

Abstract
Zeta-associated protein of 70 kD (ZAP70) is a recognized adverse prognostic marker in chronic lymphocytic leukaemia (CLL) associated with enhanced B-cell receptor signalling, significantly more aggressive disease course and poor overall survival. Zeta-associated protein of 70 kD is ordinarily expressed in T cells where it has a crucial role in T-cell receptor signalling, whereas its aberrant expression in CLL leads to enhanced B-cell receptor signalling and significantly more aggressive disease course. Although much is known about the activation of ZAP70 following engagement of T-cell receptor, there are little data on the regulation of ZAP70 gene expression in normal T cells or CLL. To understand the molecular events underpinning epigenetic regulation of ZAP70 gene in CLL, we have defined ZAP70 promoter region and outlined the regions crucial in regulating the gene activity. Following a direct comparison of ZAP70+ and ZAP70− primary CLLs, we show ZAP70 promoter in expressing CLLs to be associated with a spectrum of active histone modifications, some of which are tightly linked to aberrant DNA methylation in CLL. Cross-talk between histone modifications and reduced DNA methylation culminates in transcriptional de-repression of ZAP70. Moreover, treatment with histone deacetylase (HDAC) and DNA methylation inhibitors results in recovery of ZAP70 expression, which provides a possible explanation for the failure of HDAC inhibitors in CLL treatment and might serve as a cautionary warning for their future use in treatment of this leukaemia.
Keywords: chronic lymphocytic leukaemia, histone modifications, DNA methylation, ZAP70
Introduction
Chronic lymphocytic leukaemia is a heterogeneous disease with a variable clinical outcome. In its presentation, it ranges from a very mild, symptom and complication-free disease characterized by expression of mutated immunoglobulin variable heavy-chain region (IgVH) genes at one end of the spectrum, to an aggressive, symptomatic disease that requires urgent treatment at the other [1, 2]. In the latter, expression of unmutated IgVH genes is a key feature, widely accepted as a predictive marker of CLL with bad prognosis and poor overall survival rate. Determination of the mutational status of IgVH genes in CLL is not routinely available as a diagnostic test in laboratories. However, it has been shown in several studies that expression of zeta-chain associated protein of 70 kD (ZAP70) is closely associated with unmutated IgVH status [3, 4]. Determination of ZAP70 expression in CLL can therefore be used as surrogate marker for unmutated IgVH associated with bad prognosis.
Zeta-chain associated protein of 70 kD is an intracellular protein tyrosine kinase (PTK) that is predominantly expressed in T cells where it has a crucial role in T-cell receptor (TCR) signalling highlighted by the presence of severe combined immunodeficiency in patients harbouring gene mutation [5]. In normal B cells, the B-cell receptor (BCR) signalling utilizes a related tyrosine kinase Syk. Therefore, ZAP70 and Syk fulfil similar roles in antigen-receptor signalling in T and B cells, respectively [6, 7]. Recent studies show that ZAP70 has a functional role in CLL whereby it enhances BCR signalling, which appears to be independent of its kinase activity [6, 8–10]. This leads to stronger BCR signalling, activation of NF-κB, enhanced responses to survival signals, higher proliferation and increased resistance to apoptosis [11]. As such, ZAP70 may actively contribute to the aggressive nature of ZAP70+ CLL, and has recently been shown to be a stronger risk factor than IgVH or CD38 [12]. It is therefore imperative that we gain a greater understanding into control of ZAP70 expression in CLL as mechanistic insight may provide potential therapeutic targets as well as avoid the use of treatments that may be detrimental.
It is well accepted that regulation of gene expression can be mediated by changes in chromatin structure and DNA methylation, collectively regarded as epigenetic control mechanisms. Previous reports show ZAP70 gene expression to be tightly linked to the DNA methylation status around intron I of the gene [13, 14]. However, very little information is available on chromatin changes associated with aberrant DNA methylation or expression status of ZAP70 gene in CLL. Furthermore, a number of agents used in clinical trials for treatment of CLL can alter transcriptional activation of target genes through effects on histone modifications. For example, HDAC inhibitors (HDACi) can enhance histone acetylation at gene promoters, which can lead to re-expression of previously silent genes [15, 16]. Although used in treatment of CLL, effects of these drugs, as well as the link between histone modifications and aberrant DNA methylation, have not been detailed in control of ZAP70.
In this study, we delineate changes in chromatin structure and histone modifications found in actively transcribed versus the silent ZAP70 gene locus in cell lines and primary patient CLL. We then test the effect of certain drugs used in clinical trials for treatment of CLL on these epigenetic events. We finally demonstrate the cross-talk between DNA methylation status of the ZAP70 gene, the associated histone modification and gene expression.
Materials and methods
Ethical approval for use of human samples
All patients consented for use of their blood/peripheral blood mononuclear cell (PBMC) samples as part of the ethically approved NIHR, UK Clinical Research Network Portfolio study, LYM1 (MREC No. 06/Q2202/30).
5′ RACE
Total RNA was isolated from Jurkat, Namalwa and primary patient CLL samples (CD19/CD5+ PBMCs, >98% pure by fluorescence activated cell sorter). mRNA was purified from approximately 100 μg total RNA using oligotex mini kit (Qiagen, Crawley, UK). Rapid amplification of 5′ complementary DNA end (5′ RACE) performed with Ambion First Choice RLM-RACE Kit (Life Technologies Ltd, Paisley, UK) according to manufacturer's instructions. One microlitre of cDNA from 5′ RLM RACE reaction was used as template for PCR amplification of ZAP70. Nested primer for generation of products performed with supplied primers in combination with ZAP70 coding exon specific primers; IZAPrev-TCA CGC GTC AGG CTG CTG TGG and ZAPEX3rev-ATG CCC GCC AGC TTC AGG TGC PCR performed with TITANIUM taq (Takara-Clontech, Mountain View, CA, USA). Both first and second round of PCR contained 400 nM each primer and consisted of 35 cycles of 30 sec. at 94°C, 30 sec. at 65°C and 30 sec. at 72°C. Bands of correct size for all four templates were excised and DNA cleaned up using Qiaquick gel extraction kit (Qiagen). Each DNA sample was sequenced with ZAPEX3Rrev primer.
Western blotting
SDS-PAGE and immunoblotting were performed as previously described [17]. Antibody recognizing ZAP70 (Abcam, Cambridge, UK) was used at 1 μg/ml and β-actin antibody (Sigma-Aldrich, Dorset, UK) at 1/1000 dilution. Rabbit anti-mouse horse radish peroxidase conjugated secondary antibody was used at 1:5000 dilution.
