) for four days. After that, cells were incubated with DMOG (2 mM), AICAR (2 mM) or DMSO for three hrs, and harvested for Western blot analysis. Representative blots and densitometry data (means ± S.D.) of four independent experiments for each treatment are shown. Cardiomyocytes were infected with lentiviral CaMKKα shRNA vector (1 μg/30-mm dish, 96 hrs) or control-shRNA or incubated with transfection reagent only (polybrene (10 μg/ml), control]. Representative immunoblot of CaMKKα (E) and graph of real-time PCR for CaMKKα (F) are shown. Values represent the mean ± S.D., n = 3 independent experiments. AMPK and ACC phosphorylation has been normalized to the total protein expression, respectively.
Discussion
The prolyl hydroxylase domain proteins (PHD1-3) are oxygen-sensitive enzymes that serve as cellular oxygen sensors and provide a direct link between oxygen availability and cellular adaptations in response to hypoxia. On the other hand, AMPK acts as a critical regulator of energy homeostasis. The activation of AMPK is also considered as one of the typical cellular responses elicited by low oxygen tension. But the connection between them remains poorly understood. In the present study, we demonstrate for the first time that opening the PHD-oxygen sensing pathway using either two structurally distinct PHIs or PHD2-RNAi can activate the cellular energy sensor AMPK system in cardiomyocytes.
All three PHDs have Km values for oxygen slightly higher than tissue oxygen concentrations during normoxia, which is proposed to enable the PHDs to function as effective cellular oxygen sensors [27, 28]. Among the three different members of the PHD family, PHD2 has been shown as the most abundant and critical hydroxylase in a large variety of cultured cells studied [20, 21]. siRNA-mediated silencing of PHD2 is sufficient to stabilize HIF-1α in normoxia, indicating that this isoenzyme is the major oxygen sensor setting low steady-state levels of HIF-1α in normoxia. According to these findings one would anticipate that activation of PHD pathway could be the very first cellular event occurred when oxygen pressure drops. Based on our present finding that silencing of PHD2 could activate AMPK pathway, it is plausible that hypoxic signal could be transduced directly through PHD to cellular energy metabolism, thus providing a direct link between oxygen availability and cellular metabolic adaptations in response to hypoxia.
Full activation of the PHD pathway can be achieved using the broad-spectrum PHI. The protective effects of PHI against ischaemic/hypoxic stress have been investigated extensively in recent years. It has been demonstrated that pretreatment with PHI significantly reduced post-ischaemic infarct size in rabbit hearts [29], and systemic treatment of mice with PHI significantly increased viability and enhanced exercise performance of mice in hypoxia [30]. Similarly, in the present study, cardiomyocytes treated with PHI were shown to have enhanced survival rates under hypoxic exposure and increased glucose uptake under normoxic conditions. Furthermore, we found that the attenuated hypoxic myocyte’s death and elevated glucose uptake mediated by PHI pretreatments were abrogated substantially upon the inhibition of endogenous AMPK by DN-AMPK, suggesting a major role of AMPK pathway in PHI-induced hypoxic adaptive processes.
In this study, the increased AMPK activity induced by DMOG was in accordance with its phosphorylation at Thr172, suggesting that DMOG-induced AMPK activation occurs mainly via its phosphorylation by an AMPK upstream kinase. Two AMPK upstream kinases have been identified in the heart, i.e. tumour suppressor LKB1 [31] and CaMKK [23]. Although LKB1 has been reported to exist and function in the heart [32], we did not find obvious evidence for the contribution of LKB1 to PHI-induced AMPK activation. First, PHI treatment did not alter intracellular AMP:ATP ratio significantly. Although LKB1 is not activated directly by AMP [12], LKB1-AMPK signalling pathway is believed to be activated by an elevation of the intracellular AMP:ATP ratio [33]. Second, down-regulation of LKB1 with its DN form did not significantly block the PHI-induced AMPK and ACC phosphorylation while abrogated AICAR-induced AMPK and ACC phosphorylation. AICAR is converted into an AMP mimetic within the cell, and has previously been reported to act through LKB1 [34].
In contrast, many previous studies have indicated that AMPK activation through the CaMKK pathway is independent of the AMP:ATP ratio, but is instead induced by a rise in intracellular Ca2+ concentration [23, 24]. In this study, we are the first to demonstrate that CaMKKα is the main upstream kinase of AMPK in cardiomyocytes under DMOG treatment based on several lines of evidence. First, we found that Ca2+ ionophore was able to induce AMPK phosphorylation in cardiomyocytes and AMPK activation by DMOG was significantly abolished by Ca2+-chelating agent, suggesting that intracellular Ca2+ increase was required in PHI-induced AMPK activation. Second, CaMKK inhibitor STO-609 significantly reduced DMOG-induced increase in phosphorylation of AMPK-Thr172 and ACC-Ser79. Third, down-regulation of CaMKKα with specific shRNA significantly reduced DMOG-induced phosphorylation of AMPK and ACC. Taken together, these results strongly suggest that CaMKKα is the upstream kinase mediating AMPK activation in cardiomyocytes upon PHI treatments. It is worth noting that although we were not able to detect CaMKK by immunoblot analysis in cultured neonatal rat cardiomyocytes, our results could not exclude the possibility that CaMKKß also participates in the regulation of AMPK upon PHI treatment. In addition, neither STO-609 nor CaMKKα shRNA was able to block PHI-induced AMPK activation completely as verified by immunoblot analysis. This may reflect incomplete inhibition of enzyme activity or knockdown of CaMKK, but the contribution of another, as yet unidentified, AMPK kinase cannot be excluded.