Isolation of primary patient CLL samples and negative selection of CD2+ cells by Robosep
Sodium butyrate (NaB) was added to fresh peripheral blood to final concentration of 5 mM. This was layered onto an equal volume of Ficoll (GE Healthcare, Amersham, UK) at room temperature and centrifuged at 13.8 g for 20 min. without brake at 4°C. The PBMC layer was extracted and washed twice in 20 ml of PBS containing 5 mM NaB. Obtained cells were washed in complete media and resuspended in 250 μl Robosep buffer and set up the Robosep machine for negative selection according to manufacturer's instructions (EasySep Human CD2 Positive Selection Kit, catalogue number 18657; StemCell Technologies, Grenoble, France). The cells that did not bind the column (i.e. CD2− population) were collected, resuspended in media and used in chromatin isolation protocol.
Micrococcal nuclease digestion
Native chromatin was prepared from Nalm6 and Namalwa cell lines as described previously [18]. Following nuclei isolation, a suspension of 1.6 ml of each cell line nuclei, at a concentration of 0.5 mg/ml, was placed in a siliconized eppendorf and digested with 50 U of micrococcal nuclease (MNase)/0.5 mg/ml nuclei at 37°C over a time course of 15 min. After each minute, a 100-μl sample of digested nuclei was taken and 5 mM EDTA added to it. DNA was isolated from all samples using phenol/chloroform extraction. To verify adequate digestion of chromatin, 20 μl of each sample was loaded onto a 1.2% (w/v) agarose gel, separated by electrophoresis and detected by ethidium bromide staining under UV illumination. PCRs were performed using R1 sense (F) and R4 antisense (R) primers to assess chromatin structure around the ZAP70 promoter; R1 only primers were utilized for PCR as a control for mononucleosomes. In total, 100 ng of genomic DNA was used as template in each reaction.
Native chromatin for chromatin immunoprecipitation (ChIP) assays was prepared as outlined above except that digestion was carried out for 5 min. (unless otherwise stated) and reaction was stopped by adding EDTA to a final concentration of 5 mM. The samples were placed on ice for 5 min., then centrifuged at 16.2 g for 10 min. at 4°C, and chromatin-containing supernatant was collected and stored at −80°C until required.
Chromatin immunoprecipitation assay
Native ChIP was carried out as described elsewhere [18, 19] using 100 μg native chromatin and antibodies raised against mono/di/tri-methylated histone 3 lysine 4 (H3K4; H3K4me1/2/3), acetylated H3K14, di/tri-methylated histone 3 lysine 9 (H3K9) (all from Abcam) and acetylated H3 and H4 (Millipore, Watford, UK). Primers used for qPCR analysis of ChIP reactions were: ZAP70 R1 were sense 5′-GGC TCC CTG TGG ACT CAG CT-3′ and antisense 5′-ACC AGC TAG ACA ACC CAG AGG AG-3′, with the expected product size of 200 bp. The primers for ZAP70 R2 were sense 5′-TGG GCT GAC TCA GTG GGT TTC-3′ and antisense 5′-TCC AGC AGC AGG AAC CGT CAG-3′, with the expected product size of 236 bp. The primers for ZAP70 R3 were sense 5′-TCC CTG GAG CAC GTG GCC TGT G-3′ and antisense 5′-CGC TTA CCT GAA TGC CCC AGC AG-3′, with the expected product size of 214 bp. The primers for ZAP70 R4 were sense 5′-GGA GAC CTG GCA GAG GAT GAA-3′ and antisense 5′-AAC AGG GCA CTT GCG GGG TCT-3′, with the expected product size of 227 bp. Average values of eluates were normalized to average values of inputs. Cross-linked ChIP was carried out as previously described [19]; 100 μg cross-linked chromatin was used with antibody targeting RNApolII (Abcam).
Decitabine and HDAC inhibitor treatment
Namalwa cells were treated with 1 μM Decitabine for 24 or 48 hrs. Following the incubation, the cells were harvested and RNA and chromatin were prepared from control and treated cells. RNA was utilized in generation of cDNA, and chromatin was used in ChIP assays with anti-acetylated H3 antibody. Namalwa cells were also treated with either 1 or 10 mM valproic acid (VPA) or 1 or 10 μM MS-275 for 48 hrs. Following the incubation, the cells were harvested and RNA and protein was prepared from control and treated cells. RNA was utilized in generation of cDNA, and protein was used in western blot with anti ZAP70 or β-actin antibody.
ZAP70 promoter-luciferase reporter constructs
Constructs spanning the ZAP70 promoter and 5′ end of the gene were designed to incorporate XhoI restriction enzyme site at 5′ end and HindIII at 3′ end to facilitate cloning into pGL4.17 luciferase reporter vector (Promega, Southampton, UK). The primer sequences for constructs were: Primer 1F: 5′-ATT CTC GAG AGT TCC ACA CAG GTC TCT GAG-3′; Primer 2R: 5′-TAT AAG CTT AAT TTC CTG GAT GCG AAG ACA-3′; Primer 3R: 5′-TAT AAG CTT AAT GCC AAT GGA GAG CCG GTT-3′; Primer 4F: 5′-ATT CTC GAG ACC TGG AAC GCA TCC CTG ACA-3′; Primer 5R: 5′-TAT AAG CTT AAG GCC CTG CCG CTC ACA CTC-3′; Primer 6R: 5′-TAT AAG CTT ACT CTG CCA GGT CTC CCA GAG-3′; Primer 7R:5′-TAT AAG CTT AGC AGC AGC TCT GAG GCC CAC-3′; Primer 8F:5′-ATT CTC GAG ATG GGC TGA CTC AGT GGG TTT-3′; Primer 9F:5′-ATT CTC GAG ACA GGC TTC CTC CCT TCT GAC-3′; Primer 10F: 5′-ATT CTC GAG ACA TCA AAC ACA GCC CCC TTT-3′; Primer 11F:5′-ATT CTC GAG AGG CCA CCC CAG GGG GCT CTG-3′.
Semi-quantitative and quantitative RT-PCR
Total RNA was obtained as previously described [17]; cDNA was generated using a random hexamer primer and Moloney murine leukemia virus reverse transcriptase. We used 20 ng of cDNA in each RT-PCR reaction. Semi-quantitative RT-PCR program was 3 min. at 94°C, then 30 cycles of 20 sec. at 94°C, 30 sec. at 55°C, 30 sec. at 72°C followed by 7 min. at 72°C. Quantitative PCR program: 20 sec. at 94°C, then 40 cycles of 5 sec. at 94°C, 20 sec. at 55°C, 30 sec. at 72°C followed by ABI7500 machine (Applied Biosystems, Paisley, UK) predetermined melt curve. All reactions were normalized to the internal control and relative level of transcriptional difference was calculated using the following equation: [1/(2A)] × 100.