In summary, we establish for the first time that activation of PHD signal pathway via its pharmacologic inhibitor or PHD2-RNAi activates AMPK pathway in cardiomyocytes. In the context of PHI treatment, PHD signal to AMPK mainly via a Ca2+/CaMKK-dependent pathway in cardiomyocytes, and AMPK activation is essential for PHI to exert protective effects against hypoxic stress. Our results also imply that the PHD/Ca2+/CaMKK/AMPK pathway is present in heart cells and could be the signal pathway that leads to AMPK activation during the early stage of hypoxia, before the metabolic disturbance occurs.
Acknowledgments
This study was supported by National Natural Science Foundation of China (NSFC) program (30600649), Key Program of NSFC (30430680).
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–40. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 2.Li SH, Shin DH, Chun YS, et al. A novel mode of action of YC-1 in HIF inhibition: stimulation of FIH-dependent p300 dissociation from HIF-1 alpha. Mol Cancer Ther. 2008;7:3729–38. doi: 10.1158/1535-7163.MCT-08-0074. [DOI] [PubMed] [Google Scholar]
- 3.Ivan M, Kondo K, Yang HF, et al. HIF alpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O-2 sensing. Science. 2001;292:464–8. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 4.Kuznetsova AV, Meller J, Schnell PO, et al. von Hippel-Lindau protein binds hyperphosphorylated large subunit of RNA polymerase II through a proline hydroxylation motif and targets it for ubiquitination. Proc Natl Acad Sci U S A. 2003;100:2706–11. doi: 10.1073/pnas.0436037100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cummins EP, Berra E, Comerford KM, et al. Prolyl hydroxylase-1 negatively regulates I kappa B kinase-beta, giving insight into hypoxia-induced NF kappa B activity. Proc Natl Acad Sci U S A. 2006;103:18154–9. doi: 10.1073/pnas.0602235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elvidge GP, Glenny L, Appelhoff RJ, et al. Concordant regulation of gene expression by hypoxia and 2-oxoglutarate-dependent dioxygenase inhibition – the role of HIF-1 alpha, HIF-2 alpha, and other pathways. J Biol Chem. 2006;281:15215–26. doi: 10.1074/jbc.M511408200. [DOI] [PubMed] [Google Scholar]
- 7.Wright G, Higgin JJ, Raines RT, et al. Activation of the prolyl hydroxylase oxygen-sensor results in induction of GLUT1, heme oxygenase-1, and nitric-oxide synthase proteins and confers protection from metabolic inhibition to cardiomyocytes. J Biol Chem. 2003;278:20235–9. doi: 10.1074/jbc.M301391200. [DOI] [PubMed] [Google Scholar]
- 8.Lomb DJ, Straub JA, Freeman RS. Prolyl hydroxylase inhibitors delay neuronal cell death caused by trophic factor deprivation. J Neurochem. 2007;103:1897–906. doi: 10.1111/j.1471-4159.2007.04873.x. [DOI] [PubMed] [Google Scholar]
- 9.Sridharan V, Guichard J, Bailey RM, et al. The prolyl hydroxylase oxygen-sensing pathway is cytoprotective and allows maintenance of mitochondrial membrane potential during metabolic inhibition. Am J Physiol-Cell Physiol. 2007;292:C719–C28. doi: 10.1152/ajpcell.00100.2006. [DOI] [PubMed] [Google Scholar]
- 10.Hardie DG. AMP-activated protein kinase: the guardian of cardiac energy status. J Clin Invest. 2004;114:465–8. doi: 10.1172/JCI22683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kahn BB, Alquier T, Carling D, et al. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 12.Baron SJ, Li J, Russell RR, et al. Washington, DC: Lippincott Williams & Wilkins; 2003. Dual mechanisms regulating AMPK kinase action in the ischemic heart; pp. 337–345. [DOI] [PubMed] [Google Scholar]
- 13.Altarejos JY, Taniguchi M, Clanachan AS, et al. Myocardial ischemia differentially regulates LKB1 and an alternate 5 ‘-AMP-activated protein kinase kinase. J Biol Chem. 2005;280:183–90. doi: 10.1074/jbc.M411810200. [DOI] [PubMed] [Google Scholar]
- 14.Seko Y, Takahashi M, Tobe K, et al. Hypoxia and hypoxia/reoxygenation activate p65(PAK), p38mitogen-activated protein kinase (MAPK), and stress-activated protein kinase (SAPK) in cultured rat cardiac myocytes. Biochem Biophys Res Commun. 1997;239:840–4. doi: 10.1006/bbrc.1997.7570. [DOI] [PubMed] [Google Scholar]