Results
Effect of HDAC inhibitors on ZAP70 gene expression
Zeta-chain-associated protein of 70 kD gene expression is not only a strong prognostic marker for aggressive form of CLL but also a potential contributing factor to the higher grade disease. It is known that ZAP70 expression appears to be tightly linked to the DNA methylation status of the gene at the 5′ end. Reduction in DNA methylation is often followed by a subsequent alteration in histone modifications, in particular histone acetylation, which is, to a large extent, regulated by HDACs. A number of HDACi have been used in clinical trials for treatment of CLL [20, 21], with a number either ongoing or in the recruitment phase (please refer to http://clinicaltrials.gov; trail identifiers relating to a selection of trials—NCT00524667, NCT01090973, NCT00792831, NCT00083473, NCT00020579, NCT00431873, NCT00918723). HDACi alter chromatin structure and can cause re-expression of previously silenced genes [15, 16]. Although this is often a desirable outcome, we suggested that one possible side effect of HDACi treatment of CLL may be re-expression of ZAP70. To address this question, ZAP70− B cells were treated with VPA and MS-275, two HDACi currently used in clinical trials. We found that use of these agents at higher concentrations led to re-expression of ZAP70 gene (Fig. 1). To gain mechanistic insight into chromatin changes underpinning these events, as well as global regulation of ZAP70 gene expression, we first defined ZAP70 promoter.
Fig 1.
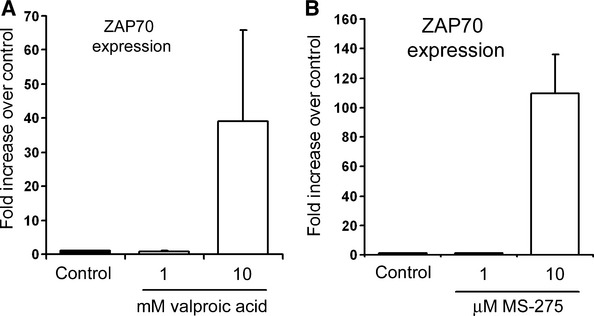
Use of HDACi leads to re-expression of ZAP70. (A) Namalwa cell line was treated with 1 or 10 mM valproic acid or vehicle control for 48 hrs; total RNA was prepared from both samples and used for RT-PCR detection of ZAP70 expression. (B) Namalwa cell line was treated with 1 or 10 μM MS-275 or vehicle control for 48 hrs; total RNA was prepared from both samples and expression of ZAP70 mRNAs was quantified and normalized to GAPDH. Results are averages ± S.E. (n = 3).
Defining the ZAP70 transcription start site and minimal active promoter
Zeta-chain-associated protein of 70 kD promoter has not previously been experimentally determined; to map the promoter region, we initially identified the transcription start site (TSS) using 5′ RACE and revealed that the transcription starts within exon 1 of ZAP70 gene (Fig. 2). ZAP70 predicted promoter was predicted in silico to encompass the region around −500 bp relative to the TSS and to extend to +90 bp into exon 1. To define the promoter, we designed an extended ZAP70 promoter/luciferase reporter construct, spanning −1000 bp to +400 bp relative to the TSS with further 10 constructs containing deletions form either 5′, 3′ or both ends of this maximal promoter (Fig. 3A). All 11 constructs were transfected into Jurkat T cells or Nalm6 B cells, both of which express ZAP70 (Fig. 3B). Activity of the promoter constructs was directly proportional to the amount of luciferase activity as detected (Fig. 3C and D). Based on the data from both Jurkat and Nalm 6 cells, promoter with optimal activity is represented by construct 4, which includes 600 bp of sequence 5′ as well as 400 bp at the 3′ to the TSS. This is therefore the ZAP70 regulatory region. We subdivided this region into four smaller fragments (termed R1, R2, R3 and R4) and designed a primer pair to distinguish each one (Fig. 3E) as tools for further experiments.
Fig 2.

Location of transcription start site (TSS) for ZAP70 in primary chronic lymphocytic leukaemia (CLL) tumour cells and lymphoid leukaemia cell lines. Sequences generated from products of RNA Ligase Mediated Rapid Amplification of cDNA 5′ Ends (5′ RLM-RACE) indicate two active TSS in CLL patient 2, Jurkat and Nalm6 and one active TSS in CLL patient 1. Both experimentally determined TSS map to within 15 nucleotides of the TSS for the NCBI Reference Sequence: NM_001079.3 positioned at 98330030 bp of chromosome 2 (GRCh37 assembly).
Fig 3.

Mapping of the ZAP70 promoter: (A) 11 promoter construct was generated and cloned into pGL4.17 luciferase vector; schematic representation shows the position and numbering of the sequence for particular constructs. (B) Expression of ZAP70 protein was determined in Jurkat T cells (J) and Nalm6 (N6) cells using a western blot. β-Actin was a loading control. Gels shown are representative of tri-replicate experiments. (C) 1 μg of each of the 11 ZAP70-promoter-luciferase reporter constructs, or empty vector, was transfected into Jurkat cells along with 100 ng of pRLTK using electroporation. The cultures were harvested 48 hrs after the transfection and luciferase assays were performed. Luciferase activities were normalized to pRLTK activity and expressed as the mean ± S.E. of four independent transfection experiments. (D) ZAP70-promoter-luciferase reporter constructs were transfected into Nalm6 cells and harvested as described in (C). (E) Schematic presentation of four primer pairs (R1–R4) designed to span ZAP70 regulatory region as determined in (C) and (D).
Assessment of chromatin structure spanning ZAP70 promoter in expressing or non-expressing B cell lines
Gene expression depends on the promoter activity, which is in turn regulated by the accessibility of DNA to the transcription factors. DNA in cells is packaged into chromatin, which can be differentially folded to either aid transcription (in opened conformation) or to prevent it (in closed conformation). There are no previous reports relating to the chromatin state at the ZAP70 regulatory region, so to initially assess the global chromatin structure, we isolated intact, native chromatin from ZAP70− Namalwa and ZAP70+ Nalm6 B cell lines (Fig. 4A). An equivalent amount of chromatin was then digested with MNase for up to 5 min. with sampling every minute (Fig. 4B). Micrococcal nuclease cleaves DNA on the basis of accessibility dictated by chromatin structure; therefore, gene regions with chromatin in relaxed conformation will be digested faster than when chromatin is in a closed, transcriptionally silenced state. DNA isolated from timed digests in both cell lines was used as template in PCRs amplifying the entire ZAP70 promoter (regions R1–R4) (Fig. 4C and D). The ability to amplify 1 kb of DNA at ZAP70 promoter diminished faster in Nalm6 cells as compared with Namalwa (Fig. 4C–E). At 2 min., there was 47% more digested DNA in Nalm6 chromatin relative to Namalwa, which reflects a more accessible, open state of chromatin in the ZAP70 expressing cell line (Fig. 4F). These data support the idea that higher order chromatin structures affect gene expression, whereas both are determined by the type of histone modification associated with the chromatin. We therefore next set out to assess histone modifications at the ZAP70 promoter.
Fig 4.

Differential global chromatin folding and accessibility in ZAP70+ versus ZAP70− cells. (A) Western blot and RT-PCR for detection of ZAP70− total RNA were prepared from Nalm6 and Namalwa cell lines and used for qRT-PCR detection of ZAP70 (top panel); 30 μg whole cell extract was separated by SDS-PAGE and immunoblotted for ZAP70 (bottom panel). Gels shown are representative of tri-replicate experiments. (B) Native chromatin was isolated from Namalwa or Nalm6 cell lines and digested with micrococcal nuclease for 5 min. with a sample taken every minute. DNA was isolated using phenol–chloroform extraction and separated on 1% agarose gel. (C) Schematic representation of ZAP70 predicted promoter sequence and the position of the primers used to amplify the 1 kb fragment (R1–R4) or the small control fragment (R1). (D) 20 ng of DNA from chromatin digests sampled at 1 min. intervals was used in RT-PCR reactions using ZAP70 R1 sense and ZAP70 R4 antisense primers to amplify R1–R4 fragment (1 kb) or ZAP70 R1 sense and antisense to generate R1 control fragment (200 bp). (E) The R1–R4 amplification as outlined in (C) was carried out using quantitative PCR and values plotted onto a graph ±S.E.M. (n = 3). (F) Difference between the rate of digestion between Nalm6 and Namalwa chromatin were calculated using the ratio between percentage amplification of R1–R4 product in two cell lines at a given time interval.
Identification of the histone modifications associated with ZAP70 expression status in ZAP70 expressing versus non-expressing B cell lines
In general terms, histone acetylation has been implicated in transcriptional activation, whereas histone methylation appears to have more diverse functions and, depending on context, can operate either as a signature for transcriptionally active or repressed chromatin [22, 23]. Histone lysine methylation signatures associated with actively transcribed chromatin include di and trimethylated lysine 4 of histone H3 (H3K4me2/3), H3K36me3 and H3K79me2/3, although the latter two are found mainly at the 3′ end of transcribed genes [24–28]. In contrast, H3K9me3 and H3K27me3 generally function as signatures for transcriptionally repressed chromatin [29–31]. A combination of ChIP and qPCR was used to investigate changes in selected, specific histone lysine acetylation and methylation modifications associated with chromatin at the ZAP70 promoter in ZAP70+ Nalm6 versus non-expressing Namalwa cell lines (Fig. 5A). The ZAP70 promoter in Nalm6 was associated with H3K4me2, a signature of active/permissive chromatin, whereas this modification was completely absent in Namalwa (Fig. 5A, antibody lane 5). We were unable to detect H3K4me1 or H3K4me3 modifications in either cell line (Fig. 5A, antibody lanes 4 and 8). However, we additionally observed a significantly lower level of acetylated H3K14 with the promoter in Namalwa versus Nalm6 cells (Fig. 5A, antibody lane 9). We also discovered enrichment of cetylated histone H4 as well as high levels of H3 acetylation at the promoter of ZAP70 expressing Nalm6 cells (Fig. 5A, antibody lanes 2 and 10). In addition to modifications of lysines in histones H3 and 4, presence of histone variant H2A.Z is a mark of actively transcribed chromatin. ChIP assay detected presence of this histone variant in ZAP70 expressing Nalm6, but not Namalwa (Fig. 5B). Interestingly, we were unable to detect any silent/permissive histone marks such as H3K9me2 and H3K9me3 (Fig. 5A, lanes 3 and 7) in either of the cell lines. The ZAP70 promoter is therefore likely to be in a transcriptionally permissive, but stalled state in ZAP70− cells. To address this possibility, we carried out RNApolII ChIP in both ZAP70+ Nalm6 and ZAP70− Namalwa cell lines. We were able to show association of RNApolII with the ZAP70 promoter in both cell lines, thus confirming our hypothesis that the promoter is found in an assembled and poised state (Fig. 5C). Taken together, these data show that lack of ZAP70 expression in Namalwa cells results from the absence of histone lysine modifications associated with transcriptionally active chromatin.
Fig 5.
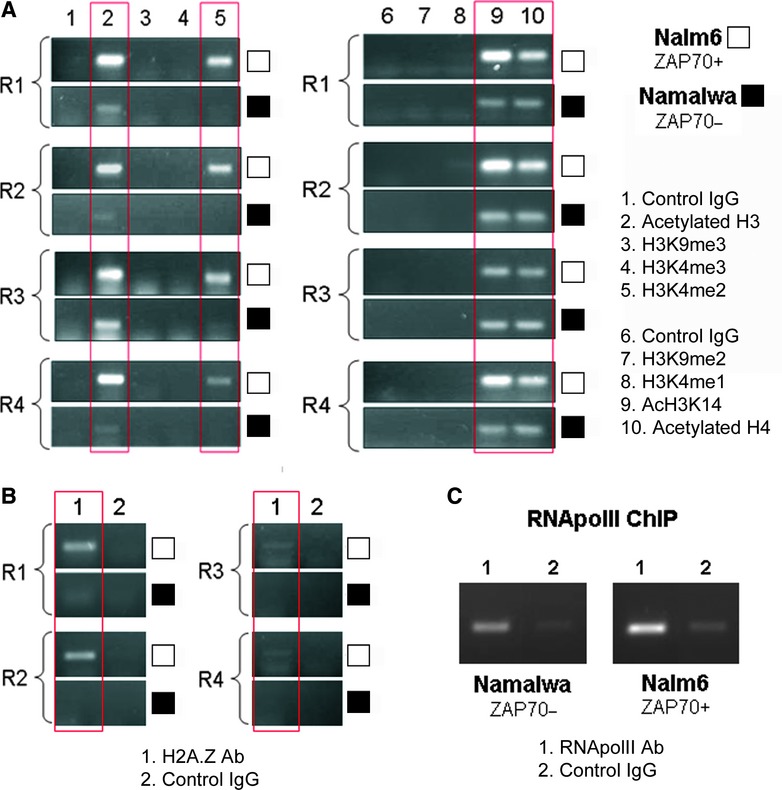
Significant differences in histone modification associated with ZAP70 promoter in ZAP70 expressing versus the non-expressing cell line. (A) 100 μg of native chromatin obtained from Nalm6 or Namalwa cell lines was incubated with 10 μg of antibodies raised against acetylated H3, H3K9me3, H3K4me3, H3K4me2, H3K9me2, H3K4me1, acetylated H3K14 and acetylated H4 or control antibody; ChIP assay was carried out and R1 to R4 ZAP70 promoter fragments were amplified. Gels shown are representative of four replicate experiments. White blocks depict Nalm6 ZAP70+ve cell samples; black blocks show Namalwa ZAP70−ve samples. (B) 100 μg of native chromatin obtained from Nalm6 or Namalwa cell lines was incubated with 10 μg of anti H2A.Z or control antibody; ChIP assay was carried out and R1 to R4 ZAP70 promoter fragments were amplified. Gels shown are representative of three replicate experiments. White blocks depict Nalm6 ZAP70+ve cell samples; black blocks show Namalwa ZAP70−ve samples. (C) 100 μg of cross-linked chromatin obtained from Nalm6 or Namalwa cell lines was incubated with 10 μg of anti RNApol II or control antibody; ChIP assay was carried out and R4 ZAP70 promoter fragment was amplified. Gels shown are representative of three replicate experiments.