- 15.Ally A, Park G. Rapid-determination of creatine, phosphocreatine, purine-bases and nucleotides (ATP, ADP, AMP, GTP, GDP) in heart biopsies by gradient ion-pair reversed-phase liquid-chromatography. J Chromatogr. 1992;575:19–27. doi: 10.1016/0378-4347(92)80499-g. [DOI] [PubMed] [Google Scholar]
- 16.Davies SP, Carling D, Hardie DG. Tissue distribution of the AMP-activated protein-kinase, and lack of activation by cyclic-AMP-dependent protein-kinase, studied using a specific and sensitive peptide assay. Eur J Biochem. 1989;186:123–8. doi: 10.1111/j.1432-1033.1989.tb15185.x. [DOI] [PubMed] [Google Scholar]
- 17.Pelletier A, Joly E, Prentki M, et al. Adenosine 5 ‘-monophosphate-activated protein kinase and p38 mitogen-activated protein kinase participate in the stimulation of glucose uptake by dinitrophenol in adult cardiomyocytes. Endocrinology. 2005;146:2285–94. doi: 10.1210/en.2004-1565. [DOI] [PubMed] [Google Scholar]
- 18.Imai K, Inukai K, Ikegami Y, et al. LKB1, an upstream AMPK kinase, regulates glucose and lipid metabolism in cultured liver and muscle cells. Biochem Biophys Res Commun. 2006;351:595–601. doi: 10.1016/j.bbrc.2006.10.056. [DOI] [PubMed] [Google Scholar]
- 19.Resende RR, Gomes KN, Adhikari A, et al. Mechanism of acetylcholine-induced calcium signaling during neuronal differentiation of P19 embryonal carcinoma cells in vitro. Cell Calcium. 2008;43:107–21. doi: 10.1016/j.ceca.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 20.Berra E, Benizri E, Ginouves A, et al. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1 alpha in normoxia. EMBO J. 2003;22:4082–90. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Appelhoff RJ, Tian YM, Raval RR, et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279:38458–65. doi: 10.1074/jbc.M406026200. [DOI] [PubMed] [Google Scholar]
- 22.Malhotra R, Brosius FC. Glucose uptake and glycolysis reduce hypoxia-induced apoptosis in cultured neonatal rat cardiac myocytes. J Biol Chem. 1999;274:12567–75. doi: 10.1074/jbc.274.18.12567. [DOI] [PubMed] [Google Scholar]
- 23.Hawley SA, Pan DA, Mustard KJ, et al. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 24.Woods A, Dickerson K, Heath R, et al. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2:21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 25.Tokumitsu H, Inuzuka H, Ishikawa Y, et al. STO-609, a specific inhibitor of the Ca(2+)/calmodulin-dependent protein kinase kinase. J Biol Chem. 2002;277:15813–8. doi: 10.1074/jbc.M201075200. [DOI] [PubMed] [Google Scholar]
- 26.Hurley RL, Anderson KA, Franzone JM, et al. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem. 2005;280:29060–6. doi: 10.1074/jbc.M503824200. [DOI] [PubMed] [Google Scholar]
- 27.Ehrismann D, Flashman E, Genn DN, et al. Studies on the activity of the hypoxia-inducible-factor hydroxylases using an oxygen consumption assay. Biochem J. 2007;401:227–34. doi: 10.1042/BJ20061151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirsila M, Koivunen P, Gunzler V, et al. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J Biol Chem. 2003;278:30772–80. doi: 10.1074/jbc.M304982200. [DOI] [PubMed] [Google Scholar]
- 29.Natarajan R, Salloum FN, Fisher BJ, et al. Hypoxia inducible factor-1 activation by prolyl 4-hydroxylase-2 gene silencing attenuates myocardial ischemia reperfusion injury. Circ Res. 2006;98:133–40. doi: 10.1161/01.RES.0000197816.63513.27. [DOI] [PubMed] [Google Scholar]
- 30.Kasiganesan H, Sridharan V, Wright G. Prolyl hydroxylase inhibitor treatment confers whole-animal hypoxia tolerance. Acta Physiol. 2007;190:163–9. doi: 10.1111/j.1748-1716.2007.01676.x. [DOI] [PubMed] [Google Scholar]
- 31.Hawley SA, Boudeau J, Reid JL, et al. Complexes between the LKB1 tumour suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sakamoto K, Zarrinpashneh E, Budas GR, et al. Deficiency of LKB1 in heart prevents ischemia-mediated activation of AMPK alpha 2 but not AMPK alpha 1. Am J Physiol Endocrinol Metab. 2006;290:E780–E8. doi: 10.1152/ajpendo.00443.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Witters LA, Kemp BE, Means AR. Chutes and ladders: the search for protein kinases that act on AMPK. Trends Biochem Sci. 2006;31:13–6. doi: 10.1016/j.tibs.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 34.Shaw RJ, Kosmatka M, Bardeesy N, et al. The tumour suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004;101:3329–35. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]