Defining the histone modifications associated with ZAP70 regulatory region in ZAP70+ and ZAP70− CLL patient samples
We next wanted to investigate if the distinct histone signatures identified at the ZAP70 promoter in B cell lines are also found between ZAP70+ and/or ZAP70− CLL patient samples. To this end, we obtained lymphocytes from blood of CLL patients; ZAP70 status of the patients was confirmed using flow cytometry. Around 98% of total lymphocytes were found to be CLLs. To eliminate possible contamination of CLLs with other cell types that express ZAP70 (primarily T and NK cells), we utilized negative selection of CD2+ cells by Robosep. We next prepared chromatin from purified ZAP70 expressing and non-expressing CLLs and carried out ChIP assays using antibodies that target active or permissive histone modifications including acetylated H3K14, di-methyl H3K4, tri-methyl H3K4, acetylated H4, acetylated H3 and di-methyl H3K9. We were able to detect the presence of active modifications in regions R1–R4 of ZAP70 promoter in both expressing and non-expressing patient CLLs (Fig. 6A–C). However, there was a significantly higher association of active marks H3K4me2 and H3K4me3 with the promoter in ZAP70+ CLL versus CLL from ZAP70− patients (Fig. 6A and B). We also found a markedly lower association of acetylated H3 in ZAP70− CLL. Therefore, using primary CLL, we were able to recapitulate key discoveries from cell line studies by showing the differential enrichment of H3K4me2 and H3K4me3 in ZAP70+ versus ZAP70− CLL; the most consistent difference in histone modifications between ZAP70 expressing and non-expressing patient samples was acetylation of histone H3, widely considered a modification associated with transcriptional activation [17, 18], which may, at least in part, explain our initial findings (Fig. 1) using HDACi. The enrichment of other active modifications was not statistically significantly different between the CLL patients. Importantly, the presence of active modifications in ZAP70− patient samples confirms a poised chromatin state around the promoter, which resembles the transcriptional state we observed in the ZAP70− Namalwa cell line.
Fig 6.
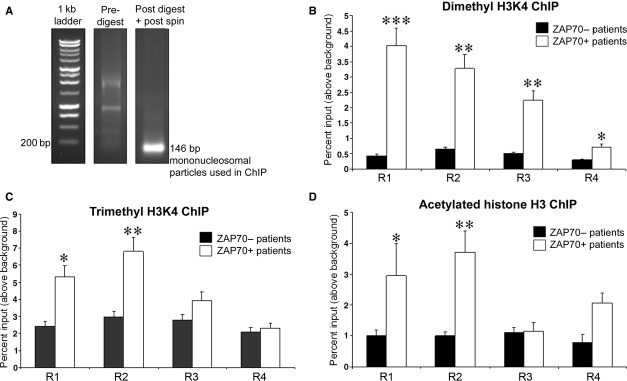
Presence of di- and tri methylated H3K4 and acetylated H3 at ZAP70 promoter is associated with gene expression. (A) Native chromatin obtained from patient samples was digested using monococcal nuclease as outlined in Materials and methods. First sample was taken before the digestion and second following a digestion and spin to remove membranes and debris. Representative picture of pre- and post-digest chromatin is shown. Obtained digested native chromatin contains only mononucleosomes. (B) 100 μg of native chromatin obtained from ZAP70 positive or negative patient CLL cells was incubated with 10 μg of anti dimethyl H3K4 or control antibody; ChIP assay was carried out and R1 to R4 ZAP70 promoter fragments were amplified, n = 8. (C) 100 μg of native chromatin obtained from ZAP70 positive or negative patient CLL cells was incubated with 10 μg of anti trimethyl H3K4 or control antibody; ChIP assay was carried out and R1 to R4 ZAP70 promoter fragments were amplified, n = 8. (D) 100 μg of native chromatin obtained from ZAP70 positive or negative patient CLL cells was incubated with 10 μg of anti acetylated H3 or control antibody; ChIP assay was carried out and R1 to R4 ZAP70 promoter fragments were amplified, n = 8. Statistical analysis – Mann U Whitney where (*), (**) and (***) indicate P < 0.05, P < 0.001 and P < 0.001 respectively.
Treatment of ZAP70− cells DNA methylation inhibitor results in re-expression of the gene and a marked increase in associated histone H3 acetylation
Others and we have previously shown that the DNA methylation status at the 5′ end of the gene controls expression of ZAP70 [10]. We were intrigued to find out whether reversal of DNA methylation at ZAP70 promoter/gene may cause re-expression of the gene by increasing histone H3 acetylation. Indeed, 48-hr treatment of ZAP70 negative cells with Decitabine induces expression of ZAP70 gene (Fig. 7A). To determine if DNA hypomethylation results in increased H3 acetylation associated with ZAP70 promoter, we isolated chromatin from Decitabine treated or control cells and carried out the ChIP assay using anti-acetylated histone H3 antibody. We show in Figure 7B and C that Decitabine treatment leads to a 7-fold increase in histone H3 acetylation at ZAP70 intron 1/exon 2 boundary, the region containing DNA methylation at C + 334 known to regulate gene expression [14]. These results suggest synergistic action of epigenetic modifying enzymes that act on DNA methylation and histones acetylation to bring about a transcriptionally active state of chromatin at the ZAP70 gene. These data confirm a role for histone H3 acetylation in the control of ZAP70 expression.
Fig 7.
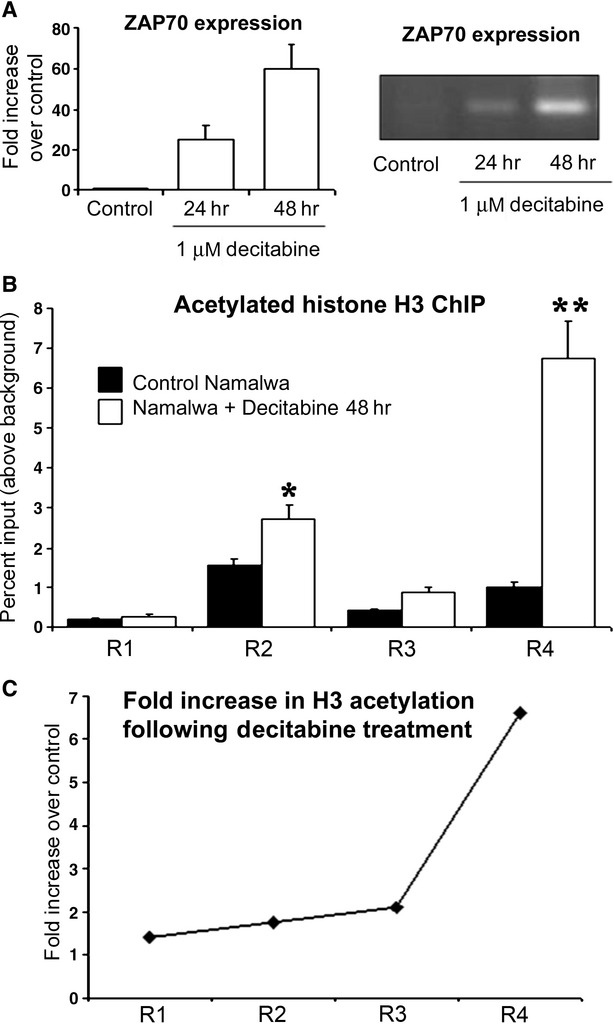
Decitabine induced reversal of DNA methylation at the ZAP70 promoter leads to an increase in acetylation of associated histone H3. (A) Namalwa cell line was treated with 1 μM Decitabine or vehicle for 24 or 48 hrs; total RNA was prepared from both samples and used for RT-PCR detection of ZAP70 expression. (B) 100 μg of native chromatin obtained from Decitabine or vehicle treated Namalwa cell lines was incubated with 10 μg of anti acetylated H3 or control; ChIP assay was carried out and R1, R2, R3 and R4 ZAP70 promoter fragments were amplified. Gels shown are representative of three replicate experiments. Statistical analysis –Mann U Whitney where (*) and (**) and indicate P < 0.05 and P < 0.01 respectively. (C) Differences between the levels of acetylated histone H3 association with the ZAP70 promoter fragments R1 to R4 were calculated using the ratio between the levels of enrichment found in chromatin from vehicle treated versus the Decitabine treated cells.
Discussion
Zeta-chain-associated protein of 70 kD, a PTK that is normally expressed in T cells, is a molecular signature of poor prognosis in CLL, possibly as a result of increased BCR signalling, which may promote a more aggressive form of the disease. ZAP70 is therefore not only a prognostic marker, but may also prove to be a therapeutic target. Epigenetic modifications, in particular, changes in DNA methylation, are associated with many human cancers and can influence disease progression by altering the expression of tumour-suppressor genes and proto-oncogenes [32]. Here, we describe evidence for alterations in histone acetylation and methylation signatures in CLL, and describe how these changes can induce transcriptional activity of ZAP70. Moreover, we provide evidence for a mechanistic link between altered DNA methylation and changes in histone modifications that underlie aberrant expression of ZAP70 in poor prognosis forms of CLL.
Expression of ZAP70 appears to be tightly linked to the presence of active histone modifications within the regulatory region of the gene, in both cell lines and patient CLL. Although H3K4me2 and acetylated H3 were enriched around the promoter of all ZAP70+ cells tested, H3K4me3 enrichment was documented only in ZAP70+ patient samples, whereas we were unable to document H3K4me3 in ZAP70+ cell lines. H3K4me2 and H3K4me3 are ordinarily found around transcriptional start sites of active promoters as well as occupying some coding regions [33–35]. H3K4me3 is associated with around 90% of active gene promoters [36], suggesting that although important for recruitment of TFIID to the promoter, it may not be crucial for active transcription [37]. Therefore, disparate H3K4me3 enrichment between ZAP70+ patient samples versus the cell lines could be a result of differing transcriptional activity between the ‘always on’ state in the cell line and ‘transiently on’ state in the primary CLL cells. Indeed, this notion is supported by a recent study by Okitsu et al., which shows that increased transcriptional activity correlates with redistribution of H3K4me3 from the promoter into the coding region of thee gene at the 5′ end [38]. This could explain why we were unable to locate H3K4me3 in the promoter of cell line Nalm6, which expresses high levels of ZAP70.
We also found no differences in the levels of silent histone marks, suggesting that repression of ZAP70 gene might be the default pathway. In support of this idea, RNApolII was associated with the transcriptional start site in both ZAP70 expressing and non-expressing cells suggestive of an assembled, but stalled transcriptional complex. Collectively, our data suggest that incorporation of active histone marks, such as H3K4me2 and H3K4me3, may lead to recruitment of additional components (possibly TFIID) to the preassembled PolII transcription complex in ZAP70− cell lines. As such, the transcriptional activity of ZAP70 locus would depend on presence of active histone marks, rather than removal of silent ones. In turn, regulation of ZAP70 expression would be dependent on activity and recruitment of histone modifying enzymes ranging from deacetylases to methyltransferases. Hence, any drugs that positively impact on presence of active marks at the ZAP70 promoter may lead to re-expression of the gene and potentially more aggressive disease.
Further to examining histone modifications associated with ZAP70 promoter, we also assessed changes in DNA methylation in the same region. All of the ZAP70 expressing primary patient CLLs used in our study lacked CpG methylation at +334 of the ZAP70 gene, which is within the extended promoter region. It has been reported that lack of DNA methylation leads to an increase in H3K4 methylation [39], which raises the intriguing possibility that hypomethylation at +334 may result in H3K4 di/trimethylation and active maintenance of ZAP70 transcription in CLL. However, the presence of H3K4 methylation can prevent de novo DNA methylation in the vicinity by preventing a non-catalytic paralogue of DNA methyltransferase (DNMT), Dnmt3L, from docking onto the nucleosome. Dnmt3L functions to recruit de novo DNMTs Dnmt3a and Dnmt3b, which then methylate DNA and induce gene silencing [40–42]. Interestingly, CLLs have significantly lower levels of Dnmt3b [43]. Taken together, the presence of H3K4me2/3 and lower level of Dnmt3b can form a positive feedback loop, which re-enforces active transcription of a gene such as ZAP70. H3K4 di- and tri-methylation appears to be distinct feature of ZAP70 expressing B cells and CLLs; it therefore may be instructive to investigate its control in primary CLL. H3K4 di/trimethylation at the ZAP70 promoter may be achieved by alterations in H3K4 methyltransferase activities. It is noteworthy that translocations, which generate H3K4 methylation defective MLL enzymes, are associated with particularly aggressive forms of acute leukaemias, which may indicate a tumour-suppressor function for H3K4 methyltransferases [44].
Increase in H3K4 di/trimethylation can lead to a greater acetylation of H3 [45], which correlates with our finding of enriched acetylated H3 in ZAP70+ CLL. However, acetylation status of H3 can also be determined by the lack of DNA methylation as the two are tightly correlated [39]. Indeed, use of HDACi leads to DNA demethylation by mechanisms that are not yet understood. The association of histone H3 acetylation with ZAP70 expression may have important consequences for CLL therapies involving HDACi. Recently, there have been a number of studies using HDACi with varied degrees of success [46–48]. Most of the studies showed little or no therapeutic effect, and those that reported positive results generally relied on HDACi to trigger some undefined event, which consequently increased the potency of secondary therapies such as TRAIL ligand [49]. One possible reason for lack of therapeutic effect of HDACi might be re-activation of ZAP70 transcription. Although some HDACi (such as VPA) show good induction of apoptosis in CLL [50], prolonged use of similar agents, along with other environmental/treatment pressures, may generate altered form of disease.
It is now well established that the presence of ZAP70 in CLL has a direct deleterious effect on the course of disease, probably through kinase-independent signalling [10]. It would certainly be worthwhile examining what effect the in vivo use of HDACi has on ZAP70 status of CLL, as such therapeutic approach may in effect be generating a more aggressive from of the disease. Techniques such as ChIP could be used to further investigate biology of primary CLL, particularly if CLL samples from patients enrolled in various clinical trials become available for such analysis.
In conclusion, epigenetic regulation of histone modifications appears to play an important role in the differential expression of ZAP70 in CLL. Our evidence suggests ‘cross-talk’ between histone modifications and DNA methylation, of which the latter has previously been shown to regulate ZAP70 expression [13, 14]. This cooperation between different types of epigenetic events may be necessary to achieve tight regulation of gene expression. With an increasingly detailed picture of ZAP70 regulation emerging, therapies targeting this kinase as well as other enzymes that regulate its expression may improve the outcome of patients with poor disease prognosis.
Acknowledgments
The authors wish to thank Tenovus Research Charity for funding this work. This work was funded by grants to D.A.M. and J.M. from the Tenovus Cancer Charity.
Conflict of interest
The authors have no financial or other conflict of interest to disclose.
References
- 1.Hamblin TJ, Davis Z, Gardiner A, et al. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94:1848–54. [PubMed] [Google Scholar]
- 2.Zanotti R, Ambrosetti A, Lestani M, et al. ZAP-70 expression, as detected by immunohistochemistry on bone marrow biopsies from early-phase CLL patients, is a strong adverse prognostic factor. Leukemia. 2007;21:102–9. doi: 10.1038/sj.leu.2404458. [DOI] [PubMed] [Google Scholar]
- 3.Rosenwald A, Alizadeh AA, Widhopf G, et al. Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. J Exp Med. 2001;194:1639–47. doi: 10.1084/jem.194.11.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wiestner A, Rosenwald A, Barry TS, et al. ZAP-70 expression identifies a chronic lymphocytic leukemia subtype with unmutated immunoglobulin genes, inferior clinical outcome, and distinct gene expression profile. Blood. 2003;101:4944–51. doi: 10.1182/blood-2002-10-3306. [DOI] [PubMed] [Google Scholar]
- 5.Elder ME, Hope TJ, Parslow TG, et al. Severe combined immunodeficiency with absence of peripheral blood CD8 + T cells due to ZAP-70 deficiency. Cell Immunol. 1995;165:110–7. doi: 10.1006/cimm.1995.1193. [DOI] [PubMed] [Google Scholar]
- 6.Zoller KE, MacNeil IA, Brugge JS. Protein tyrosine kinases Syk and ZAP-70 display distinct requirements for Src family kinases in immune response receptor signal transduction. J Immunol. 1997;158:1650–9. [PubMed] [Google Scholar]
- 7.Latour S, Chow LM, Veillette A. Differential intrinsic enzymatic activity of Syk and Zap-70 protein-tyrosine kinases. J Biol Chem. 1996;271:22782–90. doi: 10.1074/jbc.271.37.22782. [DOI] [PubMed] [Google Scholar]
- 8.Chen L, Apgar J, Huynh L, et al. ZAP-70 directly enhances IgM signaling in chronic lymphocytic leukemia. Blood. 2005;105:2036–41. doi: 10.1182/blood-2004-05-1715. [DOI] [PubMed] [Google Scholar]
- 9.Gobessi S, Laurenti L, Longo PG, et al. ZAP-70 enhances B-cell-receptor signaling despite absent or inefficient tyrosine kinase activation in chronic lymphocytic leukemia and lymphoma B cells. Blood. 2007;109:2032–9. doi: 10.1182/blood-2006-03-011759. [DOI] [PubMed] [Google Scholar]
- 10.Chen L, Huynh L, Apgar J, et al. ZAP-70 enhances IgM signaling independent of its kinase activity in chronic lymphocytic leukemia. Blood. 2008;111:2685–92. doi: 10.1182/blood-2006-12-062265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richardson SJ, Matthews C, Catherwood MA, et al. ZAP-70 expression is associated with enhanced ability to respond to migratory and survival signals in B-cell chronic lymphocytic leukemia (B-CLL) Blood. 2006;107:3584–92. doi: 10.1182/blood-2005-04-1718. [DOI] [PubMed] [Google Scholar]
- 12.Rassenti LZ, Jain S, Keating MJ, et al. Relative value of ZAP-70, CD38, and immunoglobulin mutation status in predicting aggressive disease in chronic lymphocytic leukemia. Blood. 2008;112:1923–30. doi: 10.1182/blood-2007-05-092882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chantepie SP, Vaur D, Grunau C, et al. ZAP-70 intron1 DNA methylation status: determination by pyrosequencing in B chronic lymphocytic leukemia. Leuk Res. 2010;34:800–8. doi: 10.1016/j.leukres.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 14.Corcoran M, Parker A, Orchard J, et al. ZAP-70 methylation status is associated with ZAP-70 expression status in chronic lymphocytic leukemia. Haematologica. 2005;90:1078–88. [PubMed] [Google Scholar]
- 15.Milutinovic S, D'Alessio AC, Detich N, et al. Valproate induces widespread epigenetic reprogramming which involves demethylation of specific genes. Carcinogenesis. 2007;28:560–71. doi: 10.1093/carcin/bgl167. [DOI] [PubMed] [Google Scholar]
- 16.Ou JN, Torrisani J, Unterberger A, et al. Histone deacetylase inhibitor Trichostatin A induces global and gene-specific DNA demethylation in human cancer cell lines. Biochem Pharmacol. 2007;73:1297–307. doi: 10.1016/j.bcp.2006.12.032. [DOI] [PubMed] [Google Scholar]
- 17.Oakley F, Mann J, Nailard S, et al. Nuclear factor-kappaB1 (p50) limits the inflammatory and fibrogenic responses to chronic injury. Am J Pathol. 2005;166:695–708. doi: 10.1016/s0002-9440(10)62291-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O'Neill LP, Turner BM. Immunoprecipitation of native chromatin: NChIP. Methods (San Diego, Calif.) 2003;31:76–82. doi: 10.1016/s1046-2023(03)00090-2. [DOI] [PubMed] [Google Scholar]
- 19.Mann J, Chu DC, Maxwell A, et al. MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology. 2010;138:705–14. doi: 10.1053/j.gastro.2009.10.002. 14 e1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blum KA, Advani A, Fernandez L, et al. Phase II study of the histone deacetylase inhibitor MGCD0103 in patients with previously treated chronic lymphocytic leukaemia. Br J Haematol. 2009;147:507–14. doi: 10.1111/j.1365-2141.2009.07881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Byrd JC, Marcucci G, Parthun MR, et al. A phase 1 and pharmacodynamic study of depsipeptide (FK228) in chronic lymphocytic leukemia and acute myeloid leukemia. Blood. 2005;105:959–67. doi: 10.1182/blood-2004-05-1693. [DOI] [PubMed] [Google Scholar]
- 22.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 23.Shilatifard A. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu Rev Biochem. 2006;75:243–69. doi: 10.1146/annurev.biochem.75.103004.142422. [DOI] [PubMed] [Google Scholar]
- 24.Shilatifard A. Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr Opin Cell Biol. 2008;20:341–8. doi: 10.1016/j.ceb.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sims RJ, 3rd, Reinberg D. Histone H3 Lys 4 methylation: caught in a bind? Genes Dev. 2006;20:2779–86. doi: 10.1101/gad.1468206. [DOI] [PubMed] [Google Scholar]
- 26.Yan C, Boyd DD. Histone H3 acetylation and H3 K4 methylation define distinct chromatin regions permissive for transgene expression. Mol Cell Biol. 2006;26:6357–71. doi: 10.1128/MCB.00311-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bannister AJ, Schneider R, Myers FA, et al. Spatial distribution of di- and tri-methyl lysine 36 of histone H3 at active genes. J Biol Chem. 2005;280:17732–6. doi: 10.1074/jbc.M500796200. [DOI] [PubMed] [Google Scholar]
- 28.Weake VM, Workman JL. Inducible gene expression: diverse regulatory mechanisms. Nat Rev. 2010;11:426–37. doi: 10.1038/nrg2781. [DOI] [PubMed] [Google Scholar]
- 29.Zeng W, Ball AR, Jr, Yokomori K. HP1: heterochromatin binding proteins working the genome. Epigenetics. 2010;5:287–92. doi: 10.4161/epi.5.4.11683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schuettengruber B, Chourrout D, Vervoort M, et al. Genome regulation by polycomb and trithorax proteins. Cell. 2007;128:735–45. doi: 10.1016/j.cell.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 31.Beisel C, Paro R. Silencing chromatin: comparing modes and mechanisms. Nat Rev. 2011;12:123–35. doi: 10.1038/nrg2932. [DOI] [PubMed] [Google Scholar]
- 32.Kondo Y. Epigenetic cross-talk between DNA methylation and histone modifications in human cancers. Yonsei Med J. 2009;50:455–63. doi: 10.3349/ymj.2009.50.4.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bernstein BE, Kamal M, Lindblad-Toh K, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–81. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 34.Heintzman ND, Stuart RK, Hon G, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–8. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 35.Kim TH, Barrera LO, Zheng M, et al. A high-resolution map of active promoters in the human genome. Nature. 2005;436:876–80. doi: 10.1038/nature03877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barski A, Cuddapah S, Cui K, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–37. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 37.Vermeulen M, Mulder KW, Denissov S, et al. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell. 2007;131:58–69. doi: 10.1016/j.cell.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 38.Okitsu CY, Hsieh JC, Hsieh CL. Transcriptional activity affects the H3K4me3 level and distribution in the coding region. Mol Cell Biol. 2010;30:2933–46. doi: 10.1128/MCB.01478-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- 40.Chen ZX, Mann JR, Hsieh CL, et al. Physical and functional interactions between the human DNMT3L protein and members of the de novo methyltransferase family. J Cell Biochem. 2005;95:902–17. doi: 10.1002/jcb.20447. [DOI] [PubMed] [Google Scholar]
- 41.Ooi SK, Qiu C, Bernstein E, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–7. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suetake I, Shinozaki F, Miyagawa J, et al. DNMT3L stimulates the DNA methylation activity of Dnmt3a and Dnmt3b through a direct interaction. J Biol Chem. 2004;279:27816–23. doi: 10.1074/jbc.M400181200. [DOI] [PubMed] [Google Scholar]
- 43.Kn H, Bassal S, Tikellis C, et al. Expression analysis of the epigenetic methyltransferases and methyl-CpG binding protein families in the normal B-cell and B-cell chronic lymphocytic leukemia (CLL) Cancer Biol Ther. 2004;3:989–94. doi: 10.4161/cbt.3.10.1137. [DOI] [PubMed] [Google Scholar]
- 44.Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7:823–33. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- 45.Nightingale KP, Gendreizig S, White DA, et al. Cross-talk between histone modifications in response to histone deacetylase inhibitors: MLL4 links histone H3 acetylation and histone H3K4 methylation. J Biol Chem. 2007;282:4408–16. doi: 10.1074/jbc.M606773200. [DOI] [PubMed] [Google Scholar]
- 46.Aron JL, Parthun MR, Marcucci G, et al. Depsipeptide (FR901228) induces histone acetylation and inhibition of histone deacetylase in chronic lymphocytic leukemia cells concurrent with activation of caspase 8-mediated apoptosis and down-regulation of c-FLIP protein. Blood. 2003;102:652–8. doi: 10.1182/blood-2002-12-3794. [DOI] [PubMed] [Google Scholar]
- 47.Bhalla S, Balasubramanian S, David K, et al. PCI-24781 induces caspase and reactive oxygen species-dependent apoptosis through NF-kappaB mechanisms and is synergistic with bortezomib in lymphoma cells. Clin Cancer Res. 2009;15:3354–65. doi: 10.1158/1078-0432.CCR-08-2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Inoue S, Riley J, Gant TW, et al. Apoptosis induced by histone deacetylase inhibitors in leukemic cells is mediated by Bim and Noxa. Leukemia. 2007;21:1773–82. doi: 10.1038/sj.leu.2404760. [DOI] [PubMed] [Google Scholar]
- 49.Inoue S, MacFarlane M, Harper N, et al. Histone deacetylase inhibitors potentiate TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in lymphoid malignancies. Cell Death Differ. 2004;11:S193–206. doi: 10.1038/sj.cdd.4401535. [DOI] [PubMed] [Google Scholar]
- 50.Stamatopoulos B, Meuleman N, De Bruyn C, et al. Antileukemic activity of valproic acid in chronic lymphocytic leukemia B cells defined by microarray analysis. Leukemia. 2009;23:2281–9. doi: 10.1038/leu.2009.176. [DOI] [PubMed] [Google Scholar]